

Abstract
To understand the demographic history of Arabidopsis thaliana within its native geographical range, we have studied its genetic structure in the Iberian Peninsula region. We have analyzed the amount and spatial distribution of A. thaliana genetic variation by genotyping 268 individuals sampled in 100 natural populations from the Iberian Peninsula. Analyses of 175 individuals from 7 of these populations, with 20 chloroplast and nuclear microsatellite loci and 109 common single nucleotide polymorphisms, show significant population differentiation and isolation by distance. In addition, analyses of one genotype from 100 populations detected significant isolation by distance over the entire Iberian Peninsula, as well as among six Iberian subregions. Analyses of these 100 genotypes with different model-based clustering algorithms inferred four genetic clusters, which show a clear-cut geographical differentiation pattern. On the other hand, clustering analysis of a worldwide sample showed a west–east Eurasian longitudinal spatial gradient of the commonest Iberian genetic cluster. These results indicate that A. thaliana genetic variation displays significant regional structure and consistently support the hypothesis that Iberia has been a glacial refugium for A. thaliana. Furthermore, the Iberian geographical structure indicates a complex regional population dynamics, suggesting that this region contained multiple Pleistocene refugia with a different contribution to the postglacial colonization of Europe.
THE annual wild weed species Arabidopsis thaliana is a model organism not only for molecular biology but also for ecological and evolutionary genetics, and hence, revealing the geographical structure of its genetic variation has become of paramount relevance (Mitchell-Olds and Schmitt 2006). Quantification of genetic diversity within and among populations of A. thaliana and analysis of its spatial distribution pattern across the species geographical range is the basis for elucidating demographic (historical) and ecological influences (Mitchell-Olds and Schmitt 2006). Furthermore, identification of genetic structure is relevant to mapping the causal genes responsible for the natural variation of adaptive traits by genomewide association analysis. In these assays, knowledge of the genetic structure will reduce spurious correlations between genotype and phenotype due to historical relationships affecting the genetic background (Cardon and Palmer 2003; Zhao et al. 2007).
A. thaliana shows a worldwide geographical distribution, although its native range spans mainly Europe and central Asia, while it is mostly naturalized elsewhere (reviewed in Hoffmann 2002). It remains unknown if the main A. thaliana center of origin is central Asia or Europe/North Africa, both areas showing the highest diversity of related species (Hoffmann 2002). Currently, there are wild genotypes (accessions) collected from >500 populations across its world distribution, which have been used to estimate the amount and patterns of genetic variation on a worldwide scale. These analyses have found significant population structure on a global scale, as well as long-range isolation by distance among different world regions (Sharbel et al. 2000; Nordborg et al. 2005; Ostrowski et al. 2006; Schmid et al. 2006; Beck et al. 2008). In addition, several laboratories have recently initiated the development of new A. thaliana collections for genetic variation studies on a regional scale in regions of the native distribution area such as northern Europe (Stenøien et al. 2005; Bakker et al. 2006), France (Le Corre 2005), central Asia (Schmid et al. 2006), and China (He et al. 2007), as well as in regions of presumed recent introduction and expansion such as Japan (Todokoro et al. 1996) and North America (Jørgensen and Mauricio 2004; Bakker et al. 2006). Thus far, a significant regional correlation between genetic and geographical distances has been observed only in China, which supports a progressive natural dispersal under strong anthropogenic influence. In contrast, lack of such correlations within non-native regions has been interpreted as a consequence of a recent colonization and expansion from mixed origin or a consequence of multiple colonizations (Todokoro et al. 1996; Jørgensen and Mauricio 2004; Stenøien et al. 2005).
The Iberian Peninsula (IP), located at the western border of Eurasia and very close to Africa, has received some attention in several A. thaliana genetic structure studies due to special interest in the biodiversity history of Europe (Symonds and Lloyd 2003; Nordborg et al. 2005; Schmid et al. 2006). This region is a major part of the largest biodiversity hotspot in Europe, the Mediterranean basin (Myers et al. 2000), and it has been one of the most important Pleistocene glacial refugia for numerous plant and animal species of the European subcontinent (reviewed in Hewitt 2001; Gomez and Lunt 2006). However, analyses of Iberian A. thaliana diversity have been limited mostly to small sets of genotypes collected in a single subregion of Spain (Robbelen 1965; Kuittinen et al. 2002; Beck et al. 2008). These studies have suggested that Iberia is genetically differentiated from other world regions (Symonds and Lloyd 2003; Nordborg et al. 2005; Schmid et al. 2006) and that it might be part of a Mediterranean glacial refugium for A. thaliana (Sharbel et al. 2000).
To better understand the evolutionary history of A. thaliana in its native geographical range, we have systematically studied the A. thaliana genetic structure in the Iberian Peninsula at different spatial levels. To achieve this goal, we generated a collection of 268 individuals sampled from 100 populations covering this region, and this has been used to determine the amount and spatial distribution of genetic variation. We have inferred the genetic structure by analyzing genomewide genotypes obtained with microsatellites (MSs) and single nucleotide polymorphisms (SNPs). We show that Iberian genetic diversity is geographically structured, which indicates population isolation in the past and provides evidence supporting the role of the Iberian Peninsula as a Pleistocene refugium for postglacial colonization of Europe. In addition, inference of four spatially separated Iberian genetic clusters indicates a complex regional population dynamics that suggests the occurrence of multiple glacial refugia for A. thaliana in this region.
MATERIALS AND METHODS
Plant material and sampling design:
One hundred natural populations of A. thaliana were surveyed in a region of ∼800 × 700 km of the IP and Menorca Island (Figure 1). These were spaced at an average distance of 326 ± 180 km, with a minimum and maximum of 1 and 898 km, respectively. Populations were assigned to six geographical subregions defined according to the six major Iberian mountain systems, using the largest rivers as the main subregional borders (Figure 1 and supplemental Table S1). Sampled populations cover most of the A. thaliana distribution area in the IP and were located in a wide range of habitats (supplemental Figure S1 and supplemental Table S1). Population size was roughly estimated in the field and ranged from a few individuals (<25) covering an ∼1-m2 patch to at least 1000 individuals in a 100-m tract (supplemental Table S1).
Figure 1.—

Geographical location of A. thaliana populations of the Iberian Peninsula surveyed in this work. Numbers I–VI and dotted lines indicate the six IP geographical subregions considered in spatial analyses. Red circles depict the location of the seven populations used for local population differentiation analysis.
Seven of these populations, distanced between 86 and 564 km, were chosen for analysis of local population differentiation. They show large population sizes and were extensively sampled following a transect where seeds from 20 to 32 plants were individually collected at a minimum distance of 0.5 m. Populations were named with three letters indicating the closest village or locality, followed by a different number code for each sampled individual.
Forty-three additional individuals from different local populations covering most of the rest of the A. thaliana world distribution were also analyzed. Five of them are new European individuals collected by the authors, while the rest were accessions obtained from public collections available at stock centers (supplemental Table S2).
DNA isolation and marker genotyping:
DNA was isolated from 175 individuals of the 7 extensively sampled IP populations and from one randomly chosen individual of the remaining 93 IP populations. From each sampled mother plant, a mix of leaf tissue from at least six sister plants grown from the sampled seeds was used for DNA isolation in the 7 large populations, which represented the DNA of the field mother plants. For the rest of IP and world individuals, leaf tissue was harvested from a single plant grown from sampled or stock center multiplied seeds. DNA was isolated using a previously described protocol (Bernartzky and Tanksley 1986) without mercaptoethanol.
