

Abstract
Background
Generation of physiologically active vascular beds by delivery of combinations of growth factors offers promise for vascular gene therapy.
Methods and Results
In a mesenteric model of physiological angiogenesis, combining endothelial nitric oxide synthase (eNOS) (and hence NO production) with VEGF and angiopoietin-1 overexpression resulted in a more functional vascular phenotype than growth factor administration alone. eNOS gene delivery upregulated eNOS, VEGF, and Ang-1 to similar levels as gene transfer with VEGF or Ang-1. eNOS overexpression resulted in neovascularization to a similar extent as VEGF and Ang-1 combined, but not by sprouting angiogenesis. Whereas combining Ang-1 and VEGF increased both exchange vessels and conduit vessels, neither growth factor nor eNOS alone resulted in vessels with smooth muscle cell (SMC) coverage. In contrast, combining all three generated microvessels with SMCs (arteriolar genesis) and further increased functional vessels. Use of a vasodilator, prazosin, in combination with Ang1 and VEGF, but not alone, also generated SMC-positive vessels.
Conclusion
Coexpression of eNOS, VEGF, and Ang-1 results in a more mature vascularization of connective tissue, and generates new arterioles as well as new capillaries, and provides a more physiological therapeutic approach than single growth factor administration, by combining hemodynamic forces with growth factors.
Keywords: angiogenesis, arteriogenesis, VEGF, Ang-1, eNOS, pericyte, vascular smooth muscle
Therapeutic angiogenesis remains a promising tool in the treatment of ischemic diseases, but as yet clinical trials remain largely unsuccessful.1 For ultimate efficacy of proangiogenic gene therapy, the neovessel bed generated must be functional, stable, and homeostatic. This can be achieved not only by the generation of capillaries, but also by the recruitment of pericytes and vascular smooth muscle cells (VSMCs) to the vessels, so that the vascular bed can regulate perfusion. Current treatment relying on the overexpression of individual growth factors has not generated the growth of a complete vascular tree.2 A contributing factor to this could be the focus on single growth factors, which are unable to stimulate all components of vessel growth.
Combining different growth factors that stimulate different parameters of angiogenic growth results in an additive3 or even synergistic effect on angiogenesis.4 Although a combination of the destabilizing vascular endothelial growth factor-A (VEGF) and stabilizing angiopoietin-1 (Ang-1) in the adult rat mesentery induced a greater degree of functionality than either factor alone, the vessels formed were still of capillary phenotype (ie, microvessels with no VSMC coverage). Generating arterioles that contain smooth muscle cells (arteriolar genesis) is a requirement for revascularization of poorly vascularized tissues if the revascularized tissues are to be able to regulate their perfusion through normal physiological feedback mechanisms.
Increased blood flow, for instance by inducing vasodilatation with prazosin, is known to induce angiogenesis in skeletal muscle5,6 and increased shear stress is a critical mediator of vessel wall thickness and composition.7-11 A reduction in sprouting angiogenesis in favor of capillary splitting coincides with the highest blood flow in experimental models of hindlimb ischemia.11 Taken together, increased blood flow initiates both vessel growth and remodeling of angiogenic vessels.
Nitric oxide is a potent vasodilator. In the endothelium, it is produced by catalysis of l-arginine by endothelial nitric oxide synthase (eNOS), which is a calcium dependent enzyme involved in growth factor mediated angiogenesis.12 We aimed to test the hypothesis that angiogenic blood vessels could be remodeled by increased nitric oxide, presumably acting as a vasodilator to result in arteriolar genesis (formation of muscular arterioles) as well as angiogenesis (formation of new blood vessels, mainly capillaries). Using the mesenteric angiogenesis assay, a detailed and specific analysis of the microvessel phenotype was produced and a measurement of vessel function was made. To locally increase blood flow through the vessels around the mesenteric panel, we locally overexpressed eNOS to generate a continuous gradient of NO.
Therefore, we tested the hypothesis that by inducing angiogenesis in a nonischemic and well-validated model, overexpression of eNOS will induce both angiogenic growth and the recruitment of VSMC to the neovessels.
Materials and Methods
In brief, we used the adult rat mesenteric model of angiogenesis, as previously described.3,13 In brief (see online supplement for full methods at http://atvb.ahajournals.org), adult male rats (350g) were anesthetized and a laparotomy performed under sterile conditions. The mesenteric panels were imaged intravitally. Adenoviruses expressing eGFP, VEGF, Ang-1, or eNOS were injected into the mesenteric fat pad. In experiments where prazosin was used, prazosin was administered at 50 mg/L in the rat’s drinking water. The animal was sutured and allowed to recover. Seven days later the same mesenteric panel was located and imaged. It was then fixed in vivo and staining for angiogenic markers was carried out. Imaging of angiogenic blood vessels was carried out by immunofluorescence and confocal microscopy.
