

Abstract
Pseudomonas aeruginosa is a major cause of nosocomial respiratory infections. The eradication of P. aeruginosa from the lung involves the orchestrated actions of the pulmonary epithelium and both resident and recruited immune cells. The NKG2D receptor is constitutively expressed on the surface of circulating and tissue-resident NK cells (and other cytotoxic lymphocytes), and is capable of controlling NK cell activation and production of cytokines such as IFN-γ via interactions with ligands expressed on the surface of stressed cells. Previously, we demonstrated that NKG2D mediates pulmonary clearance of P. aeruginosa. In the present study, we investigated the cellular and molecular mechanisms of NKG2D-mediated clearance of P. aeruginosa using a novel transgenic mouse model of doxycycline (DOX)-inducible conditional expression of NKG2D ligands (Raet1a) in pulmonary epithelial cells. NKG2D ligand expression in this model increased pulmonary clearance, cellular phagocytosis, and survival following P. aeruginosa respiratory infection. Additionally, NK cell sensitivity to ex vivo LPS stimulation was greater in lung cells isolated from naive transgenic mice administered DOX. We also showed that NK cells are the primary source of lymphocyte-derived IFN-γ in response to P. aeruginosa respiratory infection. Significantly, we demonstrated that NKG2D is critical to the non-redundant IFN-γ production by pulmonary NK cells following acute P. aeruginosa infection. These results represent the principal report of NKG2D-mediated activation of lung NK cells following respiratory infection with an opportunistic pathogen and further establish the importance of NKG2D in the host response against P. aeruginosa respiratory infection.
Keywords: Natural killer cells, bacterial, lung, transgenic/knockout mice
Introduction
Pseudomonas aeruginosa is a ubiquitous, gram-negative opportunistic pathogen that is a major causative microorganism of nosocomial respiratory infections (1). Importantly, immunocompromised patients are at increased risk for P. aeruginosa infection, and it is the predominant cause of morbidity and mortality in patients with cystic fibrosis (CF) (2–4). P. aeruginosa is a frequently identified pathogen in patients with ventilator-associated pneumonia (a severe complication of intensive care), and has a high mortality rate compared to other pathogens (34–48%) (5). Additionally, P. aeruginosa is associated with exacerbations of chronic obstructive pulmonary disease (COPD) (6). The pathogenesis of P. aeruginosa lung infection is complex, and because of its ubiquitous nature and ability to develop resistance to antibiotics, P. aeruginosa respiratory infections are problematic and difficult to treat.
The clearance of P. aeruginosa from the airways involves the coordinated effort of multiple cell types, including the respiratory epithelium and both resident and recruited immune/inflammatory cells. NK cells are cytotoxic lymphocytes generally recognized for their crucial role in the innate immune response against viral infections and tumors (7). Persistent bacterial infections occurring in NK cell-deficient patients underscore the clinical importance of these cells in the immune response to bacterial pathogens (8). Although the cytotoxic function of NK cells seems to be minor during bacterial infections, their production of cytokines is significant. In particular, lung NK cell-derived IFN-γ plays an important role in expunging various types of pulmonary bacterial infections (9–11). However, the role of NK cells and NK cell-derived IFN-γ in the eradication of P. aeruginosa respiratory infection is unclear.
NK cell activation is controlled by a balance of signals between activating and inhibitory receptors. The NKG2D activating receptor is constitutively expressed on the surface of circulating and tissue-resident NK cells and other cytotoxic lymphocytes (12–14), and NKG2D activation stimulates cytotoxic effects of these cells against virally infected, transformed, or stressed cells in vitro and in vivo (15). Importantly, the recognition of NKG2D ligands also induces production of several cytokines, including IFN-γ (16, 17). NKG2D binds distinct but structurally related ligands based on recognition of structural patterns (18, 19). Several families of NKG2D ligands have been identified in mice and include retinoic acid early transcript 1, α to ε (Raetl, b, c, d, e), histocompatibility 60 (H60), Ulbp1 (20, 21), and MHC I like leukocyte 1 (Mill1) (22).
Previously, we reported that NKG2D ligands are expressed on stressed airway epithelial cells (23). Our laboratory also provided the first evidence that P. aeruginosa is a potent inducer of NKG2D ligands in pulmonary epithelial cells following in vitro and in vivo infection (24). Importantly, we also showed that NKG2D receptor blockade inhibited pulmonary clearance of P. aeruginosa in mice, indicating that NKG2D effector function is required for a complete host response. In the current study, we investigated the cellular and molecular mechanisms of NKG2D-mediated pulmonary clearance of P. aeruginosa using a novel mouse model of doxycycline (DOX)-inducible conditional expression of Raet1a in pulmonary epithelial cells. Using this model, we determined a role for NKG2D ligand expression in bacterial clearance, cellular phagocytosis, and survival following acute P. aeruginosa respiratory infection. Moreover, we demonstrate that NKG2D ligand expression increases NK cell sensitivity to bacterial products, and that NKG2D is critical for IFN-γ production by NK cells following acute P. aeruginosa infection.
Materials and Methods
Mice
Ccsp-rtta transgenic mice (FVB/NJ background) were previously generated by Tichelaar and colleagues (25). Mice bearing the bearing the target (tetO)7-CMV-Raet1a transgene were generated at the University of Cincinnati Medical Center using RAET1A cDNA obtained by PCR cloning as previously described (25). Bi-transgenic Ccsp-rtta x (tetO)7-CMV-Raet1a mice (hereafter referred to as Raet1a-tg mice) were identified using PCR primers specific for each transgene as follows: the Ccsp-rtta forward primer was 5′-ACT GCC CAT TGC CCA AAC AC-3′, and the reverse primer was 5′-AAA ATC TTG CCA GCT TTC CCC-3′; the (tetO)7-CMV-Raet1a transgene forward primer was 5′-TAG TTG CCA GCC ATC TGT TGT T-3′, and the reverse primer was 5′-TCC TCC CCC TTG CTG TCC-3′. Amplification of PCR products for both Ccsp-rtta and the (tetO)7-CMV-Raet1a transgene was performed by denaturation at 95 °C for 2 min and then 35 cycles of amplification at 95°C for 45 s, 58°C for 45 s, and 72°C for 45 s, followed by extension at 72 °C for 5 min. Male and female Raet1a-tg mice administered DOX were between 8 and 10 weeks of age, and DOX was administered in the diet for 7 d prior to experimental use [TD.01306 rodent diet (2018, 625 DOX), Harlan Teklad, Madison, WI]. FVB/NJ mice (female, age 6–10 weeks) used in these studies were purchased from The Jackson Laboratory (Bar Harbor, ME), and housed in our animal facilities ≥ 1 week prior to use. All mice were housed under pathogen-free conditions in accordance with institutional guidelines, and all experimental protocols were reviewed and approved by the Institutional Animal Care and Use Committee at the University of Cincinnati College of Medicine.
RAET1 immunohistochemistry
Naïve Raet1a-tg mice were euthanized with an i.p. injection of sodium pentobarbital (Nembutal, 150–200 mg/kg, Henry Schein, Indianapolis, IN) followed by exsanguination via severing of the posterior abdominal aorta. Lung tissue was obtained, processed, and immunostained for RAET1 as described previously (24).
