

Summary
Background
A 55-year-old woman was followed over a 13-year period as part of a longitudinal study of people at risk for familial dementia. She was a member of a family with an autosomal dominant familial dementia that fulfilled consensus criteria for frontotemporal lobar degeneration. She was initially asymptomatic but developed progressive behavioral and cognitive decline characterized by early apathy, impaired emotion recognition, mixed aphasia and parietal lobe dysfunction.
Investigations
Clinical assessments, neuropsychometry, volumetric brain MRI, genetic mutation screening.
Diagnosis
Progranulin-associated frontotemporal lobar degeneration.
Management
Explanation of the patient's condition and genetic counseling for her family.
Keywords: dementia, frontotemporal dementia, progranulin, progressive aphasia
THE CASE
A 55-year-old right-handed woman was seen in a specialist cognitive disorders research unit as part of a prospective, longitudinal study of asymptomatic individuals at risk of developing dementia. She was a member of a family with an autosomal dominant history of a dementia that met consensus criteria for frontotemporal lobar degeneration (FTLD);1,2 the average age of symptom onset in the family was 57.8 years (range 54–67 years).1,2 Supplementary Figure 1 provides a pedigree of the family.
The patient was assessed at eight visits over a 13-year period (Figure 1), with each assessment involving detailed clinical and neuropsychological evaluation and volumetric brain MRI. On the initial five visits the patient was well, displaying no cognitive symptoms and scoring normally on neuropsychological assessment, apart from a slightly reduced verbal fluency score (9 ‘S’ words in 1 min, where normal is ≥10) on the Frontal Assessment Battery at visit 5. At visit 6 the patient still had no cognitive complaints; neuropsychometry revealed a decline in naming and calculation skills, but her scores remained above the 5th percentile (Supplementary Table 1).
Figure 1.

Timeline showing clinical assessments and progression of neuropsychological and neuroimaging deficits in a person at risk of frontotemporal lobar degeneration.
At visit 7 the patient complained of cognitive symptoms for the first time and reported word-finding difficulties during the previous 6 months. Verbal fluency on the Frontal Assessment Battery was further reduced (6 ‘S’ words in 1 min). Although the patient's score on the Mini Mental State Examination (MMSE) remained in the normal range (28 out of 30), her score on a graded naming test was now below the 5th percentile (10 out of 30), and further decline was evident on testing of calculation (Supplementary Table 1). Her performance had now deteriorated on a test of verbal comprehension but remained normal on tests of executive function and visuoperceptual skills. Imitation of meaningless hand positions was a little clumsy, but neurological examination at this time was otherwise normal.
By visit 8 the patient's word-finding ability had continued to deteriorate and she had developed speech production impairment with phonemic paraphasias and agrammatism. Her family reported that the patient had become more apathetic over the previous year and spent most of the time in her house. Her MMSE score was now 22 out of 30 and verbal fluency was again reduced. Mild bilateral ideomotor and ideational limb apraxia were also now evident, although the rest of the neurological examination remained normal. Naming had further deteriorated and verbal memory difficulties had now become apparent.
In order to delineate the patient's language difficulties more precisely, she underwent more detailed neurolinguistic assessment 6 months following visit 8 (2 years after symptom onset; Supplementary Table 2). Her speech contained grammatical and phonemic errors, but the flow of her speech was relatively fluent without evidence of apraxia of speech or dysarthria. She had severe word-finding difficulty with occasional circumlocutions and semantic errors. Deterioration was evident on a test of single-word comprehension, and sentence comprehension was also impaired. The patient had difficulties with both single-word and sentence repetition, and her reading was affected with evidence of phonological dyslexia (difficulty reading non-words). Writing showed evidence of agrammatism but spelling ability was relatively intact. These features are consistent with a progressive mixed aphasia. At this assessment, the patient was also noted to have a decreased forward digit span of 4 (normal >5), consistent with dominant parietal lobe involvement. There was now mild orofacial apraxia and moderate bilateral limb apraxia.
