

Abstract
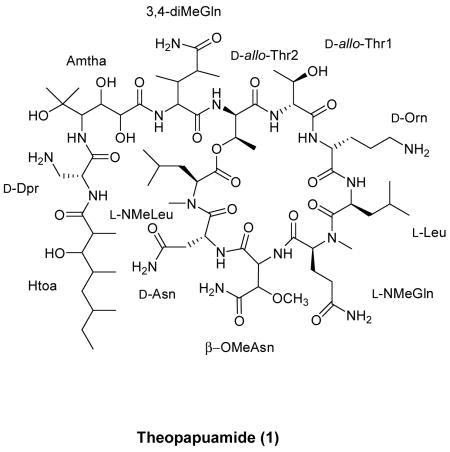
Theopapuamide (1), a new cytotoxic peptide has been isolated from the lithistid sponge Theonella swinhoei from Papua New Guinea. The structure was established by analysis of NMR, mass spectrometry and chemical methods. The undecapeptide (1) contains several unusual amino acid residues, of which the occurrence of β-methoxyasparagine and 4-amino-5-methyl-2,3,5-trihydroxy-hexanoic acid (Amtha) is unprecedented in natural peptides. Compound 1 also contains a amide linked fatty acid moiety, 3-hydroxy-2,4,6-trimethyl-octanoic acid (Htoa). Theopapuamide (1) was cytotoxic against CEM-TART and HCT-116 cell lines with EC50 values of 0.5 μM and 0.9 μM respectively.
Marine sponges have proven to be a significant source of biologically active cyclic peptides and depsipeptides.1 Among these sponge depsipeptides, callipeltin A (from New Caledonian sponge Callipelta sp.),2 neamphamide A (from a Papua New Guinea sponge Neamphius huxleyi)3 and Papuamides A-B (from Papua New Guinea sponges Theonella mirabilis and Theonella swinhoei)4 are well known for their potent HIV-inhibitory activity and their structurally unique features incorporating several modified amino acid residues. For instance, the atypical amino acid residues 3,4-dimethyl-L-glutamine and β-methoxytyrosine are common to all of the above mentioned marine depsipeptides but to date have not been described elsewhere. The rarity of these atypical amino acid residues has inspired their chemical synthesis.5 The structural diversity found among lithistid sponge metabolites (genus Theonella and Callipelta) has been attributed to symbiotic microorganisms.1
As part of our continuing studies on Theonella swinhoei from Papua New Guinea,6 the aqueous CH3CN extract of the sponge was analyzed and proved active in an in vitro anti-HIV assay. Fractionation of the active extract resulted in the isolation of a new cyclic depsipeptide, theopapuamide (1). This paper describes the isolation, structure elucidation, and stereochemical analysis of theopapuamide (1).
The crude aqueous CH3CN extract of T. swinhoei (family Theonellidae) was concentrated under vacuum and fractionated by C18 flash column chromatography. Further purification on Diaion HP-20 resin followed by CN-HPLC afforded the new cyclic undecapeptide, theopapuamide (1, 15.8 mg, 3.95 × 10−3 % yield wet wt) as an off-white amorphous solid ([α]25D −3.0, c 0.86, MeOH).
For compound 1, a molecular formula of C69H123N17O23 was established based on the divalent molecular ion [M+2H]2+ observed at m/z 779.9563 by HRESI-FTMS. The protonated molecular ion at m/z 1558.9053 [M+H]+ in the HRESI-FTMS was in agreement with a molecular weight of 1557.8980 Da (Δ = −0.2 mmu) for the neutral compound (1). The molecular formula suggested seventeen units of unsaturation. Subsequent examination of 1D-spectra of 1 reflected characteristic features of a peptide, such as an abundance of exchangeable N-H protons (δH 6.55–9.05) and carbonyl signals (δC 169.9–179.6) in the 1H and 13C NMR spectra, respectively. However, the interpretation of NMR spectra of 1 was hampered by the existence of multiple conformations in several of the common NMR solvents (MeOH-d4, CD3CN:DMSO-d6 and DMSO-d6). Attempts to improve resolution by employing acetone-d6 and DMF-d7 as solvents failed due to poor solubility of 1. Moreover, efforts to achieve a favorable conformational ratio by addition of LiCl7 (0.5–4 equi., CD3CN:DMSO-d6, 25 °C) was also ineffective. Gratifyingly, one major conformation, with improved resolution, was observed using a mixture of CD3CN:H2O (4:1) at 25 °C.
