

Abstract
Objectives
Cisplatin is a widely used chemotherapeutic agent; however, nephrotoxicity and neuropathy are obstacles for drug efficacy. Little is known about the genes or genetic variants contributing to the risk of developing these toxicities or chemotherapeutic response. Thus, we have applied a cell-based model to identify and characterize previously unknown genes that may be involved in cellular susceptibility to cisplatin.
Methods
Lymphoblastoid cell lines from 27 large Centre d'Etude du Polymorphisme Humain pedigrees were used to elucidate the genetic contribution to cisplatin-induced cytotoxicity. Phenotype was defined as cell growth inhibition following exposure of cell lines to increasing concentrations of cisplatin for 48 h.
Results
Significant heritability, ranging from 0.32 to 0.43 (P < 10−7), was found for the cytotoxic effects of each concentration (1, 2.5, 5, 10, and 20 μmol/l) and IC50, the concentration required for 50% cell growth inhibition. Linkage analysis revealed 11 genomic regions on six chromosomes with logarithm of odds (LOD) scores above 1.5 for cytotoxic phenotypes. The highest LOD score was found on chromosome 4q21.3−q35.2 (LOD = 2.65, P = 2.4 × 10−4) for 5 μmol/l cisplatin. Quantitative transmission disequilibrium tests were performed using 191 973 nonredundant single nucleotide polymorphisms (SNPs) located in the 1 LOD confidence interval of these 11 regions. Twenty SNPs, with 10 SNPs located in five genes, were significantly associated with cisplatin-induced cytotoxicity (P ≤ 1 × 10−4). Four of these 20 SNPs were found to explain over 10% of the variation in cisplatin-induced apoptosis.
Conclusions
Our results suggest that genetic factors involved in cytotoxicity also contribute to cisplatin-induced apoptosis. These cell lines provide a paradigm to identify previously unknown pharmacogenetic variants associated with drug cytotoxicity.
Keywords: apoptosis, association, Centre d'Etude du Polymorphisme Humain, cisplatin, cytotoxicity, heritability, linkage
Introduction
Platinating agents are widely used chemotherapeutic drugs having a number of platinum analogs developed to overcome cellular resistance and reduce toxicity [1,2]. Since its discovery over 40 years ago, cisplatin still remains one of the most widely used chemotherapeutic agents today [3]. The antitumor effects of cisplatin have contributed significantly to the clinical management of a variety of cancers, including ovarian, head and neck, testicular, and non-small cell lung cancers [4-6]. In particular, cisplatin treatment results in overall cure rates exceeding 90% for early-stage testicular cancer [6]. Survival rates in non-small cell lung cancer, however, are less than 30% after cisplatin treatment [7,8]. Cisplatin exerts its antitumor activity by binding preferentially to N-7 positions of adenine and guanine of DNA, resulting in the formation of intrastrand and interstrand crosslinks [6]. More specifically, 1,2-intrastrand d(GpG) crosslinks are produced by cisplatin [9].
Intrinsic and/or acquired resistance as well as toxicities associated with cisplatin are major limitations of this drug [1,4,6,10,11]. For example, about 85% of ovarian cancer patients relapse following initial response [1]. Cisplatin resistance may be multifactorial, consisting of proteins limiting the formation of DNA adducts and/or operating downstream of the interaction of drug with DNA to promote cell survival [12]. More specifically, modes of resistance include increased efflux from the cell, drug inactivation, alterations to the drug target, DNA repair, evasion of apoptosis, and drug target alteration [13].
Cisplatin-induced toxicity can also alter the outcome of treatment by limiting the dose of drug administered, with nephrotoxicity and neuropathy as the main dose-limiting toxicities [14,15]. Peripheral neuropathy is permanent in approximately 30−50% of patients receiving treatment [15]. Additionally, ototoxicity and neurotoxicity are difficult to manage and can result in the discontinuation or reduction of treatment [14]. Hence, the identification of genetic factors to better predict patients who are at risk of adverse drug reactions associated with cisplatin or relapse will be highly beneficial [10].
Deciphering dosing regimens for patients without consideration of genetic heterogeneity or heterogeneity in the disease pathogenesis is a widespread, albeit imprecise, approach [16]. For the most part, the basis of chemotherapeutic response has been studied in the context of the tumor genome and host toxicity in the context of the host genome. Successful outcomes in cancer chemotherapy, however, may rely on a delicate balance between the tumor and host genomes. For example, polymorphisms in both the tumor and host genomes might contribute to drug availability, efflux, and accessibility of the drug to the tumor [17]. Several candidate gene approaches have been implemented to identify the basis of response or toxicity to cisplatin, which have included the use of tumor cell types and model systems [18-20]. Variants in candidate genes such as glutathione S-transferases, ERCC1, ERCC2, and XRCC1 have been shown to alter response to cisplatin [21,22]. Candidate gene studies with these variant alleles, however, have also lead to inconsistent results [21]. Although variants in candidate genes may affect tumor response, they may not be good predictors of toxicity. Furthermore, focusing only on candidate genes may result in unknown genes and variants important in cisplatin-induced cytotoxicity being overlooked. To overcome these limitations, we present a comprehensive approach to identify genes and genetic variants that may be associated with human variation in response and toxicities associated with cisplatin treatment. To this end, we used lymphoblastoid cell lines (LCLs) from healthy individuals derived from large Centre d'Etude du Polymorphisme Humain (CEPH) pedigrees. We performed genome-wide linkage analysis followed by an association analysis within suggestive linkage regions at multiple drug concentrations. Significant single nucleotide polymorphisms (SNPs) associated with cytotoxicity were further interrogated for their relationship with cisplatin-induced apoptosis, providing us with a better understanding of the germline genetic influences controlling variation in cell death associated with this agent.
Materials and methods
Cell lines
Epstein–Barr virus transformed LCLs derived from 27 Caucasian Utah CEPH families of northern and western European descent (families used for cisplatin included 1334, 1340, 1341, 1344, 1345, 1346, 1349, 1350, 1356, 1358, 1362, 1375, 1377, 1408, 1418, 1420, 1421, 1424, 1444, 1447, 1454, 1459, 1463, 13291, 13292, 13293, and 13294) were purchased from the Coriell Institute for Medical Research (http://www.locus.umdnj.edu/ccr/). Cell lines were cultured in RPMI 1640 media containing 15% heat-inactivated fetal bovine serum (Hyclone, Logan, Utah, USA) and 20 mmol/l l-glutamine. Cell lines were diluted three times per week at a concentration of 300 000−350 000 cells/ml and were maintained in a 37°C, 5% CO2-humidified incubator. Medium and components were purchased from Cellgro (Herndon, Virginia, USA).
Drugs
Cisplatin was purchased from Sigma Chemical Co. (St Louis, Missouri, USA). Cisplatin was made up as a 20 mmol/l stock, filter sterilized (prepared in dimethylsulfoxide), and diluted in media immediately before the addition to cells. Final concentrations of cisplatin were 1, 2.5, 5, 10, and 20 μmol/l and exposure time to drug was 48 h. The final concentration of dimethylsulfoxide did not exceed 0.1% in wells.
