

Abstract
F2 populations are commonly used in genetic studies of animals and plants. For simplicity, most quantitative trait locus or loci (QTL) mapping methods have been developed on the basis of populations having two distinct genotypes at each polymorphic marker or gene locus. In this study, we demonstrate that dominance can cause the interactions between markers and propose an inclusive linear model that includes marker variables and marker interactions so as to completely control both additive and dominance effects of QTL. The proposed linear model is the theoretical basis for inclusive composite-interval QTL mapping (ICIM) for F2 populations, which consists of two steps: first, the best regression model is selected by stepwise regression, which approximately identifies markers and marker interactions explaining both additive and dominance variations; second, the interval mapping approach is applied to the phenotypic values adjusted by the regression model selected in the first step. Due to the limited mapping population size, the large number of variables, and multicollinearity between variables, coefficients in the inclusive linear model cannot be accurately determined in the first step. Interval mapping is necessary in the second step to fine tune the QTL to their true positions. The efficiency of including marker interactions in mapping additive and dominance QTL was demonstrated by extensive simulations using three QTL distribution models with two population sizes and an actual rice F2 population.
SIGNIFICANT progress in the development of polymorphic molecular markers has led to the intensive use of quantitative trait locus or loci (QTL) mapping in genetically segregating populations (Paterson et al. 1991; Lynch and Walsh 1998; Mackay 2001; Barton and Keightley 2002; Doerge 2002). A number of statistical methods have been developed for QTL detection and effect estimation. For regression-based methods, see Haley and Knott (1992), Martinez and Curnow (1992), Haley et al. (1994), Wright and Mowers (1994), Whittaker et al. (1996), and Feenstra et al. (2006); for maximum-likelihood-based methods, see Lander and Botstein (1989), Knott and Haley (1992), Zeng (1994), Kao et al. (1999), and Li et al. (2007, 2008); and for Bayesian model-based methods, see Satagopan et al. (1996), Ball (2001), Sen and Churchill (2001), Sillanpää and Corander (2002), Yi et al. (2003), and Bogdan et al. (2004).
For simplicity, most QTL mapping methods (here we mean linkage mapping for quantitatively inherited traits in biparental populations derived through controlled fertilization rather than association mapping in naturally mated populations) have been developed on the basis of backcross populations, doubled haploids, or recombination inbred lines derived from two parental lines (represented by P1 and P2), where two individual genotypes occur at each marker locus or QTL. F2 populations have been widely used in genetic studies of animals and plants since the rediscovery of Mendel's hybridization experiments. Relatively fewer methods have been developed on the basis of F2 populations, and dominance has sometimes been ignored (Wright and Mowers 1994; Whittaker et al. 1996; Jia and Xu 2007). Using similar principles in interval mapping (IM) as proposed by Lander and Botstein (1989), Knott and Haley (1992) investigated the maximum-likelihood methods for QTL mapping in F2 populations using simulated data. However, it is generally agreed that the mapping power of IM is low due to the lack of background control, and linked QTL cannot be properly separated (Zeng 1994). For F2 crosses between outbred lines, a mixed model was proposed to account for the variation both between and within lines (Pérez-Enciso and Varona 2000). On the basis of composite-interval mapping (CIM) (Zeng 1994), Jiang and Zeng (1995) used simulated F2 populations to demonstrate multiple-trait QTL mapping.
In populations consisting of two distinct genotypes, QTL mapping is focused on additive effects, even though the additive effect is defined differently in different populations. For example, in a backcross where P1 was used as the recurrent parent, the additive effect at a specific locus is normally defined as half of the difference between the P1 genotype and the F1 genotype (Zeng 1994). In doubled haploids or recombination inbred lines, the additive effect is defined as half of the difference between the P1 genotype and the P2 genotype. Sometimes authors claimed their methods could be extended to F2 populations (Zeng 1994). However, we report here that dominance can unexpectedly complicate the QTL mapping procedure by causing interactions between markers. As a result, the interactions detected between markers may be caused by the dominance effect of a QTL, rather than by real epistasis between interacting QTL.