Samples were genotyped at previously described microsatellite (Bell and Ecker 1994; Provan 2000; Loudet et al. 2002) and single nucleotide polymorphism loci (Törjéck et al. 2003; Nordborg et al. 2005). Sixteen nuclear microsatellites (ncMSs) and four chloroplast microsatellites (cpMSs) were analyzed (supplemental Table S3 and supplemental Figure S2). MS loci were amplified by PCR using a forward primer labeled with one of the Perkin-Elmer Applied Biosystems fluorochromes 6-FAM, NED, PET, and VIC. PCR products of four differently labeled MSs were mixed in equal amounts and the fragments of five mixes (supplemental Table S3) were separated in an ABI PRISM 3700 DNA analyzer using GeneScan-500-LIZ (Applied Biosystems) as the internal size standard. Electropherograms were visually inspected and manually scored using GeneScan 3.7 software (Applied Biosystems). Molecular sizes (in base pairs) of DNA fragments were calculated and sizes were used to estimate the number of MS motif repeats on the basis of the available sequence of Columbia accession. MS alleles were recorded and analyzed as the closest number of presumed motif repeats. MS loci showed an average frequency of missing data of 3.5%. However, nga111, nga172, and msat3.18 rendered 17.4, 16.4, and 8.0% of nonamplifying individuals, respectively, suggesting that these loci might contain null alleles or additional polymorphisms within the primer sequences. The average frequency of missing data per individual was 1.7% for cpMSs and 3.2% for ncMSs. MS error rates of the molecular size estimates were calculated by amplifying and analyzing duplicated samples of 20 genotypes, which provided an average genotyping error proportion of 0.046/locus. All but one of the ncMSs are di- or trinucleotide repeat loci (supplemental Table S3) and showed an error rate of 0.019, while all cpMSs are mononuclotide repeats showing a 0.09 error rate. Most mistyped genotypes differed in one single base pair, whereas only a 0.007 error rate per locus was due to size estimates differing in more than one nucleotide.
Two different sets of nuclear SNP loci were analyzed (supplemental Table S4 and supplemental Figure S2). Ninety-six Col/C24 SNP markers were selected because they are common polymorphisms in central Europe (Schmid et al. 2006). In addition, other 47 SNPs segregating in six Iberian accessions collected in several geographical subregions (Fei-0, Ll-0, Pro-0, Se-0, Ts-1, and Ts-5) were randomly chosen from polymorphisms described by Nordborg et al. (2005). SNPs were genotyped with the SNPlex technique (Applied Biosystem), using three mixes of 47 or 48 loci (supplemental Figure 2S) through the CEGEN genotyping service (http://www.cegen.org). Twenty-six Col/C24 SNP loci showing >25% missing data and eight other Col/C24 SNP loci that did not segregate among the individuals analyzed in this work were discarded for further analyses. The final 62 Col/C24 SNP loci had a 6.0% average frequency of missing data. The 47 IP loci showed an average missing frequency of 4.6%. Ini-0 and Nac-0 individuals failed for the SNPlex mix containing the 47 IP SNP markers and were not included in some comparisons. The SNPlex genotyping error rate was calculated by duplicated analysis of 25 individuals with the three SNPlex mixes representing 117 SNP markers, which provided an average error rate per SNP locus of <0.0004.
To integrate the genotypes generated in this work with previously published data, we first validated the SNP loci by genotyping several accessions included in other studies. Twenty-four accessions genotyped by Schmid et al. (2006) and 14 accessions analyzed by Nordborg et al. (2005) were genotyped for the Col/C24 SNPs and the IP SNP loci, respectively. All except Sf-2 and Ll-1 presented the same genotypes at all loci in both studies, indicating that markers are the same but that some accessions might be misclassified.
Population data analysis:
Three sets of multilocus genotypes were generated in this work (supplemental Table S5): (1) genotypes of 100 IP individuals, one from each of the 100 populations; (2) genotypes of 175 IP individuals from the 7 extensively sampled populations; and (3) genotypes of 43 individuals from different populations from the rest of world. In addition, SNP genotypes of IP were compared with two worldwide SNP data sets previously generated (Nordborg et al. 2005; Schmid et al. 2006). To have comparable data from IP and world samples, a single random individual was selected from each local population and from each group of individuals with similar genotype in previous world data sets. This selection did not affect greatly the analyses and comparisons presented in this work. Thus, we used two sets of 193 and 93 individuals from different world populations genotyped with 62 Col/C24 SNPs (Schmid et al. 2006) and 47 IP SNPs (Nordborg et al. 2005), respectively. Populations from outside the IP were assigned to seven world geographical regions, according to Bakker et al. (2006): northern America, western Europe, eastern Europe (limit at longitude 60° W), southern Europe and northern Africa (here referred to as the Mediterranean basin), northern Europe, Asia, and Japan.
Genetic diversity was measured as the percentage of polymorphic loci (PL), mean number of observed alleles per locus (na), mean allelic richness per locus (RS), mean private allelic richness per locus (RP), mean gene diversity (HS), and number of multilocus haplotypes (NH). These genetic parameters were estimated using the software programs FSTAT v. 2.9.3 (Goudet 1995), POPGENE v. 1.32 (Yeh et al. 1999), and HP-Rare v. 1.0 (Kalinowski 2005). Observed heterozygosities (HO) and inbreeding coefficients (FIS) were calculated for nuclear microsatellite loci using FSTAT. Outcrossing frequencies were estimated as (1 − FIS)/(1 + FIS) according to Allard et al. (1968).
Linkage disequilibrium (LD) between pairs of loci was tested using the LD exact test implemented in FSTAT v. 2.9.3 (Goudet 1995). The proportion of pairs of loci showing significant LD over the total number of possible pairs (PLD) was estimated excluding polymorphic loci that segregate only as singleton alleles.
Genetic differentiation among populations or groups of individuals was estimated by hierarchical analysis of molecular variance (AMOVA, Excoffier et al. 1992) using the program ARLEQUIN, v. 3.1 (Excoffier et al. 2005). We calculated the FST statistics analogous to the fixation index FST (Weir and Cockerham 1984) and their significances from 1000 permutations. AMOVA tests were performed using multilocus genotypes on the following data sets: (1) seven local IP populations; (2) IP and the rest of world individuals; (3) 100 IP individuals classified into four genetic groups inferred with STRUCTURE; and (4) 100 IP individuals grouped into six geographical subregions.
Genetic relationships among accessions were determined by neighbor-joining (NJ) analysis. Genetic distances between individuals were calculated as the proportion of allelic differences over the total number of alleles in the corresponding set of polymorphic loci, using the software program GGT v. 2.0 (Van Berloo 1999; http://www.plantbreeding.wur.nl/UK/software_ggt.html). NJ trees were constructed from 1000 bootstrap replicates using the software POPULATIONS v. 1.2.30 (http://bioinformatics.org/; O. Langella, unpublished results) and drawn with MEGA v. 3.1 (Kumar et al. 2004).
Phylogenetic relationships among chloroplast haplotypes (chlorotypes) were established and visualized as chlorotype frequency maps constructed with a median-joining network (Bandelt et al. 1999) using the program NETWORK v. 4.2 (http://www.fluxus-engineering.com). Given the large number of chlorotypes observed with four cpMS loci, the network presented was generated using only three markers (cp70189 was excluded; see supplemental Table S5). For clarity, chlorotypes detected in single individuals and unconnected to the network were removed from figures (11 of 36 chlorotypes corresponding to 7.1 and 10.3% of the IP and world individuals, respectively).
Genetic structure was inferred using the model-based clustering algorithms implemented in STRUCTURE v. 2.1 (Pritchard et al. 2000; Falush et al. 2003) and TESS v. 1.1 (François et al. 2006). These Bayesian approaches were applied to the following sets of genotypes: (1) 100 and 193 individuals from different populations of the IP and rest of world, respectively and (2) 100 IP individuals from different populations. For STRUCTURE analyses, we used a similar setting to that described by Nordborg et al. (2005). Basically, SNP multilocus genotypes were analyzed with a haploid setting, using the linkage model with correlated allele frequencies, and running the algorithm with 50,000 MCMC iterations of burn-in length and 20,000 after-burning repetitions for parameter estimations. Genetic positions of loci were directly obtained from the consensus A. thaliana genetic map (http://www.arabidospsis.org) or by interpolation from the physically closest loci with known genetic position. To estimate the K number of ancestral genetic populations and the ancestry membership proportions of each individual in these clusters, the algorithm was run 10 times for each K value from 2 to 15. Differences between the data likelihood of successive K values were tested using the nonparametric Wilcoxon test for two related samples. The final K was estimated as the largest K value with significantly higher likelihood than that from K − 1 runs (two-sided P < 0.005). Similarity between runs was estimated using the symmetric similarity coefficient (Nordborg et al. 2005) and the extent of membership in a single cluster was measured using the clusteredness coefficient (Rosenberg et al. 2005). These parameters and the average matrix of cluster membership proportions of the 10 runs were computed using a Structure-sum R-script (Ehrich 2006).
