

Abstract
Injured axons of the adult CNS undergo lengthy retraction from the initial site of axotomy after spinal cord injury. Macrophage infiltration correlates spatiotemporally with this deleterious phenomenon, but the direct involvement of these inflammatory cells has not been demonstrated. In the present study, we examined the role of macrophages in axonal retraction within the dorsal columns after spinal cord injury in vivo and found that retraction occurred between days 2 and 28 after lesion and that the ends of injured axons were associated with ED-1+ cells. Clodronate liposome-mediated depletion of infiltrating macrophages resulted in a significant reduction in axonal retraction; however, we saw no evidence of regeneration. We used time-lapse imaging of adult dorsal root ganglion neurons in an in vitro model of the glial scar to examine macrophage–axon interactions and observed that adhesive contacts and considerable physical interplay between macrophages and dystrophic axons led to extensive axonal retraction. The induction of retraction was dependent on both the growth state of the axon and the activation state of the macrophage. Only dystrophic adult axons were susceptible to macrophage “attack.” Unlike intrinsically active cell line macrophages, both primary macrophages and microglia required activation to induce axonal retraction. Contact with astrocytes had no deleterious effect on adult dystrophic axons, suggesting that the induction of extensive retraction was specific to phagocytic cells. Our data are the first to indicate a direct role of activated macrophages in axonal retraction by physical cell–cell interactions with injured axons.
Keywords: spinal cord injury, dystrophic, inflammation, growth cone, proteoglycan, immunosuppression
Introduction
The environment of a spinal cord lesion is extremely complex. Components of the glial scar, such as highly sulfated proteoglycans, Ephs, slits, and myelin membrane fragments (Filbin, 2003; Silver and Miller, 2004; Harel and Strittmatter, 2006; Yiu and He, 2006; Busch and Silver, 2007; Xie and Zheng, 2008), as well as the process of neuroinflammation (Donnelly and Popovich, 2008) all contribute to regeneration failure. As inflammatory cells accumulate within the lesion, the local architecture of the CNS becomes further disrupted (Fitch et al., 1999). Astrocytes move away from the center of the lesion, become hypertrophic, and dramatically upregulate production of inhibitory chondroitin sulfate proteoglycans (CSPGs) that cause the formation of dystrophic endbulbs on the severed fibers (Tom et al., 2004).
Although the inhibitory effects of proteoglycans and myelin on axonal growth have been well established, the role of neuroinflammation in regeneration failure remains highly controversial (Popovich and Longbrake, 2008). Several studies report beneficial effects of macrophages after injury (Prewitt et al., 1997; Rapalino et al., 1998; Schwartz et al., 1999; Yin et al., 2003); however, others suggest that invading immunological cells are detrimental to CNS regeneration (Fitch et al., 1999; McPhail et al., 2004; Donnelly and Popovich, 2008). Numerous studies in which macrophages have been depleted or inactivated after spinal cord injury have reported neuroprotection, increased regeneration, and improvements in motor, sensory, and autonomic function (Oudega et al., 1999; Popovich et al., 1999; McPhail et al., 2004; Stirling et al., 2004).
Injured axons within the CNS withdraw from the site of axotomy during a period of hours to weeks after an initial injury. Long-distance retraction or “resorption” of the proximal axon was first described by Ramon y Cajal (1928), who observed that severed axons appear increasingly farther away from the initial lesion site over time after injury. He concluded that axonal retraction was a phenomenon intrinsic to the neurons themselves. It has been suggested that proximal retraction is similar to Wallerian degeneration of the distal axon and the protracted axonal degeneration observed in many neurodegenerative diseases (Raff et al., 2002). Since the original observations of Ramon y Cajal, there have been differing reports as to the nature of axonal retraction, its cause, extent, and timing, as well as discussion of whether it is a passive or active process.
In the present study, we observed a close correlation between ED-1+ cells and dystrophic retraction clubs at the ends of severed axons after a dorsal column crush spinal cord injury. We examined the effect of macrophage depletion on axonal retraction in vivo using clodronate liposomes (Popovich et al., 1999) and observed a significant reduction in axonal retraction in the clodronate liposome-treated animals. We hypothesized that ED-1+ cells could mediate axonal retraction and examined the interactions between these cell types using an in vitro model of the glial scar (Tom et al., 2004; Steinmetz et al., 2005) in real time. Our data indicate, for the first time, that ED-1+ cells are directly responsible for retraction of injured axons through physical cell–cell interactions.
Materials and Methods
Dorsal column crush lesion model.
Thirty-three adult female Sprague Dawley rats (250–300 g) were used for in vivo studies. Rats were anesthetized with inhaled isoflurane gas (2%) for all surgical procedures. A T1 laminectomy was performed to expose the dorsal aspect of the C8 spinal cord segment. Durotomies were made bilaterally 0.75 mm from midline with a 30 gauge needle. A dorsal column crush lesion was then made by inserting Dumont no. 3 jeweler's forceps into the dorsal spinal cord at C8 to a depth of 1.0 mm and squeezing the forceps, holding pressure for 10 s and repeating two additional times. Completion of the lesion was verified by observation of white matter clearing. The holes in the dura were then covered with gel film. The muscle layers were sutured with 4-0 nylon suture, and the skin was closed with surgical staples. On closing of the incision, animals received Marcaine (1.0 mg/kg) subcutaneously along the incision as well as buprenorphine (0.1 mg/kg) intramuscularly. Postoperatively, animals were kept warm with a heating lamp during recovery from anesthesia and allowed access to food and water ad libitum. Animals were killed at 2, 4, 7, 14, or 28 d after lesion (N = 3 per group). All animal procedures were performed in accordance with the guidelines and protocols of the Animal Resource Center at Case Western Reserve University.
Macrophage depletion.
Animals received intraperitoneal injections of liposome-encapsulated clodronate or empty liposome control on the day before the dorsal column crush injury and also 1 d after lesion for the 2 d time point, on days 1 and 3 after lesion for the 4 d time point, and on days 1, 3, and 5 after lesion for the 7 d time point (N = 3 per group). Clodronate was a gift from Roche Diagnostics. Clodronate was encapsulated in liposomes as described previously (Van Rooijen and Sanders, 1994).
Axon labeling.
The dorsal columns were labeled unilaterally with Texas Red-conjugated 3000 molecular weight (MW) dextran 2 d before perfusion for all time points, which provided sufficient time for the tracer to be transported to the lesion site. Briefly, the sciatic nerve of the right hindlimb was exposed and crushed with Dumont no. 3 forceps for 10 s and repeated two additional times. A total of 1.0 μl of 3000 MW dextran–Texas Red 10% in sterile water was then injected via a Hamilton syringe into the sciatic nerve at the crush site. The muscle layers were closed with 4-0 nylon suture and the skin with surgical staples. On closing of the incision, animals received Marcaine (1.0 mg/kg) subcutaneously along the incision as well as buprenorphine (0.1 mg/kg) intramuscularly. Postoperatively, animals were kept warm with a heating lamp during recovery from anesthesia and allowed access to food and water ad libitum. Animals were killed 2 d after labeling with an overdose of isofluorane and perfused with PBS followed by 4% paraformaldehyde (PFA). Tissue was harvested and postfixed in 4% PFA and processed for immunohistochemistry.
Immunohistochemistry.
Tissue was postfixed in 4% PFA overnight and then submersed in 30% sucrose overnight, frozen in OCT mounting media, and cut on a cryostat into 20 μm longitudinal sections. Tissue was then stained with anti-GFAP (Accurate Chemical & Scientific) and anti-ED-1 (Millipore), incubated with Alexa Fluor 405 or Alexa Fluor 488 (Invitrogen), respectively, and then imaged on a Zeiss Axiovert 510 laser-scanning confocal microscope.
In vivo axonal retraction quantification.
Three consecutive sections starting at a depth of 200 μm below the dorsal surface of the spinal cord were analyzed per animal to quantify axonal retraction. The lesion center was identified via characteristic GFAP and ED-1 staining patterns. The distance between the ends of the labeled axons and the center of the lesion were measured using Zeiss LSM 5 Image Browser software. The measurements from all sections from all animals in a group were averaged to yield the average distance of retraction per time point.
The technique we used to trace injured fibers labeled axons located very superficially within the dorsal columns. Also, the numbers of labeled fibers varied because of the extent of fasciculation of the sciatic nerve at the level at which the tracer was injected. The linear extent of the lesion increased at deeper levels of the dorsal columns. Therefore, axons located deeper within the spinal cord encountered a much larger lesion than those at more superficial levels. We quantified a specific population and location of labeled axons that could be consistently examined and accurately quantified in all animals. This depth consistently contained labeled fibers in all animals, whereas some animals did not have labeled fibers at deeper depths. Quantification of retraction was performed at similar locations of the lesion to allow for accurate comparison between animals and groups.
DRG dissociation.
DRGs were harvested as previously described (Tom et al., 2004; Davies et al., 1999). Briefly, DRGs were dissected out of adult female Sprague Dawley rats (Zivic-Miller Laboratories; Harlan). Both the central and peripheral roots were removed and ganglia were incubated in a solution of collagenase II (200 U/ml; Worthington Biochemicals) and dispase II (2.5 U/ml; Roche) in HBSS. The digested DRGs were rinsed and gently triturated in fresh Ca2+/Mg2+-free HBSS (HBSS-CMF) three times followed by low-speed centrifugation. The dissociated DRGs were then resuspended in Neurobasal A media supplemented with B-27, Glutamax, and penicillin/streptomycin (all from Invitrogen) and counted. DRGs were plated on Delta-T dishes (Thermo Fisher Scientific) at a density of 3000 cells/ml for a total of 6000 cells/dish.