Due to the lack of suitable QTL mapping methods for epistasis, some authors have used two-way ANOVA between markers to gain a rough idea of the importance of epistasis (Yu et al. 1997; Hua et al. 2003). More recently, Bayesian models have been widely investigated for mapping epistasis (Ball 2001; Broman and Speed 2002; Yi et al. 2003; Baierl et al. 2006). ANOVA between marker classes at one marker locus or two marker loci and some Bayesian model-based QTL mapping methods are valid under the assumption that QTL are completely linked with markers. Therefore, if QTL are located between marker intervals, false interacting QTL caused by the dominance effect may be detected by using these methods.
In this study, we report an inclusive linear model that includes interaction variables between two flanking markers, capable of completely absorbing both additive and dominance effects of QTL. On the basis of the linear model, we propose the inclusive composite-interval mapping (ICIM) suitable for QTL studies using F2 populations. Simulations were conducted to compare ICIM with CIM, and an actual F2 population was used to investigate QTL affecting plant height in rice (Oryza sativa L.).
MATERIALS AND METHODS
One-QTL model in F2 populations:
For one QTL (Q and q are the two alleles) in F2 populations, the genotypic value of an individual with a known QTL genotype, i.e., QQ, Qq, or qq, is written by
 |
(1) |
where  is the mean of the two homozygous genotypes QQ and qq, a is the additive genetic effect, d is the dominance effect, and w and v are indicators for QTL genotypes valued at 1 and 0 for QQ, 0 and 1 for Qq, and −1 and 0 for qq.
is the mean of the two homozygous genotypes QQ and qq, a is the additive genetic effect, d is the dominance effect, and w and v are indicators for QTL genotypes valued at 1 and 0 for QQ, 0 and 1 for Qq, and −1 and 0 for qq.
For two codominant markers (A-a and B-b) flanking the QTL, nine marker classes can be found in F2 (Table 1). In F2 populations, two indicators (represented by x and y, respectively) occur for each marker locus, similarly defined as indicators w and v for a QTL in model (1). The expectations of w and v, i.e., E(w) and E(v), can be calculated from the frequencies of the three QTL genotypes in each marker class (Table 1). In QTL mapping, the QTL genotype of an individual is usually unknown, but the marker type or the class of its flanking markers is known. In general, we can define the expected genotypic value (the last column in Table 1) of an individual with known marker types as
 |
(2) |
where x1 and y1 are the indicators for the left marker, x2 and y2 are the indicators for the right marker, x1 and x2 have similar values to w, and y1 and y2 have similar values to v. Similar to two genes, we can define the additive effects of the two markers, i.e., (a)A1 and (a)A2, dominance effects of the two markers, i.e., (d)D1 and (d)D2, and various interactions between the two markers, i.e., (d)AA12, AD12, DA12, and (d)DD12 in Equation 3, where  is the mean of the four homozygous marker classes (Table 1):
is the mean of the four homozygous marker classes (Table 1):
 |
(3) |
TABLE 1.
Coefficients of additive and dominance effects for each marker class
| Marker class | No. of samples | Indicators for markers
|
Expectation of w under each marker class, i.e., 
|
Expectation of v under each marker class, i.e., 
|
Genetic mean of each marker class | ||||
|---|---|---|---|---|---|---|---|---|---|
| Frequency | x1 | x2 | y1 | y2 | |||||
| AABB | n1 |  |
1 | 1 | 0 | 0 |  |
 |
 |
| AABb | n2 |  |
1 | 0 | 0 | 1 |  |
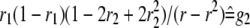 |
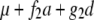 |
| AAbb | n3 |  |
1 | −1 | 0 | 0 |  |
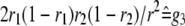 |
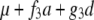 |
| AaBB | n4 |  |
0 | 1 | 1 | 0 |  |
 |
 |
| AaBb | n5 |  |
0 | 0 | 1 | 1 | 0 |  |
 |
| Aabb | n6 |  |
0 | −1 | 1 | 0 |  |
 |
 |
| aaBB | n7 |  |
−1 | 1 | 0 | 0 |  |
 |
 |
| aaBb | n8 | 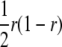 |
−1 | 0 | 0 | 1 |  |
 |
 |
| aabb | n9 |  |
−1 | −1 | 0 | 0 |  |
 |
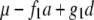 |
One QTL (Q and q are the two alleles) was assumed to be located between two marker loci (A and a and B and b are the marker alleles). r, r1, and r2 are recombination frequencies between the two flanking markers, between QTL and the left marker locus, and between QTL and the right marker locus, respectively. Assuming there is no crossover interference,  . It can be easily seen that
. It can be easily seen that  and
and  .