In contrast to STRUCTURE, the TESS algorithm incorporates spatial population models assuming geographical continuity of allele frequencies by including the interaction parameter 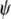 , which defines the intensity of two neighbor individuals belonging to the same genetic cluster. In addition, TESS treats K as a variable to be estimated. Haploid multilocus genotypes were analyzed with TESS using the MCMC method, with the F-model and a
, which defines the intensity of two neighbor individuals belonging to the same genetic cluster. In addition, TESS treats K as a variable to be estimated. Haploid multilocus genotypes were analyzed with TESS using the MCMC method, with the F-model and a 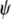 value of 0 (which assumes a noninformative spatial prior) as well as with the admixture model and
value of 0 (which assumes a noninformative spatial prior) as well as with the admixture model and 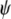 values between 0.5 and 0.7. For each model, the algorithm was run 200 times, each run with a total of 70,000 sweeps and 50,000 burn-in sweeps. K was estimated from the 10–20% runs with highest data likelihood. Similarity coefficients between runs and the average matrix of ancestry membership were calculated using CLUMPP v. 1.1 (Jakobsson and Rosenberg 2007).
values between 0.5 and 0.7. For each model, the algorithm was run 200 times, each run with a total of 70,000 sweeps and 50,000 burn-in sweeps. K was estimated from the 10–20% runs with highest data likelihood. Similarity coefficients between runs and the average matrix of ancestry membership were calculated using CLUMPP v. 1.1 (Jakobsson and Rosenberg 2007).
Estimated average matrices of membership proportions were graphically represented using DISTRUCT software (Rosenberg et al. 2002). Geographical distribution of ancestry matrices were represented by Kriging methods using the R-script available at http://www-timc.imag.fr/Olivier.Francois/admix_display.html (O. François, unpublished results).
The relationship between genetic distance and Euclidean geographical distance among population pairs of the Iberian Peninsula was determined by Mantel correlation test (Mantel 1967; Smouse et al. 1986) using ARLEQUIN (Excoffier et al. 2005) and the isolation by distance (IBD) web service v. 3.13 (http://ibdws.sdsu.edu/∼ibdws/) (Jensen et al. 2005). Genetic and geographical distances were log transformed prior to analysis and the significance of correlations was calculated with 1000 randomizations. We estimated genetic distance between population pairs in two ways: when using local populations or groups of individuals, we calculated the Slatkin's linearized FST expressed as D = FST/(1 − FST) (Slatkin 1995) with ARLEQUIN; when using populations represented by one randomly chosen individual, we computed the proportion of pairwise allelic differences as described above. Since Mah is an island population located >300 km apart from the IP (Figure 1), this genotype was dropped from these analyses. Isolation by distance analyses were carried out on the following sets of genotypes: (1) seven Iberian local populations; (2) 99 IP individuals from different populations; (3) 99 IP individuals grouped into six geographical subregions, using the average geographical distance among all pairs of individuals from different subregions; and (4) 15 subgroups of the 99 IP individuals, corresponding to all different pair combinations of the six geographical subregions.
RESULTS
General genetic diversity in the Iberian Peninsula:
To estimate A. thaliana genetic diversity in the IP, we sampled 100 local populations in a region of 800 × 700 km (Figure 1 and supplemental Table S1). A randomly chosen individual from each population was genotyped with four sets of markers corresponding to 16 ncMSs, 4 cpMSs, 62 Col/C24 SNP, and 47 IP SNP loci (Table 1 and materials and methods). As expected, genetic diversity estimates at MS loci were consistently larger than those at SNP loci. In addition, the two sets of microsatellites differed significantly, cpMSs showing considerably lower diversity values than ncMSs (Table 1). Twelve of the Col/C24 SNP loci (19.4%) were monomorphic while only one of the IP SNP markers (2.1%) was not polymorphic in the 100 individuals. IP SNP loci showed a slightly higher average minor allele frequency (MAF) (0.19 ± 0.16) than Col/C24 SNPs (0.14 ± 0.15) (supplemental Figure S3) and larger diversities were estimated with IP SNPs than with Col/C24 loci (Table 1). All 100 individuals showed different multilocus genotypes on the basis of any of the two sets of SNP loci or on the ncMSs, while cpMSs distinguished a total of 34 different chlorotypes. On average, we estimated that pairs of A. thaliana individuals collected in different natural populations differed in 55, 81, 23, and 26% of the polymorphic cpMSs, ncMSs, Col/C24, and IP SNP loci, respectively.
TABLE 1.
Genetic diversity of A. thaliana in the Iberian Peninsula and the rest of world
| Marker set | Region | N | PL | na | RS | RP | HS | NH |
|---|---|---|---|---|---|---|---|---|
| cpMSs | IP | 100 | 100 | 6.50 ± 2.52 | 5.74 ± 1.86 | 1.35 ± 1.78 | 0.55 ± 0.18 | 34 |
| RW | 43 | 100 | 5.75 ± 2.22 | 5.75 ± 2.22 | 1.35 ± 1.54 | 0.57 ± 0.20 | 23 | |
| ncMSs | IP | 100 | 100 | 18.69 ± 8.38 | 15.91 ± 7.20 | 5.72 ± 3.89 | 0.82 ± 0.18 | 100 |
| RW | 43 | 100 | 14.44 ± 5.30 | 14.35 ± 5.29 | 4.15 ± 1.42 | 0.83 ± 0.23 | 43 | |
| Col/C24 SNPs | IP | 100 | 80.6 | 1.81 ± 0.40 | 1.80 ± 0.39 | 0.01 ± 0.04 | 0.19 ± 0.18 | 100 |
| RW | 193 | 100 | 2.00 ± 0.00 | 1.96 ± 0.11 | 0.18 ± 0.35 | 0.22 ± 0.15 | 193 | |
| IP SNPs | IP | 98 | 97.9 | 1.98 ± 0.15 | 1.98 ± 0.15 | 0.09 ± 0.29 | 0.27 ± 0.18 | 98 |
| RW | 93 | 91.3 | 1.91 ± 0.28 | 1.91 ± 0.28 | 0.02 ± 0.15 | 0.26 ± 0.18 | 92 |
RW, rest of the world; N, sample size; PL, percentage of polymorphic loci; na, number of observed alleles; RS, allelic richness; RP, private allelic richness; HS, gene diversity; NH, number of multilocus haplotypes. na, RS, RP, and HS are mean values ±SD estimated from four cpMSs, 16 ncMSs, 62 Col/C24, and 47 IP SNP loci.
Genetic diversity and differentiation of local populations:
To determine the genetic variation within Iberian local populations and their differentiation, we genotyped 175 individuals collected from seven populations (20–32/population) with the same four marker sets (Figure 1, Table 2). All microsatellites segregated among populations but only 46 loci from the 62 Col/C24 SNPs and 42 from the 47 IP SNP loci were polymorphic. A two- to fourfold difference in genetic diversity was found among populations, depending on the parameter (supplemental Table S6). However, gene diversity estimates (HS) of ncMSs and of the two sets of SNP loci were highly correlated (N = 7; r > 0.81; P < 0.026), indicating that both types of nuclear loci detected the same patterns of genetic variation. On average, pairs of A. thaliana individuals collected in the same local population differed in 38, 58, 16, and 13% of the polymorphic cpMSs, ncMSs, Col/C24, and IP SNP loci, respectively.
TABLE 2.
