

Abstract
Knockdown or inhibition of SIRT2 enhances biological stress-tolerance. We extend this phenotype showing that SIRT2 knockdown reduces anoxia-reoxygenation injury in H9c2 cells. Gene array analysis following SIRT2 siRNA knockdown identifies 14-3-3 ζ as the most robustly induced gene. SIRT2 knockdown evokes induction of this chaperone, facilitating cytosolic sequestration of BAD with a corresponding reduction in mitochondrial BAD localization. Concurrent siRNA against SIRT2 and 14-3-3 ζ abolishes the SIRT2 depleted cytoprotective phenotype. SIRT2 functions to moderate cellular stress tolerance, in part, by modulating the levels of 14-3-3 ζ with the concordant control of BAD subcellular localization.
Keywords: SIRT2, BAD, 14-3-3 ζ, H9c2 cells, anoxia-reoxygenation
1. Introduction
Sirtuins, through NAD+-dependent deacetylation of target proteins, generally augment stress tolerance and lifespan extension and are thought to function in part by augmenting DNA repair and anti-oxidant defenses [1]. The functions of sirtuins have predominantly been explored in the context of caloric restriction; however the ameliorative effects of these proteins have been explored in response to more robust biological stressors including ischemic and oxidative stress in the heart and cardiomyocytes. SIRT1 and SIRT7 attenuate oxidative and genotoxic stress in cardiac myocytes [2,3], protect the intact heart against oxidative stress [4] and prevent aging-associated cardiac dysfunction [3].
In contrast, evidence is beginning to demonstrate that SIRT2 functions as a negative regulator of biological stress [5,6]. The mechanisms governing this regulation is beginning to be explored [7]. To further explore this we investigated the role of SIRT2 in cardiac-derived H9c2 cells. We show that SIRT2 is induced by anoxia-reoxygenation, and that SIRT2 overexpression increased susceptibility to cell death via its deacetylase activity. Conversely SIRT2 siRNA enhances anoxia-reoxygenation tolerance. As SIRT2 depletion enhances oxidative stress-tolerance, we employed gene array profiling to screen for putative adaptive candidate genes. We identified adaptor protein 14-3-3 ζ as the most highly induced gene. 14-3-3 ζ protein was concordantly induced and associated with the sequestration of BAD in the cytoplasm. Knockdown of 14-3-3 ζ prevents SIRT2 depletion mediated anoxia-reoxygenation tolerance. These results identify SIRT2 as a negative regulator moderating tolerance to anoxia-reoxygenation in H9c2 cells, in part, by modulating 14-3-3 ζ and identify BAD as a cytosolic ‘substrate’ of 14-3-3 ζ.
2. Materials and methods
2.1 Cell Culture and Transfection
H9c2 rat cardiac-derived myoblasts (ATCC) were cultured in DMEM containing 10% FBS, 1% penicillin/streptomycin at 37°C, 5% CO2. Cells transfected with negative control siRNA (Invitrogen), SIRT2 siRNA (Dharmacon – Cat. No.M-08072-00), 14-3-3 ζ siRNA (Qiagen – Cat. No.SI02055536), pcDNA3.1 plasmid (Invitrogen), pcDNA-FLAG/HA-14-3-3ζ (Addgene), pcDNA3.1-Myc/His-SIRT2 construct (Biochain) or with the deacetylase mutant SIRT2 construct pcDNA-SIRT2 (N168A) [8] using a Nucleofector II Device (Amaxa) were cultured for 48 h prior to experiments.
2.2 Cell Viability
Transfected cells were exposed to 17-h anoxia in DMEM media (no glucose, pyruvate, or antibiotics), using a GasPak EZ system (Becton-Dickenson). Following anoxia, fully-constituted DMEM was added and cells were incubated for an additional 2 h. Cellular injury was determined using a LDH cell viability assay (CytoTox 96, Promega) spectrophotometrically and confirmed by measuring propidium iodine (PI- BD Pharmingen) uptake using flow cytometry (BD FACSCalibur).