.
By resolving the above linear equations, the relationship between marker effects and QTL effects can be identified; i.e.,
 |
(4) |
Clearly, the additive QTL effect (a) causes only additive marker effects, i.e., (a)A1 and (a)A2, but the dominance QTL effect (d) causes additive-by-additive and dominance-by-dominance marker interactions, i.e., (d)AA12 and (d)DD12, as well as dominance marker effects, i.e., (d)D1 and (d)D2. The genetic model used in Equations 1 and 3 is usually called the  model (Zeng et al. 2005). It has been proved that the use of other models such as the F2 model or the G2A model (Zeng et al. 2005) cannot eliminate the influence of the dominance effect on the interactions between markers either (results not shown).
model (Zeng et al. 2005). It has been proved that the use of other models such as the F2 model or the G2A model (Zeng et al. 2005) cannot eliminate the influence of the dominance effect on the interactions between markers either (results not shown).
In Equation 4, f1, f2, f3, g1, g2, g3, g4, and g5 defined in Table 1 (similar to Table 1 in Haley and Knott 1992) are functions of recombination frequencies and independent of QTL effects. Denote
 |
(5) |
The expectations of w and v under each marker class can be proved as
 |
(6) |
and
 |
(7) |
Equation 6 has been widely used in mapping QTL with additive effects regardless of the statistical method, e.g., regression analysis, maximum likelihood, or Bayesian models (for examples see Zeng 1994; Whittaker et al. 1996; Kao et al. 1999; Ball 2001; Sen and Churchill 2001; Sillanpää and Corander 2002; Yi et al. 2003; Bogdan et al. 2004; Feenstra et al. 2006; Li et al. 2007). However, we have not seen Equation 7 used in QTL mapping studies of F2 populations.
Using Equations 6 and 7, the genotypic value of an F2 individual with known marker class can be represented by marker variables and two-marker interactions as
 |
(8) |
where  , representing the mean of the four homozygous marker classes (i.e., AABB, AAbb, aaBB, and aabb in Table 1).
, representing the mean of the four homozygous marker classes (i.e., AABB, AAbb, aaBB, and aabb in Table 1).
For clarity, we added the symbols of QTL effects to various marker effects in Equations 3, 4, and 8. For example,  is the additional mean contributed by QTL dominance,
is the additional mean contributed by QTL dominance,  is the additive effect of the left marker caused by QTL additive effect,
is the additive effect of the left marker caused by QTL additive effect,  is the additive-by-additive effect between the left and right markers caused by QTL dominance effect, and so on. Model (8) is a completely fitted model, and coefficients in it contain all the information regarding QTL location and effects. In other words, the additive and dominance effects of the flanked QTL are completely absorbed by the six variables in model (8). The nonzero marker interactions (d)AA12 and (d)DD12, caused by the dominance effect, indicate that marker variables by themselves cannot completely absorb the effects of QTL located between the two markers.
is the additive-by-additive effect between the left and right markers caused by QTL dominance effect, and so on. Model (8) is a completely fitted model, and coefficients in it contain all the information regarding QTL location and effects. In other words, the additive and dominance effects of the flanked QTL are completely absorbed by the six variables in model (8). The nonzero marker interactions (d)AA12 and (d)DD12, caused by the dominance effect, indicate that marker variables by themselves cannot completely absorb the effects of QTL located between the two markers.