The aliphatic nature of the peptide was supported by the absence of any sp2-hybridized carbon resonances in the region between 110–150 ppm, as well as localization of resonances in the upfield region of the 13C NMR spectrum of 1. Due to the extensive overlap of aliphatic resonances in 1, the assembly of individual amino acid residues required combined analysis of 1H-1H-COSY, gHSQC, gHSQC-TOCSY, 1D-TOCSY and z-DIPSI-tocsy8 spectra. In conjunction with NMR analysis, the gross structure elucidation of 1 was guided by standard amino acid analysis,9 which revealed molar concentrations of ~1:2:1 for Asx, Thr and Leu respectively. The presence of two N-methylated amino acid residues were suggested based on the characteristic 1H and 13C chemical shifts of the N-methyl groups at δH 2.81 (δC 30.8) and δH 2.88 (δC 31.8). HMBC correlations were used to identify these N-methylated amino acids as NMeLeu and NMeGln, respectively. Additionally, the presence of a methoxy-bearing amino acid residue was suggested by the characteristic 1H and 13C chemical shifts of the O-methyl group at δH 3.34 (δC 60.3). Based on HMBC correlations, the methoxy-bearing amino acid residue was identified as β-OMeAsn. An ester linked threonine residue was suggested from a typical ~1.0 ppm downfield shift of the β-hydroxymethine proton (δH 5.55). A subsequent 15N-HSQC experiment showed four pairs of signals for the primary amide protons of 3,4-diMeGln, Asn, β-OMeAsn and NMeGln residues. However, the 1H-15N-correlations for the primary amino protons of Orn and Dpr could not be observed under the given experimental conditions [CD3CN-H2O (4:1), 25 °C].
The connectivity between amino acid residues was established based on careful analysis of HMBC, WATERGATE-NOESY and WATERGATE-ROESY10 spectra. Further inspection of HMBC data supported formation of an 8-residue ring moiety in 1 via an ester linkage between the C-3-hydroxyl group of Thr (δH-3 5.55) and the C-1-carboxylic acid of NMeLeu (δC-1 171.5). Additionally, a NOESY cross peak between Thr (δH-3 5.55) and NMeLeu (δH-2 5.07) was in agreement with such a macrocyclic ring formation. The constituents of the macrocycle, identified based on sequential α-NH/NH and α-NH/NCH3 NOESY correlations, consisted of NMeLeu (δCH3 2.81), Asn (δH 8.18), β-OMeAsn (δH 6.69), NMeGln (δCH3 2.88), Leu (δH 7.20), Orn (δH 8.00), and two residues of Thr (Thr1 δH 8.25, Thr2 δH 8.89). Furthermore, based on HMBC and NOESY data, the α-NH (δH 8.89) of the ester linked Thr was attached to a 4-residue sequence of 3,4-diMeGln, 4-amino-5-methyl-2,3,5-trihydroxy-hexanoic acid (Amtha), 2,3-diaminopropionic acid (Dpr) and 3-hydroxy-2,4,6-trimethyl-octanoic acid (Htoa), forming the linear portion of the peptide. Of the component amino acid residues identified in undecapeptide 1, 5 of the residues were also described in both callipeltin A and neamphamide A (3,4-diMeGln, Thr (×2), Leu, and NMeGln).2,3 A Htoa dimer, bourgeanic acid has been isolated from several Ramalina sp. lichens.11
Concurrent attempts to sequence the peptide by Edman degradation12 were unsuccessful signifying a concealed or chemically modified N-terminus in 1. Additionally, theopapuamide (1) was resistant to digestion by a variety of proteases (trypsin, thermolysin and pepsin)13 likely due to collective effects of N-methylated amino acid residues, peptide bonds involving D-amino acids and the cyclic nature of 1. Initial attempts to clarify the amino acid sequence by MS/MS (ESI and MALDI) analysis of the intact cyclic depsipeptide (1) was not informative, and yielded random cleavage products. Subsequently, the ester linkage in theopapuamide (1) was subjected to base hydrolysis (1N KOH, r.t., 2 h) to generate the acyclic peptide 2 [m/z 1576.9 (M + H)+]. As anticipated, the acyclic peptide (2) was amenable to tandem MS approach.14 Accordingly, successful sequence analysis of 2 was carried out on the basis of fragment ion spectra generated by SORI-CID ESI-FTMS/MS (Figure 1).
Figure 1.