From the 5th visit onwards the patient was tested on her ability to recognize simple and complex facial emotions. On a test of basic facial emotion recognition, based on the Ekman emotional faces stimulus set,3 the patient scored 19 out of 24 (age-matched and sex-matched normal range: 20–24) and her score continued to deteriorate over the next three visits (Supplementary Table 1). By 18 months after symptom onset the patient scored only 11 out of 24 on this test; her performance was at chance level (3 out of 16) for recognizing negative emotions (fear, disgust, anger, sadness) but was normal (8 out of 8) for recognizing positive emotions (happiness, surprise). On a task evaluating complex facial emotion recognition,4 the patient's performance fell from an initial score of 26 out of 36 (age-matched and sex-matched normal range: 24–34) to a score of 22 at visit 6 (Supplementary Table 1). Eighteen months after symptom onset her score on this task had fallen to 18 out of 36.
T1-weighted MRI brain volumes were acquired at each visit on a 1.5T scanner (General Electric, Milwaukee, WI). A semi-automated technique of brain segmentation was performed for each scan followed by an affine (12 degrees of freedom) registration in order to align the repeat scan onto the baseline image (Figure 2 and Supplementary video 1). Over the first four scans there were no clinically significant changes beyond normal ageing. Between the 4th and 5th scans, however, there was a marked decrease in brain volume (Figures 2 and 3). The 5th scan (18 months before symptom onset) showed asymmetrical frontal, temporal and parietal lobe atrophy that predominantly affected the left cerebral hemisphere (Figure 2B). Progressive atrophy in a similar distribution was present on the 6th scan (6 months before the onset of symptoms; Figure 2C). The distribution of volume change in the left hemisphere that occurred between the 6th and 7th scans (spanning the onset of symptoms) was further analyzed by use of a fluid registration technique to produce a voxel compression map.5 Progressive regional atrophy during this time period again involved the left frontal, temporal and parietal lobes (Figure 3A). In the frontal lobes there was atrophy of the medial superior frontal and frontopolar regions, and involvement of the anterior cingulate gyrus. There was marked atrophy of the left temporal pole. The left middle and inferior temporal and fusiform gyri were particularly affected, with some atrophy of the left amygdala, hippocampus and superior temporal gyrus. In the parietal lobes there was relatively selective atrophy of the left angular gyrus. There was also evidence of left caudate, pallidal and thalamic atrophy. Registration of the 7th and 8th scans showed a similar pattern of atrophy but also involvement of the right hemisphere (Figure 3B). The most severe change across the scans involved prefrontal and inferior parietal areas, as well as orbitofrontal and inferior temporal areas, in the left hemisphere. Quantification of longitudinal regional atrophy confirmed that involvement of the left hemisphere preceded that of the right by a number of years: Supplementary Figure 2 shows the volume changes in the whole brain, left and right hemispheres and ventricles in graphical form
Figure 2.

Series of five registered T1-weighted MRI images of a patient with progranulin-associated frontotemporal lobar degeneration. (A) Eight and a half years before symptom onset. (B) Eighteen months before symptom onset. (C) Six months before symptom onset. (D) Six months after symptom onset. (E) Eighteen months after symptom onset.
Figure 3.
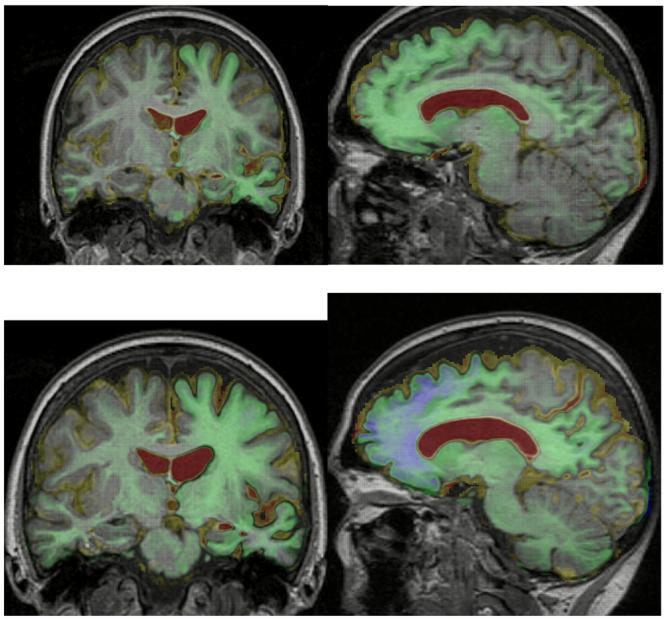
Sagittal and coronal MRI images of a patient with progranulin-associated frontotemporal lobar degeneration, with voxel-compression-mapping overlay over the time periods (A) from 6 months before to 6 months after symptom onset, and (B) 6–18 months after symptom onset. Red represents 20% or greater expansion of voxels and blue represents 20% or greater contraction of voxels.
