

Abstract
Ffh and FtsY are GTPase components of the signal recognition particle co-translational targeting complex that assemble during the SRP cycle to form a GTP-dependent and pseudo two-fold symmetric heterodimer. Previously the SRP GTPase heterodimer has been stabilized and purified for crystallographic studies using both the non-hydrolysable GTP analog GMPPCP and the pseudo-transition state analog GDP:AlF4, revealing in both cases a buried nucleotide pair that bridges and forms a key element of the heterodimer interface. A complex of Ffh and FtsY from T. aquaticus formed in the presence of the analog GMPPNP could not be obtained, however. The origin of this failure was previously unclear, and it was thought to have arisen from either instability of the analog, or, alternatively, from differences in its interactions within the tightly conscribed composite active site chamber of the complex. Using insights gained from the previous structure determinations, we have now determined the structure of the SRP GTPase targeting heterodimer stabilized by the non-hydrolysable GTP analog GMPPNP. The structure demonstrates how the different GTP analogs are accommodated within the active site chamber despite slight differences in the geometry of the phosphate chain. It also reveals a K+ coordination site at the highly conserved DARGG loop at the N/G interdomain interface.
The Signal Recognition Particle (SRP) mediates signal peptide recognition and co-translational targeting of secreted and membrane proteins to the membrane translocon (Walter and Johnson, 1994; Keenan et al., 2001). SRP is a phylogenetically conserved ribonucleoprotein that comprises, in prokaryotes, Ffh, the SRP GTPase, and the 4.5S RNA (Luirink and Dobberstein, 1994). The primary structure of Ffh includes three domains, the N and G domains, and the M domain (Bernstein et al., 1989; Römisch et al., 1989). The M domain provides sites for signal sequence recognition and for interaction with the RNA (Zopf et al., 1990; Luirink et al., 1992; Lütcke et al., 1992). The N and G domains of the SRP GTPase, together the ‘NG’ domain, form a structural and functional unit (Freymann et al., 1997). The membrane associated receptor for SRP is also phylogenetically conserved, and its primary structure includes an NG GTPase as well (Montoya et al., 1997). The two SRP NG GTPases interact directly with each other forming a GTP-dependent heterodimeric targeting complex that plays a central role in co-translational protein targeting to the membrane (Powers and Walter, 1995; Powers and Walter, 1997; Rapiejko and Gilmore, 1997; Song et al., 2000; Mandon et al., 2003).
The structure of the SRP GTPase complex in the GMPPCP and GDP:AlF4 stabilized forms have been determined (Egea et al., 2004; Focia et al., 2004; Focia et al., 2006). The structure is a remarkably pseudo-symmetric heterodimer, in which the two NG domains assemble across their respective GTP binding sites to generate a composite active site chamber that is shared between them. Hydrogen bonding between a γ-phosphate oxygen of each nucleotide analog and the ribose 3′ hydroxyl of the nucleotide analog across the interface means that the two nucleotides contribute directly to the interface, and in the absence of that interaction, the heterodimer does not form (Egea et al., 2004). With GDP:AlF4 bound, the interaction is with the fluorine atom of the AlF4 group (Focia et al., 2006), and the putative transition state analog is accommodated within a ‘ground state’ structure of the active site chamber. The structures of the heterodimer explain key features of the assembly of the SRP GTPase targeting complex, and have revealed coordinated structural changes that occur on assembly that provide insight into the mechanisms by which its assembly, or disassembly by GTP hydrolysis, may be regulated (Shan et al., 2004; Focia et al., 2006).
Interestingly, initial attempts to assemble the SRP GTPase heterodimer for crystallization using the commonly used non-hydrolysable GTP analog GMPPNP failed (Shepotinovskaya and Freymann, 2002), despite its being successfully used in a number of biochemical studies for trapping the interaction between the SRP GTPases (Rapiejko and Gilmore, 1992; Miller et al., 1994; Peluso et al., 2000; Shan et al., 2004). Preliminary data in our laboratory suggested that this failure to stabilize the complex arose from hydrolysis of the nucleotide analog during the long incubation time required for an endogenous proteolysis found to be essential for assembly of the stable complex to occur (Shepotinovskaya and Freymann, 2002). However, the question of whether different nucleotides or nucleotide analogs exhibit different properties for assembly of the SRP GTPase complex has not been fully explored. Interaction specific to the analog GMPPNP in a different GTPase was suggested by a structure of an EF-G complex in which a peptide flip within the P-loop directs a carbonyl oxygen towards the β–γ amido group of the analog (Hansson et al., 2005). A peptide flip of the corresponding residue of the P-loop in Ffh is observed as a minor population in structures of the Ffh NG domain determined ultra-high resolution (Ramirez and Freymann, 2006). Further, the presence of the β–γ methylene bridging group of GMPPCP (substituting for the amido and ester groups of GMPPNP or GTP, respectively) raises the possibility that the GMPPCP-stabilized complex structure might obscure polar or hydrogen bonding interactions present in the native complex. We used insight gained from the structure of the GMPPCP-stabilized T. aquaticus Ffh:FtsY complex to generate a fast-assembly deletion construct of FtsY (Focia et al., 2006; Gawronski-Salerno et al., 2006), and have exploited it here to determine the structure of the GMPPNP-stabilized SRP GTPase complex.