Confirmation of viral transfection was carried out by ELISA from mesenteric fat panels. To support in vivo data, primary human adipocytes were cultured and infected with the same adenoviruses as described previously. ELISA was used to confirm transfection and to measure changes in other angiogenic cytokine levels.
Statistical Analysis
All data presented as mean±SEM, and all groups n=5 unless explicitly stated. P<0.05 was considered statistically significant. Data were analyzed by ANOVA unless stated with posthoc Neuman Keuls tests when ANOVA showed an overall P<0.05.
Results
Increased Protein Levels After Adenovirus Injections
Consistent with other reports, injection of multiple adenoviruses into the mesenteric fat pad results in overexpression of the virus’s respective proteins as a percentage of total protein extracted 3 days post-infection.3,13 Treatment groups containing Ad-Ang-1 and Ad-eNOS all increased expression of Ang-1 and eNOS compared with control (see supplemental Table I; P<0.001). VEGF protein was also increased by Ad-eNOS, Ad-Ang-1+Ad-VEGF, and Ad-Ang-1+VEGF+eNOS. No increase in eNOS levels were observed after administration of any combination of growth factors without Ad-eNOS being present. However, eNOS overexpression did induce the expression of the other two critical determinants of blood vessel growth—VEGF and Ang-1. To determine whether this was attributable to overexpression in adipocytes or for other reasons, cultured adipocytes were transfected with GFP or eNOS virus and expression compared. Whereas eNOS virus transfection upregulated expression of eNOS protein (see supplemental Table I), doubled nitrite levels, and increased VEGF levels 46±5% (P<0.05 for each compared with control, t tests), it did not significantly affect Ang-1 expression.
ENOS Overexpression Increases Functional Vessel Area
To determine whether the increased protein levels in the mesenteric fat pad resulted in a functional change in vessel perfusion, the total area of patent vessels was measured (functional vessel area, FVA, Figure 1A). Overexpression of eGFP did not increase the FVA on day 1 compared with day 7 (Figure 1B, P=0.58) but Ang-1+VEGF (Figure 1B, P=0.002), eNOS (Figure 1B, P=0.03) and eNOS+Ang-1+VEGF (Figure 1B, P=0.0003) did significantly increase FVA. Furthermore, eNOS+Ang-1+VEGF induced the greatest response compared with eGFP (Figure 1C, P<0.001), Ang-1+VEGF (Figure 1C, P<0.001), and eNOS (Figure, 1C, P<0.001). None of the other proangiogenic groups were significantly different from each other (Figure 1C, P>0.05). In summary, overexpression of eNOS, Ang-1, or VEGF (or together) stimulates an increase in the functional vessels available for flowing blood.
Figure 1.

Intravital imaging of the rat mesentery demonstrates increased vessel area after growth factor or hemodynamic factor overexpression but not control (A, B). Comparison of the increase in FVA reveals that a combination of eNOS+Ang-1+VEGF induced the greatest response.
ENOS Overexpression Remodelled Sprouting Vessels
To determine whether the increase in FVA induced by eNOS was a result of higher blood flow (attribtuable to subsequent vasodilatation induced by nitric oxide), we investigated the phenotype of the vessels produced. Confocal microscopy of the mesenteric panel stained for isolectin IB4, antibodies to NG2 and αSMA enabled detailed analysis of the endothelial and periendothelial composition of the vessel (Figure 2). All treatments that increased the FVA also increased the microvessel density compared with control (Ang-1+VEGF P<0.05, eNOS, eNOS+Ang-1+VEGF P<0.01; Figure 3A). None of the angiogenic groups differed from each other significantly. In addition, all the angiogenic groups increased the proliferating endothelial cell (PEC) density (Figure 3B) compared with control (P<0.01), but not when compared with each other (P>0.05). Thus all three treatments resulted in an angiogenic response (increased number of capillaries and proliferation of endothelial cells).
Figure 2.
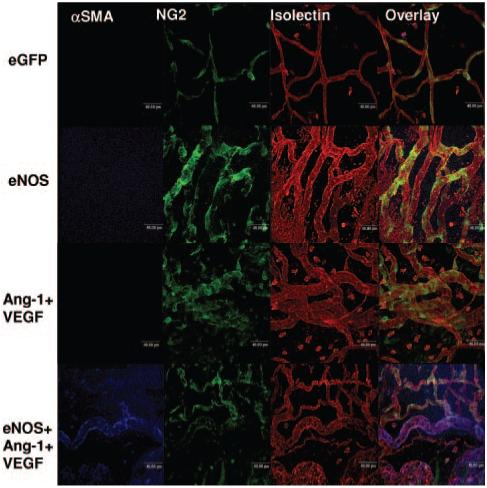
Confocal images after control or angiogenic treatment. eGFP vessels do not sprout, and pericytes wrapped around the vessel. eNOS induces radial enlargement of the vessel and increases vessel number and branch point density. Ang-1+VEGF induced a similar phenotype. eNOS+Ang-1+VEGF was the only group to increase VSMC coverage.