Cell growth inhibition
Up to 343 cell lines derived from 27 large CEPH families were treated with 1 (n = 318), 2.5 (n = 294), 5 (n = 343), 10 (n = 343), and 20 (n = 318) μmol/l cisplatin using a short-term assay to determine cell growth inhibition. Cytotoxicity was performed in the absence (control) and presence of increasing drug concentrations using a high throughput alamarBlue (Biosource International, Camarillo, California, USA) assay as previously described [23]. Drug solution (100 μl) was added 24 h after plating. Cytotoxicity measurements were performed in triplicate for each drug concentration per experiment, with two to three experiments per cell line. Final cytotoxicity values were averaged from at least six replicates taken from two separate experiments. IC50, the concentration required to inhibit 50% cell growth, was calculated for each cell line by curve fitting of each concentration in Microsoft Excel software (Redmond, Washington, USA). Test of normality of phenotypes was based on the Kolmogorov−Smirnov statistic.
Apoptosis assay
Cisplatin-induced apoptosis was evaluated for the unrelated HapMap samples using the Guava PCA machine with the Guava Nexin Kit (Guava Technologies, Hayward, California, USA). Cells were plated at 100 000 cells/ml in six well plates and treated the following day with 10 μmol/l cisplatin for 24 h. Cells were then collected, resuspended in Nexin buffer, and stained for Annexin V and 7-amino-actinomyosin D (7-AAD), where early apoptotoic cells are Annexin V( + ), 7-AAD( − ) and late apoptotic cells are Annexin V( + ), 7-AAD( + ). Final apoptosis values (early + late) were averaged from at least two replicates taken from two separate experiments. The resulting percentage apoptosis for each cell line was determined relative to untreated control.
Heritability analysis
Heritability analysis was performed using Sequential Oligogenic Linkage Analysis Routines (SOLAR) (http://www.sfbr.org/solar/) computer software to estimate the narrow sense heritability at each cisplatin concentration as previously described [23,24]. This analysis is applied to determine the proportion of cytotoxicity at each dose that can be explained by genetic factors.
Linkage analysis
Multipoint Engine for Rapid Likelihood Inference (MERLIN) [25] was used to perform nonparametric quantitative trait locus linkage analysis with 7209 SNP and microsatellite markers as previously described [23]. The physical positions of selected microsatellite and SNP markers were found using Build 36 of the UCSC Genome Browser (http://www.genome.ucsc.edu). Genetic maps were constructed based on microsatellite and SNP positions in the Marshfield map.
Association analysis for cytotoxicity
Eighty-six HapMap CEPH samples (out of 90), comprising trios, were phenotyped for cisplatin sensitivity. Three samples (GM10855, GM12716, and GM12717) were not phenotyped due to the inability to obtain ≥ 85% viability. Additionally, another sample (GM12236) was not available from Coriell at the time of phenotyping. SNP genotypes were downloaded from the International HapMap database (http://www.hapmap.org) (release 22). A total of 191 973 nonredundant SNPs (minor allele frequency > 5%, no Mendelian transmission errors) within the 1 logarithm of odds (LOD) confidence intervals of the 11 linkage regions for the cisplatin phenotypes were used for the association study in the 30 HapMap CEPH trios. A P value threshold of ≤ 10−4 was used. All 86 IC50 values were log2 transformed to obtain normally distributed data.
The quantitative transmission disequilibrium test (QTDT) was performed to identify any genotypecytotoxicity association using QTDT software [26]. Population stratification and total association between the final SNPs and percent cell survival for 1, 2.5, 5, 10, and 20 μmol/l cisplatin and the IC50 was performed using QTDT. Sex was used as a covariate to adjust for normalized percentage survival values.
Linear regression for apoptosis
Fifty-seven unrelated HapMap CEPH samples (out of 60) were phenotyped for apoptosis following cisplatin treatment. Three samples were left out of the apoptosis study; two (GM12716 and GM12717) for the reasons described above and one (GM12236) because it was not available from Coriell at the time of collecting apoptosis measurements. To determine what proportion of SNPs associated with cytotoxicity contributed to cisplatin-induced apoptosis, linear regression was performed by regressing log2 transformed apoptosis data on the SNP genotype (coded as 0, 1, 2). The adjusted R2 was estimated using each genotype/phenotype combination as the ratio of the regression sum of squares to the total sum of squares. The false discovery rate (FDR) procedure [27] was used to control for multiple testing within the phenotype using R-2.3.1 (www.r-project.org). An FDR cutoff of < 5% was used as a threshold for significance.
Results
Cisplatin-induced cytotoxicity
The mean (± SD) of cell survival pertaining to the 1, 2.5, 5, 10, and 20 μmol/l cisplatin concentrations were 75.9 ± 8.9, 64 ± 10.3, 54.7 ± 11.5, 44.3 ± 11.4, and 34 ± 10.3, respectively. IC50 was determined for 318 of the 343 cell lines treated with cisplatin because some cell lines did not have percentage survival values above or below 50% owing to extreme resistance or sensitivity. The average IC50 value for 48 h cisplatin was 7.5 ± 6.3 μmol/l with a range 1.52−58.9 μmol/l. Intrafamily and interfamily variations for all of the cisplatin concentrations are shown in the boxplots in Fig. 1.
Fig. 1.
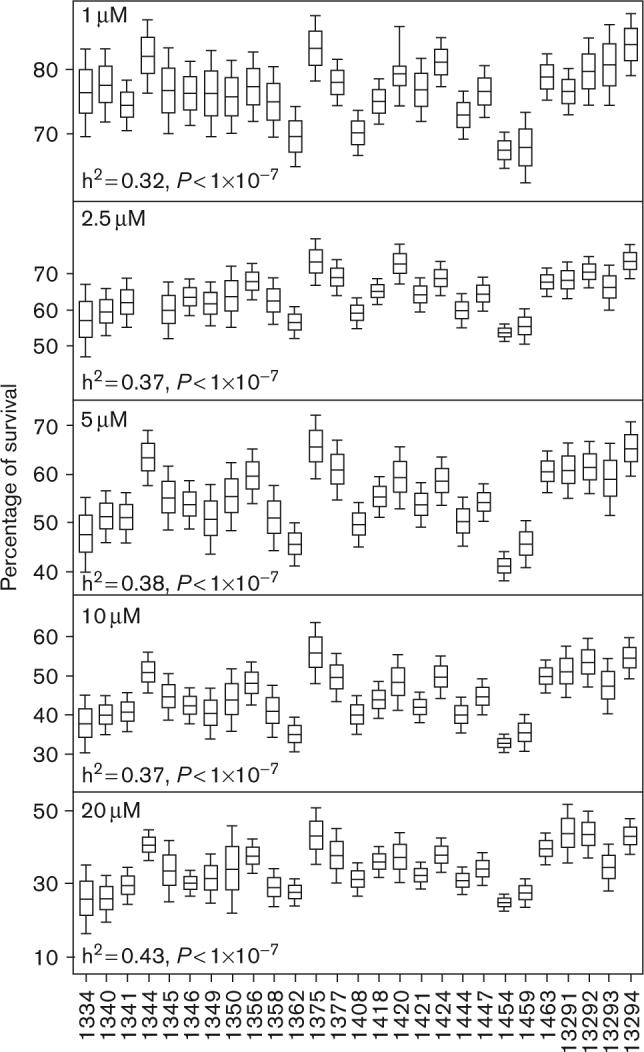
Boxplots and heritability (h2) values for 27 families are shown for 1 (top), 2.5, 5, 10, and 20 μmol/l (bottom) for cisplatin. CEPH family ID pertaining to the boxplot is located on the x-axis. The middle line within the box represents the mean of each family, whereas the box edges and whiskers represent the standard error of the mean and twice the standard error of the mean, respectively. CEPH, Centre d'Etude du Polymorphisme Humain.