P. aeruginosa inoculation
The stationary-phase P. aeruginosa strain PAO1 (26), a burn would isolate (strain SBI-N), and a PAO1 strain harboring a green fluorescent protein-expressing plasmid (PAO1-GFP) were used. Briefly, isolated single colonies grown on tryptic soy agar plates were inoculated in Luria broth (LB) followed by overnight incubation of shaken cultures at 37°C. The optical density of cultures at 600 nm were diluted with LB to 1.5, and P. aeruginosa was harvested by centrifugation (6,800 × g, 3 min) followed by three 1 ml washes and an appropriate final dilution with sterile PBS without Ca2+ and Mg2+. All mice were briefly anesthetized with isoflurane and infected intranasally with a 20 μl suspension.
Bacterial enumeration
Sixteen hours post-infection with 1 × 107 CFU PAO1 or SBI-N, Raet1a-tg mice were euthanized as described above, and the lungs were harvested and homogenized in 1 ml of PBS with a Pyrex Tenbroeck tissue grinder. Serial dilutions of lung homogenates diluted in PBS were plated onto tryptic soy agar plates, incubated overnight at 37°C, and individual colony counts in terms of log10 CFU were determined.
Cellular phagocytosis
After 2 h of infection with 5 × 108 CFU PAO1 constitutively expressing GFP, Raet1a-tg mice were euthanized as described above, and the lungs were lavaged with two 1-ml aliquots of Hanks’ balanced salt solution without Ca2+ and Mg2+ (pH 7.2, 37°C, Invitrogen, Carlsbad, CA). Total cell counts were performed with a hemacytometer using trypan blue (Invitrogen). Recovered bronchoalveolar lavage (BAL) fluid samples were then centrifuged (400 × g, 10 min, 4°C), and the cell pellets washed with 2 ml FACS buffer (0.5% BSA/0.05% sodium azide in PBS) prior to flow cytometry. Cells were re-suspended in 100 μl FACS buffer and incubated with 1 μg of purified Mouse BD Fc Block [rat anti-mouse CD16/CD32 (clone 2.4G2, BD Biosciences, Franklin Lakes, NJ)] for 10 min at 4°C. For cell surface staining, cells recovered from the bronchoalveolar lavage (i.e., BAL cells) were incubated with allophycocyanin (APC)-conjugated anti-mouse F4/80 [pan macrophage marker (clone BM8, eBioscience, San Diego, CA)] or APC-conjugated rat IgG2a isotype control (eBioscience) for 30 min on ice. Cells were washed and resuspended in 500 μl FACS buffer. Flow cytometry was performed immediately using a BD FACSCalibur system, and the data were analyzed using BD CellQuest Pro software. BAL cells were identified based on autofluorescent (AF) properties in the FL2 channel as well as cell surface expression of F4/80 as follows: F4/80+(i.e., alveolar macrophages, monocytes), AF-/F4/80- (i.e., neutrophils and lymphocytes). Phagocytosis of PAO1-GFP was detected by increased fluorescence in the FL1 channel.
Characterization of NKG2D+ lymphocytes in the lung
Naïve FVB/NJ mice were euthanized and the lungs were then voided of blood by perfusion through the right ventricle with 10 ml PBS containing 0.6 mM EDTA. Lungs were withdrawn aseptically from the chest cavity, washed with PBS, diced into pieces with a total volume ≤ 300 μl, and digested in 5 ml RPMI 1640 with 2.05 mM L-glutamine (HyClone, Logan, UT) containing 175 U/ml collagenase I-A, 0.2 U/ml pancreatic elastase, 35 U/ml hyaluronidase, 20 kU/ml DNase I (Sigma-Aldrich, St. Louis, MO), 10% fetal calf serum, 100 U/ml penicillin, and 100 μg/ml streptomycin (MP Biomedicals, Solon, OH) for 1 h at 37°C on an orbital shaker (60 rpm). The digested lungs were sheared through 19- and 21-gauge needles, and filtered through 40 μm cell strainers (BD Biosciences) to obtain a single-cell suspension. Residual RBCs were lysed with RBC Lysis Solution (Qiagen, Valencia, CA), and cells were then centrifuged in 30% Percoll (Sigma-Aldrich). Cells were then washed, resuspended in 100 μl FACS buffer, and incubated with 1 μg of purified Mouse BD Fc Block at 4°C for 10 min. The following antibodies were used for cell surface staining of lymphocytes: APC-conjugated rat anti-mouse NKG2D (clone CX5, eBioscience), PE-conjugated rat anti-mouse CD8a (clone 53-6.7, BD Biosciences), PerCP-Cy5.5-conjugated hamster anti-mouse CD3e (clone 145-2C11, BD Biosciences), FITC-conjugated rat anti-mouse CD4 (clone GK1.5, BD Biosciences), FITC-conjugated TCR γδ (clone GL3, BD Biosciences), and goat anti-mouse NKp46 (AF2225, R&D Systems, Minneapolis, MN). For cell surface staining of NKp46, cells were washed, resuspended in 100 μl FACS buffer, and incubated with APC-conjugated donkey anti-goat IgG secondary antibody (F0108, R&D Systems) for 30 min on ice. Additionally, the appropriate isotype control antibodies were utilized as follows: PE rat IgG1 (12-4301, eBioscience), FITC hamster IgG2 (clone B81-3, BD Biosciences), and PerCP-Cy5.5 hamster IgG1 (clone A19-3, BD Biosciences). After staining, cells were washed and fixed in 500 μl of 2% paraformaldehyde and flow cytometry was performed as described above.
Ex vivo NK cell activation
Naïve Raet1a-tg mice were euthanized and the lungs harvested, digested, and RBCs removed as described above. The cells were then resuspended in RPMI 1640 with 2.05 mM L-glutamine (HyClone) containing 10% FBS, 1% sodium pyruvate, 100 μg/ml kanamycin, 0.05 mM 2-mercaptoethanol, 1X non-essential amino acids (MP Biomedicals), and 20 U/ml recombinant mouse IL-2, and 5 × 105 cells/well were aliquoted into a 96-well round-bottom culture plate (Costar, Corning, NY). The cells were stimulated with 1, 10, and 100 ng/ml LPS from Escherichia coli O111:B4 (L5293, Sigma-Aldrich) or sterile PBS (unstimulated) for 22 h at 37°C in a humidified incubator containing 5% CO2. Brefeldin A (10 μg/ml, eBioscience) was added for the final 4 h of incubation. The cells were washed, re-suspended in 100 μl FACS buffer, and incubated with 1 μg of purified Mouse BD Fc Block at 4°C for 10 min. Cell surface staining of NKp46 was performed as described above. The cells were then fixed in 100 μl fixation buffer (2% paraformaldehyde) for 20 min at room temperature, and then washed with permeabilization buffer [FACS buffer containing 0.1% saponin (MP Biomedicals)]. The cells were resuspended in 100 μl permeabilization buffer containing 10% mouse serum (MP Biomedicals), and incubated for 15 min at 4°C to block. After washing, cells were again resuspended in 100 μl permeabilization buffer and stained intracellularly with FITC-conjugated anti-mouse IFN-γ (clone XMG1.2, eBioscience) or FITC-conjugated rat IgG1 isotype control (clone A110-1, BD Biosciences) for 30 min on ice. Cells were washed and fixed in 500 μl of 2% paraformaldehyde and flow cytometry was performed.
Flow cytometric analysis of NK cell recruitment
Four and 16 h post-infection with 1 × 107 CFU PAO1, FVB/NJ mice were euthanized and the lungs lavaged, harvested, digested, and the RBCs removed as described above. Cells from the BAL and whole lung compartments were stained for NKp46 as described above. The cells were washed and fixed in 500 μl of 2% paraformaldehyde and flow cytometry was performed.