2.3 RNA Isolation and Expression Levels
H9c2 total RNA was prepared using a RNeasy RNA isolation kit (Qiagen). RNA was quantified and used for gene array analyses and RT-PCR. Amplification of H9c2 RNA and gene array analysis was performed using Rat Genome 230 2.0 Array (Affymetrix) microarrays. Real-time quantitative PCR was performed with SYBR Green PCR Master Mix (Applied Biosystems) using a MJ Research DNA Engine Opticon 2 fluorescence detection system. Ribosomal 18s and Tbp1 primers were used as internal standards.
2.4 Protein Analyses
Primary antibodies: rabbit 14-3-3 ζ (Santa Cruz); mouse actin (Ambion); rabbit BAD (Cell Signaling); mouse VDAC (Invitrogen) and rabbit aldose reductase (Santa Cruz). H9c2 cells were harvested and lysed in RIPA buffer containing protease inhibitors. For BAD localization experiments, cells were subfractionated using the Qproteome Mitochondrial Isolation Kit (Qiagen). Briefly, cells were centrifuged (1000 x g, 10min, 4°C) and the cytosolic protein-containing supernatant collected. The pellet was resuspended in Qiagen Disruption Buffer and then centrifuged (1000 x g, 10min, 4°C) and the supernatant transferred to a new tube. After additional centrifugation (6000 x g, 10min, 4°C) the pellet was washed and centrifuged at 6000 x g for 20min at 4°C. The mitochondria pellet was resuspended and used for SDS-PAGE. For co-immunoprecipitation experiments equal amounts of cell lysate protein were incubated for 1 h at 4°C with primary antibody and immunoprecipitated with agarose overnight at 4°C. Samples were used for SDS-PAGE and Western immunoblotting.
2.5 Immunofluorescence Confocal Microscopy
Cells grown on glass slides were washed in PBS and fixed with 4% paraformaldehyde followed by permeabilization with 0.1% Triton X-100. Samples were blocked with 4% normal goat serum and then incubated with the rabbit polyclonal antibody to BAD (1:100) and monoclonal antibody to COXI (MitoSciences, 1:100) for 1 hr followed by incubation with Alexa Fluor 488 conjugated goat anti-rabbit IgG and 594 conjugated goat anti-mouse IgG (1:250, Molecular Probes). Nuclei were counterstained with 4′6′-diamidino-2 phenylindole (DAPI, Molecular Probes). The images were collected using a Zeiss LSM 510 confocal microscope.
2.6 Statistical Analysis
Gene array expression levels were analyzed after segregation to exclude gene probesets employing a false discovery rate (FDR) of < 20% and a fold change > 1.2. Differences between groups were evaluated for significance using the Student t-test. Analyses to assess cell death were evaluated for significance using ANOVA. P < 0.05 was considered statistically significant, and data are expressed as mean ± SEM.
3. Results
3.1 Anoxia-reoxygenation stress tolerance in response to modulation of SIRT2
In keeping with a detrimental regulatory role of SIRT2 in response to biological stress we find a temporal induction of SIRT2 transcript levels in response to anoxia-reoxygenation (Fig. 1A). To functionally explore whether SIRT2 modulation alters stress tolerance we utilized anoxia-reoxygenation stress in H9c2 cells as previously described [9]. Transient overexpression of SIRT2 resulted in an 8-fold increase in SIRT2 gene expression and conversely the mRNA levels of SIRT2 were blunted in excess of 75 ± 3% following siRNA transfection (Suppl. Fig. 1A and 1B). SIRT2 overexpression and gene depletion did not alter basal cell viability with normoxia (Fig. 1B). In contrast, the induction of SIRT2 enhanced cellular damage in response to 17 hours of anoxia and reoxygenation and the genetic depletion of SIRT2 enhanced H9c2 viability, as measured by cellular LDH release following reoxygenation. Interestingly, the overexpression of a deacetylase mutant SIRT2 construct enhanced tolerance above that achieved with the empty vector. To confirm this ameliorative phenotype in response to the reduction in SIRT2 we assessed the cellular uptake of propidium iodide comparing cells with basal and diminished SIRT2 levels. In parallel with the LDH release assay, a greater proportion of SIRT2 siRNA-treated cells were viable in response to anoxia-reoxygenation injury compared to controls (Fig. 1C).