Time-lapse dish preparation.
Delta-T cell culture dishes were prepared similarly to Tom et al. (2004). Briefly, a single hole was drilled through the upper one-half of the side wall of each dish with a number 2 bit to create a port for the addition of cells to the cultures during time-lapse microscopy. The hole created was sufficiently large enough to allow passage of a 20 gauge needle although small enough to prevent media leakage. Dishes were then rinsed with sterile water and coated with poly-l-lysine (0.1 mg/ml; Sigma-Aldrich) overnight at room temperature, rinsed with sterile water, and allowed to dry. Aggrecan gradient spots were created by pipetting 2.0 μl of aggrecan solution (2.0 mg/ml; Sigma-Aldrich; in HBSS-CMF; Invitrogen) onto the culture surface and allowed to dry. Six spots were placed per dish. After the aggrecan spots dried completely, the entire surface of the dish was bathed in laminin solution (10 μg/ml; BTI) in HBSS-CMF for 3 h at 37°C. The laminin bath was then removed immediately before plating of cells. Dishes containing a laminin-only substrate were prepared as above with only the laminin bath and no aggrecan. It is important to note that the concentrations of substrates used here differ from those used by Tom et al. (2004). We found that we can increase the clarity of our microscopy by eliminating the nitrocellulose from our dish preparation protocol. However, to compensate for the difference in substrate binding to the dish surface, we have had to recalibrate the concentrations of the substrates used to those listed above.
After time-lapse imaging, DRGs were fixed in 4% PFA and immunostained with anti B-tubulin-type III (1:500; Sigma-Aldrich) and anti-chondroitin sulfate (CS-56; 1:500; Sigma-Aldrich).
Cell line macrophage cultures.
NR8383 cells (ATCC CRL-2192; American Type Culture Collection), an adult Sprague Dawley alveolar macrophage cell line, were cultured as described by Yin et al. (2003). Briefly, cells were cultured in uncoated tissue culture flasks (Corning) in F-12K media (Invitrogen) supplemented with 15% FBS, Glutamax, Pen/Strep (Invitrogen), and sodium bicarbonate (Sigma-Aldrich), and fed two to three times per week. This cell line formed a mixed culture of adherent and suspended cells and was passed by collecting and replating floating cells at the time of feeding. NR8383 macrophages are intrinsically active, with a high level of ED-1 expression, a rounded phagocytic morphology, and are highly motile. To prepare the cell line macrophages for time-lapse microscopy experiments, cells were harvested with 0.5% trypsin/EDTA (Sigma-Aldrich), washed three times with serum-free F-12K, and plated in uncoated tissue culture flasks at a density of 1.0 × 106/ml in serum-free F-12K. Before use in time-lapse experiments the following day, the cultured cell line macrophages were harvested with EDTA and a cell scraper and resuspended in Neurobasal A supplemented as above with the addition of HEPES (50 μm; Sigma-Aldrich) at a density of 2.5 × 105/70 μl.
Primary bone marrow-derived macrophage cultures.
Bone marrow progenitor cells were harvested using a previously established protocol (Tobian et al., 2004). Briefly, femurs were removed from adult female Sprague Dawley rats (225–275 g; Harlan). The ends of the femurs were removed, a syringe containing cold DMEM supplemented with 10% FBS, Glutamax, Pen/Strep, β-mercaptoethanol, and HEPES (Invitrogen) (D10F) was inserted into the femur, and the bone marrow was flushed out and collected. The resulting cell mixture was then passed through a 70 μm filter and centrifuged. Supernatant was then removed, the resulting cell pellet resuspended in AKT lysing buffer (Lonza Walkersville) to lyse red blood cells, and centrifuged. The supernatant was removed, and the pellet containing bone marrow precursor cells was resuspended and plated in DMEM additionally supplemented with 20% LADMAC cell line-conditioned media (generous gift from Dr. Clifford Harding, Case Western Reserve University, Cleveland, OH) to induce differentiation into macrophages. Cells were fed on days 5, 7, and 9, and harvested for experimentation on day 10 in culture. One day before time-lapse experiments, primary macrophages were harvested with trypsin/EDTA, washed three times with D10F, and plated in uncoated Petri dishes (BD Biosciences Discovery Labware) in D10F at a density of 1.0 × 106/ml. The following day, the primary macrophages were harvested with EDTA and a cell scraper and resuspended in Neurobasal A plus HEPES at a density of 5.0 × 105/70 μl for time-lapse microscopy experiments. In experiments involving “activated primary macrophages,” cells were stimulated with 2 ng/ml interferon-γ (generous gift from Dr. Clifford Harding) and 20 μg/ml lipopolysaccharide (LPS) (Sigma-Aldrich) or interferon-γ alone for 1 d before use in time-lapse to elevate the activation state of the cells.
Cortical astrocyte preparation.
Cortical astrocytes were harvested from the cortices of postnatal day 0 (P0) to P1 rats. Cortices were finely minced and then treated with 0.5% trypsin in EDTA. Cells were seeded in DMEM/F12 (Invitrogen) with 10% FBS (Sigma-Aldrich) and 2 mm Glutamax on T75 flasks coated with poly-l-lysine and shaken after 4 h to remove nonadherent cells. Astrocytes were allowed to mature in culture for at least 28 d. Astrocytes were harvested with EDTA and a cell scraper and resuspended in Neurobasal A plus HEPES at a density of 5.0 × 105/70 μl for time-lapse microscopy experiments.
Cortical microglia preparation.
Cortical microglia were harvested by removing the cortices of P0–P1 rats. Cortices were finely minced and then treated with 0.5% trypsin in EDTA. Cells were plated in DMEM/F12 (Invitrogen) with 20% FBS (Sigma-Aldrich) and 2 mm Glutamax on T75 flasks coated with poly-l-lysine for 5–7 d. One day before time-lapse experiments, flasks were agitated to remove less adherent cells, and these cells were plated in uncoated Petri dishes (BD Biosciences Discovery Labware) in D10F at a density of 1.0 × 106/ml. The following day, the primary microglia were harvested with EDTA and a cell scraper and resuspended in Neurobasal A plus HEPES at a density of 5.0 × 105/70 μl for time-lapse microscopy experiments.
Time-lapse microscopy studies.
DRG neurons were incubated at 37°C for 48 h before time-lapse imaging. Neurobasal A media with HEPES (50 μm; Sigma-Aldrich) was added to the culture before transfer to a heated stage apparatus. Time-lapse images were acquired every 30 s for 3 h with a Zeiss Axiovert 405M microscope using a 100× oil-immersion objective. Growth cones were chosen that extended straight into the spot rim and had a characteristic dystrophic morphology. Neurons were observed for 30 min to determine baseline behavior before the addition of additional cell types (N = 6 for all groups except primary macrophage, N = 3). Growth cones were observed for 150 min after cell addition. We tracked extension, retraction, and rate of growth with MetaMorph software.
Statistical analysis.
Data were analyzed by one- or two-way ANOVA or general linear model, where appropriate, and Tukey's post hoc test with Minitab 15 software.
Results
Ascending dorsal column sensory axons retract extensively after spinal cord injury
We hypothesized that the infiltration of activated macrophages could play a direct role in axonal retraction. Within subacute and chronic spinal cord lesions, we found that there was a close association between activated macrophages and the ends of regenerating axons, allowing for the possibility of direct physical interactions between these two cell types (Fig. 1). We then characterized the extent of sensory axon retraction after dorsal column crush injury and its correlation with the infiltration of macrophages into the lesion. The dorsal columns of adult female Sprague Dawley rats were crushed at the level of C8 and a population of injured sensory neurons was traced over a period of 2 d via dextran–Texas Red labeling of the sciatic nerve. Spinal cord tissue was harvested at 2, 4, 7, 14, and 28 d after lesion and the distances between the ends of the labeled fibers and the center of the lesions were measured (Fig. 2). By 2 d after lesion, axons had already retracted an average distance of 343 ± 46.92 μm (mean ± SD) (Fig. 2 C); however, this early retraction was most likely attributable to intrinsic mechanisms within the neurons themselves (Kerschensteiner et al., 2005). It is important to note that, at 2 d after injury, the lesion was composed of mainly reactive astrocytes (GFAP+) and a few ED-1+ cells, which were most likely resident microglia (Fig. 2 A). Between days 2 and 7 after lesion, there was a dramatic increase in the number of ED-1+ cells within the lesion (Fig. 2 B,C), the vast majority of which were most likely infiltrating macrophages (Popovich et al., 1997; Donnelly and Popovich, 2008). The second phase of retraction of ascending sensory fibers within the dorsal columns occurred most rapidly over the first week, and then progressively over the next few weeks. By 28 d after lesion, the axons had retracted to an average distance of 774 ± 70.26 μm from the lesion epicenter (Fig. 2 C). These data show that the timing of ascending sensory axon retraction corresponds spatiotemporally with the infiltration and accumulation of ED-1+ cells within the lesion.
Figure 1.

