

Abstract
Arsenicals are known to induce ROS, which can lead to DNA damage, oxidative stress, and carcinogenesis. A human urothelial cell line,, UROtsa, was used to study the effects of arsenicals on the human bladder. Arsenite [As(III)] and monomethylarsonous acid [MMA(III)] induce oxidative stress in UROtsa cells after exposure to concentrations as low as 1 μM and 50 nM, respectively. Previous research has implicated ROS as signaling molecules in the MAPK signaling pathway. As(III) and MMA(III) have been shown to increase phosphorylation of key proteins in the MAPK signaling cascade downstream of ErbB2. Both Src phosphorylation (p-Src) and cyclooxygenase-2 (COX-2) are induced after exposure to 50 nM MMA(III) and 1 μM As(III). These data suggest that ROS production is a plausible mechanism for the signaling alterations seen in UROtsa cells after acute arsenical exposure. To determine importance of ROS in the MAPK cascade and its downstream induction of p-Src and COX-2, specific ROS antioxidants (both enzymatic and non-enzymatic) were used concomitantly with arsenicals. COX-2 protein and mRNA was shown to be much more influenced by altering the levels of ROS in cells, particularly after MMA(III) treatment. The antioxidant enzyme superoxide dismutase (SOD) effectively blocked both As(III)-and MMA(III)-associated COX-2 induction. The generation of ROS and subsequent altered signaling did lead to changes in protein levels of SOD, which were detected after treatment with either 1 μM As(III) or 50 nM MMA(III). These data suggest that the generation of ROS by arsenicals may be a mechanism leading to the altered cellular signaling seen after low-level arsenical exposure.
Keywords: arsenite [As(III)], monomethylarsonous acid [MMA(III)], ROS, ROS scavengers, MAPK
Introduction
Arsenical exposure may induce reactive oxygen species (ROS), which can lead to toxicity, DNA damage, oxidative stress, and carcinogenesis (Kitchin et al., 2003; Huang et al., 2004; Shi et al., 2004a,b). Previous studies were performed with concentrations of arsenite [As(III)] greater than 10 μM, but recent studies have emerged confirming the presence of ROS and oxidative stress associated with low-levels of As(III) as well as its metabolite, monomethylarsonous acid [MMA(III)] (Lantz et al., 2006; Cooper et al., 2007;Wang et al., 2007).
Toxic ROS, including superoxide (O2·-), hydroxyl radicals (OH·), and hydrogen peroxide (H2O2), are generated from normal cellular respiration and aerobic metabolism or by exposure to exogenous oxidants such as arsenicals, resulting in oxidation of nucleic acids, proteins, and membrane lipids. Wang et al., (2007) found that 0.2 μMof the trivalent arsenicals, As(III), MMA(III), and DMA(III) were more potent inducers of oxidative damage in lipids than the pentavalent arsenical species. As(III) was shown to generate detectable amounts of O2·- in U937 cells, human promonocytic cells, at concentrations ranging from 1-10 μM (Wang et al. , 2007). DNA damage in HaCat cells, a human keratinocyte cell line, was detected at levels of As(III) less than 5 μM (Shi et al., 2004b). This DNA damage could be blocked by the addition of the ·OH scavenger, sodium formate. This study found that As(III) first generated O2·- which was converted to H2O2 by SOD and further converted to the highly reactive ·OH by Fe2+ (Shi et al.,2004b). These studies suggest that exposure to low-concentration trivalent arsenicals [(0-10 μM As(III); 0.2 μM MMA(III)/DMA(III)] induce oxidative stress in multiple cell types.
Upon investigating the role of oxidative stress associated with increased cellular proliferation after low-concentration As(III) treatment, Yang et al. (2007) found that 0.5 μM As(III) stimulated cellular proliferation of human lung embryonic fibroblasts (HELF), while higher concentrations (5-10 μM) actually inhibited cell growth. There was a significant positive correlation between the ROS level and the concentration of As(III) administered. For low-concentration As(III), SOD activity was significantly increased over controls, but for high-concentration, SOD activity was inhibited (Yang et al., 2007). This provides evidence that there is a concentration-dependent relationship between the level of arsenical and the amount of ROS produced. These data suggest that O2·- plays a role the stimulation of cellular proliferation following exposure of HELF cells to 0.5 μM As(III).
O2· appears to serve as a growth signal in different cells via activation of the rac/ras-MAPK signaling pathway (Kumagai et al., 2007; Buetler et al., 2004). Over expression of SOD has been shown to inhibit proliferation, whereas inhibition of SOD by pharmacologic means increased O2·- levels and stimulated cellular proliferation. Down regulation of SOD by antisense oligonucleotides blocked apoptosis when associated with a moderate increase in O2·-. A large increase in O3·- led to apoptosis, suggesting that a graded increase in the O2·- level may be the deciding factor between growth and differentiation of cells and apoptosis (Buetler et al., 2004).
Low-concentrations of arsenicals have also been shown to stimulate growth and differentiation of cells by activating specific signaling pathways. In PC12 cells, a rat tumor cell line, As(III) was shown to activate c-Jun N-terminal kinase, p38, and extracellular signal regulated kinase (ERK). This activation could be blocked by the addition of N-acetyl cysteine, suggesting a role for oxidative stress in the increased signaling observed. It has also been suggested that As(III) can bind cysteine rich areas of epidermal growth factor receptor (EGFR) leading to Ras and subsequent ERK activation. The activation of the EGFR-ERK pathway is important in mediating gene expression related to the regulation of cellular proliferation (Simeonova et al., 2002; Luster et al., 2004).
MAPK signaling is altered in multiple types of cancers, including bladder (Wadhwa et al., 2005; Eschwège et al., 2003;. It is important to investigate if As(III) and MMA(III) can induce the MAPK pathways in bladder cells and determine if the induction of the MAPK pathway plays a role in the malignant transformation of cells exposed chronically to As(III) or MMA(III). Numerous studies support the effect of arsenicals on MAPK signaling. Exposures to 0.1-5 μM trivalent arsenicals led to an increased AP-1 DNA binding activity, increased phosphorylation of ERK, but not increased phosphorylation of c-jun or p38 kinases (Drobna et al., 2002). He et al. (2007) found that low concentration As(III) (0.1 and 0.5 μM) stimulated cellular proliferation in HELF cells via activation of JNK and ERK 1/2. Studies performed utilizing pharmacologic inhibitors suggest that EGFR/ErbB2, Src activation, ERK 2 phosphorylation, and PI3K activation to play important roles in the increased expression of COX-2 following 50 nM MMA(III) exposure in UROtsa cells (Eblin et al., 2007).
In the present study, an immortalized, non-tumorigenic human urothelial cell line (UROtsa) was utilized to study the effects of arsenical induced ROS generation and the subsequent activation of the MAPK signaling pathway. The contribution of specific ROS to the increased MAPK signaling was investigated utilizing both enzymatic and non-enzymatic antioxidants. Although the MAPK pathway has been shown to be activated by both ROS and arsenicals, the correlation between ROS increase caused by arsenicals leading to the activation of the MAPK pathway has not been made. In addition, little research has been undertaken to determine if there is a difference in ROS generated between the arsenical species, As(III) or MMA(III), and what roles these differences play in MAPK activation.