Genetic diversity of seven A. thaliana local populations from the Iberian Peninsula
| Agu | Cdc | Leo | Mar | Pra | Qui | San | Average | |
|---|---|---|---|---|---|---|---|---|
| N | 21 | 32 | 20 | 30 | 24 | 27 | 21 | 25.0 ± 4.8 |
| NG | 14 | 30 | 18 | 25 | 19 | 17 | 19 | 20.3 ± 5.4 |
| NH | 8 | 24 | 8 | 16 | 11 | 5 | 16 | 12.6 ± 6.5 |
| HS | 0.13 ± 0.18 | 0.25 ± 0.26 | 0.26 ± 0.25 | 0.19 ± 0.24 | 0.33 ± 0.27 | 0.13 ± 0.22 | 0.28 ± 0.27 | 0.22 ± 0.27 |
| PLD | 12.72 | 5.58 | 22.85 | 14.43 | 31.36 | 62.34 | 8.71 | 22.6 ± 19.6 |
| HO | 0.05 ± 0.06 | 0.02 ± 0.02 | 0.03 ± 0.06 | 0.03 ± 0.04 | 0.01 ± 0.02 | 0.01 ± 0.02 | 0.00 ± 0.01 | 0.02 ± 0.02 |
| FIS | 0.87 ± 0.13 | 0.97 ± 0.04 | 0.96 ± 0.08 | 0.94 ± 0.06 | 0.99 ± 0.02 | 0.96 ± 0.05 | 0.99 ± 0.02 | 0.96 ± 0.04 |
| OR | 0.07 ± 0.07 | 0.02 ± 0.02 | 0.02 ± 0.04 | 0.03 ± 0.03 | 0.01 ± 0.01 | 0.02 ± 0.03 | 0.00 ± 0.01 | 0.03 ± 0.02 |
N, sample size; NG, number of multilocus genotypes detected from all polymorphic MS and SNP loci; NH, number of multilocus haplotypes estimated from SNP loci; HS, mean gene diversity estimated from MS and SNP loci; PLD, percentage of pairs of nuclear loci showing significant LD. Mean observed heterozygosity (HO), inbreeding coefficient (FIS), and outcrossing rates (OR) were estimated from 16 ncMS loci.
In total, 32–130 different genotypes were found, depending on the marker set, and the combined analysis of all nuclear loci resulted in the same 130 genotypes being detected with ncMSs (supplemental Table S6). No identical multilocus genotype was found in different populations with any set of nuclear markers, while four chlorotypes were detected in several populations. The combined analysis of both sets of SNP loci identified 88 different multilocus genotypes. From the joint analysis of all nuclear loci, we estimated that within the IP populations, on average, 50% of the individuals show haplotypes differing in at least one SNP locus and several ncMSs, 26% are genetically identical to other individuals, and 24% have nearly identical genotypes differing in one to three ncMS loci. In total, we found 28 pairs of individuals from the same population differing in a single ncMS locus. From this, we estimated a frequency of ∼1% nearly identical individuals per ncMS locus in an otherwise similar genetic background. This proportion was similar to the estimated ncMS error rate (see materials and methods). Therefore, we concluded that most nearly identical individuals detected with ncMSs in the same local population do not carry de novo ncMS mutations, but probably they are identical genotypes bearing MS genotyping errors.
Genomewide analysis of LD between pairs of nuclear loci in each population indicated that 5.6–62.3% of all pairwise combinations of loci present significant LD (Table 2). On the other hand, local populations showed a mean observed heterozygosity per ncMS locus (HO) of 0.02 and a mean outcrossing frequency of 2.5% (Table 2). However, substantial variation was found among populations since outcrossing frequencies ranged between 0.3 and 7.5%. In total, 12, 7, 4, and 4 individuals appeared heterozygous for one, two, three, and five ncMS loci, respectively. Together, these individuals carried 58 heterozygous ncMS data points, 46 involving alleles already segregating in the corresponding population. Therefore, most heterozygous MS loci are probably generated by outcrossing.
Neighbor-joining analysis of the 175 multilocus genotypes showed that most individuals from the same population, but not all, group together (supplemental Figure S4). In addition, AMOVA estimates of genetic differentiation among the seven populations indicated that 33.6% of the genetic variation from all loci is present among populations. Similar average FST values were estimated from the various sets of markers (FST = 0.31–0.36; P < 0.0001). Moreover, the seven populations differed genetically from each other since all FST values between pairs of populations were significant (FST = 0.12–0.66; P < 0.0001). Therefore, Iberian populations are genetically differentiated, although more genetic variation is found within than among populations.
Comparison between the Iberian Peninsula and the rest of world:
Genetic diversities of the IP and the rest of world distribution were compared by joint analyses of the 100 IP genotypes and 43–193 worldwide genotypes, depending on the marker type (Table 1 and materials and methods). Both groups of samples showed rather similar HS values for all sets of MS and SNP loci. However, Col/C24 SNP loci presented a lower percentage of polymorphic loci and mean allelic richness in the IP than in the rest of world, while the opposite behavior was observed for the IP SNP markers (Table 1). These results indicate that IP and the rest of world differ in their SNP allele frequencies and therefore diversity estimates are differently biased for the two sets of SNP loci. Since the SNP markers were selected from genotypes of Iberia and central Europe (materials and methods), IP and the rest of world are biased toward higher diversity for the IP and the Col/C24 SNP loci, respectively. Diversity estimates described for the two sets of SNP loci throughout the text reflect such marker ascertainment bias.
AMOVA analyses of genetic differentiation between the IP and the rest of world indicated that <5% of the variation distinguishes both groups (FST = 0.019–0.041; P < 0.05). This limited differentiation was mostly due to 1, 3, 12, and 11 of the cpMSs, ncMSs, Col/C24, and IP SNP loci, respectively, showing significantly different allele frequencies in the two samples (P < 0.01).
On the other hand, the relationships among chlorotypes of the IP and the rest of world was compared by frequency network analysis of 143 individuals (Figure 2 and supplemental Table S5). In total, 36 chlorotypes were detected with three cpMSs, which were arranged in a complex phylogenetic network (see materials and methods). The six most frequent chlorotypes, showing frequencies >4%, were found in both samples, while specific IP or world chlorotypes presented frequencies <4%. However, IP and world samples differed in the frequency distribution of chlorotypes (χ2 = 60.56; P < 0.001) and in the most common chlorotype (CH01 and CH18 for the IP and the rest of world, respectively). In addition, eight IP-specific chlorotypes appeared to be derived by single mutational steps from the commonest IP chlorotype, suggesting that this is the oldest Iberian chlorotype.
Figure 2.—

Chlorotype networks of A. thaliana individuals from the Iberian Peninsula and the rest of world. (A) Network of chlorotypes present in the IP and in the rest of world. Pie sizes are proportional to the total chlorotype frequency, while shaded and solid sectors correspond to relative frequencies in the IP and in the rest of world, respectively. (B) Network of Iberian chlorotypes. Pie sizes are proportional to IP frequency and the four colored sectors correspond to relative frequencies in the IP genetic groups inferred with STRUCTURE. Yellow, blue, green, and red depict the IP genetic groups 1, 2, 3, and 4, respectively. Each branch corresponds to one mutational step between chlorotypes. Nonobserved mutational steps between chlorotypes are indicated by perpendicular dashes. See materials and methods and supplemental Table S5 for details.
Finally, the overall structure of the IP genetic variation was inferred and compared to that of the rest of world by clustering analysis using the algorithm implemented in STRUCTURE. Four different genetic clusters were detected from the analysis of 293 multilocus genotypes obtained with the 62 polymorphic Col/C24 SNP loci (Figure 3 and supplemental Table S7). On average, individuals from outside the IP showed the largest membership fractions in clusters 1 and 2, while clusters 3 and 4 presented the largest proportions in the Iberian genotypes. When individuals from outside the IP were classified in seven geographic regions, two spatio-longitudinal gradients of ancestry membership frequencies, with opposite directions, were detected in Eurasia. Cluster 2 showed the highest mean fraction in samples from Asia, its frequency decreasing in eastern and western Europe. In contrast, cluster 3 showed the highest frequency in the IP, and this decreases in eastern Europe and Asia (Figure 3). In addition, cluster 1 appeared as the most frequent in western Europe but the least common in IP, whereas cluster 4 shows a high frequency in the IP and the Mediterranean basin but the lowest frequency in western Europe. Individuals from North America and Asia showed at least two genetic clusters with average membership fractions <10%, indicating that samples from these regions contain less genetic variation than samples from Japan.