Colocalization of the ends of injured ascending sensory axons with activated macrophages and microglia within a spinal cord lesion. Images of a 20 μm longitudinal section of dorsal column crush spinal cord injury 14 d after lesion. The orientation of sections in A and B is such that caudal is on the left side of the image, and rostral is on the right. A, A 10× single-plane image showing a colocalization of dorsal root ganglion neuron axons labeled with Texas Red-conjugated dextran 3000 MW (Dex-TR) (red) and ED-1+ activated macrophages and microglia (green). B, A 63× confocal z-stack three-dimensional reconstruction of the inset in A showing extensive association of activated macrophages and microglia (green) with the dystrophic axons (red). C, A 90° rotation of B about its y-axis, yielding a side view of the three-dimensional reconstruction. This image provides additional evidence that labeled axon retraction clubs (red) are surrounded by activated macrophages and microglia (green). Scale bars: A, 100 μm; B, 25 μm.
Figure 2.
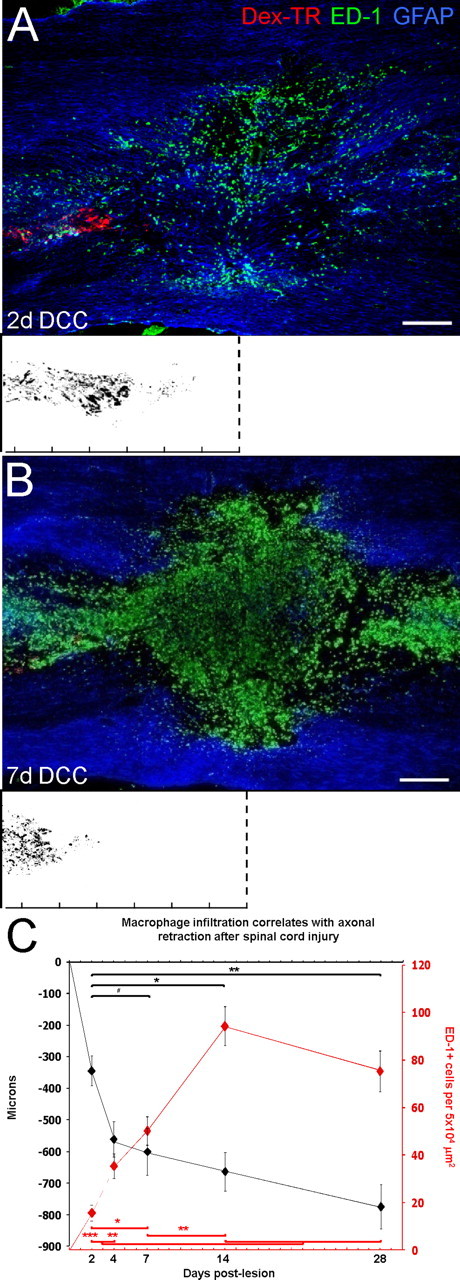
Extensive retraction of ascending sensory axons over time after spinal cord injury. A, B, Shown are 10× single-plane image montages of 20 μm longitudinal sections of a dorsal column crush (DCC) spinal cord injury 2 d (A) and 7 d (B) after lesion. Dex-TR, Texas Red-conjugated dextran 3000 MW. The orientation of the sections is such that caudal is on the left side of the image and rostral is on the right. The white boxes below represent axonal position with respect to the lesion center (dotted lines) with superimposed fiber tracings of multiple sections from one animal at each time point. The ruler tick marks indicate 200 μm increments. A, At 2 d after lesion, dorsal root ganglion axons (red) have retracted a short distance from the initial site of axotomy at the lesion center, marked by GFAP+ reactive astrocytes (blue). There are a few ED-1+ cells (green) within the lesion, which are most likely activated microglia. B, By 7 d after lesion, injured axons (red) have retracted extensively from the lesion center. The lesion and surrounding tissue are now filled with ED-1+ cells (green), which are predominantly infiltrating macrophages, whereas reactive astrocytes (blue) have vacated the lesion core. C, Graph indicating average axonal retraction over time. The majority of retraction occurred during the first 7 d after lesion; however, retraction did continue up to 28 d after lesion, the length of time studied. Axonal retraction (black graph) is as follows: day 2 is significantly different from days 7, 14, 28 (one-way ANOVA, F (4,40) = 6.50, p < 0.001; Tukey's post hoc test, # p < 0.05, *p < 0.01, **p < 0.001). Macrophage depletion (red graph) is as follows: day 2 is significant from days 7, 14, and 28; day 4 is significant from 14 and 28; day 7 is significant from days 2 and 14 (one-way ANOVA, F (4,40) = 22.83, p < 0.001; Tukey's post hoc test, *p < 0.01, **p < 0.001, ***p < 0.0001). Error bars indicate SEM. Scale bars: A, B, 250 μm.
Depletion of activated macrophages reduces axonal retraction in vivo
The majority of the second phase of axonal retraction in vivo occurs in the ascending dorsal column sensory axons between 2 and 7 d after lesion corresponding temporally to the period of macrophage infiltration. To further implicate macrophages in axonal retraction in vivo, we treated animals with clodronate liposomes to deplete circulating monocytes/macrophages (van Rooijen et al., 1997; Popovich et al., 1999). Animals received injections of clodronate liposomes every other day starting treatment 1 d before injury to deplete circulating monocytes/macrophages. Animals were then assessed for axonal retraction at 2, 4, and 7 d after lesion (Fig. 3 A,B,D). Animals that received injections of clodronate liposomes displayed a significant reduction of retraction at 4 and 7 d after lesion (402 ± 81.85 and 439 ± 46.33 μm, respectively) compared with those receiving control liposomes at 4 and 7 d after lesion (586 ± 42.89 and 806 ± 62.71 μm, respectively) (Fig. 3 D). The reduction in retraction correlated with significantly reduced numbers of ED-1+ cells within the lesion in clodronate-treated animals compared with empty liposome controls (Fig. 3 C). Clodronate liposome treatment also resulted in an increase of GFAP+ astrocytes in the lesion core, corroborating previous observations that macrophage depletion leads to a decrease in cavitation (Popovich et al., 1999). Importantly, there was no difference in the amount of retraction exhibited in the clodronate-treated and control liposome-treated animals at 2 d after lesion (Fig. 3 D). Macrophage infiltration has not yet occurred at this time, indicating that the first stage of axonal retraction is macrophage independent, and perhaps attributable to endogenous neuronal mechanisms or, potentially, interactions with activated resident microglia. Clodronate-mediated depletion of circulating macrophages/monocytes did prevent axonal retraction normally observed at 4 and 7 d after lesion, indicating that this second phase of retraction was caused by the actions of infiltrating macrophages. There was no evidence of significant regeneration (i.e., axonal elongation beyond the center of the lesion) in the clodronate-treated animals.
Figure 3.

Clodronate liposome-mediated macrophage depletion prevents the second phase of axonal retraction after injury. A, B, Shown are 10× single-plane image montages of 20 μm longitudinal sections of a dorsal column crush (DCC) spinal cord injury treated with clodronate liposomes (A) and control liposomes (B) at 7 d after lesion. The orientation of the sections is such that caudal is on the left side of the image, and rostral is on the right. The white boxes below represent axonal tracings with respect to the lesion center (dotted lines). The ruler tick marks indicate 200 μm increments. A, At 7 d, clodronate liposome treatment resulted in a significant reduction in the number of ED-1+ cells (green) within the lesion. Astrocytes (blue) continue to occupy the lesion core, and dorsal root ganglion axons (red) have retracted only a short distance from the lesion, similar to the level of retraction normally observed at 2 d after lesion. Dex-TR, Texas Red-conjugated dextran 3000 MW. B, Treatment with control liposomes resulted in no reduction in the number of ED-1+ cells (green) at 7 d, and the lesion core is devoid of reactive astrocytes (blue). In the presence of normal numbers of ED-1+ macrophages and microglia, injured axons (red) have retracted extensively from the lesion center. C, Graph showing the average numbers of ED-1+ cells in the lesion center. Clodronate significantly reduced the number of ED-1+ cells at both 4 and 7 d after lesion. Seven day liposome control is significant from all other time points. Four day liposome control is significant from 2 d clodronate and 7 d clodronate (two-way ANOVA, F (2,48) = 45.73, p < 0.001; Tukey's post hoc test, # p < 0.05, *p < 0.01, ***p < 0.0001). D, Graph indicating average axonal retraction over time. Clodronate treatment significantly reduced axonal retraction at both 4 and 7 d after lesion. Four day liposome control and 7 d liposome control are significantly different from each other and from all other groups (two-way ANOVA, F (2,264) = 22.65, p < 0.0001; Tukey's post hoc test, *p < 0.01, **p < 0.001, ***p < 0.0001). Error bars indicate SEM. Scale bars: A, B, 250 μm.