Thus, the goal of this study was to determine if it is the parent chemical As(III), the metabolite MMA(III), or the secondary generation of ROS that leads to the increased MAPK signaling seen after low-concentration arsenical exposure in human bladder cells. Cells were exposed to sub-cytotoxic, environmentally relevant levels of arsenicals [1 μM As(III) (74 ppb) and 50 nM MMA(III) (4.5 ppb)]. In arsenical exposed human populations in Romania, 50 nM MMA(III) has been detected in the urine (Aposhian et al., 2000). In addition to being environmentally relevant, these concentrations were specifically chosen since both 1 μM As(III) and 50 nM MMA(III) have been shown to malignantly transform UROtsa cells (Sens et al., 2004; Bredfeldt et al., 2006). By utilizing these concentrations, these acute studies can be linked with studies into the malignant transformation that occurs in UROtsa cells following chronic exposure to the same concentrations of arsenicals used in this study.
Methods
Chemicals
Sodium arsenite, protease inhibitor cocktail, epidermal growth factor (EGF), insulin, peg-superoxide dismutase, peg-catalase, melatonin, and potassium iodide were purchased from Sigma Chemical Company (St. Louis, MO). Dulbecco’s Modified Eagle Medium (DMEM), Dulbecco’s Modified Eagle Medium: Ham’s F12 (DMEM:F12), fetal bovine serum (FBS), antibiotic-antimycotic, and 1X trypsin-EDTA (0.25%) were acquired from Gibco Invitrogen Corporation (Carlsbad, CA). Diiodomethylarsine (MMA(III) iodide, CH3AsI2) was prepared by the Synthetic Chemistry Facility Core (Southwest Environmental Health Sciences Center, Tucson, AZ) using the method of Millar et al. (1960). Water used in studies was distilled and de-ionized. Ras Assay Reagent consisting of Raf-1 RBD and agarose, Anti-Ras clone RAS10, Mg2+ Lysis/Wash Buffer, GTPγS (10 mM), and GDP (100 mM) were all purchased as part of the Ras Activation Assay Kit (Upstate, Temecula, CA).
Cells
UROtsa cells were a generous gift from Drs. Donald and Maryann Sens (University of North Dakota). Cell culture conditions were previously described by Bredfeldt et al., (2004). Prior to experimentation, cells were fed a serum-free growth medium made up of 1:1 mixture of DMEM and Ham’s F-12 supplemented with insulin (5 μg/ml), hydrocortisone (36 ng/ml), and epidermal growth factor (10 ng/ml).
MTT assay for cytotoxicity of ROS antioxidants (non-enzymatic)
This colorimetric assay was derived from Wang et al.,(2007). Antioxidant-induced toxicity was measured by comparing treated cells with untreated control cells, measuring the ability of mitochondrial reductase enzymes to catalyze the reduction of the yellow 3-(4, 5 dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) compound to a purple formazan. Healthy control cells would have the highest activity of enzymes, so comparison between treatment groups can be performed. Mitochondria are a location of ROS generation, so this specific assay was chosen to monitor the health of the mitochondria following the various treatments. For the MTT assay of UROtsa cells, approximately 8 × 103 cells per well were seeded in a 96-well plate and incubated overnight. Cells were treated with several concentrations of ROS inhibitors for 24 h followed by the addition of 20 μl of 2 mg/ml MTT directly into the medium. After incubation (37 °C for 0.5-3 h), the plate was centrifuged and the medium removed. 100 μl of isopropanol/HCl was added into each well and crystals were dissolved by shaking the plate at room temperature. Absorbance was measured by a plate reader at 570 nm. Triplicate wells were used for each sample and the experiments were repeated at least three times to get means and standard deviations.
Trypan blue exclusion assay for cytotoxicity of ROS antioxidants and arsenicals in combination
This assay was derived from a previously described method by Bredfeldt et al., (2006). Cells were plated in 6-well plates at a density of 2 × 105 cells per well and allowed to grow for 24 h. To determine toxicity associated with these compounds, 5 mM KI, 2.5 mM melatonin, 200 units/ml peg-SOD, or catalase were added to a well for 2 h. Next, 1 μM As(III) or 50 nM MMA(III) was added to each well for a period of 24 h. Cell density for UROtsa cells treated with ROS antagonists was obtained via trypan blue exclusion assay.
Detection of ROS in UROtsa cells exposed to As(III), MMA(III) and ROS antioxidants
This method was adapted from Eblin et al., (2006). UROtsa cells were seeded at 1 × 106 cells per bioptechs dish overnight in serum-free growth medium made up of 1:1 mixture of DMEM and Ham’s F-12 supplemented with insulin (5 μg/ml), hydrocortisone (36ng/ml), and epidermal growth factor (10 ng/ml). Cells that were to be used to show antioxidant decreases in ROS were pretreated with KI (2.5 mM), melatonin (1 mM), peg-SOD (200 U/ml), or peg-catalase (200 U/ml). The cells were then stained with CM-H2DCFDA (25 μM) (Molecular Probes/Invitrogen, Carlsbad, CA) for 15 min at 37°C, then washed with 1x PBS. Upon placement on confocal microscope, cells were treated with 500 nM MMA(III) or 10 μM As(III). Images were captured with an upright Zeiss LSM 510 confocal microscope (Carl Zeiss Microimaging Inc., Thornwood, NY) with a 40x “dipping” lens. CM-H2DCFDA was excited at 488 nm and emission was collected with a 515 nm long pass filter. Quantification of immunofluorescence intensity was done using the confocal microscope.
Western Blot analysis
This method was adapted from Eblin et al., (2007). After exposure to arsenicals and/or pretreatment with antioxidants, cells were scraped into RIPA (Radio-Immunoprecipitation Assay) Buffer with protease inhibitor cocktail (Sigma, St. Louis, MO). Protein concentrations were determined by the BCA assay (Sigma, St. Louis, MO). Cellular protein (30 μg) was separated via SDS-PAGE with Mini-Protean II (BioRad, Hercules, CA) and transferred to PVDF membranes (Amersham Pharmacia Biotech, Inc/ GE Healthcare, Piscataway, NJ). Immunoblotting for proteins of interest (COX-2, phosphorylated-Src, SOD1, SOD2, catalase, and GAPDH) was achieved with monoclonal, HRP-conjugated secondary antibodies (Cayman Chemical, Ann Arbor, MI; Cell Signaling, Beverly, MA; Biodesign, Saco, ME; Calbiochem, La Jolla, CA) and fresh enhanced chemiluminescent (ECL) solution (Pierce, Rockford, IL).
Isolation of Nucleic Acids
UROtsa cells were plated 6 × 105 cells per well in 6-well plates (Falcon) and grown according to Eblin et al., (2007) in serum-free media. After exposure to arsenicals and/or antioxidant, cells were rinsed twice with PBS. Nucleic acids were isolated as previously described (Oshiro et al., 2005). Total RNA was isolated from all cells using the RNeasy Mini kit (Qiagen, Valencia, CA). All samples were quantified using absorbance at 260 nm on the NanoDrop 1000 Spectrophotometer (NanoDrop, Wilmington, DE).