Intracellular cytokine staining of NK cell-derived IFN-γ
Four hours post-infection with 1 × 107 CFU PAO1, FVB/NJ mice were euthanized and the lungs harvested, digested, the RBCs removed, and aliquoted into 96-well round-bottom culture plates as described above. The cells were maintained at 37°C overnight in a humidified incubator containing 5% CO2, and brefeldin A (10 μg/ml, eBioscience) was added for the final 4 h of incubation. Lung NK cell-derived IFN-γ was determined by flow cytometry as described above. Cells were washed and fixed in 500 μl of 2% paraformaldehyde and flow cytometry was performed.
Anti-NKG2D administration
NKG2D receptor function was blocked by administration of functional grade purified anti-mouse NKG2D (clone CX5, eBioscience) as described previously (24, 27). The CX5 antibody blocks NKG2D ligand-binding and inhibits NK cell function in vitro and in vivo (27, 28). Briefly, 16 h prior to infection with 1 × 107 CFU PAO1 or SBI-N, FVB/NJ mice were given an i.p. injection of 100 μg anti-NKG2D or functional grade purified rat IgG1 isotype control (16-4301, eBioscience). Lung NK cell-derived IFN-γ was determined by flow cytometry.
Statistical analyses
Significant differences among groups were identified by analysis of variance (ANOVA)wherever appropriate, and individual comparisons between groups were confirmed by a post hoc Tukey test. For the survival study, a Peto-Peto test was utilized to assess a significant difference in survival between groups. For all analyses, a p value < 0.05 was considered statistically significant.
Results
Tramgenic expression of NKG2D ligands
To characterize the in vivo pulmonary response elicited by overexpression of NKG2D ligands following acute P. aeruginosa infection, we generated a transgenic mouse model that allowed for the conditional expression of Raet1a in pulmonary epithelial cells. We expressed Raet1a in pulmonary epithelial cells under the control of DOX administration using the Ccsp-rtta transgenic system previously described (25). We established three separate transgenic Raet1a-tg mouse lines (Lines 20, 22, and 32) bearing the target (tetO)j-CMV-Raet1a transgene. Line 22 Raet1a-tg mice exhibited robust Raet1a transgene induction, and these mice were used in all the studies presented here. Immunohistochemical staining of lung tissue from Line 22 Raet1a-tg mice confirmed RAET1 expression was not evident in mice that did not receive DOX (Figure 1, A and C), but is induced throughout the airway and alveolar epithelium following DOX administration (Figure 1, B and D).
Figure 1.
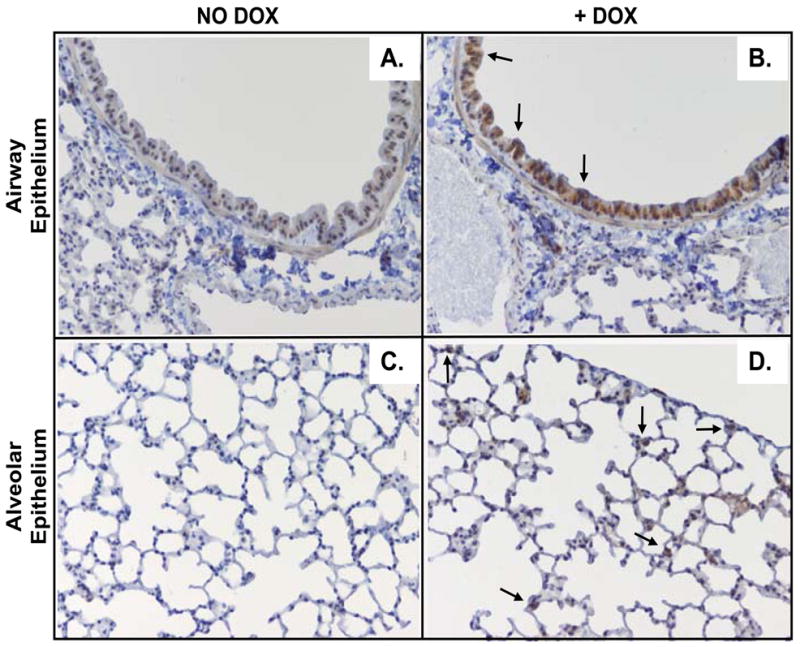
Conditional transgenic expression of the NKG2D ligand Raet1a in the lungs of mice. Immunohistochemical analysis of RAET1 was performed using sections of paraffin-embedded lungs from naive Raet1a-tg mice treated with or without doxycycline (DOX). Photomicrographs are of large airways of mice not administered doxycycline (DOX) (A) or administered DOX (B), as well as the lung parenchyma of mice not administered DOX (C) or administered DOX (D). Photomicrographs are representative of 5 mice/group (original magnification 200X).
NKG2D ligand expression increases pulmonary clearance and survival following P. aeruginosa respiratory infection
We have previously shown that NKG2D receptor blockade inhibits pulmonary clearance of P. aeruginosa (24). To assess whether overexpression of Raelta ameliorates the eradication of P. aeruginosa from the lungs, Raet1a-tg mice treated with or without DOX were intranasally infected with 1 × 107 CFU PAO1 or SBI-N. At 16-h post-infection, Raet1a-tg mice administered DOX exhibited significantly greater clearance of PAO1 than mice that did not receive DOX (Figure 2A). Similar to PAO1, Raet1a-tg mice treated with DOX exhibited significantly greater clearance of SBI-N than mice that did not receive DOX (Figure 2B). For control purposes, single transgenic mice that were positive for the (tetO)7-CMV-Raet1a transgene but did not express the Ccsp-rtta transgene were also treated with or without DOX. In these studies, no differences were observed between DOX treatment groups post-infection, indicating that administration of the DOX antibiotic had no effect on any endpoints in this study (data not shown). Additionally, all experiments using Raet1a-tg mice were performed on at least two occasions, and the results were reproducible across experiments.
Figure 2.

NKG2D ligand expression increases pulmonary clearance and survival following acute P. aeruginosa respiratory infection. Raet1a-tg mice treated with or without doxycycline (DOX) were intranasally infected with 1 × 107 CFU PAO1 (A) and SBI-N (B) and bacterial CFU in the lungs were assessed 16 h post-infection. Data are presented as means ± SEM (n = 5–7 mice/group). * Denotes significant difference from PAO1-infected Raet1a-tg mice not administered DOX, P< 0.05.
Raet1a-tg mice treated with or without DOX were intranasally infected with 1 × 109 CFU PAO1, and survival was monitored for up to 96 h post-infection. Raet1a-tg mice administered DOX were less susceptible and survived significantly longer than mice that did not receive DOX (p = 0.048, Figure 3). All Raet1a-tg mice that did not receive DOX succumbed to lethal PAO1 infection by 31 h (n = 9). In contrast, 2 out of 10 Raet1a-tg mice that were administered DOX were still alive after 96 h.
Figure 3.

NKG2D ligand expression increases survival following a lethal infection with P. aeruginosa. Raet1a-tg mice treated with (n = 10) or without doxycycline (DOX) (n = 9) were intranasally infected with 1 × 109 CFU PAO1, and survival times were recorded. The survival curves of mice administered DOX versus mice not administered DOX were significantly different as determined by a Peto-Peto test, P < 0.05.