Heritability analysis
Heritability analysis, which compares the covariance of each trait with the overall variance of that trait, revealed a significant genetic contribution for each concentration of cisplatin (Fig. 1). Approximately 32−43% (P < 1 × 10−7) of the variation in cisplatin cytotoxicity is due to heritable components (Fig. 1). No sex-specific heritability effects were found for the cisplatin phenotypes. The heritability for the IC50 phenotype was 0.34 (P = 1 × 10−7).
Linkage analysis
Nonparametric linkage analysis was performed on six cytotoxic phenotypes with significant heritability estimates to localize genetic determinants contributing to cisplatin cytotoxicity. A total of 7209 SNPs and micro-satellite markers with high heterozygosity were used for this analysis. As drug cytotoxicity is most likely to be a multigenic trait, a LOD threshold of > 1.5 was chosen to include genes of modest effect [23]. Eleven different genomic regions were found (containing a total of 1602 nonredundant genes) with LOD scores > 1.5 for the different cisplatin concentrations tested (Table 1, Fig. 2a). No linkage peaks with LOD > 1.5 were found for the IC50 phenotype. The highest LOD score of 2.65 was found on chromosome 4q24–q32.3 for the 5 μmol/l concentration, with 208 genes in the one LOD confidence interval region. This region also overlapped linkage regions for the 1, 10, and 20 μmol/l cytotoxic phenotypes, with LOD scores of 1.91, 2.33, and 1.97, respectively (Table 1). An inverse relationship existed between LOD scores on chromosome 4 and cisplatin concentration (Fig. 2b), suggesting a greater degree of genetic effect with lower drug concentration. Other overlapping linkage regions included: (a) chromosome 3 for the 2.5 and 20 μmol/l concentrations with associated LOD scores of 1.7 and 1.54, respectively; (b) chromosome 8 for the 2.5 and 10 μmol/l concentrations with associated LOD scores of 1.7 and 1.9, respectively; and (c) chromosome 11 for the 5 and 10 μmol/l concentrations with associated LOD scores of 2.15 and 2.5, respectively.
Table 1.
Linkage regions with LOD scores above 1.5 used in association analysis
| 1 LOD confidence interval | 1 LOD confidence interval (cM) | Maximum LOD (cM) | Phenotype (μmol/l) | No. genes in 1 LOD interval | SNPsa |
|---|---|---|---|---|---|
| 3p25.3-p24.1 | 27−51 | 1.7 (36) | 2.5 | 46 | 14 640 |
| 3p24.3-p22.1 | 43−63 | 1.54 (55) | 20 | 77 | 15 936 |
| 4p15.33-p15.1 | 28−52 | 1.99 (41) | 2.5 | 46 | 13 492 |
| 4q21.3-q35.2 | 96−167 | 2.33 (122) | 10 | 200 | 51 916 |
| 113−162 | 1.91 (117) | 1 | 217 | 35 889 | |
| 114−166 | 2.65 (141) | 5 | 230 | 37 670 | |
| 127−147 | 1.97 (142) | 20 | 57 | 10 867 | |
| 4q22.1-q35.2 | 100−203 | 1.67 (196) | 5 | 338 | 64 356 |
| 8q24.13-q24.22 | 147−170 | 1.7 (168) | 2.5 | 25 | 11 695 |
| 154−168 | 1.9 (163) | 10 | 26 | 6804 | |
| 10p13-p12.1 | 37−52 | 1.91 (44) | 1 | 60 | 10 362 |
| 10q26.3 | 163−172 | 1.64 (170) | 20 | 4 | 1816 |
| 11p15.4-q13.2 | 11−67 | 2.15 (22) | 5 | 102 | 45 007 |
| 13−55 | 2.5 (22) | 10 | 80 | 35 117 | |
| 11q24.1-q25 | 121−147 | 1.9 (129) | 20 | 95 | 11 703 |
| 16q23.1-q24.1 | 100−120 | 1.76 (112) | 20 | 38 | 7658 |
LOD, logarithm of odds; SNPs, single nucleotide polymorphisms.
The number of SNPs, located in the 1 LOD confidence intervals that were used in the association analysis.
Fig. 2.
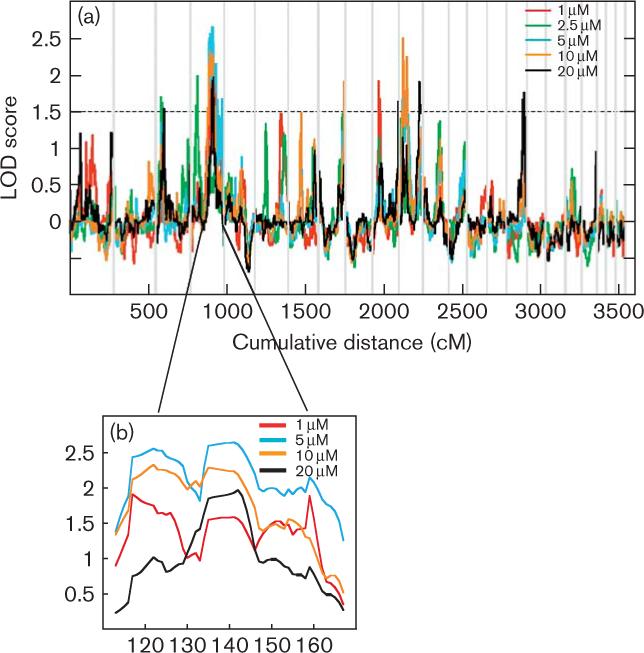
Linkage analyses for cisplatin-induced cytotoxicity. (a) Nonparametric QTL linkage analysis was performed using 27 large pedigrees for the 1, 2.5, 5, 10, and 20 μmol/l concentrations. Each vertical line indicates chromosome boundaries and the horizontal lines indicate LOD scores. Peaks above the dotted line (LOD = 1.5) were used for association analysis. (b) Overlapping linkage peaks for 1, 5, 10, and 20 μmol/l concentrations at 4q21.3–q35.2 (113−166 cM). LOD, logarithm of odds; QTL, quantitative trait locus.
Association studies
A linkage-directed association analysis, using 86 CEPH HapMap samples, was used to further narrow down the linkage regions (LOD > 1.5) contributing to cisplatin-induced cytotoxicity. This approach was also used to reduce multiple-testing issues associated with whole genome association studies. The linkage-directed association analysis resulted in significant associations in 5 of 11 linkage regions and half of the associated SNPs are in nongenic regions (Table 1; Supplementary Table 1). Twenty significant SNPs were found (10 of which are located in 5 genes) that were shown to be significantly associated with cisplatin cytotoxic phenotypes (Supplementary Table 1). Several of these SNPs are in complete linkage disequilibrium (LD) (D′ = 1, r2 = 1) including rs1026686, rs3860575, and rs10510534 on chromosome 3. SNP rs10510534 is also in complete LD with rs17018468 and rs7652737. On chromosome 4, rs17041972, rs17624452, 17041968, and rs2278782 are in complete LD and rs4834232 is in complete LD with rs6848982. On chromosome 11, rs7133868, rs7119153, and rs7949504 are in complete LD.
