

Abstract
Alginate is a major constituent of mature biofilms produced by Pseudomonas aeruginosa. The penultimate step in the biosynthesis of alginate is the conversion of some β-D-mannuronate residues in the polymeric substrate polymannuronan to α-L-guluronate residues in a reaction catalyzed by C5-mannuronan epimerase. Specificity studies conducted with size-fractionated oligomannuronates revealed that the minimal substrate contained 9 monosaccharide residues. The maximum velocity of the reaction increased from 0.0018 s−1 to 0.0218 s−1 as the substrate size increased from 10 to 20 residues, and no additional increase in kcat was observed for substrates up to 100 residues in length. The Km decreased from 80 μM for substrate containing fewer than 15 residues to 4 μM for substrate containing over 100 residues. In contrast to C5-mannuronan epimerases that have been characterized in other bacterial species, P. aeruginosa C5-mannuronan epimerase does not require Ca2+ for activity, and the Ca2+-alginate complex is not a substrate for the enzyme. Analysis of purified, active enzyme by inductively coupled plasma-emission spectroscopy revealed that no metals were present in the protein. The pH dependence of the kinetic parameters revealed that 3 residues on the enzyme which all have a pKa of about 7.6 must be protonated for catalysis to occur. The composition of the polymeric product of the epimerase reaction was analyzed by 1H-NMR spectroscopy, which revealed that tracts of adjacent guluronate residues were readily formed. The reaction reached an apparent equilibrium when the guluronate composition of the polymer was 75%.
Pseudomonas aeruginosa is an opportunistic human pathogen that has been linked to certain pathological conditions of immunocompromised humans, and especially the respiratory tract infections that accompany cystic fibrosis (1) . Pulmonary infections caused by the mucoid phenotype of P. aeruginosa are almost impossible to eradicate, even with antibiotic treatment (2). The high antibiotic resistance is attributed in part to the formation of a biofilm, a complex extracellular polymeric matrix in which the cells are embedded. One of the primary constituents of the mature P. aeruginosa biofilm is the polysaccharide alginate. This cell-associated virulence factor is a high molecular weight (500-2000 kDa) linear polysaccharide comprised of residues of β-D-mannuronate (M) and its C-5 epimer α-L-guluronate (G), which are covalently linked by β-1,4 glycosidic bonds (3). The relative ratio of these building blocks in the polymer and the linear distribution of G residues strikingly alters physical properties of alginate such as viscosity and gel forming ability, and therefore plays a crucial role in the function of the biopolymer (4).
Most of the genes required for alginate biosynthesis are located in the algD operon on the P. aeruginosa chromosome (5). The first polymeric product in the pathway is polymannuronan, which is synthesized from GDP-mannuronic acid. The formation of GDP-mannuronic acid from fructose 6-phosphate occurs in the cytoplasm; the synthesis of polymannuronan is not well understood, but two cytoplasmic membrane proteins, Alg8, which is a β-glycosyl transferase-like protein, and Alg44 are believed to be involved in polymer formation (6). Polymannuronan is transported across the inner cytoplasmic membrane, and conversion of some mannuronate residues in the polymer to guluronate occurs in the periplasmic space. The reaction is catalyzed by C5-mannuronan epimerase (Scheme 1), which is a 55 kDa protein encoded by the algG gene (7). Mature alginate is acetylated at some mannuronate residues at O2 or O3 (8). The acetylated residues are not substrates for the epimerase and guluronate residues are not acetylated, so the final composition of the alginate polymer is determined by the relative activities of the epimerase and the acetyltransferases that act on alginate. Following acetylation, the polysaccharide is exported across the outer membrane by the porin-like protein AlgE (9).
Scheme 1.
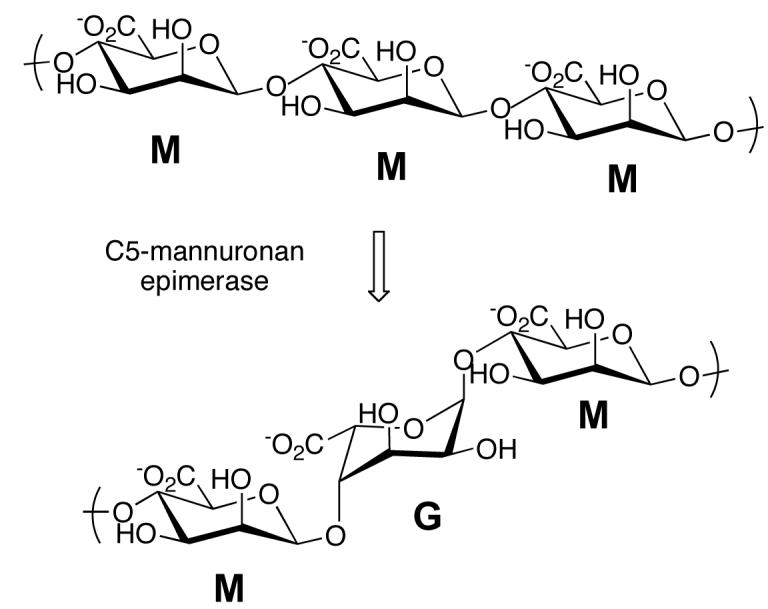
Mature alginate that is isolated from P. aeruginosa biofilms has G1 and M residues randomly distributed throughout the polymer, and homopolymeric G blocks or M blocks are absent (10). Based on the composition of P. aeruginosa alginate, it is widely believed that C5 mannuronan epimerase cannot catalyze the epimerization of adjacent residues to form poly-G blocks. In contrast, the epimerases from Azotobacter vinelandii, have been demonstrated to form poly-G blocks (11, 12). A. vinelandii contains several C5-mannuronan epimerases, which are Ca2+-dependent, and highly homologous with one another (13). P. aeruginosa mannuronan epimerase is not closely related to the A. vinelandii enzymes, and although its activity has been reported to be Ca2+-dependent, that aspect of the reaction has not been examined in detail. Sequence analysis and homology modeling of the P. aeruginosa epimerase suggests that the epimerase domain of the protein is a right-handed β-helix, which is characteristic of enzymes that utilize polysaccharides as substrates (14). A variety of kinetic studies are presented here, which provide insight into the interactions between the epimerase and alginate. The substrate size specificity and metal ion dependence of the reaction have been defined. Through 1H-NMR analysis of the product, we have investigated the sequential distribution of G residues with respect to the fractional content of the polymeric chain. Our results suggest that the enzyme does not require Ca2+ for activity, and that it is capable of forming alginate containing poly-G blocks.