Figure 3.
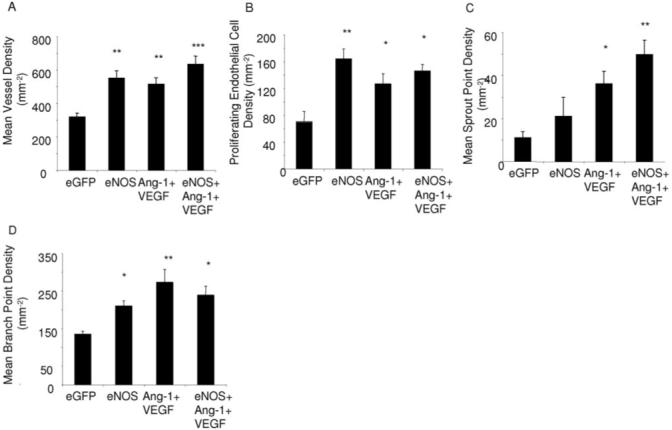
Analysis of confocal images. Growth factor and eNOS overexpression induced an angiogenic response, as indicated by an increased density of blood vessels (A) and an increase in endothelial proliferation (B). Increased blood flow induced by eNOS increased sprout point density (C) and branch point density (D).
Different angiogenic agents have previously been demonstrated to induce different vessel branching patterns, which are likely to reflect the presence or absence of sprouting angiogenesis.3,14-16 Overexpression of eNOS alone did not significantly increase sprouting in the microvasculature (figure 3C). Although overexpression of eNOS+Ang-1+VEGF induced a significantly higher sprout point density compared with control (Figure 3C, P<0.01), this sprouting was still significantly lower than the sprouting seen after VEGF overexpression (demonstrated to induce vessel sprouting) alone in the adult rat mesentery,3,13 and was no different to VEGF and Ang-1 in the absence of eNOS. Although sprouting did not appear to be stimulated by eNOS overexpression, there was also an increase in branch point formation after eNOS administration and eNOS+Ang-1-VEGF compared with control (P<0.01) and this was no different from Ang-1+VEGF. Taken together, despite VEGF and Ang-1 levels being approximately equal in all angiogenic groups measured, there appeared to be differences in how blood vessels were forming. Ang-1+VEGF appeared to remodel sprouts into vessels and eNOS overexpression appeared to induce vessel growth through a sprouting independent mechanism, as sprouting was not increased, but branch formation was.
eNOS Overexpression Induced Muscle Coated Vessel Formation
Increased blood flow is considered as a leading determinant in the arteriogenic mechanisms that underlie collateral formation and also arterialization of microvessels.17 The process of arteriogenesis results in a change in the composition of the periendothelial wall. The mesenteric connective tissue panels are perfused with small order capillaries, generally <20 μm diameter, and their periendothelium tends to be pericyte, but not smooth muscle.3 After treatment with Ad-eGFP, Ad-eNOS, or combined Ang-1 and VEGF treatment, the vessels of the mesentery remained free of smooth muscle cells. In stark contrast however, administration of combined eNOS, Ang-1, and VEGF induced a highly significant coverage of vessels with smooth muscle cells (Figures 2 and 4A; P<0.001 compared with the other groups). To determine whether the VSMC coverage was restricted to larger diameter microvessels, we investigated the density of VSMCs on conduit microvessels (16 to 35 μm diameter) and on exchange capillaries (<16 μm diameter). Although there was no significant difference between the 2 groups (P>0.05; Figure 4B) there was a trend for VSMCs to be present on the larger vessels. Approximately 30% of each vessel is covered by pericytes (fractional pericyte area [FPA]) in control vessels and this was not significantly altered after eNOS or Ang-1+VEGF treatment (Figure 4C). The pericyte coverage was significantly reduced, however, after eNOS+Ang-1+VEGF treatment compared with all other groups (P<0.05; Figure 4C). This reduced pericyte coverage matched the increase in coverage by aSMA resulting in an approximately equal mural cell coverage of blood vessels after triple adenovirus treatment (Figure 4C). There was no correlation, however, between fractional pericyte area and fractional smooth muscle cell area (Figure 4D). Although VSMCs were only present after Ad-eNOS+Ang-1+VEGF, eNOS treatment alone induced an equal frequency distribution of vessel diameters compared with control (Figure 4E; P>0.05). All other treatment groups resulted in a shift to the right in distribution, indicating a greater number of larger diameter vessels (Figure 4D; P<0.001 versus eGFP). This suggests that eNOS overexpression was sufficient to generate a “normal” distribution of vessels.
Figure 4.

Total periendothelium complement to the vessel does not change, just its composition (A, C). There is a greater degree of coverage in larger vessels (B). VSMC presence does not correspond with lack of pericyte support (D). eNOS overexpression produced a distribution equal to control treated vessels (E).