Figure 3.—

Worldwide population structure of A. thaliana. (Top) Graphical representation of ancestry membership coefficients of all individuals. Each individual is shown as a vertical line divided into segments representing the estimated membership proportions in the four ancestral genetic clusters inferred with STRUCTURE from 62 Col/C24 SNP loci. Individuals are classified into eight indicated geographical regions. Individuals within each region are arranged according to estimated cluster membership proportions. (Bottom) The number of individuals and the mean membership fractions in the four genetic clusters (C1–C4) for each region. Standard deviations of all mean membership proportions were <0.001 (not shown).
Analyses of genetic structure in the Iberian Peninsula:
To establish the genetic structure of A. thaliana Iberian populations more precisely, we first carried out NJ analyses of the 100 IP individuals (supplemental Figure S5). These analyses detected several groups of genotypes, but bootstrap support was mostly low. Therefore, a model-based clustering approach was used to determine the structure of these populations (Figure 4 and supplemental Table S7). Four genetic clusters were inferred with STRUCTURE when analyzing the multilocus genotypes obtained with all 95 polymorphic SNP loci. However, only three significant clusters were found when using 50 Col/C24 or 45 IP SNP loci (supplemental Table S7). Cluster membership coefficients distinguished different but overlapping groups of individuals in the three analyses, illustrating the synergistic and complementary behavior of both sets of SNP loci (data not shown). Basically, cluster C4 differentiated with the Col/C24 SNP loci in the worldwide analysis was split into two clusters when including the IP SNPs (for comparison, red IP individuals with high C4 membership fraction in the worldwide analysis of Figure 3 closely correspond to the red individuals in the K = 2 analysis of Figure 4), while the remaining clusters C1–C3 were assigned to two other clusters. Most individuals showed an estimated major membership proportion >0.6 and therefore could be classified into four distinct genetic groups according to their largest ancestry membership fractions (Figure 4). This classification was in agreement with NJ analyses since individuals assigned to the same genetic group tend to cluster together in NJ trees (supplemental Figure S5). Nevertheless, it has been argued that in populations with continuous spatial distribution of genetic diversity, clusters detected by STRUCTURE might be influenced by uneven geographical distribution of samples (Rosenberg et al. 2005; François et al. 2006). Hence, clustering analyses were also performed using the algorithm implemented in TESS (see materials and methods). Four genetic clusters were also inferred in TESS analyses using different assumptions of the spatial distribution of genetic clusters (supplemental Figure S6). Most individuals showed the same major ancestry membership coefficient as that estimated with STRUCTURE, supporting the robustness of the inferred clusters. However, similarity coefficients among runs of the same TESS model were considerably lower than those estimated among STRUCTURE runs (supplemental Table S7).
Figure 4.—

Population structure of A. thaliana in the Iberian Peninsula. Genetic relationships among 100 IP individuals from different populations were estimated with STRUCTURE using 95 polymorphic SNP loci. Each individual is depicted as a vertical rectangle divided into segments representing the estimated membership proportions in the ancestral genetic clusters (K) fitted in the model. Individuals are arranged according to estimated cluster membership proportions. Arrows at the bottom indicate individuals from the seven populations used for local population differentiation analyses.
Genetic diversities of the four STRUCTURE groups show a twofold variation among groups, with groups 1 and 2 being consistently more diverse than groups 3 and 4 (supplemental Table S8). AMOVA analyses using all loci showed an average FST differentiation of 0.11 over the four groups, FST values being significant for all sets of markers (supplemental Table S9). The lowest differentiation was observed between groups 1 and 2, while the largest differentiations were estimated between group 3 and the remaining groups (supplemental Table S9).
We further analyzed the four genetic groups for their chlorotype diversity (Figure 2B). Each genetic group carried numerous chlorotypes and no clearly distinct maternal origin of any group was observed. However, groups 1 and 2 bore at least four chlorotypes also present outside Iberia and located throughout the network. In contrast, groups 3 and 4 showed more restricted geographical and evolutionary chlorotype variation. Most individuals of group 3 carried different IP-specific chlorotypes, whereas group 4 contained the smallest number of IP-specific chlorotypes. These results suggest that group 3 is an Iberian-specific group that has remained rather isolated from other world regions, whereas seeds of groups 1, 2, and 4 might have migrated between IP and other world regions.
Analyses of geographical structure in the Iberian Peninsula:
To determine if A. thaliana genetic variation is spatially structured in Iberia, we first tested isolation by distance among the 100 genotypes collected from different local populations. Mantel tests showed that genetic distances are positively correlated with geographical distances, r values ranging between 0.1 and 0.23, depending on the marker set (P < 0.004). This correlation was maximum when using the genetic distances estimated from all loci (r = 0.28; P < 0.001). The IBD pattern of geographical structure was also analyzed in the seven local populations extensively sampled (Figure 5A), with the Mantel test showing significant correlations for SNP loci (r = 0.55–0.60; P < 0.04) and marginal significances for MS markers (r = 0.40–0.53; P = 0.06).
Figure 5.—

Geographical structure of A. thaliana in the Iberian Peninsula. (A) Correlation between geographical and genetic distances in seven local populations extensively sampled. (B) Correlation between geographical and genetic distances among six IP geographical subregions. In A and B, genetic distances are estimated from all 115 polymorphic MS and SNP loci. (C) Geographical location and genetic composition of 100 individuals from different populations. Each population is shown as a pie chart representing membership proportions in the four genetic clusters inferred with STRUCTURE (colors of clusters are as in Figure 4: cluster 1, yellow; cluster 2, blue; cluster 3, green; cluster 4, red). Asterisks indicate the seven populations used for local population differentiation analyses.
A. thaliana spatial structure in the IP was further evaluated by classifying the 100 populations in six geographical subregions (Figure 1 and supplemental Table S1). Genetic diversities were rather similar in the six subregions (supplemental Table S10), but AMOVA analyses indicate significant differentiation among all of them for three sets of loci (supplemental Table S9). As shown in Figure 5B, a strong significant correlation was found between geographical and genetic distances among the six IP subregions (r = 0.64; P = 0.014). This significant regional isolation by distance was in agreement with the clustering obtained by NJ analysis, where individuals from the same subregion trend to cluster together (supplemental Figure S5). To identify geographical barriers that might limit genetic flow, we also performed Mantel tests with the individual genotypes from each of the 15 pair combinations of the six geographical subregions. All pair comparisons of subregions showed significant positive correlation between geographical and genetic distances (P < 0.005). The lowest values corresponded to comparisons involving pairs of subregions I–IV (r = 0.21–0.26; P < 0.005). In contrast, comparisons of subregions V and VI with the rest of the subregions showed considerably larger correlations (r = 0.30–0.62; P < 0.001), indicating that the northeastern subregions have been more isolated from the rest of Iberia.
Finally, geographical structure of A. thaliana genetic variation was also inferred from the four genetic clusters previously established by model-based approaches. As shown in Figure 5C, genetic clusters derived with STRUCTURE were not evenly distributed across Iberia, but instead appear restricted mostly to particular subregions. Overall, clusters 1, 2, 3, and 4 are located mainly in the northwestern, northeastern, central/southeastern, and southwestern areas, respectively. Genetic clusters inferred from a TESS model assuming noninformative spatial prior showed nearly the same geographical distribution as STRUCTURE clusters (supplemental Figure S6, A and B). Results from both algorithms differed mainly in the frequencies of clusters 1 and 2, with TESS analysis leading to higher and lower frequencies of these groups, respectively. In addition, genetic clusters inferred with TESS using an interaction parameter value of 0.7 also show considerable spatial overlapping with STRUCTURE clusters (supplemental Figure S6C). However, the latter TESS model estimated a more restricted geographical distribution of clusters 2 and 3 and a broader distribution of cluster 4.
DISCUSSION
A. thaliana is widely distributed as a native species in the Iberian Peninsula. In this region, A. thaliana appears not only in agricultural fields and other relatively anthropogenic habitats but also in a wide range of naturally disturbed habitats, from mesic and xeric grasslands to Atlantic and Mediterranean forests (supplemental Figure S1). In this work, we have developed an Iberian collection of 268 individuals sampled from 100 populations, which correspond to 181 distinct genotypes as estimated from presumed neutral cpMSs, ncMSs, and nuclear SNP loci.