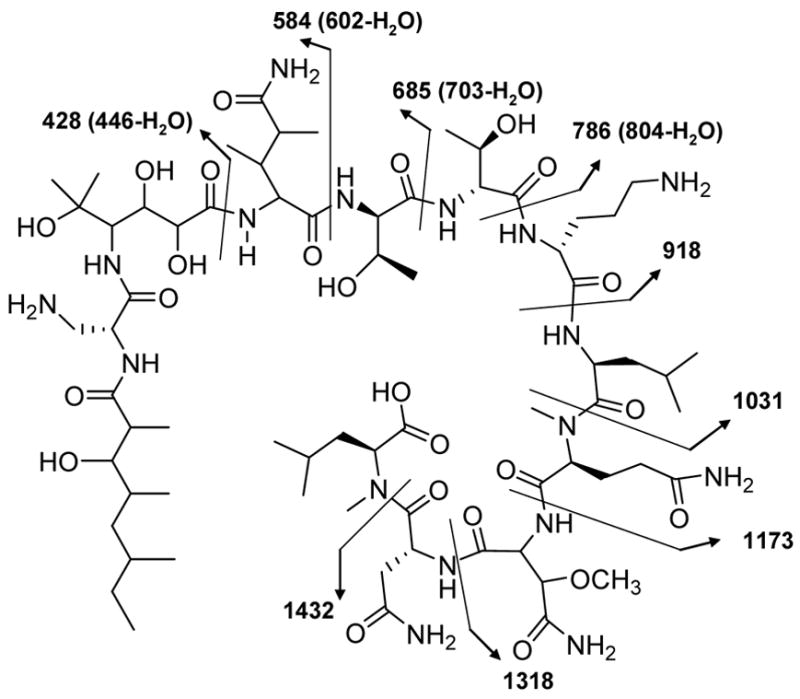
SORI-CID MS/MS fragmentations of the acyclic peptide (2)
The absolute configurations of the amino acid constituents of theopapuamide (1) were determined by acid hydrolysis of 1 (6N HCl, 110 °C, 12 h), followed by chiral HPLC analysis of the hydrolysate, and RP-HPLC analysis of FDAA derivatives.15 By chiral HPLC [Chirex phase 3126(D), iPrOH-2mM CuSO4 (5:95)], diagnostic peaks were observed for L-NMeLeu, D-Asp, L-Leu, L-NMeGlu, D-Orn and D-Dpr residues. The chrial HPLC analysis failed to give sufficient separation of the D/L-allo-Thr standards [D/L-allo-Thr (6.3)], but ruled out the presence of D/L-Thr in the hydrolysate of 1. Subsequently, acid hydrolysate of 1 was derivatized with FDAA and analyzed by RP-HPLC, which allowed for the assignment of D-configuration for the two allo-Thr residues. To facilitate configurational analysis of the remaining stereocenters in 1, X-ray crystallographic study was attempted. Unfortunately, under variety of conditions, theopapuamide (1) only yielded fine crystals that were unsuitable for X-ray diffraction studies.
In summary, theopapuamide (1) was cytotoxic against CEM-TART and HCT-116 cell lines with EC50 values of 0.5 μM and 0.9 μM respectively. Theopapuamide (1) contains a high degree of D-amino acids and N-methylated amino acids along with several other modified amino acid residues of non-ribosomal origin. Theopapuamide (1), neamphamide A and callipeltins share the same basic structural skeleton of a 7- or 8-residue ring moiety formed by cyclization through a β-hydroxyl group of a Thr residue and a poly-substituted side chain linked to the amino terminus of a 3,4-diMeGln residue. Since the 3,4-diMeGln residue is conserved in all of the above mentioned HIV-inibitory marine depsipeptides (callipeltin A, neamphamide A, and Papuamides AB), it has been postulated by Acevedo et al.5c that the 3,4-diMeGln residue may play a role in their biological activity. However, theopapuamide (1), which also carries the atypical 3,4-diMeGln residue, failed to show any appreciable HIV-activity. The major difference between theopapuamide (1) and related HIV-inhibitory peptides is the absence of a β-methoxytyrosine residue in 1. Therefore, this work indicates the potential significance of the β-methoxytyrosine residue for biological activity.
Experimental Section
General Experimental Procedures
Optical rotations were measured on a JASCO DIP-370 digital polarimeter. UV spectra were acquired in spectroscopy grade methanol using a Hewlett Packard 8452A diode array spectrophotometer. IR spectra were recorded on a JASCO FT/IR-420 spectrometer. NMR spectra were recorded on Varian INOVA spectrometers operating at 500/600 MHz and 125/150 MHz, respectively. Chemical shifts are reported in ppm and were referenced to residual acetonitrile (δC 118.69; δH 1.49) in CD3CN:water (4:1). Low-resolution mass spectra were obtained using a PE Sciex API III mass spectrometer operating in the ESI mode. High-resolution ESIMS analyses were performed on a Bruker (Billerica, MA) APEXII FTICR mass spectrometer equipped with an actively shielded 9.4 Tesla superconducting magnet (Magnex Scientific Ltd., UK), an external Bruker APOLLO ESI source, and a Synrad 50W CO2 CW laser.14 Nanoelectrospray was employed. Typically, a 5μL sample was loaded into the nanoelectrospray tip (New Objective, Woburn, MA) and a high voltage about 1000 V was applied between the nanoelectrospray tip and the capillary. Mass spectra were internally/externally calibrated using HP tuning mix. In the FTMS/MS experiments, the precursor ions were isolated using correlated sweep and then dissociated using a Sustained Off-Resonance Irradiation with Collision Induced Dissociation (SORI-CID). Automated amino acid analysis was performed on a Hitachi L8800 amino acid analyzer. Reversed-phase flash column chromatography was performed on BakerbondTM C18, 40 μm, prep LC packing. HPLC was performed on either a Beckman System Gold or an Agilent 1100 series instrument (diode array detector). For chiral HPLC, a Phenomenex Chirex phase 3126 (D), (250 × 4.6 mm) column was utilized. All solvents were HPLC grade, purchased from Fisher Scientific. Amino acid standards were purchased from Aldrich or Fisher Scientific.