NKG2D ligand expression increases phagocytosis of P. aeruginosa in BAL cells
Phagocytosis is an important process in the clearance of bacteria. To determine whether overexpression of Raelta increased phagocytosis of P. aeruginosa, Raet1a-tg mice treated with or without DOX were infected intranasally with 5 × 108 CFU PAO1-GFP for 2 h, and phagocytosis of PAO1-GFP in BAL cells was assessed. No differences in cellularity as determined by autofluorescent (AF) properties in the FL2 channel as well as cell surface expression of F4/80 were observed between uninfected (control) Raet1a-tg mice treated with and without DOX (data not shown). Following infection with PAO1-GFP, Raet1a-tg mice administered DOX exhibited significantly greater cellular phagocytosis of PAO1-GFP than mice that did not receive DOX. Specifically, total PAO1-GFP+ cells recovered from the BAL, as well as BAL cells characterized as PAO1-GFP+/F4/80+ and PAO1-GFP+/AFF4/80−, were greater in Raet1a-tg mice administered DOX as compared to those mice that were not given DOX (Figure 4).
Figure 4.

NKG2D ligand expression increases phagocytosis of P. aeruginosa in bronchoalveolar lavage (BAL) cells. BAL cells were recovered from Raet1a-tg mice treated with or without doxycycline (DOX) intranasally infected with 5 × 108 CPU PAO1-GFP, and phagocytosis of PAO1-GFP by BAL cells was determined by flow cytometry 2 h post-infection. BAL cells were identified based on autofluorescent (AF) properties in the FL2 channel as well as cell surface expression of F4/80 (APC-conjugated Ab). Phagocytosis of PAO1-GFP was detected by increased fluorescence in the FL1 channel. * Denotes significant difference from PAOl-GFP-infected Raet1a-tg mice not administered DOX, p < 0.05.
NK cells constitute the majority of NKG2D+ pulmonary lymphocytes
NK cells, CD8+ T cells, and γδ+ T cells express NKG2D in mice (12–14). To thoroughly survey the abundance of NKG2D-expressing lymphocytes in the lungs of FVB/NJ mice, flow cytometry was performed on isolated lung lymphocytes. NK cells were defined as NKp46+ cells in the lymphocyte gate. Although CD4+ T cells > NK cells > CD8+ T cells > γδ+ T cells in terms of the relative abundance of pulmonary lymphocytes (Figure 5A), only NK cells and γδ T cells were predominantly NKG2D+ (Figure 5B). Furthermore, NK cells comprised the greatest percentage of total NKG2D+ pulmonary lymphocytes (Figure 5C), with γδ+ T cells, CD4+ T cells, and CD8+ T cells accounting for a small proportion of the NKG2D+ population.
Figure 5.

Characterization of NKG2D expression on pulmonary lymphocytes. Lung cells were harvested from naive FVB/NJ mice, and lymphocytes were isolated and cell surface stained with the following Abs: APC-conjugated NKG2D, PE-conjugated CD8a, PerCP-Cy5.5-conjugated CD3e, FITC-conjugated CD4, FITC-conjugated TCR γ δ, and unconjugated NKp46 (followed by an APC-conjugated secondary Ab to distinguish NK cells), and cells were analyzed by flow cytometry. The percentage of pulmonary lymphocytes (A), the percentage of lymphocyte populations that express NKG2D (B), and the percentage of total NKG2D+ lymphocyte populations (C) are presented as means ± SEM (n = 3 mice).
NKG2D ligand expression increases NK-cell derivedIFN-γfollowing ex vivo LPS stimulation
To determine whether overexpression of Rae1ta altered intracellular IFN-γ levels in resident lung lymphocytes, lung cells were isolated from naive Raet1a-tg mice treated with or without DOX, stimulated ex vivo in culture with 1, 10, and 100 ng/ml LPS for 4 and 22 h, and flow cytometry performed. LPS was utilized because it is a potent toll-like receptor (TLR) ligand known to induce intracellular IFN-gamma; in NK cells. There was no difference in the number of IFN-γ-producing NK cells (defined as NKp46+IFN-γ+ cells in the lymphocyte gate) between mice administered DOX as compared to mice that were not given DOX at baseline (i.e., unstimulated). After 22 h of LPS stimulation, there was a dose-dependent increase in the number of IFN-γ-producing NK cells from the lungs of mice treated with or without DOX (Figure 6). However, mice administered DOX had significantly more IFN-γ-producing NK cells compared to mice that did not receive DOX following stimulation with all 3 doses of LPS, with the greatest difference observed after stimulation with the lowest dose (1 ng/ml). LPS stimulation also induced IFN-γ in non-NK cell lymphocytes (defined as NKp46−IFN-γ+ cells in the lymphocyte gate) in a dose-dependent manner in lung cells from mice treated with DOX and untreated mice. No differences in LPS-induced non-NK cell IFN-γ production was observed between the DOX treatment groups.
Figure 6.

NKG2D ligand expression increases intracellular IFN-γ in NK cells following ex vivo stimulation with LPS. Lung cells were harvested from naïve Raet1a-tg mice treated with or without doxycycline (DOX) and stimulated with 1.0, 10, or 100 ng/ml LPS or sterile PBS (unstimulated) for 22 h, and the presence of IFN-γ-producing NK cells was assessed. For analysis by flow cytometry, cells underwent cell surface staining with an unconjugated NKp46 Ab and an APC-conjugated secondary Ab (to distinguish NK cells) followed by intracellular staining with a FITC-conjugated IFN-γ Ab. Numbers in the upper right quadrant are percentage of NK cells that are IFN-γ+ (NKp46+IFN-γ+). Numbers in the lower right quadrant are percentage of non-NK cell lymphocytes that are IFN-γ+ (NKp46−IFN-γ+). Dot plots shown are gated on lymphocytes and are representative of 2 mice per group from 4 independent experiments.
Lung NK cell recruitment following P. aeruginosa respiratory infection
FVB/NJ mice were intranasally infected with 1 × 107 CFU PAO1 (or left uninfected for control purposes), and the lungs were lavaged and then digested 4 and 16 h post-infection (n = 3 mice/group). Flow cytometry was then performed to determine the presence of NK cells (defined as NKp46+ cells in the lymphocyte gate) in the BAL and whole lung compartments. The total numbers of NK cells in the whole lung compartment were not increased over uninfected control values at either time point post-infection (Figure 7A). The total number of NK cells in the BAL compartment were not increased over control 4 h post-infection, but were significantly increased 16 h after infection (Figure 7B). However, NK cells in the BAL compartment at this time point post-infection represent only approximately 3.7% of the total number of NK cells in the entire lung.
Figure 7.

NK cell recruitment into the lung following acute P. aeruginosa respiratory infection. FVB/NJ mice were intranasally infected with 1×107 CFU PAO1, and total numbers of NK cells were assessed in the whole lung (A) and bronchoalveolar lavage (B) compartments 4 and 16 h post-infection. For analysis by flow cytometry, cells underwent cell surface staining with an unconjugated NKp46 Ab and an APC-conjugated secondary Ab to distinguish NK cells in the lymphocyte gate. Data are presented as means± SEM (n = 3 mice/group). * Denotes significant difference from uninfected (control) mice, p < 0.05.