Dystrophic growth cones retract extensively after contact with macrophages in an in vitro model of the glial scar
Our observation of the close association of ED-1+ cells with injured axons in vivo suggested that interactions between these cell types may play a role in axonal retraction. Previous studies have shown that Aplysia hemocytes, the equivalent of vertebrate macrophages, and neurons can associate in coculture (Farr et al., 2001), and that the presence of macrophages at high concentrations can decrease the number of neurites over time and induce neuronal apoptosis (Arantes et al., 2000), although the cellular and molecular mechanisms underlying these effects were unknown. We focused our attention on the interactions between adult sensory neuron axons and macrophages in an in vitro model of the glial scar in which dystrophic endbulbs form at the tips of the regenerating adult sensory axons as their forward growth stalls for extended periods (but, importantly, they do not tend to retract) on a substrate consisting of opposing gradients of the growth-promoting protein laminin and the inhibitory proteoglycan aggrecan (Tom et al., 2004; Longbrake et al., 2007). After a 30 min period of baseline observation, NR8383 macrophages, which are intrinsically active, were added to the cultures and their interactions with dystrophic axons were monitored. Direct cell–cell contacts were frequently observed between dystrophic axons and macrophages (Figs. 4 A, 5; supplemental Movie S1, available at www.jneurosci.org as supplemental material). These contacts were of an extended duration and, when coupled with migration of the macrophage, led to dramatic manipulations of the axon that resulted in forceful bending and lifting of the axon from the substrate (Fig. 5 A,D; supplemental Movie S1, available at www.jneurosci.org as supplemental material). It was evident that strong, long-lasting adhesions could be made between the two cell types because lengthy trailing processes connecting the two cells often remained after the macrophages migrated away from the axons (Figs. 4 A, 5 B). However, macrophage-induced retraction did not permanently prevent regrowth of the axon, because some axons that lost macrophage contact after retraction were able to extend until they again became dystrophic (data not shown). Direct cell–cell contact between these two cell types eventually led to extensive retraction of the axon 100% of the time (Figs. 4, 9 A–C). Therefore, macrophage contact induced retraction of dystrophic axons in an in vitro model of the glial scar.
Figure 4.

Macrophages induce extensive retraction of dystrophic adult dorsal root ganglion axons in an in vitro model of the glial scar. A, Six-panel montage of single-frame images from a time-lapse movie in which NR8383 macrophages were added to a culture of dystrophic adult dorsal root ganglion neurons growing on an inverse spot gradient of the growth-promoting extracellular matrix molecule laminin and the potently inhibitory chondroitin sulfate proteoglycan aggrecan. Times for each frame are given in the bottom right of each image, and an arrow marks the central domain of the growth cone. An asterisk marks a consistent point in the culture as a reference for growth cone position during frame shifts. Initial macrophage–growth cone contact was made immediately after macrophage addition at 30 min (30′). Physical contacts are observed between a second macrophage and the dystrophic axon at 61′. Additional macrophages physically altered the axonal trajectory as retraction began at 110′. The growth cone is obscured by multiple macrophages and has retracted nearly out of the frame at 150′. The entire movie can be viewed in supplemental Movie S1 (available at www.jneurosci.org as supplemental material). Scale bar, 20 μm. B, Positional graph tracking the growth cone for entire time-lapse movie in A. Each point represents the position of the central domain of the growth cone for a single frame (every 30 s). The axon underwent extensive retraction of ∼100 μm after macrophage contact. C, Positional graph from another representative time-lapse experiment.
Figure 5.

Macrophages physically interact with dystrophic axons in an in vitro model of the glial scar. A, Select frames from a time-lapse movie in which macrophages physically contact a dystrophic axon. Before retraction occurred, the growth cone was still attached while the axon was lifted from the substrate and severely bent (arrows). B, A higher magnification image of the third image from Figure 3 A. Several adhesive contacts were made between a macrophage and a dystrophic axon. The arrows indicate membrane processes that formed from these contacts as the macrophage moved away from the axon. C, A 40× confocal z-stack three-dimensional reconstruction of a culture of adult DRG neurons (red) 2.5 h after macrophage (green) addition. A macrophage is observed in direct contact with the dystrophic growth cone. D, A 90° rotation of C about the x-axis yielding a side view of the three-dimensional reconstruction. The arrow indicates a neuronal process (red) that has been lifted from the substrate by the adjacent macrophage (green). Scale bars: A, B, 20 μm; C, 50 μm.
Figure 9.
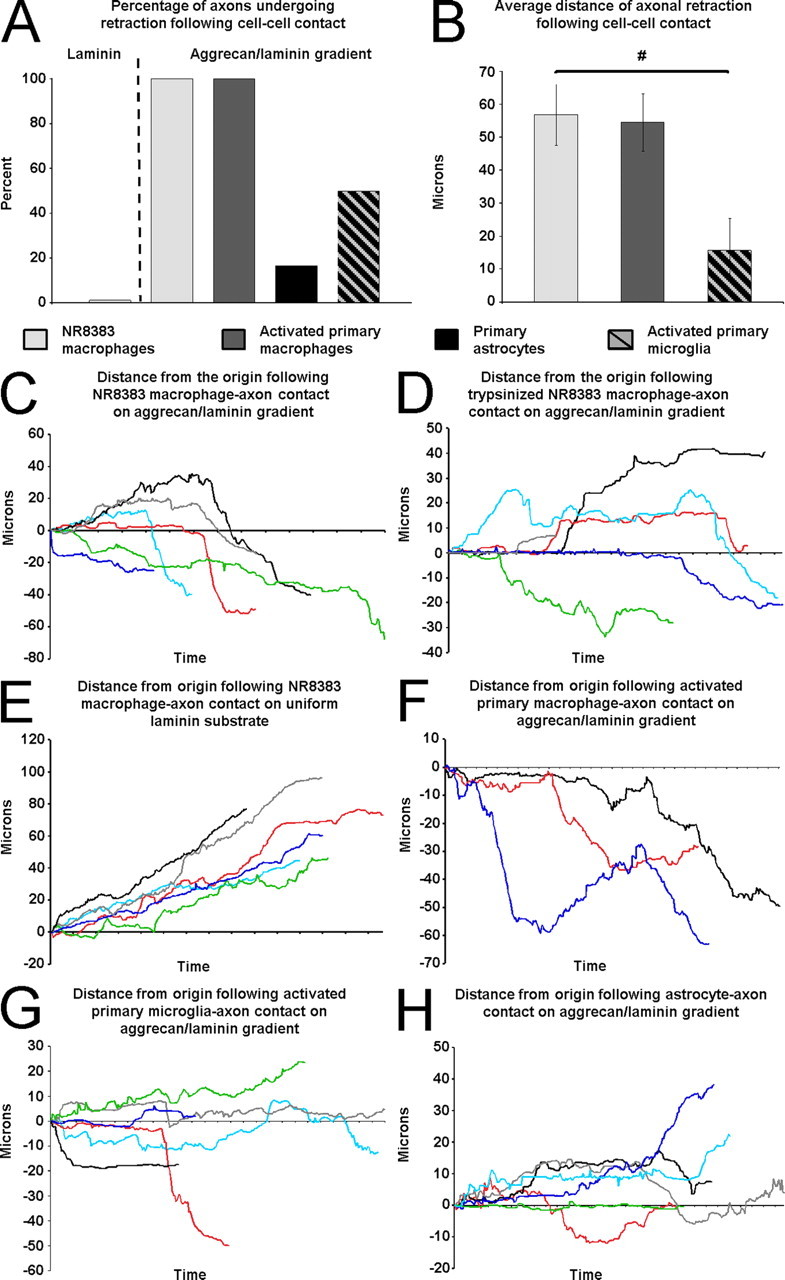
Analyses of time-lapse experiments. A, The percentage of axons in each group that retracted >20 μm after cell–cell contact. B, The average distance of retraction after cell contact. Cell line macrophages are significantly different from activated primary microglia (general linear model, F (2,12) = 5.97, p = 0.016; Tukey's post hoc test, # p < 0.05). Error bars indicate SEM. C, The results of six experiments showing that six dystrophic axons on the aggrecan/laminin gradient retract after contact with NR8383 macrophages. D, The results of six experiments showing the responses of six dystrophic axons on the aggrecan/laminin gradient after contact with trypsinized NR8383 macrophages. E, The results of six experiments showing that axons continue to extend on a uniform laminin substrate after contact with NR8383 macrophages. F, The results of three experiments showing that three dystrophic axons on the aggrecan/laminin gradient retract after contact with activated primary bone marrow-derived macrophages. G, The results of six experiments showing that axons on the aggrecan/laminin gradient have mixed responses to microglia contact. H, The results of six experiments showing that dystrophic axons on the aggrecan/laminin gradient maintain their position after astrocyte contact.
Direct physical cell–cell interactions are required for macrophage-induced retraction
We next sought to determine whether physical interactions between axons and macrophages were required to induce axonal retraction, or whether macrophage-derived factors were sufficient. During the course of our study, there were numerous instances in which macrophages were observed to migrate very close to, but not touch, dystrophic axons, and axonal retraction was not observed in these cases (data not shown). To further explore this issue, we lightly treated macrophages with trypsin to remove extracellular proteins before their addition to the DRG cultures (supplemental Movie S2, available at www.jneurosci.org as supplemental material). Pretreatment of macrophages with trypsin still allowed for extensive macrophage mobility and multiple collisions with axons. However, the treatment largely prevented the macrophages from physically tethering to the axons, and consequently far fewer retractions were observed in the absence of long-lasting direct cell–cell contacts (see Fig. 9 D). Although we have only observed axonal retraction after macrophage contact, it was possible that macrophages were secreting a factor(s) that induced retraction. To test this hypothesis, we added macrophage-conditioned media to dystrophic axons in vitro and examined its effect. Macrophage-conditioned media did not induce retraction (data not shown). Therefore, the mere presence of macrophages, or their secreted factors, in the vicinity of dystrophic axons was not sufficient to induce retraction in the absence of physical interactions.