Crystallization of the GMPPNP-stabilized complex
The crystal structure of the SRP GTPase heterodimer revealed the nature of a 20 amino acid N-terminal truncation of T. aquaticus FtsY that removed an apparent barrier to formation of a stable complex (Shepotinovskaya and Freymann, 2002). The uncharacterized proteolysis of FtsY occurred slowly, over days, but it was necessary for the successful purification (and crystallization) of the complex (Shepotinovskaya and Freymann, 2002; Shepotinovskaya et al., 2003; Egea et al., 2004; Focia et al., 2004). A construct of T. aquaticus FtsY in which the first 20 amino acids were deleted (FtsY NGd20) was subsequently expressed and purified, and it allowed us to readily assemble and purify the SRP GTPase complex stabilized using GMPPNP. Ffh NG and FtsY NGd20 were purified as described previously (Shepotinovskaya et al., 2003; Focia et al., 2006). Formation of the FtsY NGd20:Ffh NG complex was assayed using gel filtration over a Sephadex 75 HR 10/30 column, monitoring the A280/A260 ratio to distinguish the nucleotide-bound from unbound species, and was found to be essentially complete within minutes, rather than days (Shepotinovskaya and Freymann, 2002). For crystallization trials the complex was purified by ion exchange chromatography from a reaction mix of 16 μM FtsY NGd20, 21 μM Ffh NG, 1 mM GMPPNP (CalBiochem) in 50 mM HEPES pH 7.5, 2 mM MgCl2, and 50 mM NaCl, incubated at 37°C for 60′. The mix was desalted to remove unbound nucleotide and the complex purified over Q Sepharose using 50 mM Tris, pH 8.0 and a linear gradient to 0.5 M NaCl; subsequently the eluted complex peak was desalted and purified from unbound protein as the flow-through fraction over an SP Sepharose column in 10 mM HEPES 7.5. The purified complex was concentrated to 6 mg/ml using a YM 30 Centricon, yielding 73% of the starting material in the GMPPNP-stabilized heterodimer. The protein was used directly for crystallization trials in a Nextal PEGS screen, using 1μl drops equilibrated against 100μl mother liquor wells assembled with an Apogent Discoveries Hydra II Microdispenser. Blade-like crystals were obtained under the condition: 0.2 M KI, 20% PEG 3350.
A 100 μ × 250 μ crystal was harvested to mother liquor supplemented by 15% ethylene glycol, mounted using a nylon loop, and frozen in LN2. Data were measured at SBC beamline 19ID, using a wavelength of 1.011 Å, and an exposure time of 12s/0.7° oscillation for 175° rotation at a distance of 160 mm. Data were integrated using HKL2000 (Otwinowski and Minor, 1997), yielding an overall Rsym of 0.067 and 99.2% completeness to 1.97 Å (Table 1). The crystal had space group P21, with two Ffh:FtsY NG heterodimers in the asymmetric unit. The structure was determined by molecular replacement with PHASER (Storoni et al., 2004), using the structure of the GMPPCP-stabilized complex (1OKK) with all ligands and waters removed as the search model. Water molecules were built using ARP/wARP (Lamzin et al., 2001) and the model subsequently refined with REFMAC (Murshudov et al., 1997). The final model has an Rcryst of 18.4% and Rfree 22.7% (Table 1), with 98.7% of residues in the favored region of the Ramachandran plot (Richardson et al., 2003). The N-terminal four residues of both Ffh molecules in the asymmetric unit are disordered, as was observed previously (Focia et al., 2004), due to displacement by the C-terminal helix on formation of the complex. For both FtsY molecules the linker between the N and G domains (residues 79–95) could not be built; however, the linker was not cleaved during complex formation (as there was no long incubation (Shepotinovskaya and Freymann, 2002)) suggesting that it is disordered in this structure. Persistent negative peaks at 28 (of 112 total) glutamate residues were interpreted as decarboxylation caused by radiation damage and were modeled by setting the terminal carboxylate occupancies to 0.5 (Burmeister, 2000). Similarly, radiation damage was found to affect the terminal methylthio groups of two (of 20) methionine sidechains; in addition, the sidechain of Ffh Met39 exhibited continuous density suggesting a covalent modification, but the nature of the modification was unclear and it was modeled as an ‘unknown’ atom (data not shown). Finally, the four Mg2+GMPPNP groups, and several well defined solute features - six molecules of ethylene glycol, sixteen I− atoms and two K+ ions (see below) - could readily be interpreted from the electron density map. A ribbon diagram of the overall heterodimer structure is shown in Fig. 1A.