P. aeruginosa preferentially induces IFN-γin NK cells following respiratory infection
NK cell-derived IFN-γ is important in the host response and eradication of many pathogens. To analyze the capacity of lung NK cells to produce IFN-γ following P. aeruginosa respiratory infection, FVB/NJ mice were intranasally infected with 1 × 107 CFU PAO1 for 4 h (or left uninfected for control purposes), and total lung cells were isolated and analyzed by flow cytometry to determine the presence of IFN-γ-producing NK cells (defined as NKp46+IFN-γ+ cells in the lymphocyte gate). Significantly more lung NK cells from PAO1-infected mice produced IFN-γ than similar cells from uninfected control mice (Figure 8). Furthermore, the number of IFN-γ-producing NK cells was five times greater than other IFN-γ-producing non-NK cell lymphocytes (defined as NKp46−IFN-γ+ cells in the lymphocyte gate) following infection.
Figure 8.

Preferential induction of IFN-γ n NK cells following acute P. aeruginosa respiratory infection. Lung cells were harvested from uninfected FVB/NJ mice (A) and mice intranasally infected with 1 × 107 CPU PAO1 for 4 h (B), and the presence of IFN-γ-producing NK cells was assessed. For analysis by flow cytometry, cells underwent cell surface staining with an unconjugated NKp46 Ab and an APC-conjugated secondary Ab (to distinguish NK cells) followed by intracellular staining with a FITC-conjugated IFN-γ Ab. Numbers in the upper right quadrant are percentage of NK cells that are IFN-γ+ (NKp46+IFN-γ+). Numbers in the lower right quadrant are percentage of non-NK cell lymphocytes that are IFN-γ+ (NKp46−IFN-γ+). Dot plots shown are gated on lymphocytes and are representative of 2–3 mice per group from 3 independent experiments.
NKG2D is critical to NK cell-derived IFN-γ production following P. aeruginosa respiratory infection
Since P. aeruginosa induces NIC-cell derived IFN-γ following respiratory infection and NKG2D ligand expression increases the number of NK cells producing IFN-γ following ex vivo stimulation with LPS, we hypothesized that blockade of NKG2D can abate IFN-γ production by lung NK cells in P. aemginosa-infected mice. FVB/NJ mice were administered an NKG2D blocking antibody 16 h prior to intranasal infection with 1 × 107 CFU PAO1 or SBI-N [or left uninfected for control purposes (Figure 9A)], and total lung cells were isolated 4 h post-infection and analyzed by flow cytometry. No differences in NK cell derived IFN-γ were observed between uninfected mice administered the anti-NKG2D antibody or between infected mice administered the IgGl isotype control antibody (data not shown). Significantly more lung NK cells from PAO1- and SBI-N-infected mice produced IFN-γ than NK cells from uninfected control mice (Figure 9, B and D). Mice pretreated with the anti-NKG2D antibody had significantly fewer IFN-γ-producing lung NK cells compared to mice that did not receive the antibody following respiratory infection with PAO1 (Figure 9, B and C). Compared to PAO1, mice pretreated with the anti-NKG2D antibody also had fewer IFN-γ-producing lung NK cells compared to mice that did not receive the antibody following respiratory infection with SBI-N (Figure 9, D and E).
Figure 9.

NK cell-derived IFN-γ production following P. aeruginosa respiratory infection is dependent upon NKG2D. Lung cells were harvested from uninfected FVB/NJ mice (A), mice intranasally infected with 1 × 107 CFU PAO1 (B) or SBI-N for 4 h (D) and mice administered an NKG2D blocking antibody (100 mg of CX5) 16 h prior to intranasal infection with 1 × 107 CFU PAO1 (C) or SBI-N for 4 h (E). The presence of IFN-γ-producing NK cells was assessed. For analysis by flow cytometry, cells underwent cell surface staining with an unconjugated NKp46 Ab and an APC-conjugated secondary Ab (to distinguish NK cells) followed by intracellular staining with a FITC-conjugated IFN-γ Ab. Numbers in the upper right quadrant are percentage of NK cells that are IFN-γ+ (NKp46+IFN-γ+). Numbers in the lower right quadrant are percentage of non-NK cell lymphocytes that are IFN-γ+ (NKp46−IFN-γ+). Dot plots shown are gated on lymphocytes and are representative of 2–3 mice per group from 3 independent experiments.
Discussion
Pulmonary epithelial cells are the initial line of defense against exposure to inhaled pathogens. However, our understanding of the role of these cells in signaling to resident mucosal immune cells is limited. We have previously established that NKG2D ligand expression is induced on pulmonary epithelial cells following acute P. aeruginosa respiratory infection, and NKG2D blockade inhibits the eradication of P. aeruginosa from the lungs (24). In the current study, we show that NKG2D-mediated pulmonary epithelial cell-lymphocyte interactions prior to respiratory infection enhance the host defense against P. aeruginosa. Furthermore, we demonstrate a critical role for NKG2D in the non-redundant production of IFN-γ by resident lung NK cells following acute P. aeruginosa infection.
Pulmonary epithelial cell-specific induction of NKG2D ligands enhanced the antibacterial defense of the lung as evidenced by increased bacterial clearance and cellular phagocytosis in our model of P. aeruginosa lung infection, and those outcomes could account for the observed increase in survival in DOX-treated Raet1a-tg mice infected with a lethal dose of PAO1. Because NK cells constitute the majority of resident NKG2D+ cells in the pulmonary submucosa and parenchyma of naïve FVB/NJ mice (Figure 4C), we initiated investigations on the role of NKG2D ligand expression on NK cell activation to determine the cellular and molecular mechanisms of NKG2D-mediated host defense against P. aeruginosa. In naïve Raet1a-tg mice, expression of NKG2D ligand by itself did not induce NK cell-derived IFN-γ. Thus, our ex vivo results demonstrate that resident lung NK cells are primed by NKG2D ligand expression to produce greater amounts of IFN-γ in response to pathogenic stress. We hypothesize that the priming of resident NK cell-derived IFN-γ can augment the early microbial effector functions of resident alveolar macrophages (e.g., phagocytosis) following acute P. aeruginosa lung infection. Our data implicate that pulmonary epithelial cell-NK cell interactions are vital in directing production of NK cell-derived IFN-γ at the mucosal interface, and these interactions within the local microenvironment are important in the early host defense against acute P. aeruginosa respiratory infection. Furthermore, priming of NK cell-activation by NKG2D ligand expression prior to pathogenic stress could also be clinically significant as a potential therapeutic strategy useful in the treatment of P. aeruginosa respiratory infection.
In addition to conditional expression of Raet1a in pulmonary epithelial cells prior to infection, there are likely alternative sources of NKG2D ligand production that are generated following acute P. aeruginosa respiratory infection in our Raet1a-tg model. Immunohistochemical analysis previously revealed that RAET1A was strongly induced in the conducting airway epithelium, alveolar epithelium, and alveolar macrophages of mice 24 h after P. aeruginosa lung infection (24). Thus, it is plausible that P. aeruginosa-induced NKG2D ligand expression on alveolar macrophages and epithelial cells may contribute to persistent NKG2D-mediated production of IFN-γ from resident NK cells (and perhaps those that infiltrate into the alveolar space) following respiratory infection. The induction of the NKG2D receptor-ligand system by P. aeruginosa could lead to sustained NKG2D-mediated upregulation of the microbial effector functions of infiltrating inflammatory cells at later time points post-infection, resulting in the facilitation of pulmonary clearance.