Real-Time RT-PCR
Total RNA (250 ng) was converted to cDNA (Applied Biosystems, Foster City, CA). Converted cDNA was added to Universal PCR Master Mix (Applied Biosystems, Foster City, CA) and gene-specific Taqman Primer/Probe (Applied Biosystems, Foster City, CA) and was then subjected to Real-Time PCR analysis using the ABI 7500 Real-Time detection system (Applied Biosystems, Foster City, CA). Results were calculated using the Delta Ct method normalizing to GAPDH expression for each sample. ABI Assay ID numbers: Hs00166575_m1, Hs00167309_m1, Hs00153133_m1, Hs00156308_m1.
Statistics
Graphs and data analysis were generated in Microsoft Office Excel and the data analysis package (Microsoft Corp., Redmond, WA). Data from the confocal imaging, MTT assay, trypan blue proliferation assay, densitometry, and real-time RT-PCR are expressed as the average of three experiments. These data are represented as the mean ±SEM. Statistical significance was determined using Student’s t-test for comparison of samples and statistical significance was marked by either an asterisk (*) or a cross (†). Statistical significance was determined if p< 0.05. The western blots are representative of three experiments. Densitometry was performed using Total Lab image analysis solution software and analyzed as described above (Nonlinear Dynamics, UK).
Results
Cytotoxicity studies of ROS scavengers in UROtsa cells
Previous work established the efficacy and concentrations of enzymatic antioxidants, SOD and catalase, necessary to decrease arsenical-induced oxidative stress (Eblin et al., 2006). In addition, the non-enzymatic ROS antagonists chosen, KI and melatonin, have been used as scavengers of ·OH /H2O2 and 1O2/·OH, respectively (Ahmad et al. 2004; Kashimoto et al., 1999; Sofic et al., 2005; Miyachi et al., 1982). While these inhibitors are not as specific as enzymatic antioxidants, they provide an additional means to investigate the importance of ROS. Initial cytotoxicity studies with the non-enzymatic antioxidants, KI and melatonin, established the proper concentrations and treatment duration to be used (Figure 1a). To assure that the use of these compounds would not decrease cell viability when in co-treatment with As(III) or MMA(III), the percent viability of cells normalized to control levels was determined after treatment with an arsenical with or without a ROS antagonist (Figure 1b & c). Treatment of UROtsa cells with As(III) alone and in co-treatment with CAT led to a statistically significant decrease in cell viability. In contrast, the addition of melatonin, KI, or SOD led to the blockage of cytotoxicity associated with 1 μM As(III) alone. These data suggest that ROS do play a role in As(III) induced cytotoxicity (Figure 1b). MMA(III) treatment did not cause a decrease in cell viability alone or in combination with an ROS scavenger (Figure 1c). Taken together, these data suggest that there may be different mechanisms of action between these two arsenicals.
Figure 1.

MTT cytotoxicity of antioxidants, KI and melatonin. A) 24 and 48 h toxicity tests of 10.0, 5.0, and 2.5 mM KI and melatonin in UROtsa cells (N=3). B & C) Trypan blue exclusion assay to determine cytotoxicity associated with co-treatment of arsenicals with ROS scavengers in UROtsa cells (N=3). B) Co-treatment of 1 μM As(III) and ROS scavengers does not cause a decrease in cell viability in UROtsa cells after 24 hour exposure when compared to As(III) treated alone group. C) Treatment with 50 nM MMA(III) with or without ROS scavengers has no significant effect on cytoxicity in UROtsa cells after 24 hour exposure when compared to MMA(III) treatment alone. ROS scavenger concentrations (vehicle) and abbreviations as follows: CAT-100 units/ml media of catalase; GSH-5 mM glutathione (H2O); NaN3- 1 mM sodium azide (H2O); KI-1 mM potassium iodide(H2O); melatonin-1 mM melatonin (EtOH); SOD- 100 units/ml media of superoxide dismutase. Cytotoxicity was also measured after 48 h with similar results (data not shown).
Detection of ROS in UROtsa cells exposed to As(III), MMA(III) and ROS antioxidants
To ensure that the concentrations of the ROS antioxidants, both enzymatic and non-enzymatic, chosen were efficient in decreasing the amount of ROS generated by As(III) or MMA(III), ROS production was detected utilizing the fluorescent ROS indicator, CM-H2DCFDA. All of the antioxidant concentrations chosen were efficacious in decreasing significantly the amount of ROS generated by 10 μM As(III) and 500 nM MMA(III) (Figure 2a and 2b). Although the remainder of the studies are performed with the lower concentrations of 1 μM As(III) and 50 nM MMA(III), the higher concentrations of 10 μM As(III) and 500 nM MMA(III) were used in this study as they induced a significant amount of ROS when compared to control background levels of fluorescence, so a noticeable and statistical decrease could be shown when the antioxidants were used as a pretreatment prior to arsenical exposure.
Figure 2.
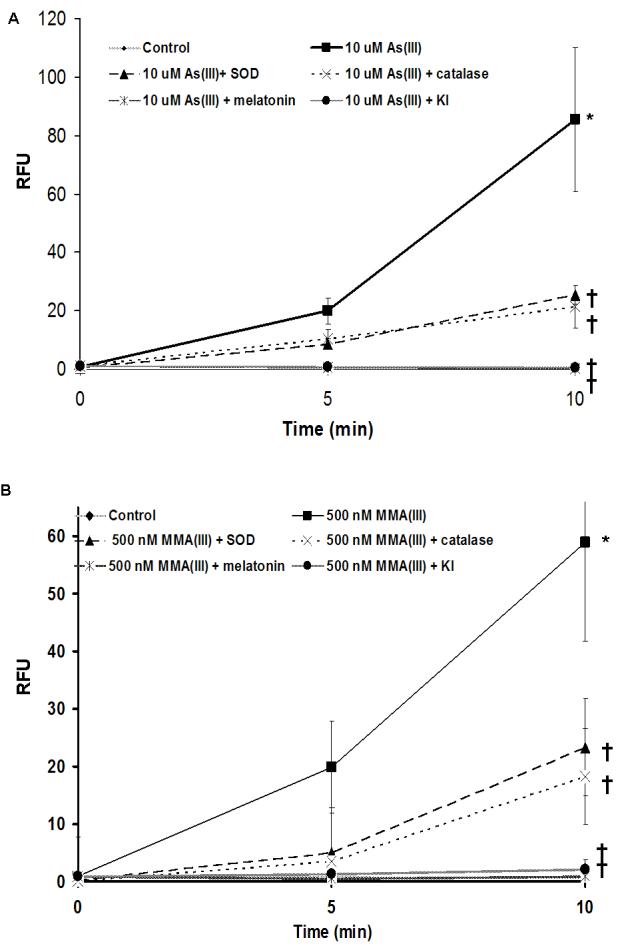
Antioxidants abrogate the ability of arsenicals to induce ROS A) Quantitative comparison of relative DCFDA fluorescence following 10 μM As(III) treatment alone or after pretreatment with 2.5 mM melatonin, 5 mM KI, peg-SOD, or peg-catalase (N=3). B) Quantitative comparison of relative DCFDA fluorescence following 500 nM MMA(III) treatment alone or after pretreatment with 2.5 mM melatonin, 5 mM KI, peg-SOD, or peg-catalase (N=3). (*) Marks statistically significant increase in relative fluorescence when compared with control UROtsa (p≤0.05). (†) Marks statisticallysignificant decrease in relative fluorescence when compared with arsenical-treated UROtsa (p≤0.05).