Experimental Procedures
Cloning, overexpression and purification of C5-mannuronan epimerase
The algG gene was successfully amplified from P. aeruginosa PAO1 genomic DNA and inserted into the pET-14b expression vector. The PCR protocol used for amplification of DNA from the GC-rich genome of P. aeruginosa has been described (15). E. coli strain JM109 was used for cloning and maintaining the plasmid. For protein production, the recombinant plasmid was transformed into E. coli BL21(DE3)pLysS cells. Cells were grown in LB medium supplemented with ampicillin (100 μg/mL) and chloramphenicol (34 μg/mL) at 30°C with rotary shaking until the OD600 reached 0.8. Subsequently, 400 μM IPTG was added to induce expression of C5-mannuronan epimerase, and growth of the cells was continued at 30 °C for 16 hrs. Cells were harvested by centrifugation (6500×g, 15 min), yielding 25 g of cell paste, and resuspended in 130 mL lysis buffer containing 100 mM MOPS, pH 7.5, 100 mM NaCl, 1 mM β-mercaptoethanol and 10 mM imidazole. Bacterial lysis was achieved by repeated freeze/thaw cycles of the resuspended cells in the presence of 0.5 mM PMSF and 0.5 mM TLCK to inhibit protease activity. To lower the viscosity of the cell-free extract, 1.3 mg DNAse was added, along with 2 mM CaCl2 and 2 mM MgCl2. After the insoluble cell debris was removed by centrifugation (12200×g, 60 min), the cell-free extract was loaded onto a Ni-NTA affinity column (30 ml, Qiagen). The resin bound His6-tagged fusion protein was washed with 30 column volumes of buffer containing 100 mM MOPS, pH 7.5, 300 mM NaCl, 1 mM β-mercaptoethanol and 10 mM imidazole, followed by 30 column volumes of buffer containing 100 mM MOPS, pH 7.5, 300 mM NaCl, 1 mM β-mercaptoethanol and 50 mM imidazole. C5-Mannuronan epimerase was eluted from the column with a buffer containing 100 mM MOPS, pH 7.5, 300 mM NaCl, 1 mM β-mercaptoethanol and 500 mM imidazole. The (His)6 affinity tag was cleaved from the fusion protein by thrombin digestion, which was conducted at 4 °C using 1 unit of protease for every 5 mg epimerase at an epimerase concentration of 3 mg/mL. Under these conditions the reaction was complete within 48 hrs. Subsequently, thrombin was removed by passing the solution over a p-aminobenzamidine-agarose column (Sigma Chemical Company). The purified C-5 mannuronan epimerase at a concentration of 2.5–3 mg/mL was dialyzed against the following buffers in succession: 100 mM MOPS, pH 6.8, 100 mM NaCl, 250 mM imidazole, 1 mM β-mercaptoethanol, and 1 mM EDTA (12 hrs); 1 L 100 mM MOPS, pH 6.8, 100 mM NaCl, 1 mM β-mercaptoethanol, and 1 mM EDTA (12 hrs) and 1 L 10 mM MOPS, pH 6.8, 100 mM NaCl, 1 mM β-mercaptoethanol, and 1 mM EDTA (12 hrs). The epimerase was concentrated to 5 mg/mL by ultrafiltration. Glycerol was added to the protein to a final concentration of 10% (v/v) and aliquots were stored at −80 °C.
Overexpression and purification of L-guluronate lyase
The plasmid containing the gene for L-guluronate lyase, designated pRC5, was the generous gift of Prof. Dennis E. Ohman (Virginia Commonwealth University). The plasmid pRC5 contains the aly gene from Klebsiella aerogenes. For expression of guluronate lyase, E. coli BL21(DE3)pLysS cells were transformed with pRC5 and the cells were grown at 37 °C in 6 L of LB supplemented with ampicillin (100 μg/mL). Cells were harvested by centrifugation (6800×g, 15 min) yielding 16.9 g cell paste, which was resuspended in buffer containing 100 mM MOPS, pH 7.3 and 1 mM DTT. Cell lysis was achieved by digestion with lysozyme (0.2 mg/g cell pellet) in the presence of 0.5 mM PMSF, 0.5 mM TLCK and DNase (10 μg/mL). Nucleic acids were removed from the cell-free extract by adding protamine sulfate to a final concentration of 0.7 mg/mL. Ammonium sulfate was added to 40% saturation and the proteins that precipitated were discarded. The concentration of ammonium sulfate in the supernatant was raised to 80% saturation and the suspension was centrifuged (12200×g, 20 min). The pelleted proteins, including guluronate lyase, were resuspended in buffer containing 100 mM MOPS, pH 7.3, 1 mM DTT and 10% (v/v) glycerol. The enzyme was stored at −80 °C at a concentration of 20 mg/mL.
Production and purification of poly-D-mannuronan
The substrate for C5-mannuronan epimerase, polymannuronan was isolated from Pseudomonas aeruginosa strain FRD462, also the generous gift of Dr. Dennis Ohman. In order to maintain the P. aeruginosa in the mucoid form, frozen glycerol stocks were initially subcultured in media containing 1 g sodium gluconate, 1 g sodium glutamate, 0.15 g Na2HPO4 and 0.015 g MgSO4•7H2O per 50 mL at pH 7.0. After an 18 hr incubation period at 30 °C with rotary shaking, agar plates with media containing 10 mM MgSO4, 16.8 mM K2HPO4, 7.5 mM Na2HPO4, and 100 mM sodium glutamate overlaid with 250 μl 40% (v/v) glycerol per plate were inoculated with mucoid cells (100 μl- 500 μl) and incubated at 37 °C for 48 hr. Colonies were scraped from the plates and the solid material was suspended in 500 mL of 100 mM NaCl and 20 mM EDTA buffer at neutral pH. The bacterial cells were pelleted by centrifugation at 1700×g for 5 min. The supernatant was added to an equal volume of ethanol to precipitate the exopolysaccharide. The crude product was deacetylated in 5 mM NaOH solution at 95 °C for 0.5 hr and then refluxed in 0.4 N acetic acid for 18 hr. The typical yield per MAP plate was 10-20 mg of polymannuronan with a final dp ranging from 1-30.
Purification of poly-D-mannuronan from alginate
25 g alginic acid (Sigma Chemical Company) was suspended in 500 mL of 10 mM Tris, pH 8.5, containing 100 mM NaCl and 20 mg L-guluronate lyase. The heterogeneous mixture was stirred for 48 hr at room temperature and then centrifuged at 6800×g for 30 min to obtain a clear supernatant, which was mainly composed of homopolymeric M- and G-blocks. Poly-M and poly-G blocks were obtained by fractional precipitation as follows. 1 N HCl was pipetted into the supernatant to lower the pH by 1 unit intervals, and after each acidifying step any precipitated polysaccharide was pelleted by a 30 min centrifugation at 6800×g. The white solid material (dry weight, 2.2 g) obtained by precipitation between pH 3.0 and 2.0 was 90% pure poly-M, and was further purified by refluxing the material in 1 N NaOH for 20 minutes. Under basic conditions the contaminating homopolymeric guluronan was decomposed, while poly-M remained intact. Mild acid hydrolysis in 0.3 N acetic acid at 80 °C overnight, followed by ethanol precipitation yielded 2.1 g pure poly-D-mannuronan with dp 10-200.