Biological Material
The marine sponge Theonella swinhoei Gray (family Theonellidae) was collected by hand using SCUBA from Milne Bay, Papua New Guinea (S 10° 21.55, E 150° 44.70) in 2001. A voucher specimen of the sample PNG01-5-051 is held at the University of Utah.
Extraction and Isolation
The frozen sponge (400 g wet wt) was cut into small pieces and soaked in CH3CN-H2O (1:1 v/v, 3 × 1000 mL). The aqueous CH3CN extract was evaporated to dryness under vacuum, and the resulting crude material was applied onto a C18-column pre-equilibrated with aqueous CH3CN (~90% H2O). The column was eluted with a step gradient of CH3CN-H2O (0–100% CH3CN) containing 0.05% TFA. The 20% CH3CN eluate was chromatographed on Diaion HP-20 resin using an aqueous acetone solvent gradient of 20%–100% acetone to yield eight fractions. Fractions two through five were combined and further purified on HPLC (Phenomenex Luna 5 μm CN, 250 × 10 mm, flow rate 2.5 mL/min, detection 210 and 230 nm) using a gradient of 10%–100% CH3CN in H2O (0.05% TFA) over 30 min to yield four fractions. The third fraction was re-chromatographed, on a C8-HPLC column (ZORBAX Eclipse XDB 5 μm, 150 × 4.6 mm, flow rate 0.5 mL/min, detection 210 and 230 nm) using a gradient of 50%–100% MeOH in H2O (0.05% TFA). Pure theopapuamide (1) was obtained as an off-white amorphous solid (15.8 mg, 3.95 × 10−3 % yield wet wt).
Theopapuamide (1): off-white amorphous solid, [α]25D −3.0 (c 0.86, MeOH); UV (MeOH) λmax (log ε) 270 (3.28), 210 (4.12) nm; IR (film) νmax 3305, 2915, 2850, 1650, 1525, 1450, 1195, 1130 cm−1; 1H and 13C NMR data, see Table 1; HRESI-FTMS m/z 1558.90536 (M + H)+ (calculated for C69H124N17O23, 1558.90505).
Table 1.
1H and 13C NMR (500/125 MHz) Data for theopapuamide (1), in CD3CN:H2O (4:1)
| No | δC | δH | mult. (J in Hz) | HMBC (H→C) | wgnoesya,b |
|---|---|---|---|---|---|
| N-Methylleucine (NMeLeu) | |||||
| 1 | 171.5 | ||||
| 2 | 55.3 | 5.07 | dd (12.1,4.4) | 171.5, 30.8 | 2.81, 1.76, 1.52, 1.39, 0.76, 5.55 |
| 3 | 36.9 | 1.76 | m | 2.81, 5.07 | |
| 1.52 | 2.81, 5.07 | ||||
| 4 | 25.4 | 1.39 | m | 2.81, 5.07 | |
| 4-CH3 | 23.4 | 0.87 | d (6.6) | ||
| 5 | 21.3 | 0.76 | d (6.6) | 2.81, 5.07, 2.69 | |
| N-CH3 | 30.8 | 2.81 | s | 172.2, 55.3 | 0.76, 1.39, 1.76, 1.52, 8.18 |
| Asparagine (Asn) | |||||
| 1 | 172.2 | ||||
| 2 | 48.0 | 5.20 | m | 37.7 | 8.18 |
| 3 | 37.7 | 2.69 | dd (15.3,8.9) | 174.9, 48.0 | 6.27, 8.18 |
| 2.41 | dd (15.3,4.4) | 174.9, 172.2, 48.0 | 8.18 | ||
| 4 | 174.9 | ||||
| 4-NH2 | 6.27 | m | 2.69 | ||
| 7.04 | |||||
| NH | 8.18 | d (9.0) | 169.9 | 6.69, 5.20, 4.89, 2.81, 2.69, 2.41 | |
| β-Methoxyasparagine (β-OMeAsn) | |||||
| 1 | 169.9 | ||||
| 2 | 56.2 | 4.89 | dd (9.0,1.7) | 169.9, 80.5, 171.8 | 8.18, 4.41, 3.34 |