Two significant SNPs (rs4834232 and rs11944754) were associated with the 5 μmol/l concentration and two significant SNPs (rs7683488 and rs6848982) were associated with the 20 μmol/l concentration, which are located in the chromosome 4 overlapping linkage region located at 127−140 cM (Supplementary Table 1). The CC genotype of intronic SNP rs4834232, located in LARP2, is associated with a greater sensitivity to 5 μmol/l cisplatin. Three intronic SNPs (rs17041972, rs17624452, and rs17041968) were in strong LD in PITX2, which were associated with the 2.5 μmol/l concentration. One intronic SNP (rs2278782) in PITX2 was associated with the 20 μmol/l concentration in the chromosome 4 overlapping linkage region (116 cM). The GG genotype of rs17041972 is associated with sensitivity to 2.5 μmol/l cisplatin. Additionally, rs17624452 is located in the most highly conserved region across multiple species. The CC genotype of rs2278782, also located in PITX2, confers an increased sensitivity to 20 μmol/l cisplatin (Supplementary Table 1). A nongenic SNP associated with the 1 μmol/l phenotype, rs7119153, was found to be in the most highly conserved region across species.
Linear regression
Cytotoxicity is a broad phenotype encompassing cells arrested in the cell cycle, cells damaged but undergoing DNA repair, and cells undergoing cell death through apoptosis, necrosis, or some other process. To further study which of the associated SNPs with cytotoxicity contributed to apoptosis, linear regression was performed between those SNPs and apoptosis in 57 unrelated CEPH HapMap samples. The average percentage apoptosis in these cells was 31.4% (± 7.8%). Four of the 20 SNPs associated with cytotoxicity individually accounted for ≥ 10% and nine explained ≥ 1% of the variation in degree of early plus late apoptosis with FDR values less than 5% (Table 2 and Supplementary Table 1). The most highly correlated SNP with apoptosis was rs7131224 (R2 = 0.16, FDR = 0.4%) with the CC geno-type of this SNP correlated to higher levels of apoptosis and increased sensitivity to cisplatin-induced cytotoxicity. We further evaluated whether these genetic variants were important for either early or late apoptosis. More than 18% of the variation in early apoptosis was explained by rs11944754 with the TT genotype of this SNP associated with increased sensitivity to the cytotoxic effects of drug (Fig. 3a) and increased levels of early apoptosis (Fig. 3b). The highest level of variation (11%) in late apoptosis was explained by rs6848982 with the GG genotype associated with lower survival following treatment with cisplatin (Fig. 4a) and increased levels of late apoptosis (Fig. 4b).
Table 2.
SNPs associated with cisplatin-induced cytotoxicity and explaining at least 10% of the variation in apoptosis
| Cytotoxicity phenotype (μmol/l) | SNPa | Chromosome (cM) | SNP P valueb | Adjusted R2c | FDR (%) |
|---|---|---|---|---|---|
| 20 | rs6848982 | 4 (128.9) | 1 × 10−4 | 0.13 | 1 |
| 5 | rs4834232 | 4 (128.9) | 1 × 10−4 | 0.10 | 1 |
| 5 | rs11944754 | 4 (140.5) | 1 × 10−4 | 0.15 | 1 |
| 1 | rs7131224 | 11 (31.7) | 1 × 10−4 | 0.16 | 0.4 |
FDR, false discovery rate; QTDT, quantitative transmission disequilibrium test; SNP, single nucleotide polymorphism.
Only one SNP, rs4834232, is located within a gene (LARP2).
P values from QTDT association analysis.
R2 values from linear regression between SNP genotype and log2 apoptosis data.
Fig. 3.
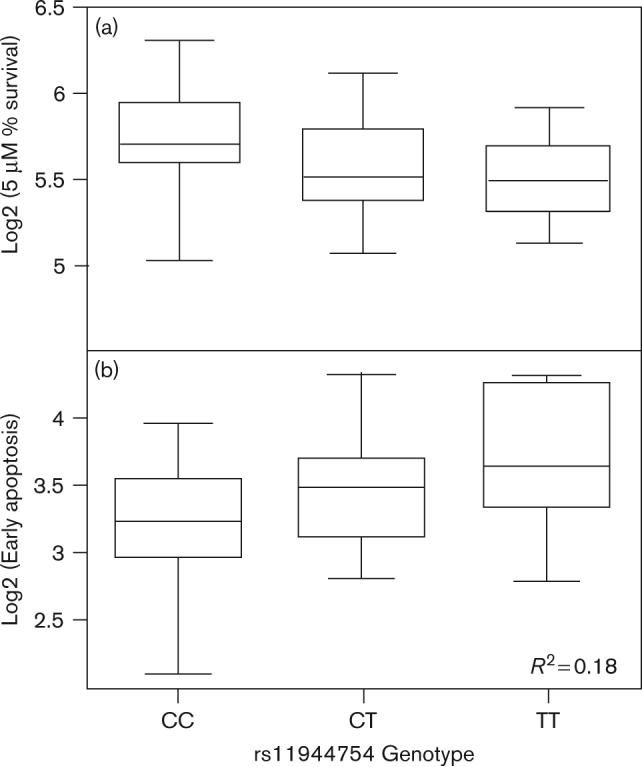
Genotypes for rs11944754 associated with cisplatin-induced cytotoxicity and apoptosis. (a) Genotype versus log2 transformed percentage survival following treatment with 5 μmol/l cisplatin. (b) Genotype versus log2 transformed early apoptosis (FDR = 1%). This SNP explains over 18% of variation in early apoptosis. FDR, false discovery rate; SNP, single nucleotide polymorphism.
Fig. 4.
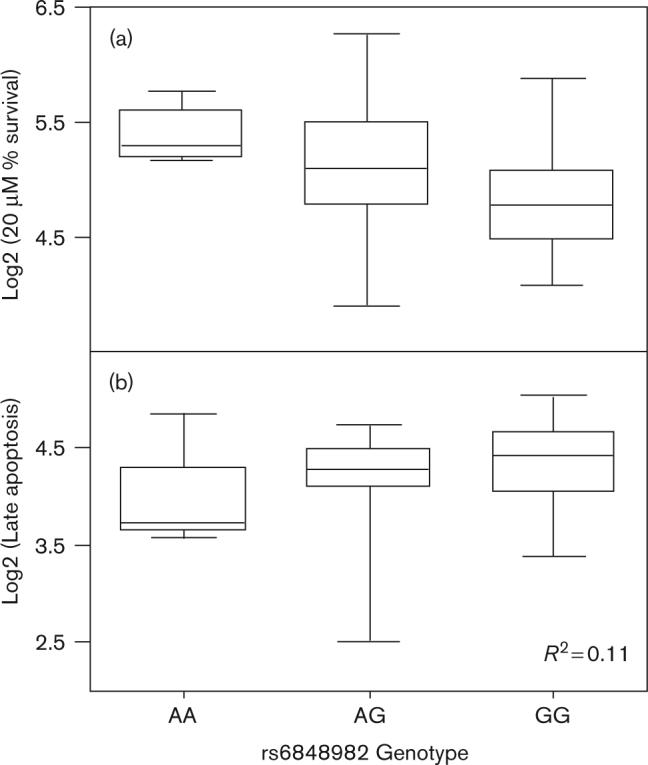
Genotypes for rs6848982 associated with cisplatin-induced cytotoxicity and apoptosis. (a) Genotype versus log2 transformed percentage survival following treatment with 20 μmol/l cisplatin. (b) Genotype versus log2 transformed late apoptosis (FDR = 1%). This SNP explains over 10% of variation in late apoptosis. FDR, false discovery rate; SNP, single nucleotide polymorphism.