The inclusive linear model for multiple QTL:
For succinctness, we assume there are m QTL located in m intervals defined by m + 1 markers on one chromosome. The genotypic value of an F2 individual is defined as
 |
(9) |
where wj and vj are the indicators for genotypes at the jth QTL. By using Equations 6 and 7, the genotypic value of an F2 individual with known marker types can be reorganized as
 |
where
 |
and
 |
 |
Therefore, the inclusive linear model simultaneously containing all markers and phenotyping errors is
 |
(10) |
where P is the phenotypic value of the trait of interest, and  is the random environmental error.
is the random environmental error.
It can be seen that coefficients in model (10) are affected only by neighboring QTL. In other words, QTL effects will be completely absorbed by the six variables of the two closest markers. Model (10) is suitable for QTL mapping in F2 populations, as it completely explains both additive and dominance variations. In some studies, marker interactions were not included (for examples see Jiang and Zeng 1995; Kao et al. 1999; Jia and Xu 2007), which may bias the QTL mapping results and be problematic when extending to epistatic mapping.
ICIM in F2 populations:
Assume there are n individuals in an F2 population. Similar to QTL mapping for other populations (Li et al. 2007, 2008), we adopted a two-step mapping strategy. In the first step, stepwise regression was used to estimate the parameters in model (10). Coefficients of those variables not retained by stepwise regression were set at 0. However, we did not exclude the possibility that other model selection methods (Miller 1990; Piepho and Gauch 2001) may achieve similar or better performance in model selection than stepwise regression. In the second step, traditional interval mapping (Lander and Botstein 1989) was conducted on adjusted phenotypic values; i.e.,
 |
(11) |
where k and k + 1 represent the two flanking markers of the current testing position,  represents each F2 individual, and the circumflex means “estimated.” Under the condition of isolated QTL (Whittaker et al. 1996), adjusted values in Equation 11 contain all the location and effect information of QTL in the current interval, but at the same time, QTL in other chromosomal intervals have been completely controlled. At a testing position in the interval [k, k + 1], phenotypes of the three QTL genotypes QQ, Qq, and qq were assumed to be normally distributed as
represents each F2 individual, and the circumflex means “estimated.” Under the condition of isolated QTL (Whittaker et al. 1996), adjusted values in Equation 11 contain all the location and effect information of QTL in the current interval, but at the same time, QTL in other chromosomal intervals have been completely controlled. At a testing position in the interval [k, k + 1], phenotypes of the three QTL genotypes QQ, Qq, and qq were assumed to be normally distributed as  , where k = 1, 2, 3, representing the three QTL genotypes, respectively. The two hypotheses used to test the existence of QTL at the scanning position are
, where k = 1, 2, 3, representing the three QTL genotypes, respectively. The two hypotheses used to test the existence of QTL at the scanning position are
 |
vs.
 |
The logarithm likelihood under HA is, therefore,
 |
where  denotes individuals belonging to the
denotes individuals belonging to the  marker class (j = 1, 2, … , 9; Table 1),
marker class (j = 1, 2, … , 9; Table 1),  (k = 1, 2, 3) is the proportion of the
(k = 1, 2, 3) is the proportion of the  QTL genotype in the
QTL genotype in the  class, and
class, and  is the density function of the normal distribution
is the density function of the normal distribution  .
.
Most individuals in marker classes 1, 5, and 9 have QTL genotypes QQ, Qq, and qq, respectively. Hence, the initial parameters used in the EM algorithm (Dempster et al. 1977; Li et al. 2007) can be defined as
 |
 |
and
 |
In the E-step, the posterior probabilities of an individual belonging to the three QTL genotypes were calculated as
 |
where  . In the M-step, parameters in the maximum-likelihood equation were updated by
. In the M-step, parameters in the maximum-likelihood equation were updated by
 |
and
 |
The genetic effects in model (1) were therefore estimated by
 |
Under the null hypothesis, the three QTL genotypes follow the same normal distribution, denoted by  . Parameters under H0 were calculated as
. Parameters under H0 were calculated as
 |
from which the maximum likelihood under H0 and the LOD score between HA and H0 can be calculated. Additional hypotheses can be built to further test if the additive or the dominance effect is significant; this is not discussed in detail in this article.