Estimating genetic diversity, differentiation, and structure with microsatellites and single nucleotide polymorphisms:
As expected from the different molecular nature and mutation rate of MS and SNP loci, genetic diversities were higher when based on microsatellite than on SNP loci. Consistently, lower genetic differentiation values were often found with MS than with SNP loci. In addition, ncMS loci did not enable inference of genetic clusters when analyzed with Bayesian-model-based clustering algorithms. This was not exclusively due to the low number of ncMS loci considered because the combination of ncMS and SNP markers showed reduced clustering power compared to SNP loci alone (data not shown). Probably, this is also a consequence of the high variability of the ncMS analyzed in A. thaliana, which is predicted to generate a large proportion of molecular convergence (homoplasy) in small populations evolving with high mutation rates (Estoup et al. 2002). Thus, SNP loci were more useful than microsatellites for A. thaliana genetic structure analyses. However, population genetic parameters estimated from SNP loci were also biased because SNP markers were ascertained from small sets of genotypes with uneven geographical distribution. Selection of SNPs from small panels of genotypes biases the sets of SNP loci toward polymorphisms with intermediate allele frequencies (reviewed in Brumfield et al. 2003). Accordingly, we analyzed mainly common sequence polymorphisms since only 27–44% of the SNP loci show MAF <5% (supplemental Figure S3), compared with 55% of synonymous SNPs presenting MAF <5% in random samples (Nordborg et al. 2005). Such distortion increases overall diversity estimates and decreases differentiation of recent branches of genealogical trees (Brumfield et al. 2003). However, due to the detected geographical structure, selection of SNP loci from genotypes of particular regions, such as Iberia or central Europe, biases mainly population parameter estimates in those regions. It is expected that the joint analysis of two sets of SNP loci selected from different world regions reduces SNP ascertainment bias and increases the capability of inferring genetic clusters.
It must be emphasized that as discussed for human populations (Rosenberg et al. 2005), A. thaliana genetic clusters detected by STRUCTURE algorithm are probably due to small IP geographical discontinuities of allele frequencies, and such clusters represent only a small fraction of the total genetic variation (average FST differentiation among IP clusters is 0.11). The clustering capacity of STRUCTURE is affected by several factors, such as the number of loci (Rosenberg et al. 2005). The two sets of SNP loci used in this work were selected to contain nearly 50 loci, since this is the minimum number estimated for consistent inference of genetic clusters (Rosenberg et al. 2005). Furthermore, it has been argued that uneven spatial sampling in the experimental design might also affect the STRUCTURE clustering patterns (Rosenberg et al. 2005). As proposed by François et al. (2006), the robustness of STRUCTURE clusters detected in this work has been tested with the TESS algorithm, which incorporates spatial models for geographical continuity of allele frequencies. A large concordance was observed among the results obtained with both algorithms. Therefore, uneven geographical distribution of samples and the specific bias of MS and SNP loci are not expected to affect any major conclusion of this work.
Genetic diversity within local populations of A. thaliana:
The seven Iberian local populations analyzed in this work contain a substantial amount of genetic variation, since all of them include several chlorotypes and ∼50% of the individuals of each population show distinct nuclear multilocus genotypes. Average FST values indicate that 66% of the genetic variation is segregating within these populations. Different proportions of within-population diversity have been found in other world regions, ranging from 77 to 36% in North America (Bergelson et al. 1998; Jørgensen and Mauricio 2004), 57% in Europe (Bakker et al. 2006), 54% in China (He et al. 2007), 41% in France (Le Corre 2005), 12% in Norway (Stenøien et al. 2005), and 0% in Japan (Todokoro et al. 1995). In agreement with Beck et al. (2008), the large fraction of genetic variation segregating within Iberian populations at MS and SNP loci suggests a south–north latitudinal gradient of local population diversity in Europe. However, caution must be taken when comparing with previous studies because genetic differentiations have been estimated with different types of markers. Moreover, the genetic diversity of local populations might depend on population size, age, and habitat. The Iberian populations analyzed in this work are large populations of thousands of individuals, permanent for several years, and growing in mostly natural habitats. Such populations might contain a larger amount of genetic variation than smaller and recent populations collected in more homogeneous habitats. Accordingly, large coordinated studies avoiding differences due to type and number of molecular markers, kind of local populations, and sample size are necessary to compare the distribution of genetic diversity within and among local populations in different world regions.
Iberian populations differ considerably in their genetic diversity. In agreement with previous studies (Stenøien et al. 2005; Bakker et al. 2006), several results indicate that migration, outcrossing, and de novo mutation differentially contribute to the variation within populations. First, chloroplast and nuclear haplotype analyses show that most populations contain not only related but also genetically unrelated individuals (supplemental Figure S4), suggesting that seed migration is an important factor contributing to within-population diversity. Second, considerable variation is found among populations for the proportion of pairs of loci showing significant LD, which indicates a different contribution of cross-fertilization and recombination or of demographic factors affecting LD. In agreement, a large variation among populations was also found for outcrossing rate estimates. Iberian populations presented an average outcrossing frequency of 2.5%, which is slightly larger than previous MS-based estimates (Le Corre 2005; Stenøien et al. 2005; Bakker et al. 2006). This is probably due to the larger diversity of Iberian populations and not to mistyping errors because the three main sources of MS scoring errors (stutter bands, large-allele dropout, and null alleles) lead to underestimations of heterozygosity (reviewed in Dewoody et al. 2006). Moreover, in contrast to previous MS studies, we estimated heterozygosity of field plants instead of individuals raised from field seed, which suggests that heterozygous individuals derived from outcrossing might have higher fitness under natural conditions. Finally, the presence of some individuals heterozygous for ncMS alleles that otherwise are undetected in the same local population suggests that part of this heterozygosity might be generated by de novo ncMS mutations. However, true detection of individuals carrying new or homoplasic MS alleles generated by de novo mutation requires exhaustive genomewide genotyping with an extremely low error rate, which cannot be achieved by the standard methods of MS analysis used in this work.
Inference of A. thaliana diversity centers and genetic refugia from the worldwide geographical structure:
As discussed above, the broad diversity of habitats occupied by A. thaliana in the Iberian Peninsula, together with the large genetic variation observed within Iberian populations, suggests a longer demographic history of A. thaliana in Iberia than in other world regions. Furthermore, several results suggest that the Iberian Peninsula was a diversity center and a European glacial refugium for A. thaliana. First, Iberia contains a similar amount of neutral genetic diversity per locus as the rest of the world distribution area. Second, Iberia is genetically differentiated from the rest of world as indicated by allele frequencies at single SNP and MS loci, as well as by multilocus chlorotype frequencies. Third, the global-scale geographical structure of genetic variation inferred by model-based clustering analysis shows a pattern of genetic differentiation of Iberia and the rest of Eurasia that further supports this hypothesis (Figure 3). Two main conclusions are drawn from the joint analysis of genotypes from IP and the rest of world. On one hand, the Iberian Peninsula appears populated by several distinct genetic lineages and not by a single homogenous genetic group of populations, as previously described from the analysis of a limited number of accessions (Symonds and Lloyd 2003; Schmid et al. 2006). On the other hand, two longitudinal gradients of cluster frequencies are detected in Eurasia, suggesting a double postglacial colonization of central Europe, in agreement with spatial gradients and IBD patterns previously found (Sharbel et al. 2000; Nordborg et al. 2005; Ostrowski et al. 2006; Schmid et al. 2006). The presence of a west–east frequency decrease of the major IP cluster (C3) and an east–west frequency decrease of the main Asian genetic cluster (C2) points to a postglacial colonization of central Europe from Iberia and Asia. However, given the high western European frequency of another cluster inferred in this analysis (C1 in Figure 3), it is very likely that other Eurasian refugium(a) contributed to its colonization. Furthermore, the low genetic diversity found in Asian accessions with SNP markers (this work and Schmid et al. 2006) does not support the idea that this region was the primary center of diversity and origin of A. thaliana. In agreement, it has been recently suggested that A. thaliana arose in the Caucasus region (Beck et al. 2008). However, the limited number and distribution of genotypes studied from Asia and southern Europe, together with their restricted genotyping, precludes solid conclusions on the origin and expansion of A. thaliana in Eurasia.