Table 1.
Crystallographic statistics
| Data collection | ||
| Spacegroup | P21 | |
| Unit Cell | 51.03 Å, 129.56 Å, 88.96 Å, β=91.66° | |
| Resolution | 50.00-1.97 Å | (2.04-1.97) |
| Rsym | 0.067 | (0.411) |
| Redundancy | 3.7 | (3.7) |
| Completeness | 99.2 | (98.9) |
| I/σ(I) | 17.1 | (2.8) |
| Refinement | ||
| Rcryst | 0.184 | (0.240) |
| Rfree | 0.227 | (0.290) |
| Protein atoms (#, <B>) | 8781 | 29.1 Å2 |
| GMPPNP atoms (#, <B>) | 128 | 17.7 Å2 |
| Potassium atoms (#, <B>) | 2 | 29.7 Å2 |
| Water molecules (#, <B>) | 536 | 36.3 Å2 |
| rms Bonds | 0.011Å | |
| rms Angles | 1.486° | |
There are two Ffh:FtsY NG heterodimers in the asymmetric unit. Data in parentheses are for the high resolution shell. <B> is the mean isotropic temperature factor for the atom set.
Rsym = Σ|Ih−<Ih>|/ΣIh, where <Ih> is the average intensity over symmetry equivalents
Rcryst = Σ|Fo−Fc|/ΣFo. Rfree is for 5% of the data omitted from the refinement.
Figure 1. Comparison of the active site structure in Ffh NG:FtsY NG complexes.
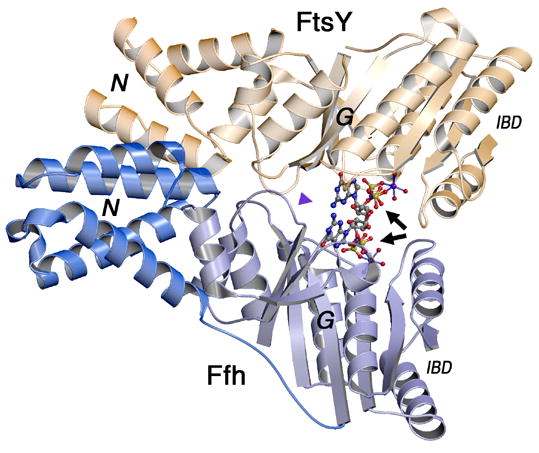
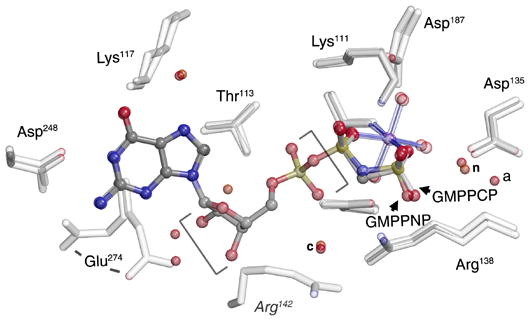
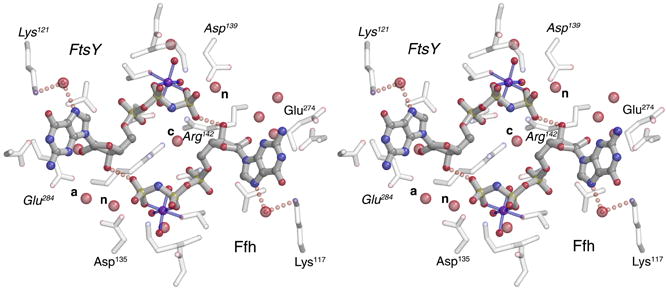
(A) Overall structure of the GMPPNP-stabilized SRP GTPase heterodimer. The two non-hydrolysable GTP analogs are buried within a composite active site chamber at the interface of the two proteins (arrows). The ‘N’ and ‘G’ domains of Ffh and FtsY are indicated. The extensive ‘latch’ interface is at left of the nucleotide pair, and much of the catalytic machinery is provided by the IBD subdomain, at right. The blue arrowhead indicates the point of view in (C). (B) The nucleotide analogs bound to Ffh within the active site chambers of the GMPPNP- and GMPPCP- stabilized complexes are superimposed (over the ribose and α-phosphate groups, bracketed). There are only small relative shifts of the two analogs, although the orientations of the terminal phosphate moieties change due to the different β–γ bridging group (-CH2- or -NH-). In neither structure do the bridging methylene or amido groups make direct interactions with sidechain or mainchain atoms or with water molecules. (C) Stereo diagram of the sidechain and water interactions of the buried GMPPNP pair. All buried waters and all sidechains that interact with the nucleotides are shown - note that the sidechains are exclusively arranged on the ‘IBD’ face of the nucleotide pair, as all interactions on the opposite face are mediated by mainchain atoms (and not shown in the figure). The nucleophilic (n), auxiliary (a), and shared central (c) waters are indicated. Figures were made with O, Molscript and Raster3D (Jones et al., 1991; Kraulis, 1991; Merritt and Bacon, 1997).