Determining the role of ROS in arsenical-induced MAPK signaling
To determine the importance of ROS in the altered MAPK signaling seen after arsenical treatment, COX-2 upregulation and the phosphorylation of Src were chosen as molecular markers. While there are many important proteins in the MAPK signaling cascade to study, Src and COX-2 were chosen because both have been shown to be important in anchorage-dependent growth of transformed UROtsa cells. In addition, they have both been shown to be up regulated in human bladder cancer, reinforcing the importance of studying these key proteins after acute exposure to arsenicals (Eblin et al. 2007). These acute changes could lead to chronic alterations in the proteins that play a role in cellular transformation.
Initial studies were performed with higher concentrations of both arsenicals (500 nM MMA(III) and 10 μM As(III); data not shown), but the lower concentrations of 1 μM As(III) and 50 nM MMA(III) were more environmentally relevant. Previous research has established that COX-2 maximal induction occurs after 4 h of exposure to 1 μM As(III) and 50 nM MMA(III) (Eblin et al. 2007). These data was reinforced by the statistically significant increase in COX-2 protein after 4 h exposure to 1 μM As(III) (Figure 3a). SOD and melatonin decreased this induction of COX-2 protein associated with 1 μM As(III) treatment back to below the levels of control (Figure 2a). This suggests that O2·- and 1O2/or ·OH likely play a role in As(III)-induced MAPK signaling. KI and SOD decreased the levels of COX-2 protein associated with 50 nM MMA(III) treatment to the level of control or below (Figure 3b). This provides evidence that ·OH, 1O2, and O2-· play a role in MMA(III)-induced MAPK signaling. Another point to note is that all of the ROS scavengers and antioxidant enzymes blocked some of the COX-2 induction caused by 50 nM MMA(III) treatment. These data suggest that ROS likely play a role in the increased COX-2 protein levels seen after exposure to arsenicals.
Figure 3.


The effects of ROS scavengers and anti-oxidant enzymes on arsenical-induced MAPK signaling in UROtsa cells. A) Densitometry analysis of COX-2 protein induction (72 kDa) seen after treatment with 1 μM As(III) alone, or in co-treatment with ROS scavengers or anti-oxidant enzymes for 4 h. B) Densitometry analysis of COX-2 protein induction (72 kDa) seen after treatment with 50 nM MMA(III) alone, or in co-treatment with ROS scavengers or anti-oxidant enzymes for 4 h. C) Densitometry analysis of phosphorylated-Src (60 kDa) protein induction seen after treatment with 1 μM As(III) alone, or in co-treatment with ROS scavengers or anti-oxidant enzymes for 1 h. D) Densitometry phosphorylated-Src protein induction seen after treatment with 50 nM MMA(III) alone, or in co-treatment with ROS scavengers or anti-oxidant enzymes for 1 h. Values are normalized to GAPDH. All levels are taken relative to control UROtsa with no treatment. (*) marks statistically significant decrease in protein level when compared with arsenical treated group (p≤0.10). (†) Marks statistically significant increase in protein level when compared with control UROtsa (p≤0.10). (N=3) for all treatment groups.
Previous research has established that Src was necessary to induce activation of the EGFR-Erk pathway following As(III) exposure (Simeonova et al. 2002, Eblin et al. 2007). The activation of Src protein by phosphorylation is increased significantly after As(III) exposure (Figure 3c). The phosphorylation of Src by As(III) was decreased following SOD treatment. MMA(III) also caused an increase in Src phosphorylation. Both catalase and KI decreased Src phosphorylation by MMA(III) to below controls levels (Figure 3d). Again these data support the role of ROS in changes seen in cells after acute arsenical treatment.
Real-time RT-PCR confirmation of COX-2 induction by arsenicals
To determine if the ROS induced protein signaling was preceded by changes in COX-2 transcript levels, real-time RT-PCR was performed. COX-2 mRNA steady-state levels increased after a 4 hour treatment with 1 μM As(III), with the levels returning close to control levels after 24 h (Figure 4a). Catalase, KI, melatonin, and SOD all blocked this increase seen in COX-2 levels when co-administered with As(III), suggesting a role for ROS in the increase. MMA(III) also significantly increased the levels of steady-state COX-2 mRNA after 4 h of exposure (Figure 4b). Again, catalase, KI, melatonin, and SOD all blocked the increase in COX-2 mRNA seen after 4 h MMA(III) treatment. These data suggest that the arsenical-induced ROS may result in altered transcriptional regulation of COX-2.
Figure 4.
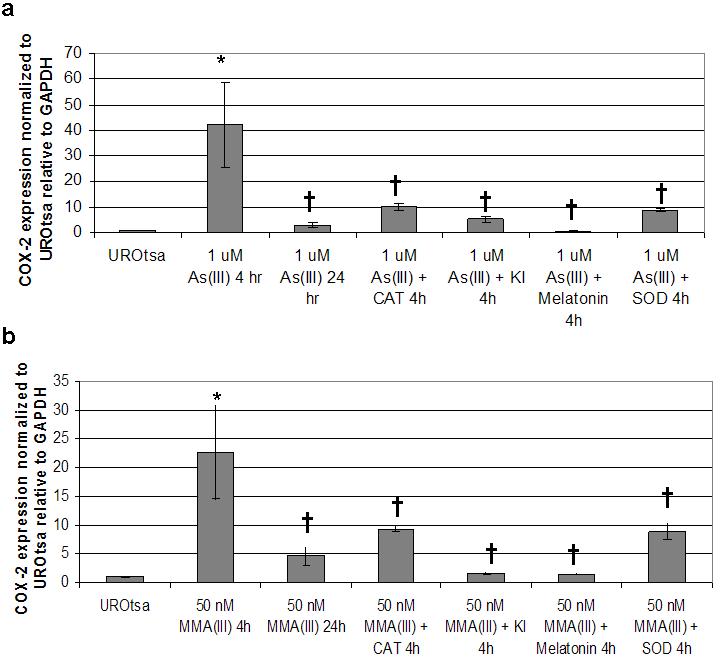
Quantitative real-time RT-PCR expression of COX-2 mRNA after treatment with arsenicals and ROS scavengers in UROtsa cells. COX-2 mRNA is increased after arsenical treatment and that increase can be blocked by the addition of ROS scavengers. A) COX-2 expression relative to GAPDH and normalized to UROtsa after 1 μM As(III) for 4 or 24 h, or after 4 h co-treatment with ROS scavengers. B) COX-2 expression relative to GAPDH and normalized to UROtsa after 50 nM MMA(III) for 4 or 24 h, or after 4 h co-treatment with ROS scavengers. Asterisks (*) denote statistically significant changes between As(III) or MMA(III) treated UROtsa levels and those treated with ROS scavengers (p<0.05). (†) Marks statistically significant increase in transcript level when compared with control UROtsa (p≤0.05). (N=3) for all treatment groups.