Role of the substrate in macrophage-induced retraction
To determine whether the substrate plays a role in axonal retraction, we cultured adult sensory neurons on a uniform growth-promoting laminin substrate that does not produce dystrophic growth cones. Growth cones on laminin were flattened with numerous filopodia and lamellapodia and overall axon extension occurred at a constant rate (Fig. 6). When macrophages were added to these cultures, direct cell–cell contact with axons was, indeed, observed. Importantly, however, these contacts were transient and quickly broken, not as extensive, and did not result in axonal retraction (Figs. 6 A, 9 A,E; supplemental Movie S3, available at www.jneurosci.org as supplemental material). Remnants of membrane contact points were resorbed rapidly back into the neurons, and the growth cones continued to extend across the substrate unimpeded (Fig. 6; supplemental Movie S3, available at www.jneurosci.org as supplemental material). Therefore, macrophage-induced axonal retraction was substrate dependent; neurons in an active growth state on the permissive substrate laminin were not susceptible to macrophage contact unlike those in a state of dystrophy induced by a CSPG gradient.
Figure 6.

Macrophages do not induce extensive retraction of actively growing adult dorsal root ganglion axons. A, Six-panel montage of single-frame images from a time-lapse movie in which NR8383 macrophages were added to a culture of actively growing adult dorsal root ganglion neurons on a uniform laminin substrate. Times for each frame are given in the bottom right of each image, and an arrow marks the central domain of the growth cone. An asterisk marks a consistent point in the culture as a reference for growth cone position during frame shifts. Initial macrophage–growth cone contact was made at 45 min (45′). Physical contacts were rapidly broken and no remnant processes were observed between the macrophages and axon at 60′ and 150′. The entire movie can be viewed in supplemental Movie S3 (available at www.jneurosci.org as supplemental material). Scale bar, 20 μm. B, Positional graph tracking the growth cone for entire time-lapse movie. Each point represents the position of the central domain of the growth cone for a single frame (every 30 s). The axon continued to extend ∼40 μm after macrophage contact. C, Positional graph from another representative time-lapse experiment.
Activated primary macrophages also induce axonal retraction
It was critical to verify that primary macrophages interacted with dystrophic axons in the same manner as the NR8383 macrophage cell line in vitro. We harvested progenitor cells from the bone marrow of adult Sprague Dawley rats and differentiated them into macrophages in vitro, yielding a culture of >80% ED-1+ cells. This particular population of macrophages has been shown to retain the phenotypic, morphological, and functional characteristics of macrophages found in spinal cord lesions unlike populations harvested from other bodily sources (Longbrake et al., 2007). The ability of primary macrophages to induce axonal retraction was then assayed in our model. Unstimulated primary macrophages were not capable of inducing retraction (data not shown). When added to the spot gradient neuronal culture, these macrophages adhered to the substrate but were not motile, displaying characteristics of macrophages in a resting state. Contacts with axons occurred only when macrophages settled directly onto dystrophic axons. Neither the macrophages nor their cell–cell interactions exhibited any of the physical characteristics previously observed with the cell line macrophages, i.e., no tugging, no signs of physical attachment via cell processes, etc.
We hypothesized that macrophages must be in an activated state to interact with dystrophic axons. We stimulated the primary macrophages with the activating cytokine interferon-γ in culture before addition to the time-lapse culture dishes. Although these macrophages exhibited a moderate state of activation and a slightly rounded morphology, they were still not motile and did not form strong attachments with dystrophic axons and, consequently, did not induce axonal retraction (data not shown). We then stimulated the primary macrophages further with a combination of interferon-γ and LPS before addition to the DRG cultures. These macrophages displayed the morphology and behavior of activated macrophages: rounded, phagocytic shape, and highly motile (Fig. 7; supplemental Movie S4, available at www.jneurosci.org as supplemental material). These activated macrophages induced retraction of dystrophic axons as frequently as cell line macrophages (Figs. 7, 9 A,B,F; supplemental Movie S4, available at www.jneurosci.org as supplemental material). They displayed vigorous physical interactions with dystrophic axons, resulting in strong adhesions between cells and physical grasping, tugging, and lifting of axons from the substrate. Primary macrophages, when in an activated state, induced retraction of dystrophic axons in vitro, validating the use of cell line macrophages in our study of axonal retraction in vitro. We therefore chose to conduct the majority of our experiments with the NR8383 macrophage cell line because it constituted a pure population of cells that were in a constant state of activation, similar to macrophages found within spinal cord lesions, without the requirement for additional stimulation.
Figure 7.

Activated primary macrophages induce extensive retraction of dystrophic adult dorsal root ganglion axons in an in vitro model of the glial scar. A, Six-panel montage of single-frame images from a time-lapse movie in which interferon-γ- and LPS-stimulated primary bone marrow-derived macrophages were added to a culture of dystrophic adult dorsal root ganglion neurons growing on an inverse spot gradient of the growth-promoting extracellular matrix molecule laminin and the potently inhibitory chondroitin sulfate proteoglycan aggrecan. Times for each frame are given in the bottom right of each image, and an arrow marks the central domain of the growth cone. An asterisk marks a consistent point in the culture as a reference for growth cone position during frame shifts. Initial macrophage-growth cone contact was made at 35 min (35′). An additional macrophage contacted with axon at 42′. The growth cone had retracted by ∼65′. A retraction fiber formed at the tip of the growth cone as the axon retracted at 70′ and 80′. The entire movie can be viewed in supplemental Movie S4 (available at www.jneurosci.org as supplemental material). Scale bar, 20 μm. B, Positional graph tracking the growth cone for entire time-lapse movie. Each point represents the position of the central domain of the growth cone for a single frame (every 30 s). The axon underwent extensive retraction of ∼70 μm after macrophage contact. C, Positional graph from another representative time-lapse experiment.
Activated microglia are moderately capable of inducing axonal retraction in vitro
Although macrophages do not typically invade the injured spinal cord until 3 d after lesion, resident microglia within the CNS respond to injury immediately (Watanabe et al., 1999). Microglia within a lesion become activated and phagocytic, much like activated macrophages. We found a limited number of ED-1+ cells within the lesion at 2 d after injury (Fig. 2 A,C), before typical macrophage infiltration, which were most likely resident microglia. We felt it was important to assess the potential contribution of microglia to axonal retraction using our in vitro model. Cortical microglia were harvested from P1 Sprague Dawley rats and matured in vitro before their addition to time-lapse cultures. Similar to primary macrophages, primary microglia had to be stimulated with interferon-γ and LPS to become activated in culture. Unstimulated microglia failed to adhere to the laminin/aggrecan spot gradient substrate, which prevented them from interacting with dystrophic axons in our model (data not shown). However, stimulated microglia did adhere and physically interact with axons, inducing retraction 50% of the time (see Fig. 9 A,B,G); however, the contacts between activated microglia and dystrophic axons were not as strong as those of macrophages. Therefore, microglia activated experimentally can also play a role in the induction of axonal retraction.
Astrocytes, another CNS cell type, fail to induce axonal retraction in vitro
We next wanted to establish that the induction of retraction of dystrophic axons in vitro is specific to phagocytic cell types normally found within a lesioned spinal cord and not merely the result of the interactions of dystrophic axons with any other cell type. Astrocytes are an integral component of the glial scar after injury to the CNS. They are present in high numbers and extensively contact regenerating axons. We harvested cortical astrocytes from P1 Sprague Dawley rats and allowed them to mature in vitro before addition to DRG cultures. Astrocytes adhered to the substrate and contacted dystrophic axons extensively (Fig. 8; supplemental Movie S5, available at www.jneurosci.org as supplemental material). It is important to note that, once bound to the substrate, astrocytes migrated rapidly down the aggrecan gradient, away from the rim. Astrocyte processes spread out over axons, sometimes resulting in lateral displacement of the axon. However, these contacts did not lead to retraction of the contacted axon (Figs. 8, 9 A,H). Therefore, the induction of retraction was specific to interactions with ED-1+ phagocytic cells and not merely physical interactions with any other cell type.
Figure 8.

Astrocytes do not induce retraction of dystrophic adult dorsal root ganglion axons in an in vitro model of the glial scar. A, Six-panel montage of single-frame images from a time-lapse movie in which primary astrocytes were added to a culture of dystrophic adult dorsal root ganglion neurons growing on an inverse spot gradient of the growth-promoting extracellular matrix molecule laminin and the potently inhibitory chondroitin sulfate proteoglycan aggrecan. Times for each frame are given in the bottom right of each image, and an arrow marks the central domain of the growth cone. An asterisk marks a consistent point in the culture as a reference for growth cone position during frame shifts. Initial astrocyte–growth cone contact was at 75 min (75′). Extensive physical contacts were observed between multiple astrocytes and the dystrophic axon throughout the remaining frames. Astrocytes were observed to migrate down the proteoglycan gradient in frames 95′, 120′, and 130′. The entire movie can be viewed in supplemental Movie S5 (available at www.jneurosci.org as supplemental material). Scale bar, 20 μm. B, Positional graph tracking the growth cone for entire time-lapse movie. Each point represents the position of the central domain of the growth cone for a single frame (every 30 s). The axon extended ∼40 μm after astrocyte contact. C, Positional graph from another representative time-lapse experiment.
Discussion
We have shown for the first time conclusive evidence that macrophages induce retraction of dystrophic adult axons through direct physical contact. The induction of retraction was dependent on both the growth state of the neurons and the activation state of the macrophages. Primary bone marrow-derived macrophages required stimulation with interferon-γ and LPS to reach a state of activation similar to the cell line macrophages and macrophages within a spinal cord lesion to induce axonal retraction in vitro. This indicates that the behavior of macrophages in vivo is state dependent and corresponds with previous work showing that macrophage infiltration only correlates with axonal retraction in the presence of myelin degeneration (McPhail et al., 2004). We found that adult sensory neurons were only susceptible to macrophage-induced retraction when they were in a dystrophic state of stalled growth induced by a gradient of inhibitory CSPG. Contacts between activated macrophages and adult neurons in an active state of growth on a uniform laminin substrate were rapidly broken and the neurons did not retract.