Crystal structure of FfhNG:FtsYNGd20:GMPPNP
The structure of the GMPPNP stabilized complex is very similar to that of the GMPPCP complex (PDB 1OKK), and comprises a pseudo-twofold interaction between the N and GTPase domains of Ffh and FtsY that buries a large interface between them that includes the two bound nucleotides (Fig. 1A) (Focia et al., 2004). The nucleotides form the center of a tripartite interaction surface - to one side (left in Fig. 1A) the ‘latch’ region is formed by extensive van der Waals and hydrogen bonding interactions that extend from the N domains of each protein to the adjacent loops of their G domains; on the other side (right in Fig. 1A) the IBD subdomain, an extension of the core GTPase fold unique to the SRP GTPases that contributes much of the catalytic machinery, packs against its partner across the interface and interacts with the bound nucleotide pair. When superimposed over the whole complex, the rmsd between the GMPPNP and GMPPCP stabilized complexes is 0.65 Å for 548 Cαs, with all large deviations located at the distal loops of the N domain (which tend to be disordered). When superimposed over either the G domain of Ffh or the G domain of FtsY (i.e. the GTPase fold that circumscribes the buried catalytic chamber) the rmsd over Cαs for both is only ~0.22 Å.
Despite the slight differences in configuration of the terminal groups of the two nucleotide analogs GMPPNP and GMPPCP, the structure of the nucleotide binding subunits is essentially identical. The bond and angle parameters for GMPPNP and GMPPCP were monitored carefully during the refinement, based on data from very high resolution protein and small molecule structures (Saenger, 1984; Scheidig et al., 1999). The target β–γ bridging (P-x-P) bond angles and lengths are slightly different (GMPPNP: 122°, 1.70Å; GMPPCP 109.5°, 1.81Å), but these differences result in no striking change in the structures in the active site, or in the adjacent water structure (Fig. 1B). The remarkable symmetric hydrogen bonding interaction between nucleotides across the interface is present (Focia et al., 2004), with symmetric 2.6–2.7Å H-bonds formed between each ribose 3′ hydroxyl and the γphosphate oxygen of GMPPNP bound across the heterodimer interface (Fig. 1C). The water structure between the two is essentially identical as well - the shared (and completely sequestered) ‘central’ water is present, as are the two corresponding ‘nucleophilic’ waters at each active center (Focia et al., 2004) (Fig. 1B, 1C). However, the position of the Glu274 sidechain is different between the two structures, which introduces an asymmetry in the water structure at the edge of the active site chamber.
As with the GMPPCP complex (Focia et al., 2004), the GMPPNP-stabilized structure is very similar to that of the GDP:AlF4 complex (PDB 2CNW) (overall rmsd 0.72 Å for 549 atoms, G-domain rmsds of ~0.25 Å over both Ffh and FtsY), although there is also a very slight rotational shift between the two domains along their interface, perhaps to accommodate the larger AlF4 group and opening of the P-loop buried at the center of their interaction (Focia et al., 2006).
A potassium ion binding site at a conserved loop
The crystal was obtained in the presence of 0.2 M KI, and sixteen iodine atoms were located in the structure, generally associated with nearby arginine and lysine sidechains. The iodine density peaks were typically large, poorly defined, and nonspherical, and so were modeled using anisotropic temperature factors, and their occupancies adjusted manually as necessary to minimize residual negative density. None affects the structure of the latch interface or active site chamber. Two K+ ions were readily identified in the map by their residual positive density, the coordination bond lengths (~2.8 Å), and the identity of the coordinating groups (i.e. three carbonyl oxygens, three water molecules) (Harding, 2002) (Fig. 2A). The K+ site is adjacent to the functionally important DARGG loop at the interface between the N and G domains of FtsY (Fig. 2B) and occurs in both of the non-crystallographic symmetry-related FtsY monomers in the asymmetric unit. Both the coordination bond distances and the residual difference density allow us to exclude Mg2+ ion at the position, and, indeed, the arrangement is quite typical of potassium - K+ is generally observed in 6, 7, or 8 coordinate configurations with the most common coordinating group being backbone carbonyl oxygens at a coordination distance of 2.84Å (Harding, 2002). All water molecules in the structure were screened for evidence of similar coordination geometry, but no other K+ ions could be identified.
Figure 2. Coordination of the DARGG loop carbonyl crown.