Genetic screening of the patient revealed a c.90_91insCTGC (C31LfsX35) mutation in exon 1 of the progranulin (GRN) gene. The mutation was also discovered in other symptomatic family members, confirming this mutation as the cause of dementia in the family.
DISCUSSION OF DIAGNOSIS
FTLD is a clinically, pathologically and genetically heterogeneous group of degenerative disorders that are characterized by atrophy of the frontal and temporal lobes.6 Three canonical subtypes are described by consensus criteria:1 frontotemporal dementia (also referred to as behavioral variant FTLD), in which patients have changes in personality and behavior; progressive nonfluent aphasia, a disorder of speech production impairment; and semantic dementia, in which patients have loss of semantic knowledge, presenting with a fluent aphasia, anomia and difficulties with single-word comprehension. However, these three syndromes may overlap with each other and with other degenerative disorders, namely motor neuron disease, corticobasal syndrome and progressive supranuclear palsy. There are few epidemiological studies of FTLD, but prevalence is probably between 3 and 15 people per 100,000.7 The condition affects both sexes equally; symptom onset is usually between the ages of 45 and 65 years.7
A clinical FTLD phenotype with an autosomal dominant pattern of inheritance should prompt screening for common disease-causing mutations accompanied by appropriate genetic counseling. Around 30–50% of patients with FTLD have a family history of the disorder. However, until recently, only mutations in the microtubule-associated protein tau (MAPT) gene were known to cause familial FTLD; these mutations account for less than 5% of cases.7 Patients with MAPT mutations commonly present with behavioral variant FTLD and/or with parkinsonism.8 Patients who present with parkinsonism may have features of corticobasal syndrome or progressive supranuclear palsy. Although patients may become anomic or develop a paucity of speech output, true semantic dementia or progressive nonfluent aphasia syndromes have not been described in association with MAPT mutations. Two other genes (encoding chromatin-modifying protein 2B [CHMP2B] and valosin-containing protein [VCP]) are rare causes of FTLD, but a fourth gene (GRN) has been shown to account for 5–10% of all cases of FTLD and about a quarter of cases with a family history of FTLD.9,10 Over 50 GRN mutations have now been described in association with FTLD.11 Patients with such a mutation can present with behavioral variant FTLD, but, importantly (in contrast to other FTLD-causing mutations), GRN mutations can also produce familial progressive aphasia. Rarely, a corticobasal syndrome is seen.12
The present case illustrates the important roles of neuropsychology and brain imaging in guiding the differential diagnosis of patients who present with cognitive impairment (Table 1). Neuropsychometric testing helps to better characterize the clinical syndrome, whilst brain imaging is useful to rule out treatable pathologies and to define a profile of atrophy. The current patient had a mixed aphasia with features of both semantic dementia and progressive nonfluent aphasia,13 as well as early parietal lobe impairment. Such a mixed aphasia and early parietal lobe impairment have both been shown to be associated with GRN mutations.12 Furthermore brain imaging revealed early, strikingly asymmetric cerebral damage, a feature that often characterizes GRN-associated FTLD.12 In individuals at risk for an inherited dementia, neuropsychometry can be useful at detecting presymptomatic cognitive and behavioral deficits that may not be apparent on routine clinical assessment. This patient had presymptomatic deficits on neuropsychological testing that included impairment of emotion processing, which is seldom evaluated in the clinic but can lead to substantial morbidity in FTLD.14 Brain imaging can also potentially detect the onset of disease a number of years before clinical symptoms develop as demonstrated by this case. The role of imaging in this setting will become more important if and when disease-modifying treatments become available for these conditions.