The induction of retraction potentially involves multiple intrinsic and extrinsic mechanisms. We have shown that retraction did not occur without direct cell–cell contact between macrophages and dystrophic neurons. The addition of trypsin-treated macrophages or macrophage-conditioned media alone was insufficient to induce retraction. This does not mean that soluble macrophage-derived factors are not playing any role in the induction of axonal retraction, but they may be in limited abundance and only effective when lengthy close encounters ensue. When macrophages physically contact axons, they may locally secrete ample amounts of proteases to disrupt adhesion of the axon. Macrophages did not have to contact dystrophic axons specifically at the growth cone to induce retraction. Contact with activated macrophages may trigger signaling pathways within the axon distant from its dystrophic ending, which could lead to disassembly of the cytoskeleton and loss of its already minimal adherence to the substratum through activation of the RhoA GTPase (Gross et al., 2007). In addition to potential intracellular changes within the neuron, there is a strong, purely physical aspect to the induction of retraction in vitro. It will be interesting to determine whether such strong physical interactions also occur in vivo by using modern live-animal imaging techniques (Misgeld et al., 2007). The contact between macrophages and dystrophic axons leading to retraction was vigorous, often resulting in the pulling and stretching of axonal segments. The physical interactions of microglia with axons in vitro were not as robust as those of macrophages; however, microglia could play a role in the induction of axonal retraction in vivo, in the early stages before macrophage infiltration (Koshinaga and Whittemore, 1995; Watanabe et al., 1999).
There are several candidate binding partners by which macrophages and neurons may physically identify one another and interact. Macrophages may use αv and β1 integrin receptors to recognize and bind to axonal vitronectin (Sobel et al., 1995) and macrophage adhesion to degenerating peripheral nerve is partially attenuated by blocking β1 integrin (Brown et al., 1997). After injury to the optic nerve, axons express ephrinB3, which is recognized by the EphB3 receptor present on macrophages (Liu et al., 2006). Sialoadhesin, a macrophage-specific receptor, can bind sialic acid present on neuronal cell membranes (Kelm et al., 1994; Tang et al., 1997). Macrophages also recognize phosphatidylserine exposed on the outer membrane surface of cells undergoing apoptosis (De et al., 2002; De Simone et al., 2004), which may flag injured neurons for endocytosis. Additionally, fractalkine is a chemokine expressed predominantly on the surface of CNS neurons, whereas its receptor, CX3CR1, is found on macrophages (Zujovic et al., 2000; Umehara et al., 2001). Additional studies must be done to determine which, if any, of these molecules are expressed or upregulated on the surfaces of dystrophic adult neurons targeting them for macrophage recognition.
We confirmed that the ascending dorsal column pathway undergoes retraction (Borgens et al., 1986; McPhail et al., 2004; Stirling et al., 2004; Baker and Hagg, 2005; Baker et al., 2007) and the timing corresponds to the infiltration of macrophages. Axonal retraction has been examined in other pathways within the spinal cord including the descending cortical spinal tract (Fishman and Kelley, 1984; Iizuka et al., 1987; Hill et al., 2001; Seif et al., 2007), bulbospinal tract (Houle and Jin, 2001), and rubrospinal tract (Schwartz et al., 2005; Cao et al., 2008). It is important to consider that there are two distinct phases of axonal retraction. A recent study that imaged axotomized ascending sensory axons of the adult mouse in vivo showed axonal retraction of ∼300 μm within the first few hours of injury followed by axon stabilization for the first 3 d after lesion (Kerschensteiner et al., 2005). Therefore, our focus here has been on the second, later phase of axonal retraction that is influenced by activated macrophages.
The effect of macrophage infiltration in CNS injury is highly controversial. Numerous studies have indicated that macrophage infiltration results in increased lesion size, decreased growth of regenerating fibers, and increased death of neurons spared by the initial lesion (Donnelly and Popovich, 2008). Inflammation increases CSPG production in the CNS (Fitch and Silver, 1997); however, there is also evidence that inflammation can have neuroprotective effects (Prewitt et al., 1997; Hohlfeld et al., 2007). We found that treatment with clodronate liposomes before injury and throughout the normal retraction period prevented the second phase of axonal retraction between 2 and 7 d after lesion. Previous work has shown that clodronate liposome-mediated macrophage depletion results in reduced lesion volume and increased neuronal survival (Popovich et al., 1999; van Rooijen and van Kesteren-Hendrikx, 2002). Although these authors stress the axon regenerative effect of macrophage depletion, our data suggest that the increased axonal content of the lesions of clodronate liposome-treated animals may be attributable, at least in part, to an attenuation of axonal retraction. This contradicts previous studies that have described beneficial effects of macrophage transplants into the injured spinal cord (Rapalino et al., 1998; Bomstein et al., 2003). However, in these studies macrophages were stimulated with degenerated peripheral nerve or skin before transplantation, which could perhaps induce a different state of activation than a combination of interferon-γ and LPS. Our results strongly suggest that infiltrating activated macrophages act as another barrier to regeneration by inducing retraction of the already struggling tip of the severed axon and increasing the distance the axon will have to regenerate.
Our results support the hypothesis that the state of activation of macrophages underlies their detrimental effects within the CNS (Popovich et al., 2002). Treatments that deplete macrophages or alter their activity may be therapeutic, but the effects may be limited to the retraction phenomenon rather than overt regeneration. Methylprednisolone treatment has been shown to reduce retraction of vestibulospinal axons, but not those of the corticospinal tract (Oudega et al., 1999). Studies have shown that deactivation of macrophages using minocycline (McPhail et al., 2004; Stirling et al., 2004), cyclosporine, FK506 (Hayashi et al., 2005; López-Vales et al., 2005), vaccinia virus complement control protein (Kulkarni et al., 2004; Reynolds et al., 2004), or silica (Blight, 1994) have had similar effects to macrophage depletion. A mouse strain with a diminished immune response exhibits an increase of axonal growth in the lesion after contusive spinal cord injury (Ma et al., 2004). Others have shown that increasing macrophage/microglial activation with leukemia inhibitory factor, a proinflammatory molecule, leads to a decrease in axonal regeneration (Kerr and Patterson, 2004). The dual nature of the effects of macrophages on CNS regeneration is complicated (Popovich and Longbrake, 2008) and suggests that the best approach for treatment after spinal cord injury may be to modify the state of macrophage activation rather than deplete the macrophages altogether (Blight, 1992; Jones et al., 2005; Hohlfeld et al., 2007).
The idea that macrophages play a role in regeneration failure and axonal retraction within the CNS has been established in the field. However, the prevailing theory has been that the detrimental effects of macrophages stem indirectly from their alteration and removal of cell and extracellular substrates upon which regenerating axons are dependent, or via their enhanced breakdown of myelin and consequent release of oligodendrocyte-associated growth inhibitors (Frisén et al., 1994; Fitch et al., 1999; McPhail et al., 2004). We are not discounting the effect of macrophage substrate modification, but our data suggest that it is only part of the mechanism underlying axonal retraction. We believe that we have shown for the first time that direct cell–cell interactions between activated macrophages and axons play a critical role in the induction of retraction through physical means. We have investigated these physical interactions and we have established an in vitro model of macrophage-mediated axonal retraction within the injured spinal cord. This model will facilitate future study of the basic biology of the extraordinarily complex environment of the postinjury CNS.
Footnotes
This work was supported by National Institute of Neurological Disorders and Stroke Grant NS25713 (J.S.), The Brumagin Memorial Fund, and National Institutes of Health Training Grant T32 AG00271. We extend our thanks to Hongmei Hu and Catherine Doller for technical assistance, Maryanne Pendergast for assistance with imaging, Reggie Gray for assistance with primary macrophage culture, and Catherine Rottkamp for helpful discussion of this manuscript.