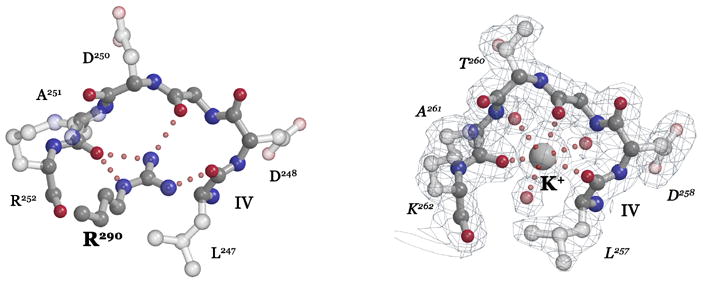
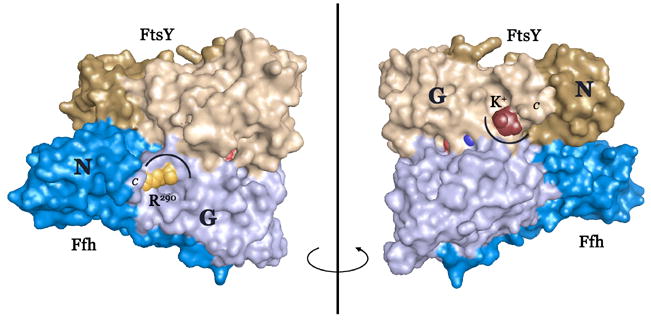
(A) In Ffh (left), Arg290 forms an extensive set of hydrogen bonding interactions with the carbonyl oxygens of the Ffh DARGG loop (which has the sequence Asp250AlaArgGlyGly254 in T. aquaticus Ffh). In FtsY (right), a potassium ion (large ball) is coordinated by the carbonyl crown contributed by motif IV/DARGG residues Leu257, Gly259 and Ala261, and by three water molecules (small red balls). The 2Fo−Fc electron density map is shown with the backbone atoms of residues 257–262 in ball-and-stick. The ‘DARGG’ loop has the sequence Thr260AlaLysGlyGly264 in T. aquaticus FtsY. (B) The context of the DARGG loop interactions in (A) are shown in two orientations of a surface representation of the heterodimer. The interactions are at the junction of the N and G domains of each protein, and in Ffh, couple to the position of the C-terminal helix (which in the intact protein is linked to the signal sequence recognition subunit, the M-domain). The C-terminus is indicated (C), and the DARGG loop indicated in each case by an arc. The figure was made with PYMOL (DeLano, 2002).
The location of the potassium coordination interaction is interesting, because the sequence of the DARGG loop is highly conserved in both Ffh and FtsY and, although it adopts various conformations in the monomeric proteins (Ramirez and Freymann, 2006), on assembly of the complex it adopts one conformation in both Ffh and FtsY (Fig. 2A). The loop links GTPase motif IV, which mediates nucleotide binding specificity (Freymann et al., 1997), to the α4 interface helix, which mediates the interaction between the N and G domains (Ramirez et al., 2002) (Fig. 2B). In Ffh (but not FtsY) the DARGG loop acquires a very well defined interaction with a completely conserved arginine sidechain that extends from the C-terminal helix and that may play a role in positioning the helix on assembly of the complex (Gawronski-Salerno et al., 2006) (Fig. 2A). In FtsY there is no such interaction; however, that the particular conformation of the loop generates a unique and stable (i.e. crystallizable) K+ coordination site suggests that there may be a functional significance to the interaction there. The crystal structure of the complex obtained after substituting NaI for KI during the crystallization is similar (rmsd over the heterodimer of 0.34 Å, 550 Cαs, over G-domains, ~0.17Å for both proteins), and in that structure there is no coordination of the FtsY DARGG loop (data not shown).
Implications of the structure
The structure of the GMPPNP complex of the SRP GTPases Ffh and FtsY highlights two issues. First, we now have obtained a similar heterodimer structure using three different GTP or transition state analogs - GMPPNP, GMPPCP, and GDP:AlF4 - and in each case the GTPase heterodimer accommodates the analog pair without significant disruption. This suggests that the stability of the heterodimer is driven by the protein:protein interaction across the interface, particularly along the extensive ‘latch’ interface that bridges the bound nucleotide pair and the N domains of the two proteins. The KD measured for both GMPPCP and GMPPNP-mediated assembly of the T. aquaticus Ffh NG:FtsY NGd20 heterodimer is ~10 nM (Focia et al., 2006). Further, there is sufficient plasticity in the interface to readily accommodate each of the nucleotide analogs, despite small shifts in the positions of the γphosphate group (to ~0.25 Å between GMPPNP and GMPPCP). We see no evidence for interaction with the β–γ bridging group of the bound nucleotide analogs in this or in previous structures, suggesting that if interactions with the bridging group arise, they must be mediated by the catalytic machinery contributed by the IBD at a subsequent step, during disengagement of the complex rather than its assembly. The only moieties that after slight rotation might be in position to interact with the β–γ bridging groups are the buried sidechains of the conserved arginine pair Arg138/Arg142 (Fig. 1B, 1C), and the sidechain of Ffh Gln107 (but not the corresponding Asn111 in FtsY). There is as of yet no structural evidence that allows us to understand the roles of these residues in the two GTPases.