| 3 | 80.5 | 4.41 | d (1.7) | 173.4, 169.9, 60.3, 56.2 | 6.85, 7.19, 6.69, 4.89, 3.34 |
| 3-OCH3 | 60.3 | 3.34 | s | 80.5 | 4.89, 6.69, 5.19 |
| 4 | 173.4 | ||||
| 4-NH2 | 6.85 | m | 80.5 | 4.41 | |
| 7.19 | 4.41 | ||||
| NH | 6.69 | d (9.0) | 171.8 | 8.18, 2.88, 5.19, 4.89, 2.01 | |
| N-Methylglutamine (NMeGln) | |||||
| 1 | 171.8 | ||||
| 2 | 57.8 | 5.19 | m | 171.8 | 6.69, 2.88 |
| 3 | 22.9 | 2.32 | m | ||
| 1.78 | 2.88 | ||||
| 4 | 31.2 | 2.18 | m | 2.88 | |
| 2.01 | 22.9 | 6.91, 2.88 | |||
| 5 | 175.1 | ||||
| 5-NH2 | 6.42 | m | |||
| 6.91 | 2.01 | ||||
| N-CH3 | 31.8 | 2.88 | s | 177.7, 57.8 | 7.20, 5.19, 2.18, 6.69, 2.01, 1.78, 2.32 |
| Leucine (Leu) | |||||
| 1 | 177.7 | ||||
| 2 | 50.7 | 4.55 | dd (8.8, 3.0) | 7.20, 0.95 | |
| 3 | 39.0 | 2.00 | m | 7.20 | |
| 1.51 | 7.20 | ||||
| 4 | 25.5 | 1.83 | m | 7.20 | |
| 4-CH3 | 23.6 | 1.00 | d (6.6) | 39.0 | |
| 5 | 20.9 | 0.95 | d (6.6) | 39.0 | 2.88, 7.20, 4.55 |
| NH | 7.20 | 174.1, 50.7 | 8.00, 4.55, 1.83, 2.00, 2.88, 1.51 | ||
| Ornithine (Orn) | |||||
| 1 | 174.1 | ||||
| 2 | 52.0 | 4.54 | m | 8.00, 1.38, 1.97, 1.64,1.53 | |
| 3 | 27.7 | 1.97 | m | 8.00, 4.54 | |
| 1.38 | 8.00, 4.54 | ||||
| 4 | 24.4 | 1.64 | m | 8.00, 2.85 | |
| 1.53 | 8.00, 2.85 | ||||
| 5 | 40.1 | 2.85 | m | 1.53, 1.64 | |
| 5-NH2 | na | ||||
| NH | 8.00 | d (9.7) | 171.2 | 4.54, 1.53, 1.64, 8.25, 1.38, 7.20, 1.97, 3.92c | |
| Threonine-1 (Thr1) | |||||
| 1 | 171.2 | ||||
| 2 | 63.5 | 3.92 | m | 171.2 | 4.29, 1.24, 8.25, 8.00 |
| 3 | 67.2 | 4.29 | m | 171.2 | 3.92 |
| 3-OH | na | ||||
| 4 | 19.8 | 1.24 | d (6.6) | 67.2, 63.5 | 3.92 |
| NH | 8.25 | d (8.8) | 3.92, 8.89, 8.00 | ||
| Threonine-2 (Thr2) | |||||
| 1 | 172.5 | ||||
| 2 | 55.7 | 5.15 | d (3.3) | 8.89, 5.55 | |
| 3 | 71.2 | 5.55 | dd (6.6, 3.3) | 171.5 | 5.15, 1.17, 8.89, 5.07 |
| 4 | 14.3 | 1.17 | d (6.6) | 71.2, 55.7 | |
| NH | 8.89 | d (10.0) | 175.3 | 8.25, 5.55, 5.15, 4,22, 1.17 | |
| 3,4-Dimethylglutamine (3,4-diMeGln) | |||||
| 1 | 175.3 | ||||
| 2 | 58.7 | 4.22 | dd (9.9,5.5) | 37.1, 175.3 | 9.05, 8.89, 7.07, 6.55 |
| 3 | 37.1 | 2.17 | m | 9.05 | |
| 3-CH3 | 14.3 | 1.04 | d (6.7) | 58.7, 42.0, 37.1 | 2.64 |
| 4 | 42.0 | 2.64 | m | 9.05, 7.07, 2.17, 1.13, 1.04, 6.55 | |
| 4-CH3 | 14.7 | 1.13 | d (6.6) | 179.9, 42.0, 37.1 | 2.64 |
| 5 | 179.9 | ||||
| 5-NH2 | 7.07 | m | 2.64 | ||
| 6.55 | 2.64 | ||||
| NH | 9.05 | br | 176.2 | 4.22, 2.64, 2.17, 3.86 | |
| 4-Amino-5-methyl-2,3,5-trihydroxy-hexanoic acid (Amtha) | |||||
| 1 | 176.2 | ||||
| 2 | 71.2 | 4.12 | d (8.3) | 72.6, 176.2 | 1.23, 1.09, 3.86 |
| 2-OH | na | ||||
| 3 | 72.6 | 3.86 | dd (8.3,3.2) | 71.2, 56.4 | 9.05, 7.79, 4.12 |
| 3-OH | na | ||||
| 4 | 56.4 | 3.96 | m | 74.5 | 7.79, 1.09, 1.23 |
| 4-NH | 7.79 | d (10.0) | 171.3 | 3.86, 4.68 | |