Muscle Coated Vessel Formation Was Also Induced by Combined Growth Factor and Vasodilatation
To determine whether the induction of a mature vasculature that resulted in higher blood flow and more new vessel formation was attributable to the vasodilatorary effects of eNOS transduction, we repeated the experiment using a pharmacological vasodilator, prazosin. Rats were treated with eGFP virus or both Ang1 and VEGF as above, and prazosin (50 mg/L) was administered in drinking water for the duration of the experiment.18 Rats treated with prazosin and eGFP virus increased functional vessel area to 349±43% compared with day1. This was significantly greater than eGFP (P<0.0043), but significantly less than eNOS alone (P>0.001), and indicates that the prazosin resulted in an increase in blood flow, presumably attributable to vasodilatation. However, it did not increase FVA in conjunction with Ang-1 and VEGF compared with the 2 growth factors alone (P>0.05), in contrast with eNOS (Figure 5A). Estimation of angiogenic parameters, such as sprout point density, indicated that there was no significant angiogenic response with prazosin alone (Figure 5B; P>0.05). Moreover, it did not enhance FVA when combined with VEGF and Ang1 compared with the two growth factors alone (P>0.05). This was also true for branch point density, vessel density, and pericyte coverage (P>0.05) In contrast with this lack of angiogenic response, however, prazosin did induce smooth muscle cell coverage of the microvessels when combined with Ang1 and VEGF (although not alone, Figure 5C). This response was predominantly limited to the larger (>16 μm) diameter vessels (Figure 5D). Thus arteriolar genesis, but not angiogenesis, in this model was enhanced by a vasodilator in the presence of VEGF and Ang1.
Figure 5.
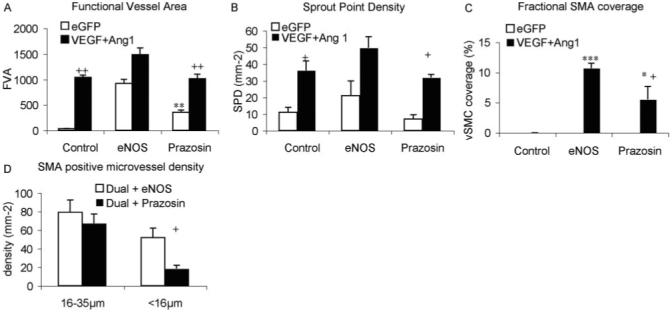
Prazosin treatment caused vasodilation, but did not enhance the effect of Ang1 and VEGF (A) or increase angiogenesis (B) but did result in smooth muscle cell coverage in the presence of Ang1 and VEGF (C), attributable predominantly to an effect on large conduit vessels (D).
Discussion
This study determined that overexpression of multiple growth factors, in addition to the overexpression of a vasodilatating agent, resulted in both an angiogenic response (as determined by an increased number of capillaries) and an arteriogenic response as demonstrated by an increase in VSMC covered vessels.
Increased Blood Flow Induces a Different Angiogenic Phenotype Despite Approximately Equal Protein Levels Compared With Ang-1+VEGF
Injection of Ad-VEGF and Ad-Ang-1 into the mesenteric fat pad increased levels of VEGF and Ang-1, respectively, as has previously been determined.3 Furthermore, Ad-eNOS injection resulted in increased levels of eNOS protein, which is qualitatively similar to others in multiple tissue types.19-21 Of interest, however, Ad-eNOS also increased VEGF expression both in the fat pad and in cultured adipocytes. This could be a result of increased HIF-1α stabilization22,23 or via increased heme-oxygenase levels,24 both shown to upregulate VEGF in response to increased NO. In addition, Ang-1 levels were also increased in the fat pad, but not in adipocytes. Ang-1 upregulation has so far only been reported in isolated cerebral endothelial cells after NO donor treatment,25,26 so it is still unclear which cells in the fat pad provide the source of Ang-1. Furthermore, the phenotype of the vessels produced by eNOS overexpression alone largely represent those produced by a overexpression of Ang-1+VEGF coincidentally.3 The stabilizing action of NO during angiogenesis (in our setting produced by eNOS) has been previously demonstrated in NO producing tumor cell line, and also in eNOS-deficient engineered blood vessels,27 which reported enhanced perfusion and association between the EC and the periendothelium. Consequently, we suggest that in the setting of angiogenesis into a largely avascular area, increased eNOS levels (and therefore increased NO levels) are likely to mediate vessel remodeling and stabilization via VEGF and Ang-1 production. Other studies have reported that EC stimulated with VEGF result in increased eNOS levels.28-30 We did not observe such findings, possibly as we are primarily transfecting adipocytes (which do not appear to express eNOS31), or the mechanisms regulating eNOS expression are not common to both EC and adipocytes. It will be interesting to elucidate the effect of VEGF antagonists on the eNOS-mediated neovascularization, as it has been demonstrated that NO-induced angiogenesis can be inhibited by the use of VEGF-TRAP in models of skeletal muscle angiogenesis.32
Increased Blood Flow Remodels Angiogenic Vessels