Detecting cellular antioxidant responses to low-level arsenical treatment
Since there was enough ROS present to trigger MAPK pathway signaling changes and cause increases in COX-2 mRNA, it was important to determine if the cellular defenses to ROS were increased after arsenical exposure and ROS induction. In addition, adding SOD and catalase to UROtsa cells prior to arsenic exposure had different effects on the MAPK signaling seen, so determination of the levels in the cells following exposure to arsenicals may establish a basis for this difference. To determine if the arsenicals caused enough ROS to elicit alterations in oxidative stress related proteins, protein levels of catalase and SOD were analyzed.
There was no change in catalase protein levels after MMA(III) treatment, but there was a slight decrease in levels following As(III) exposure (Figure 5a). This may explain why adding additional catalase to scavenge H2O2 had little effect as there was sufficient catalase present in the cells to scavenge the ROS produced. For SOD1 (Cu/Zn SOD), both 50 nM MMA(III) and 1 μM As(III) caused an increase in protein levels until 24 h where there was a decrease back to control levels in SOD1 following MMA(III) treatment (Figure 5b). There were some slight changes in SOD2 (MnSOD) levels after As(III) treatment, in a cyclical pattern, with level increasing at 0.5 hour decreasing at 2 h and again increasing at 24 h (Figure 5c). In contrast, SOD2 protein levels were decreased greatly after 50 nM MMA(III) exposure for times as short as 2 h, suggesting that MMA(III) may be targeting the protein for degradation, or blocking its translation. In addition, these data may explain why the addition of peg-SOD decreased the COX-2 mRNA and protein levels, suggesting the increase in O2·- and the combined decrease in SOD2 could lead to increased signaling through O2·-.
Figure 5.
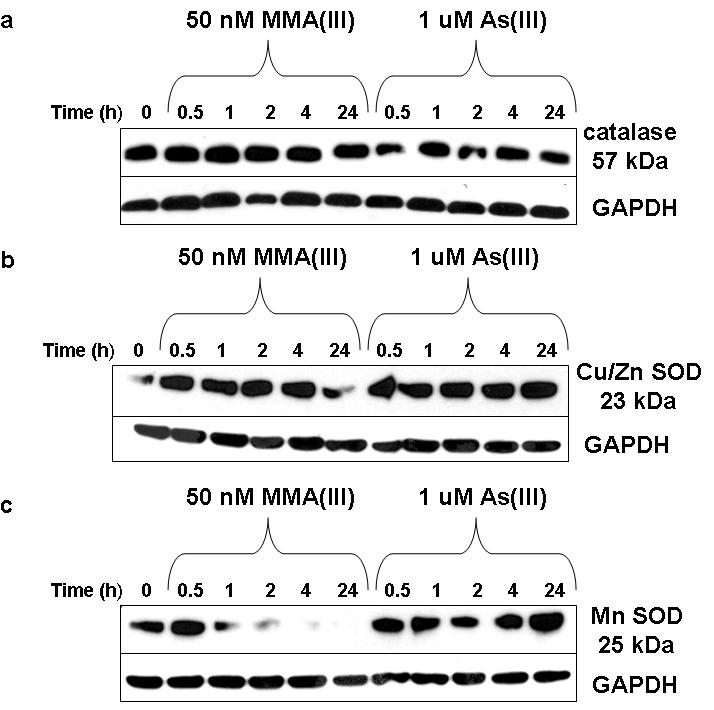
Changes in protein levels of antioxidant enzymes after acute, low-level arsenical treatment in UROtsa cells. A) Representative western blot (N≥3) shows that there is little change in catalase protein levels after either 50 nM MMA(III) or 1 μM As(III) from 0-24 h. B) Representative western blot (N≥3) shows that after either 50 nM MMA(III) or 1 μM As(III), the only changes seen is a decrease in SOD1 after 24 h of exposure to MMA(III). C) Representative western blot (N≥3) shows that there is a noteable decrease in SOD2 levels after 2 h of 50 nM MMA(III) treatment. Conversely, there is little change in SOD2 protein levels after 1 μM As(III) from 0-24 h. GAPDH serves as loading control.
Discussion
A goal of this study was to determine if the parent chemical As(III), the metabolite MMA(III), or the secondary generation of ROS led to an increase in MAPK signaling in human bladder cells. The MAPK pathway has been shown to be activated after acute treatment with either 1 μM As(III) or 50 nM MMA(III) and there is evidence for the formation of ROS at these low concentrations, so it was important to investigate if the ROS caused the increased stimulation of the mitogenic pathways in UROtsa cells after low-level As(III) or MMA(III) exposure. The link between stimulation of this pathway and increased proliferation rates in cancer makes it a relevant pathway to understand in terms of arsenical-induced perturbations. Low-level As(III) and MMA(III) can lead to an increase in oxidative stress, an increase in stress proteins (Hsp70 and MT1), and 50 nM MMA(III) can lead to an increase in 8-oxo-dG, a marker of oxidative damage to DNA. The increase in oxidative stress could be blunted through the addition of either catalase or SOD supporting the potential involvement of ROS (Eblin et al., 2006).
It is important to note that the current study was performed utilizing an environmentally relevant concentration of arsenicals. This study investigated the ability of 1 μM As(III) (74 ppb) or 50 nM MMA(III) (4.5 ppb) to induce ROS and subsequently increase MAPK signaling. It is hypothesized that inhibiting or scavenging the ROS produced by either As(III) or MMA(III) can inhibit the MAPK signaling perturbations seen after treatment. The ROS produced may be a key mechanism in the induction of permanent changes in the MAPK signaling seen after chronic exposure to MMA(III) as small but consistent changes can lead to permanent and noticeable alterations in cellular signaling (Eblin et al., 2007). These changes in MAPK signaling are important to investigate since chronic activation of MAPK can lead to hyperproliferation and potentially malignant transformation.
As this study was performed in vitro, in a cell line that lacks the normal luminal barrier to urine and its components, comparable studies in vivo would be beneficial. UROtsa cells are not fully differentiated, limiting the ability to quantitatively relate the effects seen in this system and in vivo. There is, however, evidence both in vivo and in human studies supporting the generation of oxidative stress and subsequent alterations that support findings of this study (IARC 2004; Cohen et al., 2007; Kinoshita et al., 2007; Pal et al., 2006; Pi et al., 2002; Hour et al., 2006; Mo et al., 2006; Chung et al., 2008). While keeping these limitations in mind, these cells are still very useful in identifying aberrations in signaling processes produced by arsenical exposure.