Table 1. Comparison of features in familial dementias: frontotemporal lobar degeneration, Alzheimer's disease and prion disease.
| Genetic mutation |
Presenting syndrome |
Behavioral and cognitive features | Parkinsonism | Motor neuron disease |
Other clinical features |
Neuroimaging features |
|---|---|---|---|---|---|---|
| GRN | bvFTLD PNFA CBS |
Behavioral syndrome (apathy, sweet tooth, etc.), executive dysfunction, episodic memory impairment, aphasia, parietal lobe dysfunction |
+ | Rare | Features of CBS can be present |
Often asymmetrical frontal, temporal and parietal lobe atrophy |
| MAPT | bvFTLD CBS PSP |
Behavioral syndrome (disinhibition, inappropriate social behaviour, etc.), executive dysfunction, decreased speech, anomia |
+ | − | Features of CBS or PSP can be present |
Bilateral frontotemporal lobar atrophy |
| VCP | bvFTLD | Behavioral syndrome, executive dysfunction |
− | − | Inclusion body myopathy and Paget's disease |
A few reports of frontotemporal lobar atrophy |
| CHMP2B | bvFTLD |
Behavioral syndrome (apathy, restlessness, aggression etc.), executive dysfunction, decreased speech, can have early parietal impairment |
− | Rare | None | Generalized atrophy |
|
Chr 9 FTD- MNDa |
bvFTLD MND |
Behavioral syndrome, executive dysfunction |
− | + | None | Single report of frontal lobe atrophy sparing posterior regions |
|
APP, PS1, PS215 |
AD (i.e. amnestic presentation) Atypical ADb |
Episodic memory impairment initially then global impairment |
Rare | − | Myoclonus or seizures can occur. Spastic paraparesis is seen rarely in PS1. |
Presymptomatic medial temporal lobe atrophy spreading to diffuse neocortical areas |
| PRNP16 | Dementia with neurological signs |
Highly heterogenous, ranging from rapidly progressive dementia with myoclonus and ataxia, similar to classical CJD, to much more slowly progressive syndromes involving episodic memory, executive dysfunction, dyspraxia |
+ | Rare | Ataxia, myoclonus, seizures, chorea, or dystonia |
Generalized cerebral and cerebellar atrophy typical. Rapidly progressive clinical syndromes can be associated with high signal in the caudate and putamen on T2-weighted MRI |
A locus on chromosome 9p has been associated with FTD-MND but the abnormal gene has yet to be found.
Rarely, a prominent behavioral phenotype similar to bvFTLD is seen in patients with PS1 mutations.
Abbreviations: AD, Alzheimer's disease; APP, amyloid precursor protein; bvFTLD, behavioral variant frontotemporal lobar degeneration; CBS, corticobasal syndrome; CHMP2B, chromatin-modifying protein 2B; Chr 9 FTD-MND, chromosome 9-associated frontotemporal dementia with motor neuron disease; CJD, Creutzfeldt-Jakob disease; GRN, progranulin, MAPT, microtubule-associated protein tau; PNFA, progressive nonfluent aphasia; PRNP, prion protein, PS1, presenilin 1; PS2, presenilin 2; PSP, progressive supranuclear palsy, VCP, valosin-containing protein; +, can be present; −, absent
The neuroanatomical distribution of heaviest disease burden in this case followed the pattern predicted from the neuropsychological profile: dominant frontal lobe mechanisms which mediate word retrieval and propositional speech; parietal lobe mechanisms which mediate praxis, speech repetition and calculation; and orbitofrontal–anterior temporal lobe mechanisms which are involved in emotion processing. The affected areas form part of noncontiguous but anatomically and functionally linked intrahemispheric functional networks. The progressive spread of atrophy via such networks in GRN-associated FTLD could account both for the early involvement of anatomically remote (but linked) anterior and posterior areas within a hemisphere, and for the striking asymmetry of disease burden, which can remain largely restricted to a single hemisphere for many years.
TREATMENT AND MANAGEMENT
There remains no curative treatment for any of the subtypes of FTLD. The current patient was referred for speech and language therapy, which can be useful for the development of communication strategies in patients with progressive aphasia. Family members were offered genetic counseling.