eNOS overexpression increased branch point density but did not increase sprout point density. Given that increased branching in an absence of increased sprouting is evidence of nonsprouting angiogenesis, mediated by increased blood flow,5,33,34 we therefore provide indirect evidence of intussusceptive vessel growth or vessel splitting. Increasing blood flow is known to be a critical driving force in the recruitment of contractile mural cells to the vessel wall of preexisting microvessels.17,33,35,36 eNOS is a critical mediator of vascular hemostasis12 and produces NO which is a potent vasodilator,19 which leads to increased blood flow and a consequent hyperaemia induced angiogenesis.6,18,33,37 Furthermore, eNOS has recently been demonstrated to be a critical component in the induction of arteriogenesis as eNOS-deficient mice were able to mount an angiogenic response, but failed to recruit VSMCs into the vessels, suggesting that remodeling of vessels into larger conduit vessels was not possible.38 Using the same angiogenesis model it was recently demonstrated that the mesenteric microcirculation of rats of this age and strain comprised pericyte coated vessels, with no VSMCs present,3 However, after eNOS+Ang-1+VEGF overexpression we show here that there was a significant increase in the coverage of VSMCs. Thus eNOS, in the presence of VEGF and Ang-1 expression, was able to generate new arterioles, (arteriolar genesis). Thus we demonstrate a mechanism by which arteriole formation from preexisting capillaries can occur.
It is not clear from where the VSMCs have originated. Increased blood flow is known to stimulate proliferation of fibroblasts in the mesenchyme,33 many of which had assumed a position within the vessel wall. Furthermore, enhanced shear stress increased arteriolar development, with increased VSMCs present on smaller order vessels.33 It is also possible that VSMC progenitors are present in the circulating blood—bone marrow derived circulating progenitor have been found to be increased in the periendothelium of tumor vessels.39,40 Furthermore, in models of low hypoxia, tissue resident progenitor cells appear to migrate to sites of active angiogenesis, again within the periendothelium but not expressing periendothelial markers. A long held view is that pericytes might develop into VSMCs.41 Current immunohistochemical data supports pericytes and VSMCs as not being continuous. In the tracheal and retinal microcirculations, larger macrovessels express αSMA and NG2, but NG2 is only present in the microvessels.42,43 Taken together, in studies whereby both NG2 and αSMA have been used, different cell types are identified and therefore, it is likely there are different cell populations. We saw vessels with both pericytes and VSMC, therefore we do not have evidence supporting a trans-differentiation mechanism.
Increased blood flow via prazosin (and consequently α1-adrenergic receptor blockade) leads not only to increased shear stress but also increased wall stress. In skeletal muscle beds, the expression of mature VSMCs (myosin heavy chain positive) and immature and mature VSMCs (αSMA positive) is different between terminal arterioles and higher order arterioles, and this changes through development.36 The authors interpreted the different expression patterns to demonstrate that VSMCs might originate from the higher order vessels and migrate along the vascular tree. As proof of principle, increasing blood flow with prazosin44 or via skeletal muscle extirpation33 led to quantitatively similar findings. Increasing blood flow through ligation of arterioles in the mesentery, leading to increased hemodynamic forces in the downstream and connecting vessels also leads to an increase in proliferation and presence of VSMCs.10 Interestingly, prazosin in this model led to increased muscle cell investment without contribution to angiogenesis, further suggesting that additional pathways are regulated by eNOS, which have an effect on vessel remodeling and are not limited specifically to the vasodilatatory actions of NO.
In summary we show here that coexpression of an angiogenic molecule (VEGF), an endothelial stabilizing factor (Ang-1), and the artificial increase in blood flow, mediated through NO (eNOS) results in a more robust and mature vascularization of connective tissue, and generates new arterioles as well as new capillaries. This therefore provides a mechanism for generating new vascular beds that can regulate their flow and provide homeostatic feedback for perfusion regulation. Current therapy has largely failed to generate the growth of a whole vascular tree, and we demonstrate that the combination of different aspects of vascular growth can induce such vessel remodeling. Methodologies to do so surrounding an ischemic environment may therefore lend themselves to therapeutic relevance.
Supplementary Material
Acknowledgments
Sources of Funding
This work was supported by the British Heart Foundation (FS04/22, to A.V.B.; FS06/038, to O.A.S.; and BS06/005 and BB2000003, to D.O.B.).
Footnotes
Disclosures
None.