Fig. 1. The effect of SIRT2 modulation on cell viability.

(A) Temporal regulation of SIRT2 transcript levels in response to anoxia-reoxygenation. (B) Cell viability as measured by LDH release is enhanced in cells depleted of SIRT2 while SIRT2 overexpression results in increased LDH release in response to anoxia and reoxygenation. (C) SIRT2-depleted H9c2 exposed to 17 h anoxia – 2 h reoxygenation and stained with PI exhibit decreased dead cells as measured by FACS analyses. (n = 6 per group. p < 0.05 , versus control or the scrambled siRNA or empty vector transfected control cells respectively).
3.2 Gene profiling identifies 14-3-3 ζ upregulation following SIRT2 siRNA
As the knockdown of SIRT2 enhances tolerance to anoxia-reoxygenation cell death we employed gene profiling analysis to identify putative targets that modulate this stress-tolerance following the reduction in SIRT2 levels. Only 39 genes exhibited differences using an FDR of 20% with a fold change of 1.2 (Suppl. Table 1). Confirming the efficacy of SIRT2 depletion, the most robustly downregulated gene in SIRT2 siRNA treated cells was the gene set encoding SIRT2. The most highly induced transcript encoded for the adaptor protein 14-3-3 ζ (Suppl. Table 1). The induction of this gene was confirmed using RT-PCR (Fig. 2A). To further evaluate the relationship between SIRT2 and 14-3-3 ζ, the gene levels of 14-3-3 ζ was assessed in response to SIRT2 overexpression. Here, 14-3-3 ζ transcript levels were diminished 48±5% (p=0.04) relative to empty vector controls (data not shown). We then confirmed that this gene regulatory event was mirrored at the protein level with the upregulation of the 14-3-3 ζ protein by 50 ± 4% in SIRT2 siRNA treated cells versus control cells (Fig. 2B).
Fig. 2. Confirmation of gene and protein levels of 14-3-3 ζ.
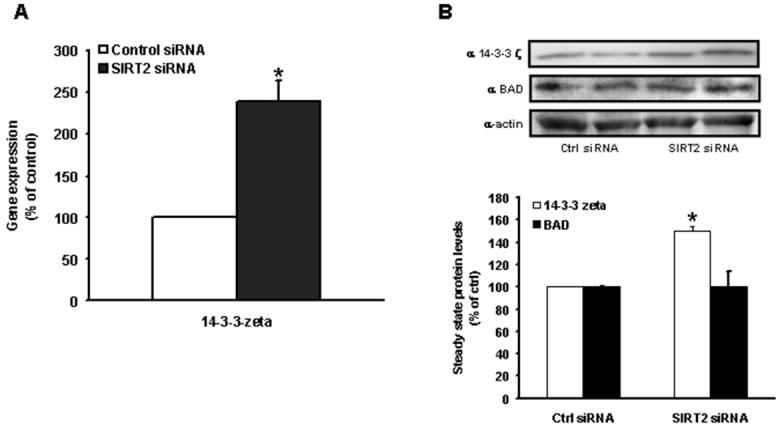
(A) RT-PCR confirms the induction of the gene encoding for 14-3-3 ζ in cells transfected with SIRT2 siRNA versus scrambled control siRNA. (B) SIRT2-depleted H9c2 exhibit increased 14-3-3 ζ protein levels compared to controls, as measured by Western immunoblotting. (n = 4 per group. p < 0.05 versus their respective controls)
3.3 SIRT2 knockdown-induced upregulation of 14-3-3 ζ enhances interaction with BAD
Interestingly, a 14-3-3 binding site consensus region has been previously shown to associate with and to sequester BAD in the cytosol thereby promoting cell survival [10]. As 14-3-3 ζ protein levels were enriched in response to SIRT2 depletion we next determined whether the interaction between the 14-3-3 ζ isoform and BAD was increased in cells with reduced SIRT2. SIRT2 knockdown did not modulate the steady state level of BAD (Fig. 2B) but evoked an increased and reciprocal interaction between 14-3-3 ζ and BAD proteins compared to control cells (Fig. 3A and 3B). To confirm that this mechanism is operational in response to anoxia-reoxygenation, we show that following anoxia the interaction between 14-3-3 ζ and BAD is enhanced in SIRT2 siRNA cells and diminished in SIRT2 over-expressing cells (Fig. 3C and 3D).