QTL distribution models in simulation:
We considered six QTL with different levels of dominance and a genome consisting of eight chromosomes in our simulation studies (Table 2). Each chromosome is of 140 cM, with 15 evenly distributed codominant markers. QTL1 has additive effect 1, without a dominance effect. QTL2 has dominance effect 1, without an additive effect. QTL3 can be viewed as completely dominant, while QTL4 is completely recessive. Both QTL5 and QTL6 show overdominance, but in different directions. No interactions between QTL were considered. Each QTL was assumed to be located in the middle of a marker interval.
TABLE 2.
Six putative QTL and their distributions in a genome consisting of eight chromosomes, each of 140 cM and evenly distributed by 15 codominant markers
| Additive effect (a) | Dominance effect (d) | Model I
|
Model II
|
Model III
|
Genotypic variation explained (%) | Phenotypic variation explained (%) | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| QTL | Chr. | cM | Chr. | cM | Chr. | cM | ||||
| QTL1 | 1 | 0 | 1 | 25 | 1 | 25 | 1 | 25 | 11.4 | 8.0 |
| QTL2 | 0 | 1 | 2 | 55 | 1 | 55 | 2 | 55 | 5.7 | 4.0 |
| QTL3 | 1 | 1 | 3 | 25 | 2 | 25 | 3 | 25 | 17.1 | 12.0 |
| QTL4 | 1 | −1 | 4 | 55 | 2 | 55 | 1 | 55 | 17.1 | 12.0 |
| QTL5 | 1 | 1.5 | 5 | 25 | 3 | 25 | 2 | 25 | 24.3 | 17.0 |
| QTL6 | 1 | −1.5 | 6 | 55 | 3 | 55 | 3 | 55 | 24.3 | 17.0 |
Genotypic and phenotypic variances explained by individual QTL were calculated for QTL distribution model I. The genetic variance of each QTL in F2 is  , and heritability in the broad sense was set at 0.7. Chr., chromosome.
, and heritability in the broad sense was set at 0.7. Chr., chromosome.
To investigate the effect of linkage on QTL mapping, we considered three QTL distribution models (Table 2). QTL were distributed on different chromosomes in model I, and two QTL were linked on each of the first three chromosomes in models II and III. In model I, QTL5 and QTL6 each explained 24.3% of genotypic variation and 17.0% of the phenotypic variance under heritability 0.7. QTL2 explained the least genotypic and phenotypic variation among the six defined QTL (Table 2).
F2 mapping populations were simulated by the genetics and breeding simulation tool of QuLine, formerly called QuCim (Wang et al. 2003, 2004). ICIM was implemented by the software QTL IciMapping, and CIM was implemented by the software QTL Cartographer (Wang et al. 2005). For CIM, we applied “Model 6: Standard Model” and “3. Forward & Backward Method” available in Cartographer. The two probabilities for entering and removing variables were set at 0.01 and 0.02. For ICIM, the same probability levels were adopted in the first step of stepwise regression. The threshold LOD score was set at 3.0 for both methods.
One F2 population in rice:
The actual F2 population used in this study consists of 180 individuals and was derived by the Rice Research Institute, Sichuan Agricultural University (Ye et al. 2005, 2007). The cross was made in Chengdu, China, in July 2002 between the indica rice variety PA64s (full name: Pei'Ai 64s) and japonica rice variety Nipponbare. Nipponbare was completely sequenced in 2002, and PA64s was partially sequenced in the same year. The F1 population was planted in Hainan, China, in December 2002, and the F2 population was planted in Chengdu, China, in April 2003 for genotyping and phenotyping. A total of 137 SSR markers were screened for building the linkage map (Ye et al. 2005), and a number of agronomic traits were investigated in the field (Ye et al. 2005, 2007). The whole genome was of 2046.2 cM, and the average marker distance was 17.1 cM. Each of the 12 chromosomes had 6–12 relatively evenly distributed markers. We used ICIM for QTL mapping of plant height, where the two probabilities for entering and removing variables in the first step of stepwise regression were set at 0.01 and 0.02, and the threshold LOD score was set at 3.0.