Causes and consequences of the geographical structure of A. thaliana in the Iberian Peninsula:
Several results demonstrate that A. thaliana neutral genetic variation is spatially structured in the Iberian Peninsula and indicate a limited genetic flow among geographical subregions. First, significant isolation by distance was detected in the entire IP and among IP subregions on the basis of pairwise comparisons of the 100 individuals and of the six geographical subregions. Global FST values indicate an average differentiation among subregions of 6.4%, which is comparable to the subregional differentiation estimated from the local populations extensively analyzed (0.7–16.6%; data not shown). Thus, local (short distance) dispersal of seeds seems an important process contributing to the founding of new populations in Iberia. Second, the geographical distribution of the genetic clusters inferred by model-based approaches shows a clear-cut spatial differentiation pattern. Consistently, the four genetic groups inferred with different population models appear distributed mainly in the four Iberian quadrants (Figure 5C and supplemental Figure S6).
The current geographical structure in four largely parapatric groups indicates isolation of genetic lineages and (meta)populations in the past. One possible scenario to generate such isolation can be speculated from the last Pleistocene glaciations, 20,000–40,000 years ago (Pérez Alberti et al. 2004). As reviewed by Gomez and Lunt (2006), the fragmented nature of suitable Iberian habitats favored the occurrence of multiple glacial refugia isolated from each other. Phylogeographic studies of many European flora and fauna species have shown strong genetic subdivision in the Iberian Peninsula, the spatially separated distribution of genetic lineages being interpreted as remnant landmarks of those Pleistocene refugia. Comparative analyses demonstrate that phylogeographic Iberian patterns of different species broadly overlap among themselves, as well as with areas of high endemism. These studies indicate the internal complexity of Iberia as a glacial refugium and have led to the proposal of several Iberian glacial refugia that are shared by multiple species (Gomez and Lunt 2006). Interestingly, the geographical distribution of A. thaliana genetic clusters inferred in this work partly overlaps with some of the refugia described for other species. Therefore, we hypothesize that Iberia was not a single A. thaliana refugium during Pleistocene glaciations but it provided several southern and northern refugia.
On the other hand, the geographical structure of A. thaliana suggests that there have been physical and/or environmental barriers limiting genetic flow throughout Iberia during the postglacial period. Mantel correlation analyses with pairs of geographical subregions suggest that the Ebro River basin (limiting subregions V and VI) is a major Iberian geographical barrier, subregion VI showing a 0.16 average FST differentiation from the rest of Iberia. In addition, the Central System mountains (stretching east–west across the IP center and separating subregions II and III) also might have limited genetic flow, since the lowest genetic differentiations were estimated between subregions I and II and subregions III and IV. Furthermore, current spatial structure might have been partly originated by other isolating factors or unknown demographic processes acting more recently in the Holocene. In addition, given the latitudinal climatic diversity of Iberia, it cannot be dismissed that this structure is also sustained by natural selection of genetic lineages adapted to different subregional climatic environments during glacial and/or postglacial isolation.
Finally, the Iberian geographical structure provides new insights into the contribution of the IP refugia to current European A. thaliana diversity. We hypothesize two different postglacial colonization waves deriving from Iberia and involving the IP genetic groups 1/2 and 3/4. Genetic groups 1 and 2 cover the northern half of the IP (Figure 5C) and are related mainly to cluster 3 of the worldwide analysis, which shows a west–east spatial gradient in Eurasia (Figure 3). Hence, groups 1 and 2 from the northern IP seem to have contributed mainly to the postglacial colonization of western and northern Europe. On the other hand, cluster 4 was identified in the worldwide analysis at a relatively high frequency only in the Mediterranean basin (Figure 3), while it shows lower frequency in the rest of the Eurasian continent. This cluster is differentiated in the IP genetic clusters 3 and 4 (Figure 4), which are found mainly in southern Iberia. Interestingly, most individuals of IP group 3 carry Iberian specific chlorotypes, suggesting that this cluster is specific for Iberia. Furthermore, IP group 3 shows the largest FST differentiations among the four IP groups, pointing to this group as the most ancient Iberian genetic lineage. These results, together with the presence of chlorotypes shared with other world regions in IP cluster 4 (Figure 2B), suggest that IP group 4 might have been differentiated from IP-specific cluster 3 and expanded later to the Mediterranean region. However, the opposite directions of differentiation and migration cannot be discarded, since it is unknown how the expansion of A. thaliana in the Mediterranean basin occurred before and after the last glaciation. Therefore, further studies of southern European peninsulas and the North Africa region are needed to elucidate the demographic history of A. thaliana in the Mediterranean basin, where other glacial refugia have been located for many European species (Hewitt 2001).
Acknowledgments
The authors thank Dorothée Ehrich for assistance with Structure-sum R-script and François Olivier for help with representation of ancestry matrixes by Kriging methods. We also thank Manuel Piñeiro (Ees, Glo, Mdd, and Nac), César Gómez-Campo (Sne), Pere Fraga (Mah), Eduardo Sánchez (Mol and Smt), José A. Aguilar (Fue), Herminia Zaldívar (Mep), Pablo Catarecha (Tor), and Ivan del Olmo (Cha) for kindly providing the samples indicated in parentheses. The authors thank Mercedes Ramiro for excellent technical assistance. This work was funded by grant BIO2004-00533 from the Ministerio de Educación y Ciencia to C.A.-B. F.X.P. was funded by grant GENOMICS/2003/12 from the European Science Foundation travel program. B.M.-V. was funded by a salary fellowship from the Fundación Para el Fomento en Asturias de la Investigación Científica Aplicada y Tecnología (Principado de Asturias, Spain).
References
- Allard, R., S. Jain and P. Workman, 1968. The genetics of inbreeding species. Adv. Genet. 14 55–131. [Google Scholar]
- Bakker, E. G., E. A. Stahl, C. Toomajian, M. Nordborg, M. Kreitman et al., 2006. Distribution of genetic variation within and among local populations of Arabidopsis thaliana over its species range. Mol. Ecol. 15 1405–1418. [DOI] [PubMed] [Google Scholar]
- Bandelt, H. J., P. Foster and A. Röhl, 1999. Median-joining network for inferring intraspecific phylogenies. Mol. Biol. Evol. 16 37–48. [DOI] [PubMed] [Google Scholar]
- Beck, J. B., H. Schmuths and B. A. Schaal, 2008. Native range genetic variation in Arabidopsis thaliana is strongly geographically structured and reflects Pleistocene glacial dynamics. Mol. Ecol. 17 902–915. [DOI] [PubMed] [Google Scholar]
- Bell, C. J., and J. R. Ecker, 1994. Assignment of 30 microsatellite loci to the linkage map of Arabidopsis. Genomics 19 137–144. [DOI] [PubMed] [Google Scholar]
- Bergelson, J., E. Stahl, S. Dudek and M. Kreitman, 1998. Genetic variation within and among populations of Arabidopsis thaliana. Genetics 148 1311–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernartzky, R., and S. Tanksley, 1986. Genetics of acting-related sequences in tomato. Theor. Appl. Genet. 72 314–324. [DOI] [PubMed] [Google Scholar]
- Brumfield, R. T., P. Beerli, D. A. Nickerson and S. V. Edwards, 2003. The utility of single nucleotide polymorphisms in inferences of population history. Trends Ecol. Evol. 18 249–256. [Google Scholar]
- Cardon, L. R., and L. J. Palmer, 2003. Population stratification and spurious allelic association. Lancet 361 598–604. [DOI] [PubMed] [Google Scholar]
- Dewoody, J., J. Nason and V. Hipkins, 2006. Mitigating scoring errors in microsatellite data from wild populations. Mol. Ecol. Notes 6 951–957. [Google Scholar]
- Ehrich, D., 2006. AFLPDAT: a collection of r functions for convenient handling of AFLP data. Mol. Ecol. Notes 6 603–604. [Google Scholar]
- Estoup, A., P. Jarne and J. M. Cornuet, 2002. Homoplasy and mutation model at microsatellite loci and their consequences for population genetics analysis. Mol. Ecol. 11 1591–1604. [DOI] [PubMed] [Google Scholar]
- Excoffier, L., P. Smouse and J. Quattro, 1992. Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131 479–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Excoffier, L., G. Laval and S. Scheneider, 2005. Arlequin ver. 3.0: an integrated software package for population genetics data analysis. Evol. Bioinform. Online 1 47–50. [PMC free article] [PubMed] [Google Scholar]
- Falush, D., M. Stephens and J. K. Pritchard, 2003. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164 1567–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- François, O., S. Ancelet and G. Guillot, 2006. Bayesian clustering using hidden Markov random fields in spatial population genetics. Genetics 174 805–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez, A., and D. Lunt, 2006. Refugia within refugia: patterns of phylogeographic concordance in the Iberian Peninsula, pp. 155–188 in Phylogeography of Southern European Refugia, edited by S. Weiss and N. Ferrand. Springer, Dordrecht, The Netherlands.