Discussion
We used genome-wide linkage analysis followed by linkage-directed association to uncover previously unknown genetic variants that may be important in cisplatin cytotoxicity and further evaluated the contribution of these variants to drug-induced apoptosis. As chemotherapeutic cytotoxicity is likely a polygenic trait, it is advantageous to use complementary genetic approaches, such as linkage and association analyses to identify previously unknown genes and variants contributing to the trait. Using cell lines derived from 27 large CEPH pedigrees, we determined that 32−43% of the variation in cellular response to cisplatin is because of genetic factors. Linkage analysis identified 1602 nonredundant genes under 11 distinct peaks with LOD scores greater than 1.5 with chromosomes 4, 8, and 11 linkage peaks overlapping multiple concentrations. Linkage-directed association analysis narrowed the list down to 20 SNPs associated with cisplatin cytotoxicity (P < 10−4) and four of those SNPs explained over 10% of variation in apoptosis. Linkage analysis revealed that sensitivity to chemotherapy is a multigenic trait with different linkage peaks associated with the cytotoxic effects of different drug concentrations. These results are consistent with earlier studies [23,28]. Linkage peaks differing at various drug concentrations imply that some genes are likely turned on at lower concentrations where the damage is not as severe and most of the cells survive, which is in contrast to higher concentrations where cells initiate the process of cell death. Masquelier et al. [29] demonstrated differences in the induction of apoptosis depending upon daunorubicin concentration, with higher doses correlating to more rapid caspase-3 induction and DNA fragmentation in leukemic cells. In another study, upregulation or downregulation of oncogenes and tumor suppressor genes depended on the concentration of drug used [30]. The IC50 phenotype is thought to be representative of the cytotoxic curve for each cell line; however, no linkage peaks greater than LOD of 1.5 were found for this phenotype. One possibility is that the IC50 phenotype may have more genes contributing to a smaller extent, thereby producing more signals but with LOD scores below the threshold used for this study. Evaluating a greater number of families may reveal previously missed peaks.
These results are an extension of an earlier study by our laboratory evaluating cisplatin-induced cytotoxicity in 10 CEPH pedigrees [28]. In the earlier study, we had suggestive linkage signals on chromosomes 1 or 12; however, this study did not result in suggestive signals on 1 or 12 for any cisplatin concentration. The reason is most likely owing to the greater number of CEPH pedigrees (27 vs. 10) and markers (SNP and microsatellite) for linkage analysis (7209 vs. 1784), allowing greater power to pick up higher resolution signals. Furthermore, this study included linkage-directed association analysis using 86 HapMap samples for cisplatin-induced cytotoxicity and interrogated a second phenotype, cisplatin-induced apoptosis.
Cytotoxicity is a broad phenotype in which the antitumor effects may include: (a) induction of apoptosis to eliminate damaged cells; (b) activation of DNA damage checkpoints and induction of cell cycle arrest to allow for repair of damaged cells; (c) transcriptional response; and (d) damage tolerance [6,31,32]. As the effect of cell cycle inhibition on cisplatin cytotoxicity has been studied by our laboratory [33], we examined which genetic variants involved in cisplatin-induced cytotoxicity are associated with cell death through an apoptotic pathway. Cisplatin is known to induce cell death through necrosis and apoptosis [6]. Necrosis is characterized by cytosolic swelling and loss of membrane integrity, whereas apoptosis is associated with cell shrinkage, loss of cell-to-cell contact, and DNA fragmentation [6,34]; however, apoptosis seems to be a central mechanism of cisplatin-induced cell death [34]. Apoptosis has also been identified as a mechanism of nephrotoxic cell death by cisplatin which has been shown to be concentration specific [35]. Thus, elucidating the genetic variants contributing to cisplatin-induced apoptosis at various concentrations may be clinically beneficial. One potential utility of evaluating SNPs specific to apoptosis is to identify genetic variants that might predict for patients at greatest risk of nephrotoxicity associated with cisplatin. Four nongenic SNPs out of the 20 examined each explained at least 10% of the variation in apoptosis with the greatest variation (16%) in early plus late apoptosis explained by a single SNP (rs7131224). Although the associated SNPs explained some variation in apoptosis, our results indicate that other cellular processes are contributing to cytotoxicity. Furthermore, the only SNPs that were evaluated for their contribution to apoptosis were those SNPs associated with cytotoxicity. Thus, there could be other SNPs that contribute a greater proportion of variance to apoptosis not evaluated in this study.
It has been shown that the type of cell death induced by cisplatin is concentration dependent [34]. For instance, primary cultures of mouse proximal tubule cells underwent necrotic death after exposure to high concentrations of cisplatin (800 μmol/l) for a few hours, whereas apoptosis was induced following exposure to lower concentrations of cisplatin (8 μmol/l) over a few days [36]. The association of nongenic SNPs with apoptosis may indicate locations of new binding sites, such as for p53, regulating cisplatin-induced apoptosis or gene expression [32]. Additionally, SNPs used in our association analysis may not have been dense enough to capture all the genotypes in known genes contributing to apoptosis. For example, caspase-6 is involved in the signaling process that determines a cell's fate to live or die and has been shown to induce cisplatin-dependent apoptosis in human osteosarcoma lines [34,37]. This gene is located in the highest LOD score (2.65) region of the linkage scan (on chromosome 4) and may warrant more in-depth analyses.
Our laboratory is involved in developing cell-based models to evaluate chemotherapeutic toxicity [23,28,38,39]. We developed a cell-based model incorporating expression to identify genetic variants important in cisplatin-induced cytotoxicity referred to as the ‘triangle approach’ [39]. This approach differs from the current approach in the following manner: (i) The earlier study focuses on gene expression in only 90 HapMap cell lines by implementing a three-way model that first correlates SNP genotypes to cisplatin IC50. Next, those significant SNPs are evaluated for association with gene expression, and a final linear correlation between gene expression and drug cytotoxicity is performed. The triangle approach identifies SNPs acting through their effect on baseline gene expression. In contrast, SNPs affecting protein function or induction of expression can be identified in the current approach; (ii) this study implements CEPH pedigrees for linkage analysis and takes the subset of HapMap trios for association analysis. Although association analysis has power to pick up alleles of modest effect, linkage analysis is useful because it is more robust to allelic heterogeneity; (iii) the triangle approach focused on genetic variants common and unique to CEPH and Yoruban populations. Yoruban cell lines from large pedigrees are not available; therefore, the examination of population differences in cytotoxicity using linkage analysis was not possible for our pedigree study; (iv) the current linkage-directed approach limited the number of nonredundant SNPs to 191 973 in the 11 LOD > 1.5 regions, in contrast to the earlier approach evaluating over 387 000 SNPs; (v) the SNPs significantly associated with cytotoxicity in this study were further interrogated by correlating them to apoptosis; and (vi) only the IC50 phenotype was evaluated in the earlier study and we did not find any linkage peaks with LOD > 1.5 for the IC50 phenotype; hence association was not performed on this phenotype in the current analysis. These differences may explain why none of the SNPs in this study overlap with the SNPs found to associate with cytotoxicity in the ‘expression focused-triangle approach’. Despite the differences in the results, both studies illustrate the importance and utility of genotyped LCLs and how different approaches can be used to better understand various cytotoxic phenotypes.