Several studies have suggested that arsenicals can activate gene expression changes via the alterations of cellular signaling pathways by modulation of intracellular phosphorylation events and MAPK activation. As(III) (50 μM) was shown to induce ligand-independent phosphorylation and activation of EGFR in UROtsa and inhibition of Src prevented this arsenic-induced activation of EGFR and subsequent ERK phosphorylation (Simeonova et al. 2002; Luster et. al 2004). Exposure of mice to 50 μg/ml As(III) in their drinking water led to an induction of c-Src protein in the urinary bladder, as well as EGFR and ERK phosphorylation in the urinary bladder (Simeonova et al. 2002; Luster et. al 2004). In addition, exposure of UROtsa cells to MMA(III) led to COX-2 over expression and ErbB2 and ERK 2 phosphorylation. It was also found that the MAPK signaling pathway, specifically EGFR and Src were upregulated in the UROtsa cells exposed acutely to 50 nM MMA(III) (Eblin et al., 2007).
In UROtsa cells treated with 1 μM As(III) or 50 nM MMA(III), the use of specific ROS scavengers inhibited the generation of ROS (Figure 2), and also established the role of ROS in the increased MAPK signaling seen. Using co-treatment of arsenicals and ROS scavengers, it was seen that As(III)-induced MAPK signaling relied on the production of O2·-, whereas MMA(III)-induced MAPK signaling relied on the production of all species of reactive oxygen studied. In addition, it appears that while ROS play a role in the induction of COX-2 at both the transcript and protein level as a result of both As(III) and MMA(III) exposure, ROS appear to have less of an effect on the phosphorylation of Src following treatment with both compounds (Figure 3c,d).
These data suggest that arsenicals activate MAPK signaling both through ROS, as well as by direct association (Figure 6). If the effect of ROS in MAPK signaling by arsenicals occurs mainly downstream of Src activation, it suggests activation of the pathway upstream of Src by direct association of the arsenical with proteins in the signaling pathways, in addition to ROS performing as a signaling molecule. Support for this idea comes from studies by Simeonova and Luster, where it was found that arsenicals may activate MAPK by binding cysteine rich areas of epidermal growth factor receptor (EGFR) leading to Ras and subsequent ERK activation (Simeonova et al., 2002; Luster et al., 2004).
Figure 6.
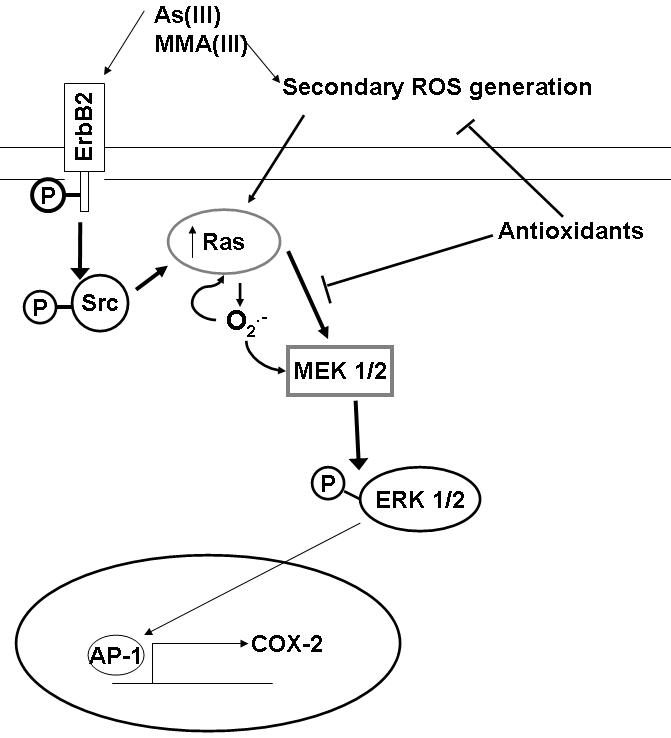
It has been suggested that cells can regulate signal transduction pathways and alter gene expression by altering ROS levels, specifically those of O2·-. The exposure of HaCat cells to 10, 30, and 50 μM As(III) led to an increase in O2·- production (Shi et al. 2004a). O2·- activated the rac/ras-MAPK signaling pathway, downstream of Src (Kumagai et al., 2007; Buetler et al., 2004). In the studies presented herein, the reliance of COX-2 mRNA and protein upregulation on the generation of ROS by arsenicals supports the importance of ROS downstream of Src, and suggests that the increased ROS affects the proteins later in the MAPK signaling cascade to a greater extent.
It is interesting to note that As(III) and MMA(III) appear to generate different ROS and to activate the MAPK pathways via different mechanisms. If the same mechanism was activated similarly by both compounds, one would expect to see the same inhibitors having the same effects. These differences could be due to the method of generation of ROS (direct generation by oxidation and reduction, or secondary generation by activation of NADPH oxidase, xanthine oxidase, or Ras) and what species of reactive oxygen are generated by these processes. These differences in ROS and mechanism of activation of MAPK signaling could give insight into the different toxicities between these two compounds. These studies open the door for future research to be performed that investigates more throroughly the differences in ROS and the actual processes that generate the ROS following As(III) and MMA(III) exposure ot human bladder cells.
As the biological effects of increased ROS after UROtsa cell exposure to arsenicals had been investigated, it was important to determine if the cell recognized the presence of the ROS. Catalase protein decreased following As(III) exposure, but remained the same after MMA(III) exposure. In further support of this idea, the protein levels of SOD2 decrease significantly after MMA(III) treatment but actually increase following As(III) exposure. This decrease in the mitochondrial SOD2 after MMA(III) exposure warrants future investigation into its effect on protective genes related to oxidative stress. This could potentially explain why MAPK signaling is more affected by ROS produced by MMA(III) than As(III) if it can cause increased degradation of the proteins in place to provide protective mechanisms to cells after ROS exposure. It is important to note that adding either SOD or catalase in co-treatment with arsenicals does block MAPK signaling induction as well as oxidative stress (Eblin at al., 2006). Therefore, these enzymes may play a role in the protection of cells from arsenical induced signaling changes, and possibly the long-term effects of constant low-level exposure to arsenicals. The differences seen in signaling following ROS inhibitor treatment warrant further investigations into the differences between these compounds and their associated toxicities.
In summary, this study identifies a relationship between arsenical exposure, secondary ROS generation, and altered MAPK signaling. Low-levels of As(III) and MMA(III) are enough to produce ROS that may lead to altered mitogenic signaling and changes in cellular oxidative stress defenses. These data also support that as chemical species, As(III) and MMA(III) behave differently and further investigation into their differences is needed. It is important to determine if this ROS generated by low-concentration arsenicals causes enough cellular changes and damage to lead to malignant transformation of bladder cells, as both 50 nM MMA(III) and 1 μM As(III) can malignantly transform cells (Sens et al., 2004; Bredfeldt et al., 2006). MAPK signaling has been implicated in the cellular transformation of UROtsa cells after chronic 50 nM MMA(III) exposure. PI3K, Src, and COX-1 and -2 were all implicated in the anchorage independent growth of UROtsa cells malignantly transformed with MMA(III) (Eblin et al. 2007). It is important to next determine if ROS are implicated in these protein changes in the transformed UROtsa. If ROS are indeed causing these permanent changes in the MAPK signaling, markers of oxidative stress could be potential biomarkers of chronic arsenical exposure and damage.