The data from our Raet1a-tg model suggests that the detection and response of pathogen by lung NK cells may involve interactions between NKG2D and TLRs. Multiple TLRs mediate P. aeruginosa recognition and signaling in vivo (29). Hamerman et al. demonstrated that TLR receptor stimulation could increase the expression of NKG2D ligands (30). Additionally, P. aeruginosa can directly induce NKG2D ligand expression on pulmonary epithelial cells in vitro(24). These observations, taken with our finding that NKG2D ligand expression increases NK cell sensitivity to LPS-induced TLR stimulation (as assessed by intracellular IFN-γ) led us to postulate that NKG2D and TLR co-stimulation creates an amplification loop following recognition of pathogen. In other words, NK cells have the capacity to detect the presence of pathogens indirectly via upregulation of NKG2D ligands, thus sensing stress via the NKG2D receptor-ligand system. This indirect signaling, in turn, could lower the threshold at which NK cells respond to the presence of pathogen directly through the engagement of TLRs. The activation threshold of non-NK cells was also lowered after LPS treatment, suggesting that the occurrence of NKG2D and TLR co-stimulation is not NK cell-specific. Our findings indicate that the NKG2D and TLR pathways operate in a coordinated effort in pathogen recognition and eradication. A more complete understanding of these pathways and the mechanisms controlling their communication is necessary.
Early, innate induction of IFN-γ is critical to immunological defense against multiple pathogens, and resident NK cells in the pulmonary submucosa and parenchyma are a prodigious source of IFN-γ. NK cell derived IFN-γ is induced in the host defense repertoire of several animal models of respiratory bacterial infection including Bordetella pertussis (9), Mycobacterium tuberculosis (10), and Shigella flexneri (11). However, compared to P. aeruginosa respiratory infection where NK cell-derived IFN-γ was the primary, non-redundant source of lymphocyte-derived IFN-γ, NK cells are not the principal source of lung lymphocyte-derived IFN-γ following B. pertussis, M. tuberculosis, and S. felxneri infections (e.g., T cells and NKT cells also produce redundant, significant amounts of IFN-γ). Thus, the early, non-redundant production of lung NK cell-derived IFN-γ is a unique feature of P. aeruginosa respiratory infection in our mouse model.
Different strains of P. aeruginosa can vary significantly in their virulence properties. For example, many clinical isolates derived from the lungs of CF patients lack the intercellular signaling system known as quorum sensing (31). Such P. aeruginosa strains are significantly impaired in murine models of burn and airway infection (32). Another significant component of P. aeruginosa virulence that involves direct contact of the bacterium to host cells is the type III secretion system (TTSS). TTSS involves a “syringe-like” apparatus that injects effector molecules (mostly toxins) into host cells (33). The type III apparatus was shown by Holder et al. to be critical for infection in a mouse burn model (34, 35). Because of the possibility of strain-specific virulence effects in our mouse model, we also employed P. aeruginosa strain SBI-N that is fully capable of both quorum sensing and TTSS to corroborate our findings with those obtained using PAO1. Raet1a-tg mice treated with DOX exhibited greater clearance of SBI-N than mice untreated mice in a manner similar to PAO1. Likewise, SBI-N-infected FVB/NJ mice pretreated with the anti-NKG2D antibody had fewer IFN-γ-producing lung NK cells compared to mice that did not receive the antibody. Compared to PAO1 respiratory infection and consistent with the increased virulence of strain SBI-N, SBI-N-infected Raet1a-tg mice exhibited a greater bacterial burden, as well as a greater NKG2D-mediated NK cell IFN-γ response in FVB/NJ mice. The findings suggest that NKG2D-regulated bacterial clearance and NK cell activation represent conserved outcomes in the host response to acute respiratory infection with different strains of P. aeruginosa that vary in their virulence characteristics in our mouse model.
NKp46 is selectively expressed by NK cells across multiple species, and creates a centralized phenotypic definition of NK cells based on NKp46 cell surface expression (36). As opposed to measuring IFN-γ in a mixed lymphocyte population or via ELISA in whole lung homogenates, we were able to specifically identify NK cells as the predominant lymphocyte producer of IFN-γ by utilizing the NKp46 cell surface maker in combination with isolated lung cells from infected mice. A great deal of support for IFN-γ in the clearance of P. aeruginosa has been previously proposed. Previously, Moser et al. showed an improved outcome of chronic P. aeruginosa respiratory infection in mice by an IFN-γ, Thl-dominated response (37). Rat models of P. aeruginosa respiratory infection show that pulmonary clearance is enhanced after pre-administration of exogenous IFN-γ via adenoviral vectors (38), and i.p. treatment with recombinant rat IFN-γ diminished the magnitude of pulmonary inflammation following P. aeruginosa infection (39). Of clinical importance, there is also mounting evidence that an IFN-γ, Thl-dominated immune response might improve the prognosis of CF patients with chronic P. aeruginosa lung infection (40, 41). Taken together, these studies support the importance of IFN-γ in protection against P. aeruginosa lung infection.
Several in vivo studies have examined the role of NKG2D in NK cell responses directly against tumors (42), as well as cells infected with an intracellular pathogen (43–45). Thus, the current paradigm for NKG2D signaling in the removal of pathogens is centered on the direct recognition of infected cells that express NKG2D ligands and their subsequent NK cell-mediated removal. In the present study, we demonstrate that NK cell activation (as assessed by IFN-γ production) is critically dependent on NKG2D signaling following acute P. aeruginosa respiratory infection. Our data represent the first observation of NKG2D-mediated activation of lung NK cells following respiratory infection with an extracellular pathogen. Significantly, our findings also expand the current paradigm of the NKG2D receptor-ligand system in that extracellular P. aeruginosa is capable of inducing NKG2D ligands on bystander cells such as pulmonary epithelial cells or alveolar macrophages, resulting in NKG2D-mediated activation of resident NK cells. In contrast to the current NKG2D signaling paradigm where activation of NK cells results in the direct NK cell-mediated removal of NKG2D ligand-expressing infected cells, we postulate that activated NK cells indirectly ameliorate the eradication of extracellular pathogen through IFN-γ-mediated augmentation of antibacterial defense mechanisms. Thus, our data indicate that NKG2D is necessary and sufficient for NK cell activation and clearance of acute P. aeruginosa in our mouse model of respiratory infection.
In the present study we show that pulmonary epithelial cell-specific NKG2D ligand expression in a transgenic mouse model increases pulmonary clearance, cellular phagocytosis, and survival following P. aeruginosa infection. Additionally, NKG2D ligand expression increases NK cell sensitivity to LPS. We unequivocally demonstrate that the non-redundant production of NK cell-derived IFN-γ following P. aeruginosa respiratory infection is dependent upon NKG2D. These findings advance our understanding of the mechanisms of interactions between the NKG2D receptor and pulmonary epithelial cell-derived ligands in the lung, and their effects on the pulmonary innate immune system. Because of the ubiquitous nature of P. aeruginosa and its ability to develop resistance to antibiotics, therapeutic strategies are limited and respiratory infections are problematic and difficult to treat. Thus, novel approaches that target NK cells, and more specifically NKG2D, may lead to alternative therapeutics designed to improve the outcome of P. aeruginosa respiratory infection.
Acknowledgments
We thank Maureen Sartor and Dr. Mario Medvedovic for statistical assistance, and Dr. Francis X. McCormack and Michael P. Davis for helpful discussion.
Footnotes
This work was supported by National Institutes of Health Grant ES015036 to M.T.B and a Pilot Project Grant to S.C.W. within the University of Cincinnati, Department of Environmental Health’s Center for Environmental Genetics ES006096.