References
- 1.Yla-Herttuala S, Markkanen JE, Rissanen TT. Gene therapy for ischemic cardiovascular diseases: some lessons learned from the first clinical trials. Trends Cardiovasc Med. 2004;14:295–300. doi: 10.1016/j.tcm.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 2.Cao Y, Hong A, Schulten H, Post MJ. Update on therapeutic neovascularization. Cardiovasc Res. 2005;65:639–648. doi: 10.1016/j.cardiores.2004.11.020. [DOI] [PubMed] [Google Scholar]
- 3.Benest AV, Salmon AH, Wang W, Glover CP, Uney J, Harper SJ, Bates DO. VEGF and angiopoietin-1 stimulate different angiogenic phenotypes that combine to enhance functional neovascularization in adult tissue. Microcirculation. 2006;13:423–437. doi: 10.1080/10739680600775940. [DOI] [PubMed] [Google Scholar]
- 4.Cao R, Brakenhielm E, Pawliuk R, Wariaro D, Post MJ, Wahlberg E, Leboulch P, Cao Y. Angiogenic synergism, vascular stability and improvement of hind-limb ischemia by a combination of PDGF-BB and FGF-2. Nat Med. 2003;9:604–613. doi: 10.1038/nm848. [DOI] [PubMed] [Google Scholar]
- 5.Egginton S, Zhou AL, Brown MD, Hudlicka O. Unorthodox angiogenesis in skeletal muscle. Cardiovasc Res. 2001;49:634–646. doi: 10.1016/s0008-6363(00)00282-0. [DOI] [PubMed] [Google Scholar]
- 6.Milkiewicz M, Brown MD, Egginton S, Hudlicka O. Association between shear stress, angiogenesis, and VEGF in skeletal muscles in vivo. Microcirculation. 2001;8:229–241. doi: 10.1038/sj/mn/7800074. [DOI] [PubMed] [Google Scholar]
- 7.Tuttle JL, Nachreiner RD, Bhuller AS, Condict KW, Connors BA, Herring BP, Dalsing MC, Unthank JL. Shear level influences resistance artery remodeling: wall dimensions, cell density, and eNOS expression. Am J Physiol Heart Circ Physiol. 2001;281:H1380–H1389. doi: 10.1152/ajpheart.2001.281.3.H1380. [DOI] [PubMed] [Google Scholar]
- 8.Unthank JL, Fath SW, Burkhart HM, Miller SC, Dalsing MC. Wall remodeling during luminal expansion of mesenteric arterial collaterals in the rat. Circ Res. 1996;79:1015–1023. doi: 10.1161/01.res.79.5.1015. [DOI] [PubMed] [Google Scholar]
- 9.Unthank JL, Nixon JC, Burkhart HM, Fath SW, Dalsing MC. Early collateral and microvascular adaptations to intestinal artery occlusion in rat. Am J Physiol Heart Circ Physiol. 1996;271:H914–H923. doi: 10.1152/ajpheart.1996.271.3.H914. [DOI] [PubMed] [Google Scholar]
- 10.Van Gieson EJ, Murfee WL, Skalak TC, Price RJ. Enhanced smooth muscle cell coverage of microvessels exposed to increased hemodynamic stresses in vivo. Circ Res. 2003;92:929–936. doi: 10.1161/01.RES.0000068377.01063.79. [DOI] [PubMed] [Google Scholar]
- 11.Rissanen TT, Korpisalo P, Markkanen JE, Liimatainen T, Orden MR, Kholova I, de Goede A, Heikura T, Grohn OH, Yla-Herttuala S. Blood flow remodels growing vasculature during vascular endothelial growth factor gene therapy and determines between capillary arterialization and sprouting angiogenesis. Circulation. 2005;112:3937–3946. doi: 10.1161/CIRCULATIONAHA.105.543124. [DOI] [PubMed] [Google Scholar]
- 12.Sessa WC. eNOS at a glance. J Cell Sci. 2004;117:2427–2429. doi: 10.1242/jcs.01165. [DOI] [PubMed] [Google Scholar]
- 13.Wang WY, Whittles CE, Harper SJ, Bates DO. An adenovirus-mediated gene-transfer model of angiogenesis in rat mesentery. Microcirculation. 2004;11:361–375. doi: 10.1080/10739680490437568. [DOI] [PubMed] [Google Scholar]
- 14.Thurston G, Rudge JS, Ioffe E, Zhou H, Ross L, Croll SD, Glazer N, Holash J, McDonald DM, Yancopoulos GD. Angiopoietin-1 protects the adult vasculature against plasma leakage. Nat Med. 2000;6:460–463. doi: 10.1038/74725. [DOI] [PubMed] [Google Scholar]
- 15.Thurston G, Wang Q, Baffert F, Rudge J, Papadopoulos N, Jean-Guillaume D, Wiegand S, Yancopoulos GD, McDonald DM. Angiopoietin 1 causes vessel enlargement, without angiogenic sprouting, during a critical developmental period. Development. 2005;132:3317–3326. doi: 10.1242/dev.01888. [DOI] [PubMed] [Google Scholar]
- 16.Thurston G, Suri C, Smith K, McClain J, Sato TN, Yancopoulos GD, McDonald DM. Leakage-resistant blood vessels in mice transgenically overexpressing angiopoietin-1. Science. 1999;286:2511–2514. doi: 10.1126/science.286.5449.2511. [DOI] [PubMed] [Google Scholar]