Second, the structure suggests that different solution ion compositions might affect the structure and biochemistry of the SRP GTPase FtsY due to differential stabilization of a functionally important DARGG loop conformation (Freymann et al., 1997; Ramirez et al., 2002). Preliminary assays for assembly of the Ffh NG/FtsY NG complex in the presence of different cations, however, have not revealed a clear difference in our laboratory (data not shown), and careful quantitative biochemistry of the solution behavior of the complex may be required to discern an effect, if such exists. We note, however, that the standard salt conditions for many reports of SRP GTPase biochemistry includes potassium acetate (Miller and Walter, 1993; Miller et al., 1993; Powers and Walter, 1995). And, interestingly, it has been shown recently that K+ ion can function as a GTPase activating element for the dimerization-dependent GTPase MnmE (Scrima and Wittinghofer, 2006). That in Ffh, in contrast to FtsY, an arginine at position 290 of the C-terminal helix is present in all sequences suggests that the residue may play a role either in stabilizing the conformation of the DARGG loop in Ffh, or in orienting the C-terminal helix on assembly of the complex. In the heterodimer, conserved Ffh sidechains Arg290, Arg286 and Arg252 are arranged as a basic ‘ladder’ along the helix, which may allow the M domain/4.5S RNA component of SRP to regulate assembly (Gawronski-Salerno et al., 2006). Similar interactions do not occur in FtsY, as there are functionally distinct packing interactions between the C-terminal helix and the N/G domain interface (Gawronski-Salerno et al., 2006).
In summary, we have reported the structure of the GMPPNP-stabilized complex of T. aquaticus Ffh and FtsY NG domains. The structure is similar to that of the GMPPCP and GDP:AlF4 stabilized complexes, but demonstrates that the GMPPNP complex can be readily obtained under certain conditions - here, the removal of the 20 amino acid N-terminal peptide of FtsY – and that the three different nucleotide analogs are readily accommodated in similar ways within the shared active site chamber of the SRP GTPase heterodimer. These structures lay the groundwork for subsequent kinetic, thermodynamic, and mutagenesis-based analyses of the mechanism of assembly and regulation of the SRP GTPase targeting heterodimer.
Database Accession
The atomic coordinates and structure factors have been deposited at the Protein Data Bank with ids: 2j7p and r2j7psf, respectively.
Acknowledgments
We thank Ivan Kruk and Pamela Focia for assistance with data collection. This work was supported by grant GM058500 from the NIH, and by support from the R.H. Lurie Comprehensive Cancer Center to the Structural Biology Facility at Northwestern University. Use of SBC beamline 19ID and the support of Norma Duke is gratefully acknowledged. The Argonne National Laboratory Structural Biology Center beamlines at the Advanced Photon Source are supported by the U.S. Department of Energy, Office of Energy Research, under Contract No. W-31-109-ENG-38.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bernstein HD, Poritz MA, Strub K, Hoben PJ, Brenner S, Walter P. Model for signal sequence recognition from amino-acid sequence of 54K subunit of signal recognition particle. Nature. 1989;340:482–486. doi: 10.1038/340482a0. [DOI] [PubMed] [Google Scholar]
- Burmeister WP. Structural changes in a cryo-cooled protein crystal owing to radiation damage. Acta Crystallogr D Biol Crystallogr. 2000;56:328–341. doi: 10.1107/s0907444999016261. [DOI] [PubMed] [Google Scholar]
- DeLano WL. The PyMOL Molecular Graphics System. San Carlos, CA, USA.: DeLano Scientific; 2002. [Google Scholar]
- Egea PF, Shan SO, Napetschnig J, Savage DF, Walter P, Stroud RM. Substrate twinning activates the signal recognition particle and its receptor. Nature. 2004;427:215–221. doi: 10.1038/nature02250. [DOI] [PubMed] [Google Scholar]
- Focia PJ, Gawronski-Salerno J, Coon VJ, Freymann DM. Structure of a GDP:AlF(4) Complex of the SRP GTPases Ffh and FtsY, and Identification of a Peripheral Nucleotide Interaction Site. J Mol Biol. 2006;360:631–643. doi: 10.1016/j.jmb.2006.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Focia PJ, Shepotinovskaya IV, Seidler JA, Freymann DM. Heterodimeric GTPase core of the SRP targeting complex. Science. 2004;303:373–377. doi: 10.1126/science.1090827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freymann DM, Keenan RJ, Stroud RM, Walter P. Structure of the conserved GTPase domain of the signal recognition particle. Nature. 1997;385:361–364. doi: 10.1038/385361a0. [DOI] [PubMed] [Google Scholar]