Fig. 3. 14-3-3 ζ and BAD protein interactions in response to SIRT2 depletion.
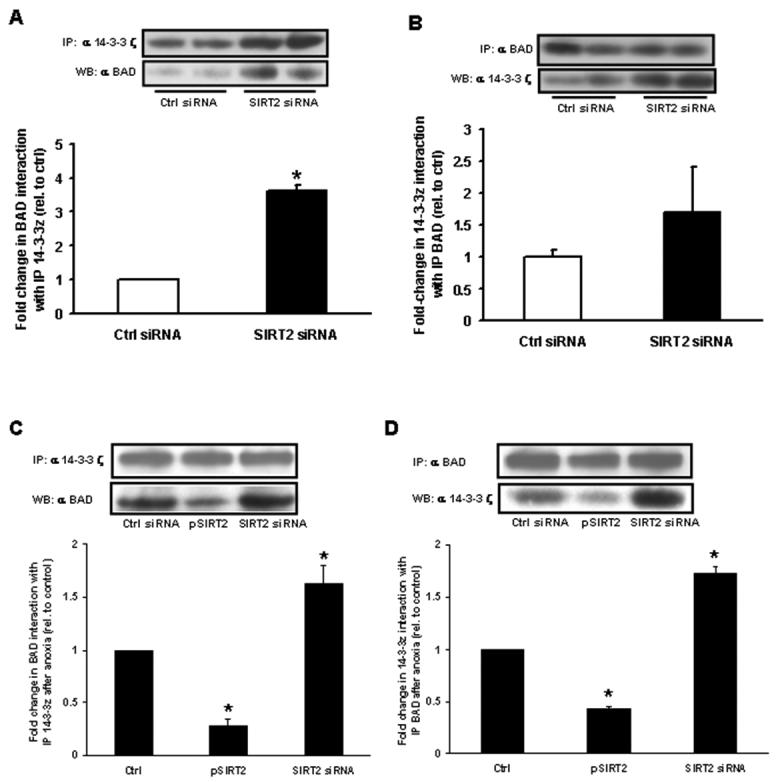
H9c2 were transfected with SIRT2 siRNA for 48 h. Results were quantified by densitometry (n = 4 per group. p < 0.05 versus scrambled siRNA transfected controls) (A) SIRT2-depleted H9c2 show increased BAD protein associated with 14-3-3 ζ relative to controls. 14-3-3 ζ protein was immunoprecipitated from cell lysates and associated BAD protein detected by Western immunoblotting. (B) SIRT2-depleted H9c2 show increased 14-3-3 ζ protein associated with BAD, relative to controls. BAD protein was immunoprecipitated from cell lysates and associated 14-3-3 ζ protein detected by Western immunoblotting. Immunoprecipitation with (C) 14-3-3 ζ and (D) BAD show increased interaction with the reciprocal protein following 14 hours of anoxia in SIRT2 siRNA cells and a diminution in this interaction in cells overexpressing SIRT2.
3.4 14-3-3 ζ siRNA abolishes SIRT2 knockdown anoxia-reoxygenation resilience
Concurrent SIRT2 and 14-3-3 ζ siRNA completely nullifies the cytoprotection of SIRT2 knockdown alone (Fig. 4). The pivotal role of 14-3-3 ζ in cytosolic sequestration of pro-apoptotic factors is further demonstrated in that 14-3-3 ζ siRNA administration exacerbates anoxia-reoxygenation injury to a similar degree to the overexpression of SIRT2 and in that 14-3-3 ζ overexpression is cytoprotective independent of SIRT2 levels (Fig. 4).
Fig. 4. SIRT2 and 14-3-3 ζ levels modulate anoxia-reoxygenation tolerance.