References
- Arantes RM, Lourenssen S, Machado CR, Blennerhassett MG. Early damage of sympathetic neurons after co-culture with macrophages: a model of neuronal injury in vitro. Neuroreport. 2000;11:177–181. doi: 10.1097/00001756-200001170-00035. [DOI] [PubMed] [Google Scholar]
- Baker KA, Hagg T. An adult rat spinal cord contusion model of sensory axon degeneration: the estrus cycle or a preconditioning lesion do not affect outcome. J Neurotrauma. 2005;22:415–428. doi: 10.1089/neu.2005.22.415. [DOI] [PubMed] [Google Scholar]
- Baker KA, Nakashima S, Hagg T. Dorsal column sensory axons lack TrkC and are not rescued by local neurotrophin-3 infusions following spinal cord contusion in adult rats. Exp Neurol. 2007;205:82–91. doi: 10.1016/j.expneurol.2007.01.018. [DOI] [PubMed] [Google Scholar]
- Blight AR. Macrophages and inflammatory damage in spinal cord injury. J Neurotrauma. 1992;9(Suppl 1):S83–S91. [PubMed] [Google Scholar]
- Blight AR. Effects of silica on the outcome from experimental spinal cord injury: implication of macrophages in secondary tissue damage. Neuroscience. 1994;60:263–273. doi: 10.1016/0306-4522(94)90220-8. [DOI] [PubMed] [Google Scholar]
- Bomstein Y, Marder JB, Vitner K, Smirnov I, Lisaey G, Butovsky O, Fulga V, Yoles E. Features of skin-coincubated macrophages that promote recovery from spinal cord injury. J Neuroimmunol. 2003;142:10–16. doi: 10.1016/s0165-5728(03)00260-1. [DOI] [PubMed] [Google Scholar]
- Borgens RB, Blight AR, Murphy DJ. Axonal regeneration in spinal cord injury: a perspective and new technique. J Comp Neurol. 1986;250:157–167. doi: 10.1002/cne.902500203. [DOI] [PubMed] [Google Scholar]
- Brown HC, Castaño A, Fearn S, Townsend M, Edwards G, Streuli C, Perry VH. Adhesion molecules involved in macrophage responses to Wallerian degeneration in the murine peripheral nervous system. Eur J Neurosci. 1997;9:2057–2063. doi: 10.1111/j.1460-9568.1997.tb01373.x. [DOI] [PubMed] [Google Scholar]
- Busch SA, Silver J. The role of extracellular matrix in CNS regeneration. Curr Opin Neurobiol. 2007;17:120–127. doi: 10.1016/j.conb.2006.09.004. [DOI] [PubMed] [Google Scholar]
- Cao Y, Shumsky JS, Sabol MA, Kushner RA, Strittmatter S, Hamers FP, Lee DH, Rabacchi SA, Murray M. Nogo-66 receptor antagonist peptide (NEP1–40) administration promotes functional recovery and axonal growth after lateral funiculus injury in the adult rat. Neurorehabil Neural Repair. 2008;22:262–278. doi: 10.1177/1545968307308550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies SJ, Goucher DR, Doller C, Silver J. Robust regeneration of adult sensory axons in degenerating white matter of the adult rat spinal cord. J Neurosci. 1999;19:5810–5822. doi: 10.1523/JNEUROSCI.19-14-05810.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De SR, Ajmone-Cat MA, Nicolini A, Minghetti L. Expression of phosphatidylserine receptor and down-regulation of pro-inflammatory molecule production by its natural ligand in rat microglial cultures. J Neuropathol Exp Neurol. 2002;61:237–244. doi: 10.1093/jnen/61.3.237. [DOI] [PubMed] [Google Scholar]
- De Simone R, Ajmone-Cat MA, Minghetti L. Atypical antiinflammatory activation of microglia induced by apoptotic neurons: possible role of phosphatidylserine-phosphatidylserine receptor interaction. Mol Neurobiol. 2004;29:197–212. doi: 10.1385/MN:29:2:197. [DOI] [PubMed] [Google Scholar]
- Donnelly DJ, Popovich PG. Inflammation and its role in neuroprotection, axonal regeneration and functional recovery after spinal cord injury. Exp Neurol. 2008;209:378–388. doi: 10.1016/j.expneurol.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farr M, Zhu DF, Povelones M, Valcich D, Ambron RT. Direct interactions between immunocytes and neurons after axotomy in Aplysia . J Neurobiol. 2001;46:89–96. [PubMed] [Google Scholar]
- Filbin MT. Myelin-associated inhibitors of axonal regeneration in the adult mammalian CNS. Nat Rev Neurosci. 2003;4:703–713. doi: 10.1038/nrn1195. [DOI] [PubMed] [Google Scholar]
- Fishman PS, Kelley JP. The fate of severed corticospinal axons. Neurology. 1984;34:1161–1167. doi: 10.1212/wnl.34.9.1161. [DOI] [PubMed] [Google Scholar]
- Fitch MT, Silver J. Activated macrophages and the blood-brain barrier: inflammation after CNS injury leads to increases in putative inhibitory molecules. Exp Neurol. 1997;148:587–603. doi: 10.1006/exnr.1997.6701. [DOI] [PubMed] [Google Scholar]
- Fitch MT, Doller C, Combs CK, Landreth GE, Silver J. Cellular and molecular mechanisms of glial scarring and progressive cavitation: in vivo and in vitro analysis of inflammation-induced secondary injury after CNS trauma. J Neurosci. 1999;19:8182–8198. doi: 10.1523/JNEUROSCI.19-19-08182.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisén J, Haegerstrand A, Fried K, Piehl F, Cullheim S, Risling M. Adhesive/repulsive properties in the injured spinal cord: relation to myelin phagocytosis by invading macrophages. Exp Neurol. 1994;129:183–193. doi: 10.1006/exnr.1994.1160. [DOI] [PubMed] [Google Scholar]
- Gross RE, Mei Q, Gutekunst CA, Torre E. The pivotal role of RhoA GTPase in the molecular signaling of axon growth inhibition after CNS injury and targeted therapeutic strategies. Cell Transplant. 2007;16:245–262. doi: 10.3727/000000007783464740. [DOI] [PubMed] [Google Scholar]
- Harel NY, Strittmatter SM. Can regenerating axons recapitulate developmental guidance during recovery from spinal cord injury? Nat Rev Neurosci. 2006;7:603–616. doi: 10.1038/nrn1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi Y, Shumsky JS, Connors T, Otsuka T, Fischer I, Tessler A, Murray M. Immunosuppression with either cyclosporine A or FK506 supports survival of transplanted fibroblasts and promotes growth of host axons into the transplant after spinal cord injury. J Neurotrauma. 2005;22:1267–1281. doi: 10.1089/neu.2005.22.1267. [DOI] [PubMed] [Google Scholar]
- Hill CE, Beattie MS, Bresnahan JC. Degeneration and sprouting of identified descending supraspinal axons after contusive spinal cord injury in the rat. Exp Neurol. 2001;171:153–169. doi: 10.1006/exnr.2001.7734. [DOI] [PubMed] [Google Scholar]
- Hohlfeld R, Kerschensteiner M, Meinl E. Dual role of inflammation in CNS disease. Neurology. 2007;68:S58–S63. doi: 10.1212/01.wnl.0000275234.43506.9b. discussion S91–S96. [DOI] [PubMed] [Google Scholar]
- Houle JD, Jin Y. Chronically injured supraspinal neurons exhibit only modest axonal dieback in response to a cervical hemisection lesion. Exp Neurol. 2001;169:208–217. doi: 10.1006/exnr.2001.7645. [DOI] [PubMed] [Google Scholar]
- Iizuka H, Yamamoto H, Iwasaki Y, Yamamoto T, Konno H. Evolution of tissue damage in compressive spinal cord injury in rats. J Neurosurg. 1987;66:595–603. doi: 10.3171/jns.1987.66.4.0595. [DOI] [PubMed] [Google Scholar]
- Jones TB, McDaniel EE, Popovich PG. Inflammatory-mediated injury and repair in the traumatically injured spinal cord. Curr Pharm Des. 2005;11:1223–1236. doi: 10.2174/1381612053507468. [DOI] [PubMed] [Google Scholar]
- Kelm S, Pelz A, Schauer R, Filbin MT, Tang S, de Bellard ME, Schnaar RL, Mahoney JA, Hartnell A, Bradfield P, Crocker PR. Sialoadhesin, myelin-associated glycoprotein and CD22 define a new family of sialic acid-dependent adhesion molecules of the immunoglobulin superfamily. Curr Biol. 1994;4:965–972. doi: 10.1016/s0960-9822(00)00220-7. [DOI] [PubMed] [Google Scholar]
- Kerr BJ, Patterson PH. Potent pro-inflammatory actions of leukemia inhibitory factor in the spinal cord of the adult mouse. Exp Neurol. 2004;188:391–407. doi: 10.1016/j.expneurol.2004.04.012. [DOI] [PubMed] [Google Scholar]
- Kerschensteiner M, Schwab ME, Lichtman JW, Misgeld T. In vivo imaging of axonal degeneration and regeneration in the injured spinal cord. Nat Med. 2005;11:572–577. doi: 10.1038/nm1229. [DOI] [PubMed] [Google Scholar]
- Koshinaga M, Whittemore SR. The temporal and spatial activation of microglia in fiber tracts undergoing anterograde and retrograde degeneration following spinal cord lesion. J Neurotrauma. 1995;12:209–222. doi: 10.1089/neu.1995.12.209. [DOI] [PubMed] [Google Scholar]
- Kulkarni AP, Kellaway LA, Lahiri DK, Kotwal GJ. Neuroprotection from complement-mediated inflammatory damage. Ann N Y Acad Sci. 2004;1035:147–164. doi: 10.1196/annals.1332.010. [DOI] [PubMed] [Google Scholar]
- Liu X, Hawkes E, Ishimaru T, Tran T, Sretavan DW. EphB3: an endogenous mediator of adult axonal plasticity and regrowth after CNS injury. J Neurosci. 2006;26:3087–3101. doi: 10.1523/JNEUROSCI.4797-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longbrake EE, Lai W, Ankeny DP, Popovich PG. Characterization and modeling of monocyte-derived macrophages after spinal cord injury. J Neurochem. 2007;102:1083–1094. doi: 10.1111/j.1471-4159.2007.04617.x. [DOI] [PubMed] [Google Scholar]