References
- 1.Diekema DJ, Pfaller MA, Jones RN, Doern GV, Winokur PL, Gales AC, Sader HS, Kugler K, Beach M. Survey of bloodstream infections due to gram-negative bacilli: frequency of occurrence and antimicrobial susceptibility of isolates collected in the United States, Canada, and Latin America for the SENTRY Antimicrobial Surveillance Program, 1997. Clin Infect Dis. 1999;29:595–607. doi: 10.1086/598640. [DOI] [PubMed] [Google Scholar]
- 2.Burns JL, Gibson RL, McNamara S, Yim D, Emerson J, Rosenfeld M, Hiatt P, McCoy K, Castile R, Smith AL, Ramsey BW. Longitudinal assessment of Pseudomonas aeruginosa in young children with cystic fibrosis. J Infect Dis. 2001;183:444–452. doi: 10.1086/318075. [DOI] [PubMed] [Google Scholar]
- 3.Emerson J, Rosenfeld M, McNamara S, Ramsey B, Gibson RL. Pseudomonas aeruginosa and other predictors of mortality and morbidity in young children with cystic fibrosis. Pediatr Pulmonol. 2002;34:91–100. doi: 10.1002/ppul.10127. [DOI] [PubMed] [Google Scholar]
- 4.West SE, Zeng L, Lee BL, Kosorok MR, Laxova A, Rock MJ, Splaingard MJ, Farrell PM. Respiratory infections with Pseudomonas aeruginosa in children with cystic fibrosis: early detection by serology and assessment of risk factors. JAMA. 2002;287:2958–2967. doi: 10.1001/jama.287.22.2958. [DOI] [PubMed] [Google Scholar]
- 5.Cook DJ, Walter SD, Cook RJ, Griffith LE, Guyatt GH, Leasa D, Jaeschke RZ, Brun-Buisson C. Incidence of and risk factors for ventilator-associated pneumonia in critically ill patients. Ann Intern Med. 1998;129:433–440. doi: 10.7326/0003-4819-129-6-199809150-00002. [DOI] [PubMed] [Google Scholar]
- 6.Murphy TF, Brauer AL, Eschberger K, Lobbins P, Grove L, Cai X, Sethi S. Pseudomonas aeruginosa in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2008;177:853–860. doi: 10.1164/rccm.200709-1413OC. [DOI] [PubMed] [Google Scholar]
- 7.Hamerman JA, Ogasawara K, Lanier LL. NK cells in innate immunity. Curr Opin Immunol. 2005;17:29–35. doi: 10.1016/j.coi.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 8.Orange JS. Human natural killer cell deficiencies and susceptibility to infection. Microbes Infect. 2002;4:1545–1558. doi: 10.1016/s1286-4579(02)00038-2. [DOI] [PubMed] [Google Scholar]
- 9.Byrne P, McGuirk P, Todryk S, Mills KH. Depletion of NK cells results in disseminating lethal infection with Bordetella pertussis associated with a reduction of antigen-specific Th1 and enhancement of Th2, but not Tr1 cells. Eur J Immunol. 2004;34:2579–2588. doi: 10.1002/eji.200425092. [DOI] [PubMed] [Google Scholar]
- 10.Junqueira-Kipnis AP, Kipnis A, Jamieson A, Juarrero MG, Diefenbach A, Raulet DH, Turner J, Orme IM. NK cells respond to pulmonary infection with Mycobacterium tuberculosis, but play a minimal role in protection. J Immunol. 2003;171:6039–6045. doi: 10.4049/jimmunol.171.11.6039. [DOI] [PubMed] [Google Scholar]
- 11.Le-Barillec K, Magalhaes JG, Corcuff E, Thuizat A, Sansonetti PJ, Phalipon A, Di Santo JP. Roles for T and NK cells in the innate immune response to Shigella flexneri. J Immunol. 2005;175:1735–1740. doi: 10.4049/jimmunol.175.3.1735. [DOI] [PubMed] [Google Scholar]
- 12.Bauer S, Groh V, Wu J, Steinle A, Phillips JH, Lanier LL, Spies T. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science. 1999;285:727–729. doi: 10.1126/science.285.5428.727. [DOI] [PubMed] [Google Scholar]
- 13.Diefenbach A, Jamieson AM, Liu SD, Shastri N, Raulet DH. Ligands for the murine NKG2D receptor: expression by tumor cells and activation of NK cells and macrophages. Nat Immunol. 2000;1:119–126. doi: 10.1038/77793. [DOI] [PubMed] [Google Scholar]
- 14.Jamieson AM, Diefenbach A, McMahon CW, Xiong N, Carlyle JR, Raulet DH. The role of the NKG2D immunoreceptor in immune cell activation and natural killing. Immunity. 2002;17:19–29. doi: 10.1016/s1074-7613(02)00333-3. [DOI] [PubMed] [Google Scholar]
- 15.Cerwenka A, Lanier LL. Natural killer cells, viruses and cancer. Nat Rev Immunol. 2001;1:41–49. doi: 10.1038/35095564. [DOI] [PubMed] [Google Scholar]
- 16.Kubin M, Cassiano L, Chalupny J, Chin W, Cosman D, Fanslow W, Mullberg J, Rousseau AM, Ulrich D, Armitage R. ULBP1, 2, 3: novel MHC class I-related molecules that bind to human cytomegalovirus glycoprotein UL16, activate NK cells. Eur J Immunol. 2001;31:1428–1437. doi: 10.1002/1521-4141(200105)31:5<1428::AID-IMMU1428>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 17.Sutherland CL, Chalupny NJ, Cosman D. The UL16-binding proteins, a novel family of MHC class I-related ligands for NKG2D, activate natural killer cell functions. Immunol Rev. 2001;181:185–192. doi: 10.1034/j.1600-065x.2001.1810115.x. [DOI] [PubMed] [Google Scholar]
- 18.McFarland BJ, Kortemme T, Yu SF, Baker D, Strong RK. Symmetry recognizing asymmetry: analysis of the interactions between the C-type lectin-like immunoreceptor NKG2D and MHC class I-like ligands. Structure. 2003;11:411–422. doi: 10.1016/s0969-2126(03)00047-9. [DOI] [PubMed] [Google Scholar]
- 19.Radaev S, Sun PD. Structure and function of natural killer cell surface receptors. Annu Rev Biophys Biomol Struct. 2003;32:93–114. doi: 10.1146/annurev.biophys.32.110601.142347. [DOI] [PubMed] [Google Scholar]
- 20.Cerwenka A, Bakker AB, McClanahan T, Wagner J, Wu J, Phillips JH, Lanier LL. Retinoic acid early inducible genes define a ligand family for the activating NKG2D receptor in mice. Immunity. 2000;12:721–727. doi: 10.1016/s1074-7613(00)80222-8. [DOI] [PubMed] [Google Scholar]
- 21.Raulet DH. Roles of the NKG2D immunoreceptor and its ligands. Nat Rev Immunol. 2003;3:781–790. doi: 10.1038/nri1199. [DOI] [PubMed] [Google Scholar]
- 22.Kasahara M, Watanabe Y, Sumasu M, Nagata T. A family of MHC class I-like genes located in the vicinity of the mouse leukocyte receptor complex. Proc Natl Acad Sci USA. 2002;99:13687–13692. doi: 10.1073/pnas.212375299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Borchers MT, Harris NL, Wesselkamper SC, Vitucci M, Cosman D. NKG2D ligands are expressed on stressed human airway epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2006;291:L222–L231. doi: 10.1152/ajplung.00327.2005. [DOI] [PubMed] [Google Scholar]