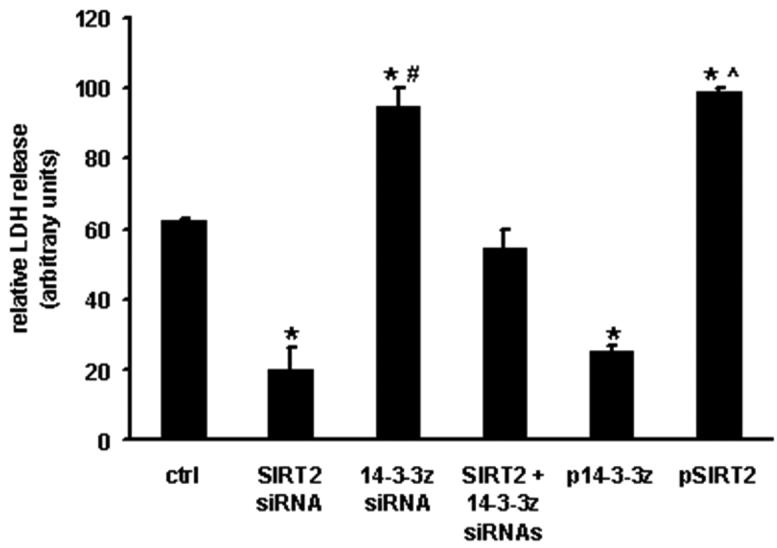
Cell viability as measured by LDH release is diminished in cells depleted of 14-3-3 ζ and conversely enhanced by the overexpression of 14-3-3 ζ. The knockdown of 14-3-3 ζ in parallel with SIRT2 abolishes the protection conferred by SIRT2 siRNA alone. (n = 3 per group. p < 0.05, * - versus the scrambled siRNA or empty vector transfected control cells, # - vs SIRT2 siRNA and ^ - versus pcDNA14-3-3 ζ respectively).
3.5 Modulation of SIRT2 determines the intracellular distribution of BAD
We hypothesized that modulating SIRT2 expression would modify the relative localization of BAD to the cytosol or mitochondria in parallel with 14-3-3 ζ expression. Using confocal microcopy and immunofluorescent antibodies directed against both BAD and a mitochondrial protein, cytochrome c oxidase I (COXI), we demonstrate diminished mitochondrial localization of BAD following SIRT2 knockdown and mitochondrial enrichment of BAD following SIRT2 over-expression (Fig. 5A). This phenotype is also operational following anoxia as shown by the subcellular localization immunoblot analysis comparing cytosolic to mitochondrial extracts (Fig. 5B and 5C).
Fig. 5. Modulation of BAD subcellular localization with varying expression of SIRT2.
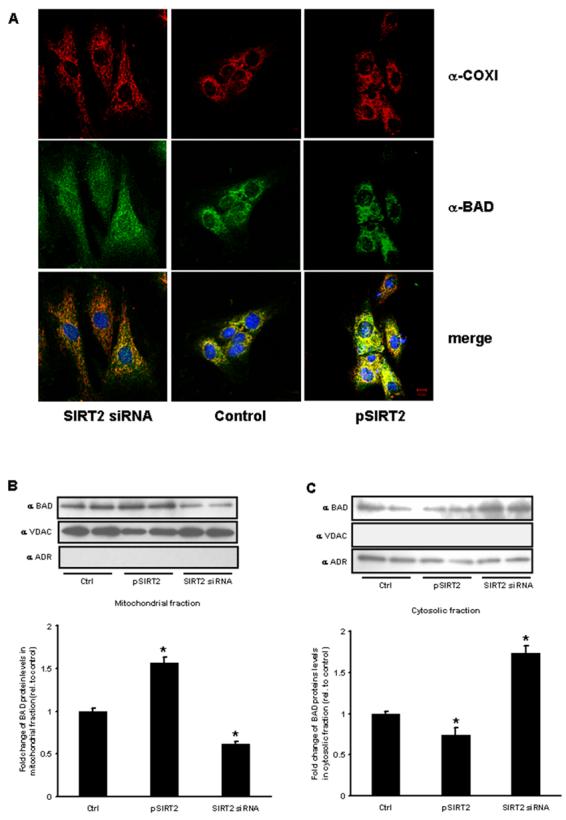
(A) SIRT2 siRNA-treated (left column), control siRNA-treated (middle column) and pSIRT2-transfected H9c2 cells (right column) were stained with anti-COXI (red) and anti-BAD (green) antibodies. The lower panels are merged images. DAPI staining (blue) is used to visualize nuclei. Scale bar, 10 μm. Subcellular fractionation for western blot analysis show reduced BAD in the mitochondrial fraction (B) and increased cytosolic levels (C) following SIRT2 siRNA and the reciprocal localization following SIRT2 overexpression. VDAC – (voltage dependent anion channel – mitochondrial enriched) and ADR – (aldose reductase is cytosolic). (n=3 per group, p<0.05 versus scrambled siRNA and empty vector controls).