- 17.Unthank JL, Sheridan KM, Dalsing MC. Collateral growth in the peripheral circulation: a review. Vasc Endovascular Surg. 2004;38:291–313. doi: 10.1177/153857440403800401. [DOI] [PubMed] [Google Scholar]
- 18.Williams JL, Cartland D, Hussain A, Egginton S. A differential role for nitric oxide in two forms of physiological angiogenesis in mouse. J Physiol. 2006;570:445–454. doi: 10.1113/jphysiol.2005.095596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alexander MY, Brosnan MJ, Hamilton CA, Downie P, Devlin AM, Dowell F, Martin W, Prentice HM, O’Brien T, Dominiczak AF. Gene transfer of endothelial nitric oxide synthase improves nitric oxide-dependent endothelial function in a hypertensive rat model. Cardiovasc Res. 1999;43:798–807. doi: 10.1016/s0008-6363(99)00146-7. [DOI] [PubMed] [Google Scholar]
- 20.Miller WH, Brosnan MJ, Graham D, Nicol CG, Morecroft I, Channon KM, Danilov SM, Reynolds PN, Baker AH, Dominiczak AF. Targeting endothelial cells with adenovirus expressing nitric oxide synthase prevents elevation of blood pressure in stroke-prone spontaneously hypertensive rats. Mol Ther. 2005;12:321–327. doi: 10.1016/j.ymthe.2005.02.025. [DOI] [PubMed] [Google Scholar]
- 21.Namba T, Koike H, Murakami K, Aoki M, Makino H, Hashiya N, Ogihara T, Kaneda Y, Kohno M, Morishita R. Angiogenesis induced by endothelial nitric oxide synthase gene through vascular endothelial growth factor expression in a rat hindlimb ischemia model. Circulation. 2003;108:2250–2257. doi: 10.1161/01.CIR.0000093190.53478.78. [DOI] [PubMed] [Google Scholar]
- 22.Kimura H, Weisz A, Ogura T, Hitomi Y, Kurashima Y, Hashimoto K, D’Acquisto F, Makuuchi M, Esumi H. Identification of hypoxia-inducible factor 1 ancillary sequence and its function in vascular endothelial growth factor gene induction by hypoxia and nitric oxide. J Biol Chem. 2001;276:2292–2298. doi: 10.1074/jbc.M008398200. [DOI] [PubMed] [Google Scholar]
- 23.Kimura H, Weisz A, Kurashima Y, Hashimoto K, Ogura T, D’Acquisto F, Addeo R, Makuuchi M, Esumi H. Hypoxia response element of the human vascular endothelial growth factor gene mediates transcriptional regulation by nitric oxide: control of hypoxia-inducible factor-1 activity by nitric oxide. Blood. 2000;95:189–197. [PubMed] [Google Scholar]
- 24.Dulak J, Jozkowicz A, Foresti R, Kasza A, Frick M, Huk I, Green CJ, Pachinger O, Weidinger F, Motterlini R. Heme oxygenase activity modulates vascular endothelial growth factor synthesis in vascular smooth muscle cells. Antioxid Redox Signal. 2002;4:229–240. doi: 10.1089/152308602753666280. [DOI] [PubMed] [Google Scholar]
- 25.Zacharek A, Chen J, Zhang C, Cui X, Roberts C, Jiang H, Teng H, Chopp M. Nitric oxide regulates Angiopoietin1/Tie2 expression after stroke. Neurosci Lett. 2006;404:28–32. doi: 10.1016/j.neulet.2006.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kashiwagi S, Tsukada K, Xu L, Miyazaki J, Kozin SV, Tyrrell JA, Sessa WC, Gerweck LE, Jain RK, Fukumura D. Perivascular nitric oxide gradients normalize tumor vasculature. Nat Med. 2008;14:255–257. doi: 10.1038/nm1730. [DOI] [PubMed] [Google Scholar]
- 27.Kashiwagi S, Izumi Y, Gohongi T, Demou ZN, Xu L, Huang PL, Buerk DG, Munn LL, Jain RK, Fukumura D. NO mediates mural cell recruitment and vessel morphogenesis in murine melanomas and tissue-engineered blood vessels. J Clin Invest. 2005;115:1816–1827. doi: 10.1172/JCI24015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Papapetropoulos A, Garcia-Cardena G, Madri JA, Sessa WC. Nitric oxide production contributes to the angiogenic properties of vascular endothelial growth factor in human endothelial cells. J Clin Invest. 1997;100:3131–3139. doi: 10.1172/JCI119868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bouloumie A, Schini-Kerth VB, Busse R. Vascular endothelial growth factor up-regulates nitric oxide synthase expression in endothelial cells. Cardiovasc Res. 1999;41:773–780. doi: 10.1016/s0008-6363(98)00228-4. [DOI] [PubMed] [Google Scholar]