- Gawronski-Salerno J, Coon JSV, Focia PJ, Freymann DM. X-ray structure of the T. aquaticus FtsY:GDP complex suggests functional roles for the C-terminal helix of the SRP GTPases. Proteins: Struct Funct Bioinfor. 2006 doi: 10.1002/prot.21200. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson S, Singh R, Gudkov AT, Liljas A, Logan DT. Crystal structure of a mutant elongation factor G trapped with a GTP analogue. FEBS Lett. 2005;579:4492–4497. doi: 10.1016/j.febslet.2005.07.016. [DOI] [PubMed] [Google Scholar]
- Harding MM. Metal-ligand geometry relevant to proteins and in proteins: sodium and potassium. Acta Crystallogr D Biol Crystallogr. 2002;58:872–874. doi: 10.1107/s0907444902003712. [DOI] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr sect A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- Keenan RJ, Freymann DM, Stroud RM, Walter P. The Signal Recognition Particle. Annu Rev Biochem. 2001;70:755–775. doi: 10.1146/annurev.biochem.70.1.755. [DOI] [PubMed] [Google Scholar]
- Kraulis PJ. Molscript - a program to produce both detailed and schematic plots of protein structures. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- Lamzin VS, Perrakis A, Wilson KS. The ARP/wARP suite for automated construction and refinement of protein models. In: Rossman MG, Arnold E, editors. International Tables for Crystallography. The Netherlands: Dordrecht, Kluwer Academic Publishers; 2001. pp. 720–722. [Google Scholar]
- Luirink J, Dobberstein B. Mammalian and Escherichia coli signal recognition particles. Mol Microbiol. 1994;11:9–13. doi: 10.1111/j.1365-2958.1994.tb00284.x. [DOI] [PubMed] [Google Scholar]
- Luirink J, High S, Wood H, Giner A, Tollervey D, Dobberstein B. Signal-sequence recognition by an Escherichia coli ribonucleoprotein complex. Nature. 1992;359:741–743. doi: 10.1038/359741a0. [DOI] [PubMed] [Google Scholar]
- Lütcke H, High S, Römisch K, Ashford AJ, Dobberstein B. The methionine-rich domain of the 54 kDa subunit of signal recognition particle is sufficient for the interaction with signal sequences. Embo J. 1992;11:1543–1551. doi: 10.1002/j.1460-2075.1992.tb05199.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandon EC, Jiang Y, Gilmore R. Dual recognition of the ribosome and the signal recognition particle by the SRP receptor during protein targeting to the endoplasmic reticulum. J Cell Biol. 2003;162:575–585. doi: 10.1083/jcb.200303143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merritt EA, Bacon DJ. Raster3D Photorealistic Molecular Graphics. Methods Enzymol. 1997;277 doi: 10.1016/s0076-6879(97)77028-9. [DOI] [PubMed] [Google Scholar]
- Miller JD, Bernstein HD, Walter P. Interaction of E. coli Ffh/4.5S ribonucleoprotein and FtsY mimics that of mammalian signal recognition particle and its receptor. Nature. 1994;367:657–659. doi: 10.1038/367657a0. [DOI] [PubMed] [Google Scholar]
- Miller JD, Walter P. A GTPase cycle in initiation of protein translocation across the endoplasmic reticulum membrane. Ciba Found Symp. 1993;176:147–159. doi: 10.1002/9780470514450.ch10. discussion 159–163. [DOI] [PubMed] [Google Scholar]
- Miller JD, Wilhelm H, Gierasch L, Gilmore R, Walter P. GTP binding and hydrolysis by the signal recognition particle during initiation of protein translocation. Nature. 1993;366:351–354. doi: 10.1038/366351a0. [DOI] [PubMed] [Google Scholar]
- Montoya G, Svensson C, Luirink J, Sinning I. Crystal structure of the NG domain from the signal-recognition particle receptor FtsY. Nature. 1997;385:365–369. doi: 10.1038/385365a0. [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of Macromolecular Structures by the Maximum-Likelihood Method. Acta Cryst. 1997;D53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. In: Carter JCW, Sweet RM, editors. Macromolecular Crystallography, part A. New York: Academic Press; 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- Peluso P, Herschlag D, Nock S, Freymann DM, Johnson AE, Walter P. Role of 4.5S RNA in Assembly of the Bacterial Signal Recognition Particle with Its Receptor. Science. 2000;288:1640–1643. doi: 10.1126/science.288.5471.1640. [DOI] [PubMed] [Google Scholar]