4. Discussion
Using anoxia-reoxygenation as a biological stressor this study expands the repertoire of stresses that is alleviated by SIRT2 depletion. We demonstrate that 14-3-3 ζ is induced in response to SIRT2 depletion and mediates SIRT2-dependent modulation of stress-tolerance in H9c2 cells. 14-3-3 ζ is moreover shown to directly interact with BAD with a parallel reduction in mitochondrial BAD localization. The functional consequence of SIRT2 depletion in enhancing tolerance to redox injury is further supported by the protective effect of overexpression of the deacetylase mutant form of SIRT2 and by the deleterious consequences of anoxia-reoxygenation following overexpression of wildtype SIRT2.
The sirtuin family of NAD+-dependent deacetylases are proposed to function through distinct targets to enhance DNA-repair and anti-oxidant defenses [1]. The most robustly characterized family member, SIRT1 has not only been shown to enhance resilience to redox stress in cardiomyocytes and the heart [2,4] but has also been shown to be necessary to maintain basal cardiomyocyte viability [2]. The modulation in expression of SIRT2 in contrast does not alter basal H9c2 myoblast cell viability. However, the SIRT2 transcript is induced by anoxia-reoxygenation stress and its genetic knockdown is cytoprotective against anoxia-reoxygenation injury. These data are consistent with SIRT2 depletion-mediated protection against hydrogen peroxide, staurosporin and puromycin mediated cell death [5,7], and where SIRT2 knockdown and inhibition diminish dopaminergic-mediated cell death in a drosophila model of Parkinson's disease [6]. Collectively, these studies implicate that SIRT2 functions as a negative regulator in response to diverse cellular stressors.
The biological targets of SIRT2 action are actively being explored and characterized [11] [7,12]. Using gene-array analysis and confirmatory RT-PCR, we identified that 14-3-3 ζ is inversely regulated by the levels of SIRT2. 14-3-3 proteins are a family of conserved regulatory phosphoserine-binding molecules which are broadly expressed. By virtue of its interactions with effector proteins, 14-3-3 can modulate diverse biological pathways including those regulating programmed cell death [13]. BAD heterodimerizes with either BCL-XL or BCL-2 at the mitochondrion and antagonizes their anti-apoptotic activities. 14-3-3 has been shown to bind proapoptotic BAD in a phosphoserine-dependent manner resulting in the sequestration of BAD in the cytosol and inhibition of cell death [14]. The anti-apoptotic role of 14-3-3 ζ has been shown with knockdown or dominant negative overexpression of this chaperone [13,15]. Our study demonstrates an increased interaction of 14-3-3 ζ and BAD, diminished BAD localization to the mitochondria as well as an increased number of viable cells in response to anoxia-reoxygenation in cells depleted of SIRT2. Whether SIRT2 depletion results in increased BAD serine phosphorylation requires further investigation.