Purification of heteropolymeric alginate
Alginic acid (6 g) was dissolved in 1000 mL boiling water and when dissolution was complete, glacial acetic acid was pipetted into the solution to a final concentration 0.3 N. The mixture was stirred at 90 °C for 18 hr, then allowed to cool. 1 N HCl was used to slowly lower the pH resulting in precipitation of M- and G-block polysaccharides, which then were removed by centrifugation. The final supernatant (pH < 2.0) contained a fraction of alginate that appeared to be soluble even at very acidic pH and dry material (1.5 g) was obtained when the aqueous solvent was removed by rotary evaporator. 1H-NMR analysis of the solid revealed that this fraction was low dp heteropolymeric alginate with repeating units of MG.
Spectrophotometric assay for C-5 epimerization
A continuous coupled assay and a fixed-time assay were developed to monitor product formation in the epimerization reaction. Spectrophotometric assays were conducted at 25 °C in a Varian CARY 50 model single-beam spectrophotometer equipped with a thermostatted cell compartment. The ternary buffer mixture used in these assays had a final composition of 20 mM Tris, 10 mM MES and 10 mM acetic acid. 100 mM sodium chloride was added to reaction mixtures to increase the solubility of oligomeric and polymeric substrates. A typical assay contained 200 μM poly-D-mannuronan, dpave, 26, in a 1 cm path length cuvette. After thermal equilibration, 10 μl L-guluronate lyase (2 mg/mL stock) and 20 μl C-5-mannuronan epimerase (4.9 mg/mL stock) were added to the cuvette and the absorbance change due to glycal formation was monitored at 235 nm. The concentration of L-guluronate lyase used in the assay was optimized empirically to make epimerization the rate-determining step in the coupled assay. In order to determine the extent of any epimerase-independent absorbance change arising from spurious D-mannuronate lyase activity in the L-guluronate lyase, the same assay was repeated in the absence of C-5-mannuronan epimerase. The rate of epimerization was then calculated by the difference of the two initial velocities. The experimentally determined maximum initial rate (abs/s) was divided by the molar absorptivity of the glycal product (6,150 M−1 cm−1 (16), and the concentration of C-5-mannuronan epimerase (1.8 μM) to obtain values for the reaction velocity expressed in units of s−1.
The fixed-time assay was conducted by periodically transferring 100 μl aliquots from a reaction mixture to glass tubes, and terminating the reaction by placing the tube in a sand bath heated to 170 °C. The L-guluronate content of each sample was determined by enzymatic endpoint assay using L-guluronate lyase. Comparable results were obtained with the continuous and fixed-time assays.
Chromatographic separation and purification of oligo-D-mannuronans
Analytical separation of oligo-uronides was carried out by high performance anion exchange liquid chromatography using a Dionex DX-500 system, equipped with a CarboPac PA-1 analytical column (4×250 mm) and a pulsed amperometric detector. Samples of 25 μl volume were injected onto the column and the individual oligomers of dp 1-30 were eluted by a binary gradient of sodium acetate between 0 and 1M at constant sodium hydroxide concentration (100 mM).
Preparative separation of oligo-uronides was achieved by anion exchange chromatography using DEAE Sephadex A-25. Poly-D-mannuronan (dp 10 – 200, 700 mg) was dissolved in 1 L buffer containing 100 mM NaCl and 5 mM sodium phosphate at pH 7.2. The sample was loaded on the column at a flow rate of 1 mL/min and eluted with a linear salt gradient from 100 mM to 600 mM NaCl over 1 L at constant sodium phosphate buffer concentration. Fractions (5.5 mL) containing poly-D-mannuronan were identified by measuring absorbance at 210 nm. The polysaccharide was precipitated by addition of an equal volume of ethanol and pelleted by centrifugation. The pelleted material was lyophilized to obtain dry material.
Determination of concentration and degree of polymerization of oligo- and poly-Dmannuronan
The molar concentration of monomeric units in the substrate was determined gravimetrically, and the concentration of reducing ends was determined by the bicinchoninic acid assay using sodium glucuronate to construct a standard curve. Briefly, the reducing end of oligo- and poly-saccharides reduces Cu2+ to Cu1+ at 60 °C, which in turn complexes to a chelator, bicinchoninic acid under mild alkaline conditions (17). The average degree of polymerization of the sample is calculated as the concentration of monomeric units divided by the concentration of reducing ends.
Characterization of the reaction product
A large scale reaction was conducted so sufficient product could be accumulated for 1H NMR analysis. A solution containing 0.5 mM poly-D-mannuronan (dpaverage, 93), 20 mM Tris, 10 mM MES, 10 mM acetic acid, and 100 mM NaCl, pH 7.27, was prepared in a total volume of 9.8 mL. The reaction was initiated by the addition of 1 mg of C5-mannuronan epimerase in a volume of 0.2 mL. The solution was incubated at 25 °C in a water bath, and aliquots were removed after 2, 17, 28, 40, 48, 64, 73, 174, 318 and 663 hours. The enzyme was precipitated from each aliquot that was removed by addition of a small volume of chloroform and removed from the sample by centrifugation. The supernatant was mixed with ethanol to precipitate alginate and after centrifugation the pellet was lyophilized overnight. The lyophilized samples were dissolved in D2O and analyzed by 1H-NMR. All NMR experiments were performed on a Bruker DRX-500 spectrometer equipped with a temperature unit. Data collection was done at elevated temperature (80 °C) and temperature stability was 0.1 °C. Calibration of the 1H-NMR spectrum at 80 °C was done by the addition of ethanol as an internal standard in near stoichiometric amount; the chemical shift of the methyl protons of ethanol (δ, 1.17 ppm), which is not sensitive to temperature, was compared to the signals of the analyte.
Ca2+ dependence of the epimerization reaction
The C5-mannuronan epimerase reaction was assayed in the presence of several different Ca2+ concentrations; the amount of poly-D-mannuronan in each assay was adjusted such that the concentration of uncomplexed poly-D-mannuronan remained constant. The appropriate amounts of Ca2+ and poly-D-mannuronan for each assay were calculated using a value of 1 mM for the dissociation constant of the binary complex (18). It was assumed that each mannuronate residue in the polymer bound a single Ca2+ atom. The concentration of free poly-D-mannuronan was 0.5 mM and the concentration of free calcium varied from 20 to 1400 μM.
Analysis of the metal ion content of C5-mannuronan epimerase
The calcium content of purified, active C5-mannuronan epimerase was determined by flame atomic absorption spectroscopy using a Model 200A spectrophotometer from Buck Scientific. The sample contained 50 μM C5-mannuronan epimerase from which the His-tag had been removed. Liquid standards and samples, used in triplicate, were aspirated and mixed with acetylene and air. The light source was a calcium hollow cathode lamp with the wavelength set at 422.7 nm and each absorbance reading was integrated over 7 seconds.