Although the ease of manipulation and dense genotypic data publicly available makes the LCLs a great resource for identifying how genetic variation affects susceptibility to drugs, there are a number of limitations with the use of LCLs. These include: (i) LCLs represent only one specific tissue type bringing into question the utility of assessing a phenotypic effect that occurs in a specific tissue (e.g. neurotoxicity). Tissue-specific regulation may be different due to polymorphisms in the promoter regions of genes that are transcribed differently in various cell types; (ii) large CEPH pedigrees are derived from Caucasian individuals of European descent, hence results may not apply to individuals of different ethnicities. Furthermore, the Caucasian samples are from healthy volunteers, not a population of Caucasian patients who have experienced severe toxicity to a chemotherapeutic agent; and (iii) Epstein–Barr virus transformation could introduce phenotypic or gene expression changes, which may affect sensitivity of the cells to drug. The ability to map the expression phenotype [40,41], however, provides some level of confidence that gene expression is genetically controlled. Despite these limitations, LCLs provide a resource to build models that identify the contribution of genetics to chemotherapeutic response and also provide genetic variant signatures worth further evaluation in a clinical setting and in other preclinical models.
The results from this study may shed light on new therapeutic targets. Until now, candidate genes, such as those involved in DNA repair, have been emphasized as key players involved in cisplatin resistance and toxicity [8]. Some commonly studied genes in relation to cisplatin resistance and toxicity are glutathione S-transferases, ERCC1, and XRCC1 [21]. Studies that have involved tumor biopsies from patients have supported the role of the glutathione metabolic pathway in acquired and inherited resistance to cisplatin [3]. Furthermore, glutathione levels were found to be 2.9 times higher in ovarian cancer cells resistant to cisplatin [42]. ERCC1 and ERCC2 play roles in the recognition of DNA damage and the removal of the damaged nucleotides [8,43]. One study found that the C/C genotype in a synonymous polymorphism in ERCC1 was associated with a favorable outcome to platinum therapy in non-small cell lung cancer patients [43]. In another study, the wild-type geno-type in ERCC1 was associated with longer survival in non-small cell lung cancer patients treated with cisplatin compared with those carrying the variant genotype [44]. Melanoma patients with the wild-type genotype treated with cisplatin, however, had shorter survival rates and worse response [45]. Additionally, it has been shown that increased variant alleles in ERCC2 and XRCC1 confer a worse survival for non-small cell lung cancer patients treated with platinum drugs [8], whereas an increasing number of variant alleles in squamous head and neck cancer treated with cisplatin had increased survival rates [22]. Even though there are a few identified genetic predictors of platinum-based treatment outcome in DNA repair genes, there is a paucity of data on genetic variants important in platinum-induced toxicity. For example, SNPs which may produce deficiencies in DNA repair genes may also increase toxicity owing to lack of repair of noncancerous cells damaged by treatment [46]. Studies focusing on somatic tissue, however, may not address the role of those germline polymorphisms involved in toxicity. The discrepancies found in the aforementioned clinical studies and the lack of studies evaluating toxicities warrant the use of unbiased whole genome approaches to identify previously unknown genes involved in chemotherapeutic toxicity. One caveat of whole genome association studies using HapMap samples, however, may be a limited power to detect rare variants contributing to the trait and/or sparse genotyping in candidate genes. Thus, an alternative strategy could be deep resequencing of candidate genes for rare variants before linkage or association analyses.
Our linkage-directed association analyses identified 10 SNPs in 5 genes and 10 nongenic SNPs showing a significant association with cellular susceptibility to cisplatin. These genes, individually or in combination, may represent pharmacodynamic targets important in cisplatin cytotoxicity. CDH13, involved in cell recognition and adhesion, is frequently associated with breast, ovarian, and lung cancers through reduced gene expression [47]. Inactivation of CDH13 expression through deletion or hypermethylation has been linked to lung cancer [48] and ovarian cancer [49]. Additionally, aberrant promoter methylation has been associated with tumor progression in a subset of diffuse large B cell lymphomas [50]. In our study, there was one intronic SNP (rs17758876) in CDH13 with the CC genotype associated with greater sensitivity to 10 μmol/l cisplatin. This gene was also significantly associated with daunorubicin-induced cytotoxicity through an intronic SNP (rs1862831) found from another linkage-directed study performed by our laboratory [23]. These two SNPs found in two different studies are not in LD and are located in two different introns (rs17758876 located in intron 6 and rs1862831 located in intron 5) of the same gene.
Other associated genes (ZNF659, LRRC3B, PITX2, and LARP2) are involved in functions such as metal–ion binding, transcription factor activity, and RNA binding. ZNF659 and LRRC3B were both associated with 10 μmol/l cisplatin, whereas PITX2 and LARP2 were associated with 2.5 μmol/l (PITX2), 5 μmol/l (LARP2), and 20 μmol/l (PITX2) cisplatin. Particular attention may be paid to conserved intronic SNPs within LRRC3B (rs7652737) and PITX2 (rs17624452), which may provide enhancer binding sites or response elements for other genes [51]. Some of the associated nongenic SNPs are found in conserved regions of the genome. One very highly conserved SNP (rs7113868) was located in a nongenic region on chromosome 11. Furthermore, the high conservation across species may indicate a functional protein binding and regulatory region [52] important in cytotoxicity.
Our whole genome linkage analysis along with a linkage-directed association approach using LCLs from non-cancerous individuals allows us to identify genes which were previously unknown [23]. This is a comprehensive model incorporating linkage analysis in large pedigrees and association analysis with HapMap trios. The results obtained with cytotoxicity were further validated by performing linear regression with cisplatin-induced apoptosis to decipher which SNPs are associated with cell death through an apoptotic pathway. This in-vitro system provides a first step toward understanding chemotherapeutic-agent-induced toxicities and response in the clinic and is likely to provide a new paradigm for investigating heritable complex traits.
Supplementary Material
Acknowledgements
This Pharmacogenetics of Anticancer Agents Research (PAAR) Group (http://pharmacogenetics.org) study was supported by NIH/NIGMS Grant GM61393 and Pharmacogenetics Research Network and Database, GM61374 (http://www.pharmgkb.org/). The authors are grateful for excellent technical support provided by Dr Jeong-Ah Kang in maintaining the cell lines. Both cytotoxicity (PS207139) and apoptosis (PS207138) data are deposited in www.pharmgkb.org
Footnotes
Supplementary data
Supplementary data are available at The Pharmacogenetics and Genomics Journal online (www.pharmacogeneticsandgenomics.com).