| 5 | 74.5 | ||||
| 5-CH3 | 27.1 | 1.09 | brs | 74.5, 56.4 | 4.12, 3.96 |
| 5-OH | na | ||||
| 6 | 27.6 | 1.23 | brs | 74.5, 56.4 | 3.96, 4.12 |
| 2,3-Diaminopropionic acid (Dpr) | |||||
| 1 | 171.3 | ||||
| 2 | 52.2 | 4.68 | brdd (7.1,6.9) | 41.0 | 7.79, 8.39 |
| 3 | 41.0 | 3.48 | m | 52.2 | 4.68, 3.31 |
| 3.31 | 52.2 | 4.68 | |||
| 3-NH2 | na | ||||
| NH | 8.39 | brd (8.0) | 2.57, 4.68 | ||
| 3-Hydroxy-2,4,6-trimethyl-octanoic acid (Htoa) | |||||
| 1 | 179.6 | ||||
| 2 | 45.1 | 2.57 | m | 13.8, 179.6 | 8.39, 0.99, 0.90 |
| 2-CH3 | 13.8 | 0.99 | d (6.7) | 179.6, 79.4, 45.1 | 1.72, 3.51, 2.57 |
| 3 | 79.4 | 3.51 | brd (10.5, 1.4) | 13.8, 17.6 | 2.57, 1.72, 0.90, 0.99 |
| 3-OH | na | ||||
| 4 | 32.2 | 1.72 | m | 3.51, 1.37 | |
| 4- CH3 | 17.6 | 0.90 | d (6.6) | 79.4, 32.2 | 2.57 |
| 5 | 36.2 | 1.18 | m | 17.6 | 2.57, 1.72c |
| 0.99 | 17.6 | 1.37c | |||
| 6 | 32.2 | 1.37 | m | 30.5, 11.4, 19.4 | 3.51c, 1.72c |
| 6- CH3 | 19.4 | 0.80 | d (6.7) | 30.5, 32.2 | 1.37c, 1.72c |
| 7 | 30.5 | 1.28 | m | 11.4, 19.4, 32.2 | |
| 1.09 | 11.4, 19.4, 32.2 | ||||
| 8 | 11.4 | 0.83 | d (7.3) | 30.5, 32.2 | |
2D [1H,1H] NOESY with watergate water suppression, using a 200 ms mixing time.
Data recorded at 600 MHz.
Correlation based on a WATERGATE-ROESY experiment using a 50 ms mixing time, at 600 MHz. na: Not assigned.
Base Hydrolysis of Theopapuamide (1)
Approximately 0.5 mg of 1 was treated with 1N KOH (200 μL) at rt for 2 h. The reaction mixture was diluted by adding ice, neutralized with 0.5 N HCl and extracted with n-BuOH (2 × 3 mL). The n-BuOH layer was evaporated to dryness and an aliquot was analyzed by LRESIMS [m/z 1576.9 (M + H)+].
Determination of Absolute Configuration
(a) Acid Hydrolysis
Theopapuamide (1), 100 μg, was dissolved in degassed 6N HCl (250 μL) and heated in a sealed glass vial at 110 °C for 12 h. The solvent was removed in vacuo and the residue was analyzed by HPLC.
(b) Chiral HPLC Analysis
The acid hydrolysate of 1 (aliquot of 10 μL) was analyzed by chiral HPLC on a Phenomenex D-penicillamine column [Chirex phase 3126 (D), (250 × 4.6 mm)]. The identity of amino acids of the acid hydrolysate were confirmed by comparison of their retention times with those of authentic standards using HPLC under the following conditions: mobile phase, iPrOH-2mM CuSO4 (5:95); flow rate, 1.0 mL/min; detection, UV 254 nm; retention times of the standards (min): L-NMeGlu (52.3), D-NMeGlu (24.1), L-Asp (20.0), D-Asp (26.9), L-Leu (18.5), D-Leu (29.0), L-NMeLeu (15.0), L-Dpr (5.8), D-Dpr (6.5), D-Orn (3.7), L-Orn (3.5), D/L-allo-Thr (6.3), D-Thr (5.8), L-Thr (5.2); retention times of the component amino acids of the hydrolysate (min): L-NMeGlu (52.4), D-Asp (26.8), L-Leu (18.5), L-NMeLeu (14.9), D-Dpr (6.5), D-Orn (3.7).