The concentration of other metal ions in the purified protein was determined by inductively coupled plasma-emission spectroscopy. The analysis was conducted at the University of Georgia with samples that contained 50 μM protein. Levels of Mg, Mn, Ca, Fe, Cu, and 20 other elements were measured.
pH rate profiles
The pH dependence of the kinetic parameters for the reaction catalyzed by C5-mannuronan epimerase was determined by means of the continuous coupled assay as well as a fixed time assay using poly-D-mannuronan that had an average degree of polymerization of 26. The pH was varied between 5 and 8.6 using a ternary buffer system composed of 20 mM Tris, 10 mM MES and 10 mM acetic acid. The kinetic parameters kcat and kcat/Km were fitted to Equation 1, where Y is either kcat or kcat/Km, and C is the pH-independent value of Y.
| (1) |
Results
Overexpression and purification of C5-mannuronan epimerase
Recombinant C5-mannuronan epimerase was purified in 100 mg quantities through metal affinity chromatography. The purified protein was 99 % homogeneous, based on Coomassie Blue-stained polyacrylamide gels. The solubility of the purified enzyme was improved by removal of the (His)6-tag at the N-terminus, which was accomplished readily by thrombin-catalyzed cleavage. Removal of imidazole by stepwise dialysis against buffer containing decreasing amounts of imidazole was found to mitigate protein precipitation. Enzyme stored at −80 °C in 10 % (v/v) glycerol retained full catalytic activity for at least two months.
Metal ion dependence of the epimerization reaction
Investigation of the Ca2+ dependence of catalytic activity was first probed by determining the Ca2+ content of purified, active C5-mannuronan epimerase. Samples containing 50 μM protein contained 1.2 μM Ca2+, as measured by atomic absorption spectroscopy, indicating that Ca2+ is not a tightly-bound, stoichiometric component of the protein. Dialysis of the enzyme against EDTA did not cause loss of activity. Furthermore, the catalytic activity of C5-mannuronan epimerase was unaffected by changes in the concentration of free Ca2+ (Figure 1A). Despite the broad range of free Ca2+ concentration in the assays (1-1400 μM), initial velocities remained the same, 0.0095 s−1, when 500 μM (total carbohydrate concentration) substrate was present. Calcium binds to poly-D-mannuronan (18), but the epimerase reaction was insensitive to the concentration of the complex, indicating that it cannot serve as a substrate (Figure 1B). Elemental analysis conducted by inductively coupled plasma-emission spectroscopy revealed that the purified protein contained no other metal ions above trace levels.
FIGURE 1.

Variation of steady-state rate with concentration of the Ca2+ complex of poly-D-mannuronan (A) and that of free Ca2+ (B). The total carbohydrate concentration of the uncomplexed poly-D-mannuronan was 500 μM. Reactions were run at pH 7.27 (25 °C).
pH rate profiles
The pH dependence of the kinetic parameters is shown in Figure 2. Both kcat and kcat/Km decrease dramatically at alkaline pH, and the data were fitted to equation 1, which assumes that deprotonation of three ionizable groups with indistinguishable pKa's leads to the decrease in kcat and kcat/Km. The kcat profile defined a pKa value of 7.87 ± 0.05 and the kcat/Km profile defined a pKa value of 7.6 ± 0.2. Alternative fits of the experimental data to functions describing ionization of 2 or 4 residues were considered less satisfactory based on visual inspection of the data (Figure 2). The values of the kinetic parameters did not vary between pH 6 and 7.5; the reaction could not be characterized below pH 6 due to precipitation of the enzyme. Between pH 6 and 7.5, using poly-D-mannuronan that had an average dp of 26, the value for kcat was 0.0218 s−1, and the value for Km was 13 μM. The pH-independent value of kcat/Km was 1.7 × 103 M−1s−1, indicating that the reaction proceeds far more slowly than the rate of diffusional encounter.
FIGURE 2.
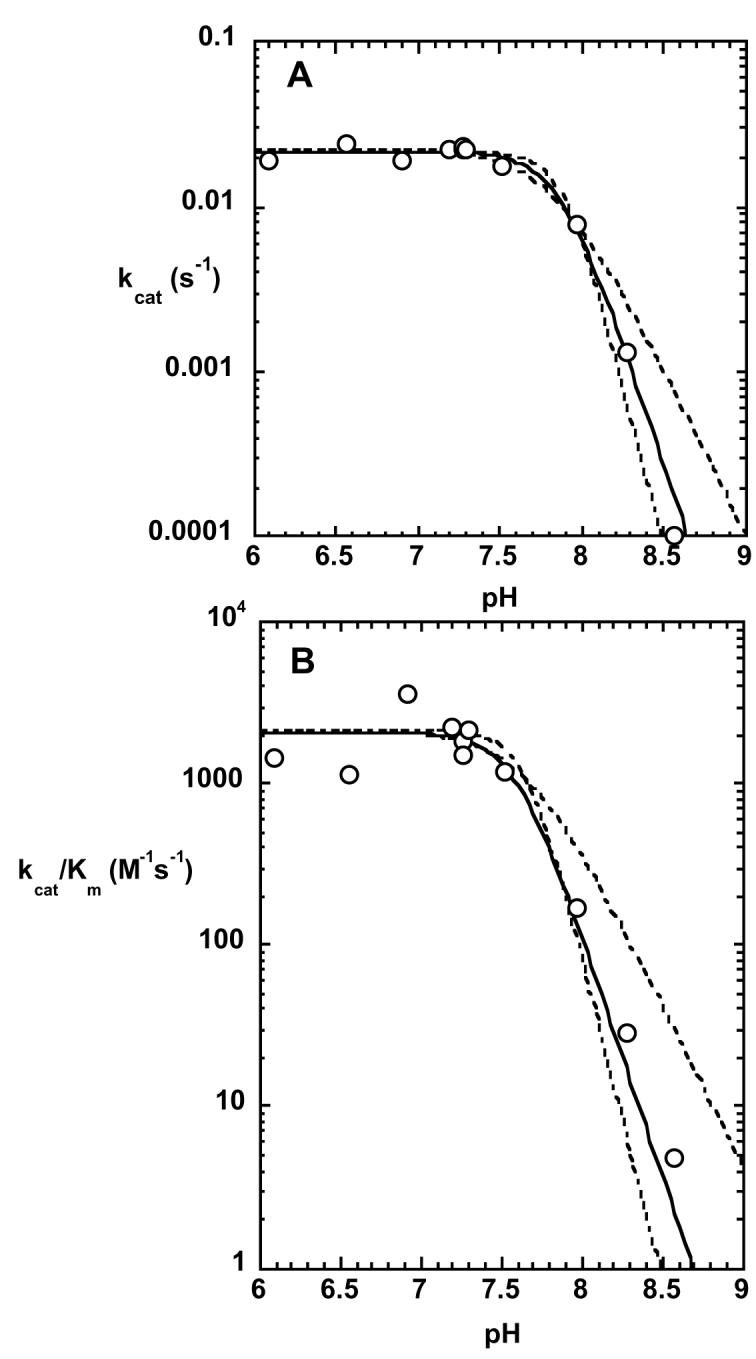
Kinetic parameters kcat and kcat/Km for the epimerase reaction as a function of pH. The reactions were conducted as described in the text and the solid lines show the fits to equation 1, which describes 3 ionizations. The dashed lines show alternative fits to equations describing 2 or 4 ionizations.