CONCLUSION
This case details the longitudinal analysis of a member of a family with a C31LfsX35 mutation in the GRN gene causing FTLD; the patient was followed up over a 13-year period from an initial asymptomatic phase through to the establishment of clinical disease. A syndrome of progressive mixed aphasia developed, heralded by presymptomatic clinical, neuropsychological and imaging changes. This case illustrates the importance of neuropsychological and radiological correlation for early detection and characterization of genetically mediated neurodegenerative diseases with multiple downstream effects.
Supplementary Material
ACKNOWLEDGEMENTS
This work was undertaken at University College London Hospital/University College London, which received a proportion of funding from the UK Department of Health's National Institute for Health Research Biomedical Research Centres funding scheme. The work was supported by the Alzheimer Research Trust and the Medical Research Council UK. JD Rohrer is supported by a Wellcome Trust Research Training Fellowship. JD Warren is supported by a Wellcome Trust Intermediate Clinical Fellowship. NC Fox holds a Medical Research Council UK Senior Clinical Fellowship. J Barnes is supported by the Alzheimer Research Trust.
Footnotes
COMPETING INTERESTS
The authors declared no competing interests.
Supplementary information in the form of two tables, two figures and a video is available on the Nature Clinical Practice Neurology website.
REFERENCES
- 1.Neary D, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51:1546–1554. doi: 10.1212/wnl.51.6.1546. [DOI] [PubMed] [Google Scholar]
- 2.Rohrer JD, et al. Parietal lobe deficits are a feature of frontotemporal lobar degeneration caused by a mutation in the progranulin gene. Arch Neurol. 2008;65(4):506–13. doi: 10.1001/archneur.65.4.506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ekman P, Friesen WV. Pictures of facial affect. Consulting Psychologists Press; Palo Alto CA: 1976. [Google Scholar]
- 4.Baron-Cohen S, et al. The “Reading the Mind in the Eyes” Test revised version: a study with normal adults, and adults with Asperger syndrome or high-functioning autism. J Child Psychol Psychiatry. 2001;42:241–251. [PubMed] [Google Scholar]
- 5.Scahill RI, et al. Mapping the evolution of regional atrophy in Alzheimer's disease: unbiased analysis of fluid-registered serial MRI. Proc Natl Acad Sci USA. 2002;99:4703–4707. doi: 10.1073/pnas.052587399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cairns NJ, et al. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol (Berl) 2007;114:5–22. doi: 10.1007/s00401-007-0237-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Neary D, et al. Frontotemporal dementia. Lancet Neurol. 2005;4:771–780. doi: 10.1016/S1474-4422(05)70223-4. [DOI] [PubMed] [Google Scholar]
- 8.van Swieten J, Spillantini MG. Hereditary frontotemporal dementia caused by Tau gene mutations. Brain Pathol. 2007;17:63–73. doi: 10.1111/j.1750-3639.2007.00052.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baker M, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442:916–919. doi: 10.1038/nature05016. [DOI] [PubMed] [Google Scholar]
- 10.Gass J, et al. Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum Mol Genet. 2006;15:2988–3001. doi: 10.1093/hmg/ddl241. [DOI] [PubMed] [Google Scholar]
- 11.Alzheimer Disease & Frontotemporal Dementia Mutation Database [http://www.molgen.ua.ac.be/FTDMutations]
- 12.Beck J, et al. A distinct clinical, neuropsychological and radiological phenotype is associated with progranulin gene mutations in a large UK series. Brain. 2008;131:706–720. doi: 10.1093/brain/awm320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rohrer JD, et al. Word-finding difficulty: a clinical analysis of the progressive aphasias. Brain. 2008;131(Pt 1):8–38. doi: 10.1093/brain/awm251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Keane J, et al. Face and emotion processing in frontal variant frontotemporal dementia. Neuropsychologia. 2002;40:655–665. doi: 10.1016/s0028-3932(01)00156-7. [DOI] [PubMed] [Google Scholar]
- 15.Ridha BH, et al. Tracking atrophy progression in familial Alzheimer's disease: a serial MRI study. Lancet Neurol. 2006;5:828–834. doi: 10.1016/S1474-4422(06)70550-6. [DOI] [PubMed] [Google Scholar]
- 16.Mead S. Prion disease genetics. Eur J Hum Genet. 2006;14:273–281. doi: 10.1038/sj.ejhg.5201544. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.