Acknowledgments
Supported by NIH grants ES04940, ES06694, ES07091
Abbreviations
- As(III)
arsenite
- CAT
catalase
- COX-2
cyclooxygenase 2
- HaCaT
human keratinocyte cell line
- HELF
human embryonic lung fibroblasts
- H2O2
hydrogen peroxide
- OH
hydroxyl radical
- mel
melatonin
- MAPK
mitogen activated protein kinase
- MMA(III)
monomethylarsonous acid
- PC12
cancer cell line derived from rat adrenal medulla
- KI
potassium iodide
- ROS
reactive oxygen species
- 1O2
singlet oxygen
- Src
protein tyrosine kinase named after the Rous sarcoma virus
- O2·-
superoxide
- SOD
superoxide dismutase
- U937
human promonocytic cells
- UROtsa
human urothelial cell line
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahmad S, Kitchin KT, Cullen WR. Plasmid DNA damage causes by methylated arsenicals, ascorbic acid, and human liver ferritin. Toxicol. Lett. 2002;133:47–57. doi: 10.1016/s0378-4274(02)00079-6. [DOI] [PubMed] [Google Scholar]
- Aposhian HV, Gurzau ES, Le XC, Gurzau A, Healy SM, Lu X, Ma M, Yip Li., Zakharyan RA, Maiorina RM, Dart RC, Tircus MG, Gonzalez-Ramirez D, Morgan DL, Avram D, Aposhian MM. Occurrence of monomethylarsonous acid in urine of humans exposed to inorganic arsenic. Chem. Res. Toxicol. 2000a;13:693–697. doi: 10.1021/tx000114o. [DOI] [PubMed] [Google Scholar]
- Bredfeldt TG, Jagadish B, Eblin KE, Mash EA, Gandolfi AJ. Monomethylarsenous acid induces transformation of human bladder cells. Toxicol. Appl. Pharmacol. 2006;216:69–79. doi: 10.1016/j.taap.2006.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buetler TM, Krauskopf A, Ruegg UT. Role of superoxide as a signaling molecule. News Physiol. Sci. 2004;19(3):120–123. doi: 10.1152/nips.01514.2003. [DOI] [PubMed] [Google Scholar]
- Chung CJ, Huang CJ, Pu YS, Su CT, Huang YK, Chen YT, Hsueh YM. Urinary 8-hydroxydeoxyguanosine and urothelial carcinoma risk in low arsenic exposure area. Toxicol. Appl. Pharmacol. 2008;226(1):14–21. doi: 10.1016/j.taap.2007.08.021. [DOI] [PubMed] [Google Scholar]
- Cohen SM, Ohnishi T, Arnold LL, Le XC. Arsenic-induced bladder cancer in an animal model. Toxicol. Appl. Pharmacol. 2007;222(3):258–63. doi: 10.1016/j.taap.2006.10.010. [DOI] [PubMed] [Google Scholar]
- Cooper KL, Liu KJ, Hudson LG. Contributions of reactive oxygen species and mitogen-activated protein kinase signaling in arsenite-stimulated hemeoxygenase-1 production. Toxicol. Appl. Pharmacol. 2007;218:119–127. doi: 10.1016/j.taap.2006.09.020. [DOI] [PubMed] [Google Scholar]
- Drobna Z, Jaspers I, Thomas D, Styblo M. Differential activation of AP-1 in human bladder epithelial cells by inorganic and methylated arsenicals. FASEB J. 2002;17:67–69. doi: 10.1096/fj.02-0287fje. [DOI] [PubMed] [Google Scholar]
- Eblin KE, Bowen ME, Cromey D, Bredfeldt TG, Mash EA, Gandolfi AJ. Arsenite and monomethylarsonous acid generate oxidative stress response in human bladder cell culture. Toxicol. Appl. Pharmacol. 2006;217:7–14. doi: 10.1016/j.taap.2006.07.004. [DOI] [PubMed] [Google Scholar]
- Eblin KE, Bredfeldt TG, Buffington SE, Gandolfi AJ. Mitogenic, pro-inflammatory signal transduction caused by monomethylarsonous acid in UROtsa cells. Toxicol. Sci. 2007;95:321–330. doi: 10.1093/toxsci/kfl160. [DOI] [PubMed] [Google Scholar]
- Eschwège P, Ferlicot S, Droupy S, Ba N, Conti M, Loric S, Coindard G, Denis I, Ferretti L, Cornelius A, Legrand A, Bedossa P, Benoît G, Jardin A, Scardino P. A histopathologic investigation of PGE(2) pathways as predictors of proliferation and invasion in urothelial carcinomas of the bladder. Eur. Urol. 2003;44(4):435–41. doi: 10.1016/s0302-2838(03)00313-0. [DOI] [PubMed] [Google Scholar]
- He XQ, Chen R, Yang P, Li AP, Zhou JW, Liu QZ. Biphasic effect of arsenite on cell proliferation and apoptosis is associated with the activation of JNK and ERK 1/2 in human embryo lung fibroblast cells. Toxicol. Appl. Pharmacol. 2007;220:18–24. doi: 10.1016/j.taap.2006.12.021. [DOI] [PubMed] [Google Scholar]
- Hour TC, Pu YS, Lin CC, Huang SW, Chen J, Chiu AW, Chen CJ, Huang CY. Differential expression of molecular markers in arsenic- and non-arsenic-related urothelial cancer. Anticancer. Res. 2006;26:375–8. [PubMed] [Google Scholar]
- Huang C, Ke Q, Costa M, Shi X. Molecular Mechanisms of arsenic carcinogenesis. Mol. Cell. Biochem. 2004;255:57–66. doi: 10.1023/b:mcbi.0000007261.04684.78. [DOI] [PubMed] [Google Scholar]
- IARC. International agency for research on cancer monographs on the evaluation of carcinogenic risk to humans . Some Drinking Water Disinfectants and Contaminants, Including Arsenic. Vol. 84. IARC Press; Lyon: 2004. Arsenic in drinking water; pp. 269–477. [Google Scholar]
- Johansson LH, Borg LAH. A spectrophotometric method for determination of catalase activity in small tissue samples. Anal. Biochem. 1998;174:331–336. doi: 10.1016/0003-2697(88)90554-4. [DOI] [PubMed] [Google Scholar]
- Kashimoto S, Kume M, Ikeya K, Ishiyama T, Kumazawa T. Effects of melatonin and superoxide dismutase on free radical formation in the postischemic reperfused heart. J. Anesth. 1999;13:23–28. doi: 10.1007/s005400050017. [DOI] [PubMed] [Google Scholar]
- Kinoshita A, Wanibuchi H, Wei M, Yunoki T, Fukushima S. Elevation of 8-hydroxydeoxyguanosine and cell proliferation via generation of oxidative stress by organic arsenicals contributes to their carcinogenicity in the rat liver and bladder. Toxicol. Appl. Pharmacol. 2007;221(3):295–305. doi: 10.1016/j.taap.2007.03.024. [DOI] [PubMed] [Google Scholar]