Substrate size specificity
The kinetic parameters for the C5-mannuronan epimerase reaction were determined using pools of oligo-mannuronate in which the average dp varied from less than 10 to more than 100. As shown in Figure 3, high performance anion exchange chromatography was used successfully to separate oligo-mannuronates up to about dp 30. The substrate pools for oligo-mannuronates of dp < 30 that were used in the kinetic assays typically had a size variation of 1-2 units. The kinetic parameters as a function of substrate size are shown in Figure 4. Km values decreased until they reached a plateau value of approximately 10 μM at dp 20. Similarly, kcat values increased with increasing substrate size until dp 20, where they reached a maximum of 0.022 s−1. No reaction could be detected for substrates that contained fewer than 9 monomeric units.
FIGURE 3.

HPAE-PAD separation of oligo-D-mannuronans (dp: 1-31). The numbers drawn above the eluting peaks represent the number of uronide residues of the individual oligomers.
FIGURE 4.

Substrate size specificity for the epimerase reaction. The figure shows variations in kcat and Km as a function of degree of polymerization of oligo-D-mannuronan. Epimerization was monitored by the continuous coupled assay at 25 °C and pH 7.27.
Product composition
C5-Mannuronan epimerase produces residues of α-L-guluronate within the substrate poly-β-D-mannuronan by inverting the configuration at C5 in the uronate monomeric unit. The composition of the product resulting from the action of C5-mannuronan epimerase can be determined by 1H-NMR analysis (19). Mannuronate residues and guluronate residues are readily distinguished by the chemical shifts of the proton on the anomeric carbon (4.6 ppm for mannuronate, and 4.95 ppm for guluronate), so these signals provide a convenient means to monitor the extent of the epimerization reaction. The chemical shift of the C-5 proton of guluronate is exquisitely sensitive to the identity of neighboring residues, so the sequence of M and G residues in the polymer to be determined. The chemical shift of the C-5 proton for the underlined residue in the triads GGM, MGM, GGG, and MGG is 4.68 ppm, 4.65 ppm, 4.39 ppm, and 4.39 ppm, respectively. The C-5 proton of mannuronate appears at 3.6 ppm and is not shown in Figure 5.
FIGURE 5.
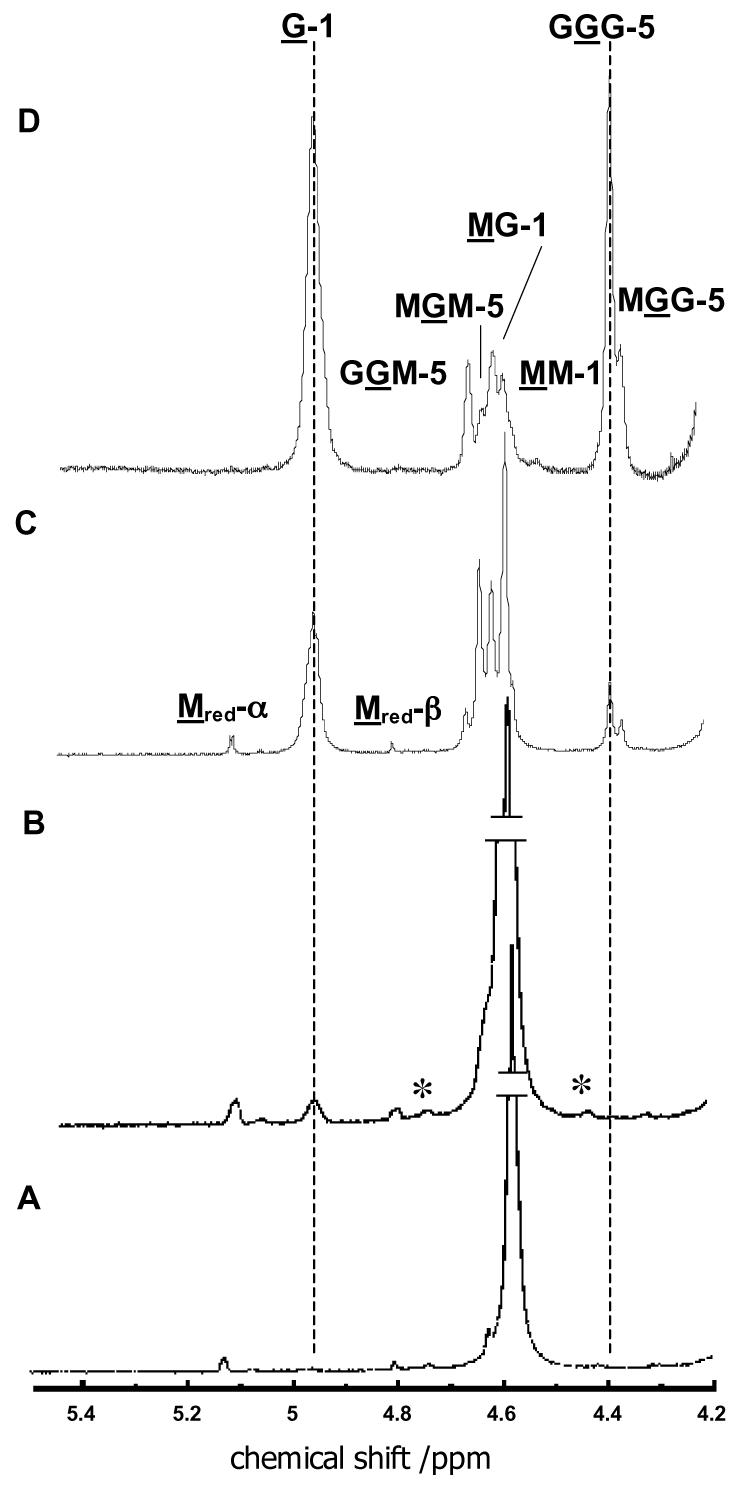
1H-NMR analysis of alginate samples obtained after partial to exhaustive epimerization of poly-D-mannuronan by P. aeruginosa C5-mannuronan epimerase. Assay conditions are discussed in the text. Spectra were recorded at 500 MHz and 80 °C in D2O. Peak assignments are indicated on panels C and D. Numbers designate the position of the H-atom in the hexose ring; the underlined letters (M and G) denote the residue that is giving rise to the indicated peak, while the letters that are not underlined specify neighboring residues in the linear polymer. Peaks labeled by asterisks are due to 13C coupling to MM-1 protons. (A) Spectrum of poly-D-mannuronan (FG = 0, FM = 1, dpaverage = 93) used as starting material in the assay. (B) Heteropolymeric alginate at very low conversion (FG = 0.02, FGG = 0). (C) Heteropolymeric alginate at moderate conversion (FG = 0.26, FGG = 0.06). (D) Heteropolymeric alginate formed as the final product by exhaustive epimerization by C5-mannuronan epimerase (FG = 0.75, FGG = 0.50). The vertical dashed line at 4.95 ppm marks the signal that arises from H-1 on guluronate and serves to indicate the extent of the reaction. The vertical dashed line at 4.40 ppm marks the signal that arises from the C-5 proton of the central guluronate residue in a GGG triad, and indicates the formation of G tracts.