- 24.Borchers MT, Harris NL, Wesselkamper SC, Zhang S, Chen Y, Young L, Lau GW. The NKG2D-activating receptor mediates pulmonary clearance of Pseudomonas aeruginosa. Infect Immun. 2006;74:2578–2586. doi: 10.1128/IAI.74.5.2578-2586.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tichelaar JW, Lu W, Whitsett JA. Conditional expression of fibroblast growth factor-7 in the developing and mature lung. J Biol Chem. 2000;275:11858–11864. doi: 10.1074/jbc.275.16.11858. [DOI] [PubMed] [Google Scholar]
- 26.Holloway BW, Krishnapillai V, Morgan AF. Chromosomal genetics of Pseudomonas. Microbiol Rev. 1979;43:73–102. doi: 10.1128/mr.43.1.73-102.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ogasawara K, Hamerman JA, Hsin H, Chikuma S, Bour-Jordan H, Chen T, Pertel T, Carnaud C, Bluestone JA, Lanier LL. Impairment of NK cell function by NKG2D modulation in NOD mice. Immunity. 2003;18:41–51. doi: 10.1016/s1074-7613(02)00505-8. [DOI] [PubMed] [Google Scholar]
- 28.Ogasawara K, Hamerman JA, Ehrlich LR, Bour-Jordan H, Santamaria P, Bluestone JA, Lanier LL. NKG2D blockade prevents autoimmune diabetes in NOD mice. Immunity. 2004;20:757–767. doi: 10.1016/j.immuni.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 29.Skerrett SJ, Wilson CB, Liggitt HD, Hajjar AM. Redundant Toll-like receptor signaling in the pulmonary host response to Pseudomonas aeruginosa. Am J Physiol Lung Cell Mol Physiol. 2007;292:L312–L322. doi: 10.1152/ajplung.00250.2006. [DOI] [PubMed] [Google Scholar]
- 30.Hamerman JA, Ogasawara K, Lanier LL. Cutting edge: toll-like receptor signaling in macrophages induces ligands for the NKG2D receptor. J Immunol. 2004;172:2001–2005. doi: 10.4049/jimmunol.172.4.2001. [DOI] [PubMed] [Google Scholar]
- 31.Schaber JA, Carty NL, McDonald NA, Graham ED, Cheluvappa R, Griswold JA, Hamood AN. Analysis of quorum sensing-deficient clinical isolates of Pseudomonas aeruginosa. J Med Microbiol. 2004;53:841–853. doi: 10.1099/jmm.0.45617-0. [DOI] [PubMed] [Google Scholar]
- 32.Passador L, Cook JM, Gambello MJ, Rust L, Iglewski BH. Expression of Pseudomonas aeruginosa virulence genes requires cell-to-cell communication. Science. 1993;260:1127–1130. doi: 10.1126/science.8493556. [DOI] [PubMed] [Google Scholar]
- 33.Yahr TL, Wolfgang MC. Transcriptional regulation of the Pseudomonas aeruginosa type III secretion system. Mol Microbiol. 2006;62:631–640. doi: 10.1111/j.1365-2958.2006.05412.x. [DOI] [PubMed] [Google Scholar]
- 34.Holder IA, Neely AN, Frank DW. PcrV immunization enhances survival of burned Pseudomonas aeruginosa-infected mice. Infect Immun. 2001;69:5908–5910. doi: 10.1128/IAI.69.9.5908-5910.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Holder IA, Neely AN, Frank DW. Type III secretion/intoxication system important in virulence of Pseudomonas aeruginosa infections in burns. Burns. 2001;27:129–130. doi: 10.1016/s0305-4179(00)00142-x. [DOI] [PubMed] [Google Scholar]
- 36.Walzer T, Blery M, Chaix J, Fused N, Chasson L, Robbins SH, Jaeger S, Andre P, Gauthier L, Daniel L, Chemin K, Morel Y, Dalod M, Imbert J, Pierres M, Moretta A, Romagne F, Vivier E. Identification, activation, and selective in vivo ablation of mouse NK cells viaNKp46. Proc Natl Acad Sci USA. 2007;104:3384–3389. doi: 10.1073/pnas.0609692104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moser C, Jensen PO, Kobayashi O, Hougen HP, Song Z, Rygaard J, Kharazmi A, HbN Improved outcome of chronic Pseudomonas aeruginosa lung infection is associated with induction of a Th1-dominated cytokine response. Clin Exp Immunol. 2002;127:206–213. doi: 10.1046/j.1365-2249.2002.01731.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lei D, Lancaster JR, Jr, Joshi MS, Nelson S, Stoltz D, Bagby GJ, Odom G, Shellito JE, Kolls JK. Activation of alveolar macrophages and lung host defenses using transfer of the interferon-gamma gene. Am J Physiol Til. 1997:L852–L859. doi: 10.1152/ajplung.1997.272.5.L852. [DOI] [PubMed] [Google Scholar]
- 39.Johansen HK, Hougen HP, Rygaard J, Hoiby N. Interferon-gamma (IFN-gamma) treatment decreases the inflammatory response in chronic Pseudomonas aeruginosa pneumonia in rats. Clin Exp Immunol. 1996;103:212–218. doi: 10.1046/j.1365-2249.1996.d01-618.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wojnarowski C, Frischer T, Hofbauer E, Grabner C, Mosgoeller W, Eichler I, Ziesche R. Cytokine expression in bronchial biopsies of cystic fibrosis patients with and without acute exacerbation. Eur Respir J. 1999;14:1136–1144. doi: 10.1183/09031936.99.14511369. [DOI] [PubMed] [Google Scholar]
- 41.Moser C, Hougen HP, Song Z, Rygaard J, Kharazmi A, Hoiby N. Early immune response in susceptible and resistant mice strains with chronic Pseudomonas aeruginosa lung infection determines the type of T-helper cell response. APMIS. 1999;107:1093–1100. doi: 10.1111/j.1699-0463.1999.tb01514.x. [DOI] [PubMed] [Google Scholar]
- 42.Cerwenka A, Baron JL, Lanier LL. Ectopic expression of retinoic acid early inducible-1 gene (RAE-1) permits natural killer cell-mediated rejection of a MHC class I-bearing tumor in vivo. Proc Natl Acad Sci USA. 2001;98:11521–11526. doi: 10.1073/pnas.201238598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lodoen M, Ogasawara K, Hamerman JA, Arase H, Houchins JP, Mocarski ES, Lanier LL. NKG2D-mediated natural killer cell protection against cytomegalovirus is impaired by viral gp40 modulation of retinoic acid early inducible 1 gene molecules. J Exp Med. 2003;197:1245–1253. doi: 10.1084/jem.20021973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vankayalapati R, Garg A, Porgador A, Griffith DE, Klucar P, Safi H, Girard WM, Cosman D, Spies T, Barnes PF. Role of NK cell-activating receptors and their ligands in the lysis of mononuclear phagocytes infected with an intracellular bacterium. J Immunol. 2005;175:4611–4617. doi: 10.4049/jimmunol.175.7.4611. [DOI] [PubMed] [Google Scholar]
- 45.Fang M, Lanier LL, Sigal LJ. A Role for NKG2D in NK Cell-Mediated Resistance to Poxvirus Disease. PloS Pathog. 2008;4:e30. doi: 10.1371/journal.ppat.0040030. [DOI] [PMC free article] [PubMed] [Google Scholar]