References
- 1.Ho YP, Au-Yeung SC, To KK. Platinum-based anticancer agents: innovative design strategies and biological perspectives. Med Res Rev. 2003;23:633–655. doi: 10.1002/med.10038. [DOI] [PubMed] [Google Scholar]
- 2.Raymond E, Faivre S, Chaney S, Woynarowski J, Cvitkovic E. Cellular and molecular pharmacology of oxaliplatin. Mol Cancer Ther. 2002;1:227–235. [PubMed] [Google Scholar]
- 3.Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer. 2007;7:573–584. doi: 10.1038/nrc2167. [DOI] [PubMed] [Google Scholar]
- 4.Chaney SG, Campbell SL, Bassett E, Wu Y. Recognition and processing of cisplatin- and oxaliplatin-DNA adducts. Crit Rev Oncol Hematol. 2005;53:3–11. doi: 10.1016/j.critrevonc.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 5.Siddik ZH. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene. 2003;22:7265–7279. doi: 10.1038/sj.onc.1206933. [DOI] [PubMed] [Google Scholar]
- 6.Wang D, Lippard SJ. Cellular processing of platinum anticancer drugs. Nat Rev Drug Discov. 2005;4:307–320. doi: 10.1038/nrd1691. [DOI] [PubMed] [Google Scholar]
- 7.Bosken CH, Wei Q, Amos CI, Spitz MR. An analysis of DNA repair as a determinant of survival in patients with non-small-cell lung cancer. J Natl Cancer Inst. 2002;94:1091–1099. doi: 10.1093/jnci/94.14.1091. [DOI] [PubMed] [Google Scholar]
- 8.Gurubhagavatula S, Liu G, Park S, Zhou W, Su L, Wain JC, et al. XPD and XRCC1 genetic polymorphisms are prognostic factors in advanced non-small-cell lung cancer patients treated with platinum chemotherapy. J Clin Oncol. 2004;22:2594–2601. doi: 10.1200/JCO.2004.08.067. [DOI] [PubMed] [Google Scholar]
- 9.Danford AJ, Wang D, Wang Q, Tullius TD, Lippard SJ. Platinum anticancer drug damage enforces a particular rotational setting of DNA in nucleosomes. Proc Natl Acad Sci U S A. 2005;102:12311–12316. doi: 10.1073/pnas.0506025102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Efferth T, Volm M. Pharmacogenetics for individualized cancer chemotherapy. Pharmacol Ther. 2005;107:155–176. doi: 10.1016/j.pharmthera.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 11.Kartalou M, Essigmann JM. Mechanisms of resistance to cisplatin. Mutat Res. 2001;478:23–43. doi: 10.1016/s0027-5107(01)00141-5. [DOI] [PubMed] [Google Scholar]
- 12.Roberts D, Schick J, Conway S, Biade S, Laub PB, Stevenson JP, et al. Identification of genes associated with platinum drug sensitivity and resistance in human ovarian cancer cells. Br J Cancer. 2005;92:1149–1158. doi: 10.1038/sj.bjc.6602447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brabec V, Kasparkova J. Modifications of DNA by platinum complexes. Relation to resistance of tumors to platinum antitumor drugs. Drug Resist Updat. 2005;8:131–146. doi: 10.1016/j.drup.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 14.Kweekel DM, Gelderblom H, Guchelaar HJ. Pharmacology of oxaliplatin and the use of pharmacogenomics to individualize therapy. Cancer Treat Rev. 2005;31:90–105. doi: 10.1016/j.ctrv.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 15.Decatris MP, Sundar S, O'Byrne KJ. Platinum-based chemotherapy in metastatic breast cancer: current status. Cancer Treat Rev. 2004;30:53–81. doi: 10.1016/S0305-7372(03)00139-7. [DOI] [PubMed] [Google Scholar]
- 16.McLeod HL, Evans WE. Pharmacogenomics: unlocking the human genome for better drug therapy. Annu Rev Pharmacol Toxicol. 2001;41:101–121. doi: 10.1146/annurev.pharmtox.41.1.101. [DOI] [PubMed] [Google Scholar]
- 17.Relling MV, Dervieux T. Pharmacogenetics and cancer therapy. Nat Rev Cancer. 2001;1:99–108. doi: 10.1038/35101056. [DOI] [PubMed] [Google Scholar]
- 18.Chang IY, Kim MH, Kim HB, Lee do Y, Kim SH, Kim HY, et al. Small interfering RNA-induced suppression of ERCC1 enhances sensitivity of human cancer cells to cisplatin. Biochem Biophys Res Commun. 2005;327:225–233. doi: 10.1016/j.bbrc.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 19.De Silva IU, McHugh PJ, Clingen PH, Hartley JA. Defects in interstrand cross-link uncoupling do not account for the extreme sensitivity of ERCC1 and XPF cells to cisplatin. Nucleic Acids Res. 2002;30:3848–3856. doi: 10.1093/nar/gkf479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Selvakumaran M, Pisarcik DA, Bao R, Yeung AT, Hamilton TC. Enhanced cisplatin cytotoxicity by disturbing the nucleotide excision repair pathway in ovarian cancer cell lines. Cancer Res. 2003;63:1311–1316. [PubMed] [Google Scholar]
- 21.Pander J, Gelderblom H, Guchelaar HJ. Insights into the role of heritable genetic variation in the pharmacokinetics and pharmacodynamics of anticancer drugs. Expert Opin Pharmacother. 2007;8:1197–1210. doi: 10.1517/14656566.8.9.1197. [DOI] [PubMed] [Google Scholar]
- 22.Quintela-Fandino M, Hitt R, Medina PP, Gamarra S, Manso L, Cortes-Funes H, et al. DNA-repair gene polymorphisms predict favorable clinical outcome among patients with advanced squamous cell carcinoma of the head and neck treated with cisplatin-based induction chemotherapy. J Clin Oncol. 2006;24:4333–4339. doi: 10.1200/JCO.2006.05.8768. [DOI] [PubMed] [Google Scholar]
- 23.Duan S, Bleibel WK, Huang RS, Shukla SJ, Wu X, Badner JA, et al. Mapping genes that contribute to daunorubicin-induced cytotoxicity. Cancer Res. 2007;67:5425–5433. doi: 10.1158/0008-5472.CAN-06-4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Almasy L, Blangero J. Multipoint quantitative-trait linkage analysis in general pedigrees. Am J Hum Genet. 1998;62:1198–1211. doi: 10.1086/301844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin–rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 26.Abecasis GR, Cardon LR, Cookson WO. A general test of association for quantitative traits in nuclear families. Am J Hum Genet. 2000;66:279–292. doi: 10.1086/302698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Benjamini Y, Yekutieli D. Quantitative trait loci analysis using the false discovery rate. Genetics. 2005;171:783–790. doi: 10.1534/genetics.104.036699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dolan ME, Newbold KG, Nagasubramanian R, Wu X, Ratain MJ, Cook EH, Jr, et al. Heritability and linkage analysis of sensitivity to cisplatin-induced cytotoxicity. Cancer Res. 2004;64:4353–4356. doi: 10.1158/0008-5472.CAN-04-0340. [DOI] [PubMed] [Google Scholar]