At very early stages of the reaction C5-mannuronan epimerase forms heteropolymeric blocks of MGM sequence. However, as the reaction progresses, the enzyme displays a preference for acting on M residues that are adjacent to G residues. Indeed, beyond 25% conversion, only the GGG and MGG triads are populated. The enzyme is unable to convert every M residue to G, however; the reaction reaches an apparent equilibrium when the polymer contains 75% G.
Discussion
Pseudomonas aeruginosa infections present severe risks to many patient populations; the pulmonary infections of cystic fibrosis patients are perhaps the most notorious (1). The presence of viscous phlegm in the lungs of cystic fibrosis patients is exacerbated by the bacterial production of alginate. The viscosity of alginate is a function of the composition of the polymer and the presence or absence of metal ions. Guluronate residues are introduced into the polymer via the action of C5-mannuronan epimerase; thus, in the context of developing strategies to treat P. aeruginosa infections, it becomes critical to characterize C5-mannuronan epimerase in detail.
The bacterium Azotobacter vinelandii produces a family of C5-mannuronan epimerases which catalyze the same reaction as the P. aeruginosa enzyme, but do not share significant sequence homology with it (20). The A. vinelandii enzymes are Ca2+-dependent, and the initial studies of the P. aeruginosa epimerase included Ca2+ as a component of the assay solution (7, 11-13). Several different roles for Ca2+ could be envisioned in the epimerase reaction. Since the epimerase reaction is likely to proceed via proton abstraction at the C5 position, Ca2+ could serve to stabilize the resulting carbanion in a fashion similar to that seen in the enolase reaction (21). The binding of Ca2+ to alginate has been well documented (18, 22), so it is possible that the true substrate for the epimerase reaction is the Ca2+-alginate complex. Finally, Ca2+ is known to be a tightly bound cofactor in some enzymes (23, 24). We therefore explored the Ca2+-dependence of the C5-mannuronan epimerase reaction.
The Ca2+ content of the purified, active enzyme was determined by atomic absorption spectroscopy. No Ca2+ above trace levels was observed, so Ca2+ is not a tightly bound cofactor in C5-mannuronan epimerase. Furthermore, dialysis of purified enzyme against EDTA did not inactivate the enzyme.
The influence of Ca2+ on the rate of the epimerization reaction was determined using a continuous spectrophotometric assay. Initial experiments indicated that Ca2+ was slightly inhibitory; however, when the depletion of free polymannuronan from the solution by complexation with Ca2+ was taken into account (18), it was evident that Ca2+ has no effect on the C5-mannuronan epimerase reaction. As shown in Figure 1, free Ca2+ does not stimulate the reaction, nor is the Ca2+-polymannuronan complex a substrate.
The absence of a requirement for Ca2+ in the catalytic reaction indicates that the presumptive carbanion intermediate in the catalytic reaction is not stabilized by a metal ion, as is the case in the reactions catalyzed by enolase superfamily members.
The substrate for C5-mannuronan epimerase in vivo is a polymer hundreds of residues long. Characterization of the size specificity of the enzyme can shed light onto how large the area of interaction is between the substrate and the enzyme, and in a practical sense, can provide information about how large potential inhibitors should be. No reaction was detected with oligomers comprised of 8 or fewer residues. The maximum velocity of the reaction increased dramatically between 10 and 20 residues, while the Km for the substrate decreased over that size range. There was little variation in either kinetic parameter for substrates containing more than 20 residues (Figure 4). Using kcat/Km as a measure of substrate specificity, the oligosaccharide containing 20 residues was favored by more than 200-fold over the substrate containing only 10 residues.
If the Km is treated as an approximation to the Kd for the substrates, it is apparent that the enzyme affinity increases with increasing substrate size up to dp 20. There is little change in Km for substrates larger than 20 residues. The simplest interpretation of these data would suggest that C5-mannuronan epimerase interacts with the substrate over a surface that encompasses up to 20 sugar residues. This represents an extensive surface area; an oligosaccharide 20 residues in length would stretch approximately 100 Å. The AlgE2 and AlgE4 mannuronan epimerases from Azotobacter vinelandii exhibit increased activity with substrates up to 2000 residues in length (25), which corresponds to a size that is clearly too large to be accomodated by a single protein molecule. These data would seem to require that the substrate polymer serves as a nucleation center to which multiple molecules of the enzyme could bind. Experiments to explore whether this occurs with the P. aeruginosa epimerase are currently underway.
The value of kcat increases for substrates between dp 10 and 20. Since kcat is determined by extrapolating to conditions of infinite substrate, the decreased affinity of the enzyme for shorter substrates does not enter into the determination of kcat. Thus, it is apparent that the increased binding energy that is available in larger oligosaccharides is used both to enhance the binding of the substrate to the enzyme in the ground state, and to increase the rate acceleration in the enzyme-substrate complex. The rate acceleration can be quantitated through the relationship ΔΔG‡ = −RTln(k20mer/k10mer) where k20mer is kcat for the reaction with the substrate containing 20 monomeric units and k10mer is kcat for the substrate containing 10 monomeric units. Using the experimental values for kcat obtained in this investigation, it can be calculated that the transition state is stabilized by an additional 1.5 kcal/mol for the dp 20 substrate compared to the dp 10 substrate. The data in Figure 4 indicate that kcat increases in a roughly linear fashion for substrates between dp 10 and dp 20, suggesting that each additional residue beyond the first 10 contributes approximately 0.15 kcal/mol of binding energy to the rate acceleration.
The pH dependence of the kinetic parameters revealed that kcat and kcat/Km both decreased dramatically at alkaline pH. Each pH profile described the ionization of three groups; because poly-D-mannuronan does not have any functional groups to which these ionizations could be assigned, they must arise from ionizable residues on the enzyme. Since the ionizations are observed in both the kcat and kcat/Km profiles, the protonation state of these ionizable groups must be critical for catalysis (26).