- López-Vales R, García-Alías G, Forés J, Udina E, Gold BG, Navarro X, Verdú E. FK 506 reduces tissue damage and prevents functional deficit after spinal cord injury in the rat. J Neurosci Res. 2005;81:827–836. doi: 10.1002/jnr.20605. [DOI] [PubMed] [Google Scholar]
- Ma M, Wei P, Wei T, Ransohoff RM, Jakeman LB. Enhanced axonal growth into a spinal cord contusion injury site in a strain of mouse (129X1/SvJ) with a diminished inflammatory response. J Comp Neurol. 2004;474:469–486. doi: 10.1002/cne.20149. [DOI] [PubMed] [Google Scholar]
- McPhail LT, Stirling DP, Tetzlaff W, Kwiecien JM, Ramer MS. The contribution of activated phagocytes and myelin degeneration to axonal retraction/dieback following spinal cord injury. Eur J Neurosci. 2004;20:1984–1994. doi: 10.1111/j.1460-9568.2004.03662.x. [DOI] [PubMed] [Google Scholar]
- Misgeld T, Nikic I, Kerschensteiner M. In vivo imaging of single axons in the mouse spinal cord. Nat Protoc. 2007;2:263–268. doi: 10.1038/nprot.2007.24. [DOI] [PubMed] [Google Scholar]
- Oudega M, Vargas CG, Weber AB, Kleitman N, Bunge MB. Long-term effects of methylprednisolone following transection of adult rat spinal cord. Eur J Neurosci. 1999;11:2453–2464. doi: 10.1046/j.1460-9568.1999.00666.x. [DOI] [PubMed] [Google Scholar]
- Popovich PG, Longbrake EE. Can the immune system be harnessed to repair the CNS? Nat Rev Neurosci. 2008;9:481–493. doi: 10.1038/nrn2398. [DOI] [PubMed] [Google Scholar]
- Popovich PG, Wei P, Stokes BT. Cellular inflammatory response after spinal cord injury in Sprague-Dawley and Lewis rats. J Comp Neurol. 1997;377:443–464. doi: 10.1002/(sici)1096-9861(19970120)377:3<443::aid-cne10>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- Popovich PG, Guan Z, Wei P, Huitinga I, van Rooijen N, Stokes BT. Depletion of hematogenous macrophages promotes partial hindlimb recovery and neuroanatomical repair after experimental spinal cord injury. Exp Neurol. 1999;158:351–365. doi: 10.1006/exnr.1999.7118. [DOI] [PubMed] [Google Scholar]
- Popovich PG, Guan Z, McGaughy V, Fisher L, Hickey WF, Basso DM. The neuropathological and behavioral consequences of intraspinal microglial/macrophage activation. J Neuropathol Exp Neurol. 2002;61:623–633. doi: 10.1093/jnen/61.7.623. [DOI] [PubMed] [Google Scholar]
- Prewitt CM, Niesman IR, Kane CJ, Houlé JD. Activated macrophage/microglial cells can promote the regeneration of sensory axons into the injured spinal cord. Exp Neurol. 1997;148:433–443. doi: 10.1006/exnr.1997.6694. [DOI] [PubMed] [Google Scholar]
- Raff MC, Whitmore AV, Finn JT. Axonal self-destruction and neurodegeneration. Science. 2002;296:868–871. doi: 10.1126/science.1068613. [DOI] [PubMed] [Google Scholar]
- Ramon y, Cajal S. Degeneration and regeneration of the nervous system. London: Oxford UP; 1928. [Google Scholar]
- Rapalino O, Lazarov-Spiegler O, Agranov E, Velan GJ, Yoles E, Fraidakis M, Solomon A, Gepstein R, Katz A, Belkin M, Hadani M, Schwartz M. Implantation of stimulated homologous macrophages results in partial recovery of paraplegic rats. Nat Med. 1998;4:814–821. doi: 10.1038/nm0798-814. [DOI] [PubMed] [Google Scholar]
- Reynolds DN, Smith SA, Zhang YP, Mengsheng Q, Lahiri DK, Morassutti DJ, Shields CB, Kotwal GJ. Vaccinia virus complement control protein reduces inflammation and improves spinal cord integrity following spinal cord injury. Ann N Y Acad Sci. 2004;1035:165–178. doi: 10.1196/annals.1332.011. [DOI] [PubMed] [Google Scholar]
- Schwartz ED, Chin CL, Shumsky JS, Jawad AF, Brown BK, Wehrli S, Tessler A, Murray M, Hackney DB. Apparent diffusion coefficients in spinal cord transplants and surrounding white matter correlate with degree of axonal dieback after injury in rats. AJNR Am J Neuroradiol. 2005;26:7–18. [PMC free article] [PubMed] [Google Scholar]
- Schwartz M, Lazarov-Spiegler O, Rapalino O, Agranov I, Velan G, Hadani M. Potential repair of rat spinal cord injuries using stimulated homologous macrophages. Neurosurgery. 1999;44:1041–1045. doi: 10.1097/00006123-199905000-00057. discussion 1045–1046. [DOI] [PubMed] [Google Scholar]
- Seif GI, Nomura H, Tator CH. Retrograde axonal degeneration “dieback” in the corticospinal tract after transection injury of the rat spinal cord: a confocal microscopy study. J Neurotrauma. 2007;24:1513–1528. doi: 10.1089/neu.2007.0323. [DOI] [PubMed] [Google Scholar]
- Silver J, Miller JH. Regeneration beyond the glial scar. Nat Rev Neurosci. 2004;5:146–156. doi: 10.1038/nrn1326. [DOI] [PubMed] [Google Scholar]
- Sobel RA, Chen M, Maeda A, Hinojoza JR. Vitronectin and integrin vitronectin receptor localization in multiple sclerosis lesions. J Neuropathol Exp Neurol. 1995;54:202–213. doi: 10.1097/00005072-199503000-00007. [DOI] [PubMed] [Google Scholar]
- Steinmetz MP, Horn KP, Tom VJ, Miller JH, Busch SA, Nair D, Silver DJ, Silver J. Chronic enhancement of the intrinsic growth capacity of sensory neurons combined with the degradation of inhibitory proteoglycans allows functional regeneration of sensory axons through the dorsal root entry zone in the mammalian spinal cord. J Neurosci. 2005;25:8066–8076. doi: 10.1523/JNEUROSCI.2111-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stirling DP, Khodarahmi K, Liu J, McPhail LT, McBride CB, Steeves JD, Ramer MS, Tetzlaff W. Minocycline treatment reduces delayed oligodendrocyte death, attenuates axonal dieback, and improves functional outcome after spinal cord injury. J Neurosci. 2004;24:2182–2190. doi: 10.1523/JNEUROSCI.5275-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang S, Shen YJ, DeBellard ME, Mukhopadhyay G, Salzer JL, Crocker PR, Filbin MT. Myelin-associated glycoprotein interacts with neurons via a sialic acid binding site at ARG118 and a distinct neurite inhibition site. J Cell Biol. 1997;138:1355–1366. doi: 10.1083/jcb.138.6.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobian AA, Canaday DH, Boom WH, Harding CV. Bacterial heat shock proteins promote CD91-dependent class I MHC cross-presentation of chaperoned peptide to CD8+ T cells by cytosolic mechanisms in dendritic cells versus vacuolar mechanisms in macrophages. J Immunol. 2004;172:5277–5286. doi: 10.4049/jimmunol.172.9.5277. [DOI] [PubMed] [Google Scholar]
- Tom VJ, Steinmetz MP, Miller JH, Doller CM, Silver J. Studies on the development and behavior of the dystrophic growth cone, the hallmark of regeneration failure, in an in vitro model of the glial scar and after spinal cord injury. J Neurosci. 2004;24:6531–6539. doi: 10.1523/JNEUROSCI.0994-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umehara H, Goda S, Imai T, Nagano Y, Minami Y, Tanaka Y, Okazaki T, Bloom ET, Domae N. Fractalkine, a CX3C-chemokine, functions predominantly as an adhesion molecule in monocytic cell line THP-1. Immunol Cell Biol. 2001;79:298–302. doi: 10.1046/j.1440-1711.2001.01004.x. [DOI] [PubMed] [Google Scholar]
- Van Rooijen N, Sanders A. Liposome mediated depletion of macrophages: mechanism of action, preparation of liposomes and applications. J Immunol Methods. 1994;174:83–93. doi: 10.1016/0022-1759(94)90012-4. [DOI] [PubMed] [Google Scholar]
- van Rooijen N, van Kesteren-Hendrikx E. Clodronate liposomes: perspectives in research and therapeutics. J Liposome Res. 2002;12:81–94. doi: 10.1081/lpr-120004780. [DOI] [PubMed] [Google Scholar]
- van Rooijen N, Bakker J, Sanders A. Transient suppression of macrophage functions by liposome-encapsulated drugs. Trends Biotechnol. 1997;15:178–185. doi: 10.1016/s0167-7799(97)01019-6. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Yamamoto T, Abe Y, Saito N, Kumagai T, Kayama H. Differential activation of microglia after experimental spinal cord injury. J Neurotrauma. 1999;16:255–265. doi: 10.1089/neu.1999.16.255. [DOI] [PubMed] [Google Scholar]
- Xie F, Zheng B. White matter inhibitors in CNS axon regeneration failure. Exp Neurol. 2008;209:302–312. doi: 10.1016/j.expneurol.2007.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y, Cui Q, Li Y, Irwin N, Fischer D, Harvey AR, Benowitz LI. Macrophage-derived factors stimulate optic nerve regeneration. J Neurosci. 2003;23:2284–2293. doi: 10.1523/JNEUROSCI.23-06-02284.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yiu G, He Z. Glial inhibition of CNS axon regeneration. Nat Rev Neurosci. 2006;7:617–627. doi: 10.1038/nrn1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zujovic V, Benavides J, Vigé X, Carter C, Taupin V. Fractalkine modulates TNF-alpha secretion and neurotoxicity induced by microglial activation. Glia. 2000;29:305–315. [PubMed] [Google Scholar]