- Kitchin KT, Ahmad S. Oxidative stress as a possible mode of action for arsenic carcinogenesis. Toxicol. Lett. 2003;137(12):3–13. doi: 10.1016/s0378-4274(02)00376-4. [DOI] [PubMed] [Google Scholar]
- Kumagai Y, Sumi D. Arsenic: Signal transduction, transcription factor, and biotransformation involved in cellular response and toxicity. Annu. Rev. Pharmacol. Toxicol. 2007;47:243–262. doi: 10.1146/annurev.pharmtox.47.120505.105144. [DOI] [PubMed] [Google Scholar]
- Lamirande E, Gagnon C. The extracellular signal related kinase(ERK) pathway is involved in human sperm function and modulated by the superoxide anion. Mol. Hum. Reprod. 2002;8(2):124–135. doi: 10.1093/molehr/8.2.124. [DOI] [PubMed] [Google Scholar]
- Lantz RC, Hays AM. Role of oxidative stress in arsenic-induced toxicity. Drug Metab. Rev. 2006;38:791–804. doi: 10.1080/03602530600980108. [DOI] [PubMed] [Google Scholar]
- Lin CL, Wang FS, Kuo YR, Huang YT, Huang HC, Sun YC, Kuo YH. Ras modulation of superoxide activates ERK-dependent fibronectin expression in diabetes-induced renal injuries. Kidney International. 2006;69:1593–1600. doi: 10.1038/sj.ki.5000329. [DOI] [PubMed] [Google Scholar]
- Luster MI, Simeonova PP. Arsenic and urinary bladder cell proliferation. Toxicol. Appl. Pharmacol. 2004;198(3):419–23. doi: 10.1016/j.taap.2003.07.017. Review. [DOI] [PubMed] [Google Scholar]
- Miyachi Y, Niwa Y. Effects of potassium iodide, colchicine and dapsone on the generation of polymorphonuclear leukocyte derived oxygen intermediates. Brit. J. Dermat. 1982;107:209–214. doi: 10.1111/j.1365-2133.1982.tb00340.x. [DOI] [PubMed] [Google Scholar]
- Mo J, Xia Y, Wade TJ, Schmitt M, Le XC, Dang R, Mumford JL. Chronic arsenic exposure and oxidative stress: OGG1 expression and arsenic exposure, nail selenium, and skin hyperkeratosis in Inner Mongolia. Environ. Health Perspect. 2006;114(6):835–41. doi: 10.1289/ehp.8723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshiro MM, Kim CJ, Wozniak RJ, Junk DJ, Munoz-Rodriguez JL, Burr JA, Fitzgerald M, Pawar SC, Cress AE, Domann FE, Futscher BW. Epigenetic silencing of DSC3 is a common event in human breast cancer. Breast Cancer Res. 2005;7(5):669–80. doi: 10.1186/bcr1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal S, Chatterjee AK. Possible beneficial effects of melatonin supplementation on arsenic-induced oxidative stress in Wistar rats. Drug Chem. Toxicol. 2006;29(4):423–33. doi: 10.1080/01480540600837993. [DOI] [PubMed] [Google Scholar]
- Pi J, Yamauchi H, Kumagai Y, Sun G, Yoshida T, Aikawa H, Hopenhayn-Rich C, Shimojo N. Evidence for induction of oxidative stress caused by chronic exposure of Chinese residents to arsenic contained in drinking water. Environ. Health. Perspect. 2002;110:331–336. doi: 10.1289/ehp.02110331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sens DA, Park S, Gurel V, Sens MA, Garrett SH, Somji S. Inorganic cadmium- and arsenite-induced malignant transformation of human bladder urothelial cells. Toxicol. Sci. 2004;79:56–63. doi: 10.1093/toxsci/kfh086. [DOI] [PubMed] [Google Scholar]
- Shi H, Hudson LG, Ding W, Wang S, Cooper KL, Liu S, Chen Y, Shi X, Liu KJ. Arsenite causes DNA damage in keritinocytes via generation of hydroxyl radicals. Chem. Res. Toxicol. 2004a;17:871–878. doi: 10.1021/tx049939e. [DOI] [PubMed] [Google Scholar]
- Shi H, Shi X, Liu KJ. Oxidative mechanism of arsenic toxicity and carcinogenesis. Mol. Cell. Biochem. 2004b;255(12):67–78. doi: 10.1023/b:mcbi.0000007262.26044.e8. Review. [DOI] [PubMed] [Google Scholar]
- Simeonova PP, Wang S, Hulderman T, Luster MI. c-Src-dependent activation of the epidermal growth factor receptor and mitogen-activated protein kinase pathway by arsenic. J. Biol. Chem. 2002;277:2945–2950. doi: 10.1074/jbc.M109136200. [DOI] [PubMed] [Google Scholar]
- Sofic E, Rimpapa Z, Kundurovic Z, Sapcanin A, Tahirovic I, Rustembegovic A, Cao G. Antioxidant effect of neurohormone melatonin. J. Neural. Transm. 2005;112:349–358. doi: 10.1007/s00702-004-0270-4. [DOI] [PubMed] [Google Scholar]
- Wadhwa P, Goswami AK, Joshi K, Sharma SK. Cyclooxygenase-2 expression increases with the stage and grade in transitional cell carcinoma of the urinary bladder. Int Urol Nephrol. 2005;37:47–53. doi: 10.1007/s11255-004-4699-z. [DOI] [PubMed] [Google Scholar]
- Wang TC, Jan KY, Wang AS, Gurr JR. Trivalent arsenicals induce lipid peroxidation, protein carbonylation, and oxidative DNA damage in human urothelial cells. Mut. Res. 2007;615:75–86. doi: 10.1016/j.mrfmmm.2006.10.003. [DOI] [PubMed] [Google Scholar]
- Wang XJ, Sun Z, Eblin KE, Gandolfi AJ, Zhang DD. Nrf2 protects human bladder urothelial cells from arsenite and monomethylarsonous acid toxicity. Toxic. Appl. Pharm. 2007;225(2):206–13. doi: 10.1016/j.taap.2007.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JQ, Buettner GR, Domann FE, Li Q, Engelhardt JF, Weydert CD, Oberley LW. v-Ha-ras mitogenic signaling through superoxide and derived oxygen species. Mol. Carcin. 2002;33:206–218. doi: 10.1002/mc.10037. [DOI] [PubMed] [Google Scholar]
- Yang P, He XQ, Peng L, Li AP, Wang XR, Zhou JW, Liu QZ. The roles of oxidative stress in hormesis induced by sodium arsenite in human embryo lung fibroblast (HELF) cellular proliferation model. J. Toxicol. Environ. Health, part A. 2007;70:976–983. doi: 10.1080/15287390701290832. [DOI] [PubMed] [Google Scholar]