The pH profiles are consistent with ionizations arising from residues that function as general acids in the catalytic reaction. The epimerization reaction requires proton abstraction from C5, followed by reprotonation from the opposite face of the intermediate. The ionizable residues observed in the pH profiles may be those that are serving as the general acids in the catalytic reaction. The data provide no evidence for the general bases that are expected to participate, but their absence from the pH profiles could simply mean that their pKa's are below the experimentally accessible pH range. It is unclear why three ionizable residues would be required for catalysis, but a formal possibility that is consistent with the data is that the active site is able to act on more than one sugar residue at a time.
Alternatively, the ionizations may arise from residues that are not functioning as general acids in the catalytic reaction. Poly-D-mannuronan is a polyanion, and so it could reasonably be expected that interactions with cationic amino acid residues are important for binding. When a complex cannot form between substrate and/or enzyme in the incorrect protonation state for reaction, the ionizations do not appear in the kcat profile (26). Thus, the protonation states of the residues whose ionizations are evident in Figure 2 are not critical for formation of an enzyme-substrate complex, but their protonation state is critical for reaction. Rather than assigning the ionizable residues to general acids at the active site, it seems more likely that they arise from residues that are important for binding of the substrate in a catalytically competent complex. Since polymannuronan is a long, rather uniform polymer which appears to interact with the enzyme over a relatively large surface area, we favor a model in which initial binding is relatively nonspecific, but then the substrate is moved into proper register for reaction by electrostatic interactions with the correctly ionized residues.
The physical and chemical properties of alginate are determined by the mannuronate and guluronate content. Adjacent mannuronate residues are joined by diequatorial glycosidic bonds, which permit more rotational freedom, and thus more flexibility. Adjacent guluronate residues are joined by diaxial glycosidic bonds, which are more restrictive, and result in a stiff polymer with dense secondary structure. Accordingly, alginate that is composed primarily of G-blocks has lower solubility, and its aqueous solution has higher viscosity than alginate containing M-blocks; furthermore, G-blocks form a rigid coordination polymer with divalent cations, such as calcium. Since C5-mannuronan epimerase introduces guluronate residues into poly-Dmannuronan, the extent to which its reaction progresses is critical to the ultimate properties of the alginate.
Alginate that is isolated from mucoid P. aeruginosa colonies is characterized by the absence of stretches of adjacent guluronate residues (G-blocks) (27). From this observation, it has been inferred that P. aeruginosa C5-mannuronan epimerase is unable to act on mannuronate residues that are adjacent to guluronate residues. The data in Figure 5 clearly belie this conclusion. It is apparent that GGG triads are readily formed; indeed, alginate that has been incubated with the epimerase exhaustively is composed of 75% guluronate and 25% mannuronate. Interestingly, the fraction of MG diads (or GM diads) is 0.5; therefore, the sequence of the final alginate product is best represented by repeating GGGM tetramers.
It is interesting that the endpoint of the reaction is reached when 75% of the uronate units are guluronates. The relative energies of mannuronate and guluronate are similar, so if the reaction were freely reversible, one would expect it to reach equilibrium when the polymer contained approximately equal amounts of mannuronate and guluronate. On the other hand, if the sugar ring conformation change that accompanies epimerization precludes binding to the enzyme, epimerization could be rendered irreversible, and one might expect the enzymatic reaction to continue until poly-G was formed. The observation that the reaction cannot proceed beyond incorporation of 75% guluronate residues suggests that the polymer composed of repeating GGGM tetrads assumes a conformation that cannot bind to the enzyme.
The apparent conflict between the composition of the alginate formed by C5-mannuronan epimerase in vitro and that isolated from mucoid cells can be resolved if the epimerization reaction takes place in competition with O-acetylation in vivo. Acetylated guluronate residues are not observed in alginate (28), so the extent of introduction of guluronate residues into the polymer will be controlled by the relative activities of the acetyltransferase and the epimerase.
The 1H-NMR data reveal another interesting feature of the C5-mannuronan epimerase reaction. Mannuronate residues adopt the 4C1 conformation, but guluronate residues are found in the 1C4 conformation, which allows the carboxyl group attached to C5 to assume the equatorial position in both sugars. The conformation change from a 4C1 hexose to a 1C4 hexose entails the movement of each equatorial substituent into an axial position, and vice versa. It is difficult to envision how this process could occur in the context of polymeric 1,4-linked sugar residues, in which one would expect there to be a significant kinetic barrier to the interconversion of the two chair forms.
One possibility is that the epimerization may occur via transient cleavage of the polymer. Such a mechanism has a number of attractive features. Stabilization of the carbanion generated by proton abstraction at C5 by formation of a glycal, 4-deoxy-L-erythro-hex-4-enopyranosyluronate, concomitant with cleavage of the 1,4-linkage would explain the absence of a metal requirement for the catalytic reaction. The transient generation of a cleaved intermediate would unlock the conformational restraint on the sugar unit undergoing epimerization and allow the intermediate to change conformation. Experiments are currently underway to examine the chemical mechanism of the epimerization reaction in more detail.
Acknowledgment
The authors are grateful to Professor Michael Henzl for assistance with the atomic absorption measurements.
Footnotes
This work was supported by a grant to P.A.T. from the National Institutes of Health (GM59653)
Abbreviations used: dp, degree of polymerization, the number of monosaccharide units in a polysaccharide or oligosaccharide; G, guluronate; M, mannuronate; poly-M, polymer of mannuronate residues; poly-G, polymer of guluronate residues; IPTG, isopropyl-ß-D-thiogalactopyranoside; PMSF, phenylmethylsulphonyl fluoride; TLCK, N-tosyl-L-lysinechloromethyl ketone; DTT, dithiothreitol.