- 30.Hood JD, Meininger CJ, Ziche M, Granger HJ. VEGF upregulates ecNOS message, protein, and NO production in human endothelial cells. Am J Physiol Heart Circ Physiol. 1998;274:H1054–H1058. doi: 10.1152/ajpheart.1998.274.3.H1054. [DOI] [PubMed] [Google Scholar]
- 31.Linscheid P, Seboek D, Zulewski H, Scherberich A, Blau N, Keller U, Muller B. Cytokine-induced metabolic effects in human adipocytes are independent of endogenous nitric oxide. Am J Physiol Endocrinol Metab. 2006;290:E1068–E1077. doi: 10.1152/ajpendo.00374.2005. [DOI] [PubMed] [Google Scholar]
- 32.Williams JL, Cartland D, Rudge JS, Egginton S. VEGF trap abolishes shear stress- and overload-dependent angiogenesis in skeletal muscle. Microcirculation. 2006;13:499–509. doi: 10.1080/10739680600785717. [DOI] [PubMed] [Google Scholar]
- 33.Hansen-Smith F, Egginton S, Zhou AL, Hudlicka O. Growth of arterioles precedes that of capillaries in stretch-induced angiogenesis in skeletal muscle. Microvasc Res. 2001;62:1–14. doi: 10.1006/mvre.2001.2308. [DOI] [PubMed] [Google Scholar]
- 34.Smith RS, Jr, Lin KF, Agata J, Chao L, Chao J. Human endothelial nitric oxide synthase gene delivery promotes angiogenesis in a rat model of hindlimb ischemia. Arterioscler Thromb Vasc Biol. 2002;22:1279–1285. doi: 10.1161/01.atv.0000026613.18742.67. [DOI] [PubMed] [Google Scholar]
- 35.Price RJ, Skalak TC. Prazosin administration enhances proliferation of arteriolar adventitial fibroblasts. Microvasc Res. 1998;55:138–145. doi: 10.1006/mvre.1997.2062. [DOI] [PubMed] [Google Scholar]
- 36.Price RJ, Owens GK, Skalak TC. Immunohistochemical identification of arteriolar development using markers of smooth muscle differentiation. Evidence that capillary arterialization proceeds from terminal arterioles. Circ Res. 1994;75:520–527. doi: 10.1161/01.res.75.3.520. [DOI] [PubMed] [Google Scholar]
- 37.Baum O, Da Silva-Azevedo L, Willerding G, Wockel A, Planitzer G, Gossrau R, Pries AR, Zakrzewicz A. Endothelial NOS is main mediator for shear stress-dependent angiogenesis in skeletal muscle after prazosin administration. Am J Physiol Heart Circ Physiol. 2004;287:H2300–H2308. doi: 10.1152/ajpheart.00065.2004. [DOI] [PubMed] [Google Scholar]
- 38.Yu J, deMuinck ED, Zhuang Z, Drinane M, Kauser K, Rubanyi GM, Qian HS, Murata T, Escalante B, Sessa WC. Endothelial nitric oxide synthase is critical for ischemic remodeling, mural cell recruitment, and blood flow reserve. Proc Natl Acad Sci USA. 2005;102:10999–11004. doi: 10.1073/pnas.0501444102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.De Palma M, Venneri MA, Roca C, Naldini L. Targeting exogenous genes to tumor angiogenesis by transplantation of genetically modified hematopoietic stem cells. Nat Med. 2003;9:789–795. doi: 10.1038/nm871. [DOI] [PubMed] [Google Scholar]
- 40.De Palma M, Venneri MA, Galli R, Sergi Sergi L, Politi LS, Sampaolesi M, Naldini L. Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell. 2005;8:211–226. doi: 10.1016/j.ccr.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 41.Nehls V, Drenckhahn D. The versatility of microvascular pericytes: from mesenchyme to smooth muscle? Histochemistry. 1993;99:1–12. doi: 10.1007/BF00268014. [DOI] [PubMed] [Google Scholar]
- 42.Baluk P, Lee CG, Link H, Ator E, Haskell A, Elias JA, McDonald DM. Regulated angiogenesis and vascular regression in mice overexpressing vascular endothelial growth factor in airways. Am J Pathol. 2004;165:1071–1085. doi: 10.1016/S0002-9440(10)63369-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hughes S, Chan-Ling T. Characterization of smooth muscle cell and pericyte differentiation in the rat retina in vivo. Invest Ophthalmol Vis Sci. 2004;45:2795–2806. doi: 10.1167/iovs.03-1312. [DOI] [PubMed] [Google Scholar]
- 44.Price RJ, Skalak TC. Chronic alpha 1-adrenergic blockade stimulates terminal and arcade arteriolar development. Am J Physiol. 1996;271:H752–H759. doi: 10.1152/ajpheart.1996.271.2.H752. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.