- Powers T, Walter P. Reciprocal stimulation of GTP hydrolysis by two directly interacting GTPases. Science. 1995;269:1422–1424. doi: 10.1126/science.7660124. [DOI] [PubMed] [Google Scholar]
- Powers T, Walter P. Co-translational protein targeting catalyzed by the Escherichia coli signal recognition particle and its receptor. Embo J. 1997;16:4880–4886. doi: 10.1093/emboj/16.16.4880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez UD, Freymann DM. Analysis of protein hydration in ultra-high resolution structures of the SRP GTPase. Ffh Acta Cryst Sect D. 2006 doi: 10.1107/S0907444906040807. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez UD, Minasov G, Focia PJ, Stroud RM, Walter P, Kuhn P, Freymann DM. Structural basis for mobility in the 1.1 A crystal structure of the NG domain of Thermus aquaticus Ffh. J Mol Biol. 2002;320:783–799. doi: 10.1016/s0022-2836(02)00476-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapiejko PJ, Gilmore R. Protein translocation across the ER requires a functional GTP binding site in the alpha subunit of the signal recognition particle receptor. J Cell Biol. 1992;117:493–503. doi: 10.1083/jcb.117.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapiejko PJ, Gilmore R. Empty site forms of the SRP54 and SR alpha GTPases mediate targeting of ribosome-nascent chain complexes to the endoplasmic reticulum. Cell. 1997;89:703–713. doi: 10.1016/s0092-8674(00)80253-6. [DOI] [PubMed] [Google Scholar]
- Richardson JS, Arendall WB, Richardson DC. New tools and data for improving structures, using all-atom contacts. Meth Enzym. 2003:385–412. doi: 10.1016/s0076-6879(03)74018-x. [DOI] [PubMed] [Google Scholar]
- Römisch K, Webb J, Herz J, Prehn S, Frank R, Vingron M, Dobberstein B. Homology of the 54K protein of signal recognition particle, docking protein, and two E. coli proteins with putative GTP-binding domains. Nature. 1989;340:478–482. doi: 10.1038/340478a0. [DOI] [PubMed] [Google Scholar]
- Saenger W. Principles of Nucleic Acid Structure. New York: Springer-Verlag; 1984. [Google Scholar]
- Scheidig AJ, Burmeister C, Goody RS. The pre-hydrolysis state of p21ras in complex with GTP: new insights into the role of water molecules in the GTP hydrolysis reaction of ras-like proteins. Structure. 1999;7:1311–1324. doi: 10.1016/s0969-2126(00)80021-0. [DOI] [PubMed] [Google Scholar]
- Scrima A, Wittinghofer A. Dimerisation-dependent GTPase reaction of MnmE: how potassium acts as GTPase-activating element. Embo J. 2006;25:2940–2951. doi: 10.1038/sj.emboj.7601171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan SO, Stroud RM, Walter P. Mechanism of association and reciprocal activation of two GTPases. PLoS Biol. 2004;2:e320. doi: 10.1371/journal.pbio.0020320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepotinovskaya IV, Focia PJ, Freymann DM. Crystallization of the GMPPCP complex of the NG domains of T. aquaticus Ffh and FtsY. Acta Crystallogr D. 2003;59:1834–1837. doi: 10.1107/s0907444903016573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepotinovskaya IV, Freymann DM. Conformational change of the N-domain on formation of the complex between the GTPase domains of Thermus aquaticus Ffh and FtsY. Biochim Biophys Acta. 2002;1597:107–114. doi: 10.1016/s0167-4838(02)00287-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W, Raden D, Mandon EC, Gilmore R. Role of Sec61alpha in the regulated transfer of the ribosome-nascent chain complex from the signal recognition particle to the translocation channel. Cell. 2000;100:333–343. doi: 10.1016/s0092-8674(00)80669-8. [DOI] [PubMed] [Google Scholar]
- Storoni LC, McCoy AJ, Read RJ. Likelihood-enhanced fast rotation functions. Acta Cryst. 2004;D60:432–438. doi: 10.1107/S0907444903028956. [DOI] [PubMed] [Google Scholar]
- Walter P, Johnson AE. Signal Sequence Recognition and Protein Targeting to the Endoplasmic Reticulum Membrane. Annu Rev Cell Biol. 1994;10:87–119. doi: 10.1146/annurev.cb.10.110194.000511. [DOI] [PubMed] [Google Scholar]
- Zopf D, Bernstein HD, Johnson AE, Walter P. The methionine-rich domain of the 54 kd protein subunit of the signal recognition particle contains an RNA binding site and can be crosslinked to a signal sequence. EMBO J. 1990;9:4511–4517. doi: 10.1002/j.1460-2075.1990.tb07902.x. [DOI] [PMC free article] [PubMed] [Google Scholar]