(c) HPLC Analysis of Marfey’s (L-FDAA) Derivatives
To the peptide hydrolysate (20 μg in 20 μL H2O) was added 6% triethylamine in H2O (10 μL) followed by a 1% solution of 1-fluoro-2,4-dinitrophenyl-5-alanine amide in acetone (L-FDAA, 20 μL). The mixture was heated at 50 °C in an oil bath for 1 h, quenched with 5% acetic acid (10 μL) and dried in vacuo. The residue was dissolved in H2O (40 μL) and aliquots (5–10 μL) were analyzed by C18-HPLC. The D- and L-amino acid standards (1 mg/mL, 50 μL) were derivatized in a similar manner, and the retention times were compared with those of the component amino acids of the peptide hydrolysate. C18-HPLC analysis conditions: column, Phenomenex μm, 250 × 4.6 mm; mobile phase, a gradient of 10%–50% CH3CN in H2O (0.05% TFA) over 30 min; flow rate, 1.0 mL/min; detection, UV 340 nm; Retention times of authentic L-FDAA-allo-Thr (min): L-allo-Thr (19.8) and D-allo-Thr (20.6); retention times of the L-FDAA-allo-Thr of the hydrolysate (min): D-allo-Thr (20.6).
Assay for T cell viability
CEM-TART cells were maintained in 80% RPMI medium, 20% fetal bovine serum supplemented with 100 units/mL penicillin, 100 μg/mL streptomycin and 0.25 μg/mL amphotericin B in a humidified incubator at 37 °C, 5% CO2. 200 μL of cells were seeded in 96-well microtiter plates at a final concentration of 30,000 cells/well. Cells were administered 3.3 ng/mL-5 μg/mL (dissolved in DMSO) of compound, each dose in quadruplicate. After 72 hours cells, 11 μL MTT (5 mg/mL) was added to each well, and plates were incubated for an additional 4 hours. The medium was aspirated, 100 μL of DMSO added to the cells to solubilize purple formazan product and the absorbance read at 570 nm using a plate reader (Bio-Rad). Average absorbance for each set of compound-treated wells was compared to the average absorbance of the control wells to determine the fractional survival at any particular drug concentration. The effective concentration 50 (EC50) is defined as the drug concentration that yielded a fractional survival of 50%. EC50 values reported are the average of three independentexperiments and were determined using GraphPad Prism. The following reagent was obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: CEM-TART from Drs. Herbert Chen, Terence Boyle, Michael Malim, Bryan Cullen, and H. Kim Lyerly.
Cell culture and MTT assay.16
The cancer cell line HCT-116 (human colon tumor) was grown at 37°C, 5% CO2 in McCoy’s 5a medium containing 10% fetal bovine serum, penicillin (50 IU/mL), streptomycin (50 μg/mL) and 2 mM L-glutamine. Cells were plated at a density of 5000 cells/well in a 96-well plate. Twenty-four hours after plating, they were exposed to medium containing compound at a concentration of 0.1 μM to 100 μM for 48 h at 37°C. After exposure of cells to compounds, 100 μL of fresh culture medium containing MTT at a final concentration of 0.3 mg/mL was added to each well and incubated for 3 h at 37°C. Formazan crystals were solubilized in 100 μL of DMSO. The absorbance of each well was measured by a microplate reader (Multi-skan labsystems) at 570 nm. The percentage cytotoxicity was calculated by comparison of the A570 reading from treated versus control cells.
Supplementary Material
1H and 13C NMR spectra and selected 2D-spectra (gHSQC, DQFCOSY, 15N-HSQC, WATERGATE-NOESY, WATERGATE-ROESY and 2D-TOCSY) of theopapuamide (1). This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
The authors wish to acknowledge the government of Papua New Guinea and PNG BioNET for permits to collect the sponge. We thank P. Krishna, University of Utah Mass Spectrometry and Proteomics Core Facility, for performing the LRESIMS experiments. The following NIH and NSF grants funded NMR instrumentation, RR06262, RR13030, DBI-0002806. This project was funded by NIH grant CA 36622.