In conclusion, our data support a negative regulatory effect of SIRT2 on tolerance to anoxia-reoxygenation. Furthermore SIRT2 downregulation promotes 14-3-3 ζ induction, the cytosolic sequestration of BAD and the attenuation of anoxia-reoxygenation induced cell death. These results identify a negative regulatory program that may be pharmacologically modulated by the novel SIRT2 inhibitor [6] as a potential cell survival promoting strategy in enhancing tolerance to anoxia-reoxygenation injury.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Nalini Raghavachari and Xiuli Xu from the NHLBI Genomics Core for assistance with the gene array experiments and analyses and J. Philip McCoy Jr. from the NHLBI Flow Cytometry Core for assistance. This work was supported by funding from the Division of Intramural Research of the NHLBI of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Haigis MC, Guarente LP. Mammalian sirtuins--emerging roles in physiology, aging, and calorie restriction. Genes Dev. 2006;20:2913–21. doi: 10.1101/gad.1467506. [DOI] [PubMed] [Google Scholar]
- 2.Alcendor RR, Kirshenbaum LA, Imai S, Vatner SF, Sadoshima J. Silent information regulator 2alpha, a longevity factor and class III histone deacetylase, is an essential endogenous apoptosis inhibitor in cardiac myocytes. Circ Res. 2004;95:971–80. doi: 10.1161/01.RES.0000147557.75257.ff. [DOI] [PubMed] [Google Scholar]
- 3.Vakhrusheva O, Smolka C, Gajawada P, Kostin S, Boettger T, Kubin T, Braun T, Bober E. Sirt7 Increases Stress Resistance of Cardiomyocytes and Prevents Apoptosis and Inflammatory Cardiomyopathy in Mice. Circ Res. 2008 doi: 10.1161/CIRCRESAHA.107.164558. [DOI] [PubMed] [Google Scholar]
- 4.Alcendor RR, et al. Sirt1 Regulates Aging and Resistance to Oxidative Stress in the Heart. Circ Res. 2007;100:1512–1521. doi: 10.1161/01.RES.0000267723.65696.4a. [DOI] [PubMed] [Google Scholar]
- 5.Grubisha O, Rafty LA, Takanishi CL, Xu X, Tong L, Perraud AL, Scharenberg AM, Denu JM. Metabolite of SIR2 reaction modulates TRPM2 ion channel. J Biol Chem. 2006;281:14057–65. doi: 10.1074/jbc.M513741200. [DOI] [PubMed] [Google Scholar]
- 6.Outeiro TF, et al. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson's disease. Science. 2007;317:516–9. doi: 10.1126/science.1143780. [DOI] [PubMed] [Google Scholar]
- 7.Wang F, Nguyen M, Qin FX, Tong Q. SIRT2 deacetylates FOXO3a in response to oxidative stress and caloric restriction. Aging Cell. 2007;6:505–14. doi: 10.1111/j.1474-9726.2007.00304.x. [DOI] [PubMed] [Google Scholar]
- 8.Finnin MS, Donigian JR, Pavletich NP. Structure of the histone deacetylase SIRT2. Nat Struct Biol. 2001;8:621–5. doi: 10.1038/89668. [DOI] [PubMed] [Google Scholar]
- 9.McLeod CJ, Aziz A, Hoyt RF, Jr., McCoy JP, Jr., Sack MN. Uncoupling proteins 2 and 3 function in concert to augment tolerance to cardiac ischemia. J Biol Chem. 2005;280:33470–6. doi: 10.1074/jbc.M505258200. [DOI] [PubMed] [Google Scholar]
- 10.Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L) Cell. 1996;87:619–28. doi: 10.1016/s0092-8674(00)81382-3. [DOI] [PubMed] [Google Scholar]
- 11.North BJ, Marshall BL, Borra MT, Denu JM, Verdin E. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol Cell. 2003;11:437–44. doi: 10.1016/s1097-2765(03)00038-8. [DOI] [PubMed] [Google Scholar]
- 12.Vaquero A, et al. SirT2 is a histone deacetylase with preference for histone H4 Lys 16 during mitosis. Genes Dev. 2006;20:1256–61. doi: 10.1101/gad.1412706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xing H, Zhang S, Weinheimer C, Kovacs A, Muslin AJ. 14-3-3 proteins block apoptosis and differentially regulate MAPK cascades. Embo J. 2000;19:349–58. doi: 10.1093/emboj/19.3.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang X, Luo C, Cai J, Pierce W, Tezel G. Phosphorylation-dependent interaction with 14-3-3 regulates Bad trafficking in retinal ganglion cells. Invest Ophthalmol Vis Sci. 2008 doi: 10.1167/iovs.07-1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Niemantsverdriet M, Wagner K, Visser M, Backendorf C. Cellular functions of 14-3-3 zeta in apoptosis and cell adhesion emphasize its oncogenic character. Oncogene. 2008;27:1315–9. doi: 10.1038/sj.onc.1210742. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.