References
- 1.Govan JRW, Harris GS. Pseudomonas aeruginosa and cystic fibrosis: unusual bacterial adaptation and pathogenesis. Micro. Sci. 1986;3:302–308. [PubMed] [Google Scholar]
- 2.Frederiksen B, Koch C, Hoiby N. Antibiotic treatment of initial colonization with Pseudomonas aeruginosa postpones chronic infection and prevents deterioration of pulmonary function in cystic fibrosis. Pediatr. Pulmonol. 1997;23:330–335. doi: 10.1002/(sici)1099-0496(199705)23:5<330::aid-ppul4>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 3.Evans LR, Linker A. Production and characterization of the slime polysaccharide of Pseudomonas aeruginosa. J. Bacteriol. 1973;116:915–924. doi: 10.1128/jb.116.2.915-924.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smidsrod O, Draget K. Chemistry and physical properties of alginates. Carbohydr. Eur. 1996;14:29–36. [Google Scholar]
- 5.Chitnis CE, Ohman DE. Genetic analysis of the alginate biosynthetic gene cluster of Pseudomonas aeruginosa shows evidence of an operonic structure. Mol. Microbiol. 1993;8:583–590. doi: 10.1111/j.1365-2958.1993.tb01602.x. [DOI] [PubMed] [Google Scholar]
- 6.Saxena IM, Brown RM, Fevre M, Geremia RA. Multidomain architecture of beta-glycosyl transferases: implications for mechanism of action. J. Bacteriol. 1995;177:1419–1424. doi: 10.1128/jb.177.6.1419-1424.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Franklin MJ, Chitnis CE, Gacesa P, Sonesson A, White DC, Ohman DE. Pseudomonas aeruginosa AlgG is a polymer level alginate C5-mannuronan epimerase. J. Bacteriol. 1994;176:1821–30. doi: 10.1128/jb.176.7.1821-1830.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davidson JW, Lawson CJ, Sutherland IW. Localization of O-acetyl groups in bacterial alginate. J. Gen. Microbiol. 1977;98:603–606. doi: 10.1099/00221287-98-1-223. [DOI] [PubMed] [Google Scholar]
- 9.Rehm BH, Boheim G, Tommassen J, Winkler UK. Overexpression of algE in Escherichia coli: subcellular localization, purification, and ion channel properties. J. Bacteriol. 1994;176:5639–5647. doi: 10.1128/jb.176.18.5639-5647.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sherbrock-Cox V, Russell NJ, Gacesa P. The purification and chemical characterization of the alginate present in extracellular material produced by mucoid strains of Pseudomonas aeruginosa. Carbohydr. Res. 1984;135:147–154. doi: 10.1016/0008-6215(84)85012-0. [DOI] [PubMed] [Google Scholar]
- 11.Ertesvag H, Doseth B, Larsen B, Skjak-Braek G, Valla S. Cloning and expression of an Azotobacter vinelandii C-5-epimerase gene. J. Bacteriol. 1994;176:2846–2853. doi: 10.1128/jb.176.10.2846-2853.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Svanem BIG, Skjak-Braek G, Ertesvag H, Valla S. Cloning and expression of three new Azotobacter vinelandii genes closely related to a previously described gene family encoding mannuronan C-5-epimerases. J. Bacteriol. 1999;181:68–77. doi: 10.1128/jb.181.1.68-77.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ertesvag H, Hoidal HK, Hals IK, Rian A, Doseth B, Valla S. A family of modular type mannuronan C5-epimerae genes controls alginate structure in Azotobacter vinelandii. Mol. Microbiol. 1995;16:719–731. doi: 10.1111/j.1365-2958.1995.tb02433.x. [DOI] [PubMed] [Google Scholar]
- 14.Douthit SA, Dlakic M, Ohman DE, Franklin MJ. Epimerase active domain of Pseudomonas aeruginosa AlgG, a protein that contains a right-handed beta-helix. J. Bacteriol. 2005;187:4573–4583. doi: 10.1128/JB.187.13.4573-4583.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Raychaudhuri A, Tipton PA. Protocol for amplification of GC-rich sequences from Pseudomonas aeruginosa. BioTechniques. 2004;37:752–756. doi: 10.2144/04375BM08. [DOI] [PubMed] [Google Scholar]
- 16.Iwamoto Y, Araki R, Iriyama K, Oda T, Fukuda H, Hayashida S, Muramatsu T. Purification and characterization of bifunctional alginate lyase from Alteromonas sp. strain No. 272 and its action on saturated oligomeric substrates. Biosci. Biotechnol. Biochem. 2001;65:133–142. doi: 10.1271/bbb.65.133. [DOI] [PubMed] [Google Scholar]
- 17.Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olsen BJ, Klenk DC. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 18.Steginsky CA, Beale JM, Floss HG, Mayer RM. Structural determination of alginic acid and the effects of calcium binding as determined by high-field n.m.r. Carbohydr. Res. 1992;225:11–26. doi: 10.1016/0008-6215(92)80036-z. [DOI] [PubMed] [Google Scholar]
- 19.Larsen B, Salem DMSA, Sallam MAE, Mishrikey MM, Beltagy AI. Characterization of the alginates from algae harvested at the Egyptian Red Sea coast. Carbohydr. Res. 2003;338:2325–2336. doi: 10.1016/s0008-6215(03)00378-1. [DOI] [PubMed] [Google Scholar]
- 20.Valla S, Li J-P, Ertesvag H, Barbeyron T, Lindahl U. Hexuronyl C5-epimerases in alginate and glycosaminoglycan biosynthesis. Biochemie. 2001;83:819–830. doi: 10.1016/s0300-9084(01)01313-x. [DOI] [PubMed] [Google Scholar]
- 21.Poyner RR, Cleland WW, Reed GH. Role of metal ions in catalysis by enolase: an ordered kinetic mechanism for a single substrate enzyme. Biochemistry. 2001;40:8009–8017. doi: 10.1021/bi0103922. [DOI] [PubMed] [Google Scholar]
- 22.Wang Z-Y, Zhang Q-Z, Konno M, Saito S. Sol-gel transition of alginate solution by the addition of various divalent metal ions: 13C-NMR spectroscopic studies. Biopolymers. 1993;33:703–711. [Google Scholar]
- 23.Hsiu J, Fischer EH, Stein EA. alpha-Amylases as calcium-metalloenzymes. II. Calcium and the catalytic activity. Biochemistry. 1964;3:61–66. doi: 10.1021/bi00889a011. [DOI] [PubMed] [Google Scholar]
- 24.Liu D, Shriver Z, Godavarti R, Venkataraman G, Sasisekharan R. The calcium-binding sites of heparinase I from Flavobacterium heparinum are essential for enzymic activity. J. Biol. Chem. 1999;274:4089–4095. doi: 10.1074/jbc.274.7.4089. [DOI] [PubMed] [Google Scholar]
- 25.Hartmann M, Holm OB, Johansen GAB, Skjak-Braek G, Stokke BT. Mode of action of recombinant Azotobacter vinelandii mannuronan C-5 epimerases AlgE2 and AlgE4. Biopolymers. 2002;63:77–88. doi: 10.1002/bip.10017. [DOI] [PubMed] [Google Scholar]
- 26.Cleland WW. The use of pH studies to determine chemical mechanisms of enzyme-catalyzed reactions. Methods in Enzymology. 1982;87:390–405. doi: 10.1016/s0076-6879(82)87024-9. [DOI] [PubMed] [Google Scholar]
- 27.Schurks N, Wingender J, Flemming H-C, Mayer C. Monomer composition and sequence of alginates from Pseudomonas aeruginosa. Int. J. Biol. Macromolecules. 2002;30:105–111. doi: 10.1016/s0141-8130(02)00002-8. [DOI] [PubMed] [Google Scholar]
- 28.Skjak-Braek G, Grasdalen H, Larsen B. Monomer sequence and acetylation pattern in some bacterial alginates. Carbohydr. Res. 1986;154:239–250. doi: 10.1016/s0008-6215(00)90036-3. [DOI] [PubMed] [Google Scholar]