- 29.Masquelier M, Zhou QF, Gruber A, Vitols S. Relationship between daunorubicin concentration and apoptosis induction in leukemic cells. Biochem Pharmacol. 2004;67:1047–1056. doi: 10.1016/j.bcp.2003.10.025. [DOI] [PubMed] [Google Scholar]
- 30.Mansilla S, Pina B, Portugal J. Daunorubicin-induced variations in gene transcription: commitment to proliferation arrest, senescence and apoptosis. Biochem J. 2003;372:703–711. doi: 10.1042/BJ20021950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Madhusudan S, Middleton MR. The emerging role of DNA repair proteins as predictive, prognostic and therapeutic targets in cancer. Cancer Treat Rev. 2005;31:603–617. doi: 10.1016/j.ctrv.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 32.Wang J, Pabla N, Wang CY, Wang W, Schoenlein PV, Dong Z. Caspase-mediated cleavage of ATM during cisplatin-induced tubular cell apoptosis: inactivation of its kinase activity toward p53. Am J Physiol Renal Physiol. 2006;291:F1300–F1307. doi: 10.1152/ajprenal.00509.2005. [DOI] [PubMed] [Google Scholar]
- 33.Fishel ML, Newell DR, Griffin RJ, Davison R, Wang LZ, Curtin NJ, et al. Effect of cell cycle inhibition on cisplatin-induced cytotoxicity. J Pharmacol Exp Ther. 2005;312:206–213. doi: 10.1124/jpet.104.073924. [DOI] [PubMed] [Google Scholar]
- 34.Gonzalez VM, Fuertes MA, Alonso C, Perez JM. Is cisplatin-induced cell death always produced by apoptosis? Mol Pharmacol. 2001;59:657–663. doi: 10.1124/mol.59.4.657. [DOI] [PubMed] [Google Scholar]
- 35.Lau AH. Apoptosis induced by cisplatin nephrotoxic injury. Kidney Int. 1999;56:1295–1298. doi: 10.1046/j.1523-1755.1999.00687.x. [DOI] [PubMed] [Google Scholar]
- 36.Lieberthal W, Triaca V, Levine J. Mechanisms of death induced by cisplatin in proximal tubular epithelial cells: apoptosis vs. necrosis. Am J Physiol. 1996;270:F700–F708. doi: 10.1152/ajprenal.1996.270.4.F700. [DOI] [PubMed] [Google Scholar]
- 37.Seki K, Yoshikawa H, Shiiki K, Hamada Y, Akamatsu N, Tasaka K. Cisplatin (CDDP) specifically induces apoptosis via sequential activation of caspase-8, -3 and -6 in osteosarcoma. Cancer Chemother Pharmacol. 2000;45:199–206. doi: 10.1007/s002800050030. [DOI] [PubMed] [Google Scholar]
- 38.Huang RS, Duan S, Bleibel WK, Kistner EO, Zhang W, Clark TA, et al. A genome-wide approach to identify genetic variants that contribute to etoposide-induced cytotoxicity. Proc Natl Acad Sci U S A. 2007;104:9758–9763. doi: 10.1073/pnas.0703736104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang RS, Duan S, Shukla SJ, Kistner EO, Clark TA, Chen TX, et al. Identification of genetic variants contributing to cisplatin-induced cytotoxicity by use of a genomewide approach. Am J Hum Genet. 2007;81:427–437. doi: 10.1086/519850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cheung VG, Spielman RS, Ewens KG, Weber TM, Morley M, Burdick JT. Mapping determinants of human gene expression by regional and genome-wide association. Nature. 2005;437:1365–1369. doi: 10.1038/nature04244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morley M, Molony CM, Weber TM, Devlin JL, Ewens KG, Spielman RS, et al. Genetic analysis of genome-wide variation in human gene expression. Nature. 2004;430:743–747. doi: 10.1038/nature02797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lewis AD, Hayes JD, Wolf CR. Glutathione and glutathione-dependent enzymes in ovarian adenocarcinoma cell lines derived from a patient before and after the onset of drug resistance: intrinsic differences and cell cycle effects. Carcinogenesis. 1988;9:1283–1287. doi: 10.1093/carcin/9.7.1283. [DOI] [PubMed] [Google Scholar]
- 43.Reed E. Platinum-DNA adduct, nucleotide excision repair and platinum based anti-cancer chemotherapy. Cancer Treat Rev. 1998;24:331–344. doi: 10.1016/s0305-7372(98)90056-1. [DOI] [PubMed] [Google Scholar]
- 44.Sarries C, Haura EB, Roig B, Taron M, Abad A, Scagliotti G, et al. Pharmacogenomic strategies for developing customized chemotherapy in non-small cell lung cancer. Pharmacogenomics. 2002;3:763–780. doi: 10.1517/14622416.3.6.763. [DOI] [PubMed] [Google Scholar]
- 45.Liu D, O'Day SJ, Yang D, Boasberg P, Milford R, Kristedja T, et al. Impact of gene polymorphisms on clinical outcome for stage IV melanoma patients treated with biochemotherapy: an exploratory study. Clin Cancer Res. 2005;11:1237–1246. [PubMed] [Google Scholar]
- 46.Suk R, Gurubhagavatula S, Park S, Zhou W, Su L, Lynch TJ, et al. Polymorphisms in ERCC1 and grade 3 or 4 toxicity in non-small cell lung cancer patients. Clin Cancer Res. 2005;11:1534–1538. doi: 10.1158/1078-0432.CCR-04-1953. [DOI] [PubMed] [Google Scholar]
- 47.Toyooka KO, Toyooka S, Virmani AK, Sathyanarayana UG, Euhus DM, Gilcrease M, et al. Loss of expression and aberrant methylation of the CDH13 (H-cadherin) gene in breast and lung carcinomas. Cancer Res. 2001;61:4556–4560. [PubMed] [Google Scholar]
- 48.Sato M, Mori Y, Sakurada A, Fujimura S, Horii A. The H-cadherin (CDH13) gene is inactivated in human lung cancer. Hum Genet. 1998;103:96–101. doi: 10.1007/s004390050790. [DOI] [PubMed] [Google Scholar]
- 49.Kawakami M, Staub J, Cliby W, Hartmann L, Smith DI, Shridhar V. Involvement of H-cadherin (CDH13) on 16q in the region of frequent deletion in ovarian cancer. Int J Oncol. 1999;15:715–720. doi: 10.3892/ijo.15.4.715. [DOI] [PubMed] [Google Scholar]
- 50.Ogama Y, Ouchida M, Yoshino T, Ito S, Takimoto H, Shiote Y, et al. Prevalent hyper-methylation of the CDH13 gene promoter in malignant B cell lymphomas. Int J Oncol. 2004;25:685–691. [PubMed] [Google Scholar]
- 51.Thornborrow EC, Patel S, Mastropietro AE, Schwartzfarb EM, Manfredi JJ. qA conserved intronic response element mediates direct p53-dependent transcriptional activation of both the human and murine bax genes. Oncogene. 2002;21:990–999. doi: 10.1038/sj.onc.1205069. [DOI] [PubMed] [Google Scholar]
- 52.Dermitzakis ET, Reymond A, Scamuffa N, Ucla C, Kirkness E, Rossier C, et al. Evolutionary discrimination of mammalian conserved non-genic sequences (CNGs). Science. 2003;302:1033–1035. doi: 10.1126/science.1087047. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.