References
- 1.(a) Bewley CA, Faulkner DJ. Angew Chem Int Ed. 1998;37:2162–2178. doi: 10.1002/(SICI)1521-3773(19980904)37:16<2162::AID-ANIE2162>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]; (b) Fusetani N, Matsunaga S. Chem Rev. 1993;93:1793–1806. [Google Scholar]
- 2.Zampella A, D’Auria MV, Paloma LG, Casapullo A, Minale L, Debitus C, Henin Y. J Am Chem Soc. 1996;118:6202–6209. [Google Scholar]
- 3.Oku N, Gustafson KR, Cartner LK, Wilson JA, Shigematsu N, Hess S, Pannell LK, Boyd MR, McMahon JB. J Nat Prod. 2004;67:1407–1411. doi: 10.1021/np040003f. [DOI] [PubMed] [Google Scholar]
- 4.Ford PW, Gustafson KR, McKee TC, Shigematsu N, Maurizi LK, Pannell LK, Williams de Silva ED, Lassota P, Allen TM, Soest RV, Andersen RJ, Boyd MR. J Am Chem Soc. 1999;121:5899–5909. [Google Scholar]
- 5.(a) Okamoto N, Hara O, Makino K, Hamada Y. Tetrahedron Assym. 2001;12:1353–1358. [Google Scholar]; (b) Liang B, Carroll J, Joullié MM. Org Lett. 2000;2:4157–4160. doi: 10.1021/ol006679t. [DOI] [PubMed] [Google Scholar]; (c) Acevedo CM, Kogut EF, Lipton MA.Tetrahedron 2001576353–6359.18159221 [Google Scholar]; (d) Çalimsiz S, Lipton MA. J Org Chem. 2005;70:6218–6221. doi: 10.1021/jo050518r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Thoen JC, Ramos M, Lipton MA. Org Lett. 2002;4:4455–4458. doi: 10.1021/ol0269852. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Zampella A, D’Orsi R, Sepe V, Casapullo A, Monti MC, D’Auria MV. Org Lett. 2005;7:3585–3588. doi: 10.1021/ol0513600. [DOI] [PubMed] [Google Scholar]; (g) Okamoto N, Hara O, Makino K, Hamada Y. J Org Chem. 2002;67:9210–9215. doi: 10.1021/jo0258352. [DOI] [PubMed] [Google Scholar]
- 6.Ratnayake AS, Davis RA, Harper MK, Veltri CA, Andjelic CD, Barrows LR, Ireland CM. J Nat Prod. 2005;68:104–107. doi: 10.1021/np049721s. [DOI] [PubMed] [Google Scholar]
- 7.(a) Köck M, Kessler H, Seebach D, Thaler A. J Am Chem Soc. 1992;114:2676–2686. [Google Scholar]; (b) Kessler H, Hehlein W, Schuck R. J Am Chem Soc. 1982;104:4534–4540. [Google Scholar]
- 8.2D [1H,1H] TOCSY using a z-DIPSI-2 isotropic mixing scheme. Cavanagh J, Rance M. J Magn Reson. 1992;96:670–678.
- 9.Standard amino acid analysis, 5.7 N HCl, 110 °C, 16 h; concentrations of the amino acids were quantified using a Hitachi model L8800 analyzer.
- 10.2D [1H,1H] NOESY with watergate water suppression. Piotto M, Saudek V, Sklenáà V. J Biomol NMR. 1992;2:661–665. doi: 10.1007/BF02192855.
- 11.(a) Bodo B, Hebrard P, Molho L, Molho D. Tetrahedron Lett. 1973;18:1631–1634. [Google Scholar]; (b) White JD, Johnson AT. J Org Chem. 1994;59:3347–3358. [Google Scholar]
- 12.(a) Edman P. Acta Chem Scand. 1950;4:283–293. [Google Scholar]; (b) Yarwood A. Chapter 6. In: Findlay JBC, Geisow MJ, editors. Protein Sequencing, a practical approach. IRL Press; Oxford: 1989. pp. 119–145. [Google Scholar]
- 13.Trypsin digestion, 37 °C, pH 7.9, 2 days; Thermolysin digestion, 37 °C, pH 8.1, 1–2 days; Pepsin digestion, 37 °C, pH 1.5, 2 days.
- 14.(a) McDonald LA, Barbieri LR, Carter GT, Kruppa G, Feng X, Lotvin JA, Siegel MM. Anal Chem. 2003;75:2730–2739. doi: 10.1021/ac0264731. [DOI] [PubMed] [Google Scholar]; (b) Palmblad M, Håkansson K, Håkansson P, Feng X, Cooper HJ, Giannakopulos AE, Green PS, Derrick PJ. Eur J Mass Spectrom. 2000;6:267–275. [Google Scholar]
- 15.(a) Bhushan R, Brückner H. Amino Acids. 2004;27:231–247. doi: 10.1007/s00726-004-0118-0. [DOI] [PubMed] [Google Scholar]; (b) B’Hymer C, Bayon MM, Caruso JA. J Sep Sci. 2003;26:7–19. [Google Scholar]; (c) Hess S, Gustafson KR, Milanowski DJ, Alvira E, Lipton MA, Pannell LK. J Chromatogr A. 2004;1035:211–219. doi: 10.1016/j.chroma.2004.02.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1H and 13C NMR spectra and selected 2D-spectra (gHSQC, DQFCOSY, 15N-HSQC, WATERGATE-NOESY, WATERGATE-ROESY and 2D-TOCSY) of theopapuamide (1). This material is available free of charge via the Internet at http://pubs.acs.org.