- Goudet, J., 1995. FSTAT: a computer program to calculate F-statistics. J. Hered. 86 485–486. [Google Scholar]
- He, F., D. Kang, Y. Ren, L. J. Qu, Y. Zhen et al., 2007. Genetic diversity of the natural populations of Arabidopsis thaliana in China. Heredity 99 423–431. [DOI] [PubMed] [Google Scholar]
- Hewitt, G. M., 2001. Speciation, hybrid zones and phylogeography—or seeing genes in space and time. Mol. Ecol. 10 537–549. [DOI] [PubMed] [Google Scholar]
- Hoffmann, M. H., 2002. Biogeography of Arabidopsis thaliana (L.) Heynh. (Brassicaceae). J. Biogeogr. 29 125–134. [Google Scholar]
- Jakobsson, M., and N. A. Rosenberg, 2007. CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23 1801–1806. [DOI] [PubMed] [Google Scholar]
- Jensen, J. L., A. J. Bohonak and S. T. Kelley, 2005. Isolation by distance, web service. BMC Genet. 6 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jørgensen, S., and R. Mauricio, 2004. Neutral genetic variation among wild North American populations of the weedy plant Arabidopsis thaliana is not geographically structured. Mol. Ecol. 13 3403–3413. [DOI] [PubMed] [Google Scholar]
- Kalinowski, S., 2005. HP-Rare: a computer program for performing rarefaction on measures of allelic diversity. Mol. Ecol. Notes 5 187–189. [Google Scholar]
- Kuittinen, H., D. Salguero and M. Aguadé, 2002. Parallel patterns of sequence variation within and between populations at three loci of Arabidopsis thaliana. Mol. Biol. Evol. 19 2030–2034. [DOI] [PubMed] [Google Scholar]
- Kumar, S., K. Tamura and M. Nei, 2004. MEGA3: integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief. Bioinformatics 5 150–163. [DOI] [PubMed] [Google Scholar]
- Le Corre, V., 2005. Variation at two flowering time genes within and among populations of Arabidopsis thaliana: comparison with markers and traits. Mol. Ecol. 14 4181–4192. [DOI] [PubMed] [Google Scholar]
- Loudet, O., S. Chaillou, C. Camilleri, D. Bouchez and F. Daniel-Vedele, 2002. Bay-0 × Shahdara recombinant inbred line population: a powerful tool for the genetic dissection of complex traits in Arabidopsis. Theor. Appl. Genet. 104 1173–1184. [DOI] [PubMed] [Google Scholar]
- Mantel, N., 1967. The detection of disease clustering and a generalized regression approach. Cancer Res. 27 209–220. [PubMed] [Google Scholar]
- Mitchell-Olds, T., and J. Schmitt, 2006. Genetic mechanisms and evolutionary significance of natural variation in Arabidopsis. Nature 441 947–952. [DOI] [PubMed] [Google Scholar]
- Myers, N., R. A. Mittermeier, C. G. Mittermeier, G. A. B. Da Fonseca and J. Kent, 2000. Biodiversity hotspots for conservation priorities. Nature 403 853–858. [DOI] [PubMed] [Google Scholar]
- Nordborg, M., T. T. Hu, Y. Ishino, J. Jhaveri, C. Toomajian et al., 2005. The pattern of polymorphism in Arabidopsis thaliana. PLoS Biol. 3 e196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrowski, M. F., J. David, S. Santoni, H. Mckhann, X. Reboud et al., 2006. Evidence for a large-scale population structure among accessions of Arabidopsis thaliana: possible causes and consequences for the distribution of linkage disequilibrium. Mol. Ecol. 15 1507–1517. [DOI] [PubMed] [Google Scholar]
- Pérez Alberti, A., M. Valcárcel Díaz and R. B. Chao, 2004. Pleistocene glaciation in Spain, pp. 389–394 in Quaternary Glaciations: Extent and Chronology, Part I: Europe, edited by J. Ehelers and P. L. Gibbard. Elsevier, Amsterdam.
- Pritchard, J. K., M. Stephens and P. Donnelly, 2000. Inference of population structure using multilocus genotype data. Genetics 155 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provan, J., 2000. Novel chloroplast microsatellites reveal cytoplasmic variation in Arabidopsis thaliana. Mol. Ecol. 9 2183–2185. [PubMed] [Google Scholar]
- Robbelen, G., 1965. The LAIBACH standard collection of natural races. Arabidopsis Inf. Ser. 2 36–47. [Google Scholar]
- Rosenberg, N. A., J. K. Pritchard, J. L. Weber, H. M. Cann, K. K. Kidd et al., 2002. Genetic structure of human populations. Science 298 2381–2385. [DOI] [PubMed] [Google Scholar]
- Rosenberg, N. A., S. Mahajan, S. Ramachandran, C. Zhao, J. K. Pritchard et al., 2005. Clines, clusters, and the effect of study design on the inference of human population structure. PLoS Genet. 1 e70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid, K. J., O. Torjek, R. Meyer, H. Schmuths, M. H. Hoffmann et al., 2006. Evidence for a large-scale population structure of Arabidopsis thaliana from genome-wide single nucleotide polymorphism markers. Theor. Appl. Genet. 112 1104–1114. [DOI] [PubMed] [Google Scholar]
- Sharbel, T. F., B. Haubold and T. Mitchell-Olds, 2000. Genetic isolation by distance in Arabidopsis thaliana: biogeography and postglacial colonization of Europe. Mol. Ecol. 9 2109–2118. [DOI] [PubMed] [Google Scholar]
- Slatkin, M., 1995. A measure of population subdivision based on microsatellite allele frequencies. Genetics 139 457–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smouse, P., J. Long and R. Sokal, 1986. Multiple regression and correlation extensions of the Mantel test of matrix correspondence. Syst. Zool. 35 627–632. [Google Scholar]
- Stenøien, H. K., C. B. Fenster, A. Tonteri and O. Savolainen, 2005. Genetic variability in natural populations of Arabidopsis thaliana in northern Europe. Mol. Ecol. 14 137–148. [DOI] [PubMed] [Google Scholar]
- Symonds, V. V., and A. M. Lloyd, 2003. An analysis of microsatellite loci in Arabidopsis thaliana: mutational dynamics and application. Genetics 165 1475–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todokoro, S., R. Terauchi and S. Kawano, 1995. Microsatellite polymorphisms in natural populations of Arabidopsis thaliana in Japan. Jpn. J. Genet. 70 543–554. [Google Scholar]
- Törjéck, O., D. Berger, R. C. Meyer, C. Mussig, K. J. Schmid et al., 2003. Establishment of a high-efficiency SNP-based framework marker set for Arabidopsis. Plant J. 36 122–140. [DOI] [PubMed] [Google Scholar]
- Van Berloo, R., 1999. GGT: software for the display of graphical genotypes. J. Hered. 90 328–329. [Google Scholar]
- Weir, B., and C. Cockerham, 1984. Estimating F-statistics for the analysis of population structure. Evolution 38 1358–1370. [DOI] [PubMed] [Google Scholar]
- Yeh, F., R. Yang and T. Boyle, 1999. POPGENE, version 1.32. Microsoft Windows-Based Freeware for Population Genetic Analysis. University of Alberta, Edmonton, Alberta.
- Zhao, K., M. J. Aranzana, S. Kim, C. Lister, C. Shindo et al., 2007. An Arabidopsis example of association mapping in structured samples. PLoS Genet. 3 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]