RESULTS
Expected effects of the flanking markers:
The expected additive, dominance, additive-by-additive, and dominance-by-dominance effects of the two nearest flanking markers associated with each defined QTL in Table 2 were calculated from Equation 4 and are shown in Table 3. When the dominance effect is zero, i.e., QTL1 (a = 1 and d = 0), the two flanking markers have only additive effects, the size of which is dependent on the QTL additive effect and its location between the two markers. In cases where the QTL was located at the center of its flanking marker interval, both markers have the same additive effect, which approximates half of the QTL additive effect (Table 3). Therefore, when the dominance effect can be ignored, or the additive effect is the only genetic effect of interest, including one marker indicator for each marker locus will allow the QTL additive effect to be absorbed. This is the case of QTL mapping in populations consisting of two individual genotypes (Li et al. 2007), where the additive is the only genetic effect of interest.
TABLE 3.
Genetic effects of each QTL on its two flanking markers
| QTL |  |
 |
 |
 |
 |
 |
 |
Interaction variation (%) |
|---|---|---|---|---|---|---|---|---|
| QTL1 | 0.000 | 0.498 | 0.498 | 0.000 | 0.000 | 0.000 | 0.000 | 0.0 |
| QTL2 | 0.253 | 0.000 | 0.000 | 0.248 | 0.248 | −0.248 | 0.243 | 21.8 |
| QTL3 | 0.253 | 0.498 | 0.498 | 0.248 | 0.248 | −0.248 | 0.243 | 5.7 |
| QTL4 | −0.253 | 0.498 | 0.498 | −0.248 | −0.248 | 0.248 | −0.243 | 5.7 |
| QTL5 | 0.379 | 0.498 | 0.499 | 0.371 | 0.371 | −0.371 | 0.364 | 9.6 |
| QTL6 | −0.379 | 0.498 | 0.498 | −0.371 | −0.371 | 0.371 | −0.364 | 9.6 |
 is the additional mean caused by dominance,
is the additional mean caused by dominance,  and
and  are the additive effects of the left and right flanking markers,
are the additive effects of the left and right flanking markers,  and
and  are the dominance effects of the two flanking markers, and
are the dominance effects of the two flanking markers, and  and
and  are the additive-by-additive and dominance-by-dominance interaction between the two flanking markers. The last column represents the percentage of interaction variation under linkage equilibrium and allele frequency 0.5.
are the additive-by-additive and dominance-by-dominance interaction between the two flanking markers. The last column represents the percentage of interaction variation under linkage equilibrium and allele frequency 0.5.
When there is no additive effect, i.e., QTL2 (a = 0 and d = 1), the two flanking markers do not have additive effects either, but they do have additive-by-additive interaction (Table 3). Obviously, this interaction was caused by the dominance effect of QTL2 and did not indicate there were two interacting QTL. When both additive and dominance effects are present, i.e., QTL3–QTL6, additive, dominance, additive-by-additive, and dominance-by-dominance effects can all occur on the two flanking markers (Table 3). The dominance effect of a QTL causes not only marker dominance effects, but also marker interactions (Equation 4 and Table 3). Under linkage equilibrium and when each marker allele had a frequency of 0.5, ANOVA indicated that marker interactions caused by QTL2 explained >20% of the variation, those caused by QTL3 and QTL4 each explained >5% of the variation, and those caused by QTL5 and QTL6 each explained ∼10% of the variation between marker classes (Table 3).
The results from Equation 4 and Table 3 clearly indicated that the dominance of a QTL could complicate the coefficients of the two markers flanking a QTL by causing interactions between markers. We used the  model, i.e., Equation 3, to illustrate the phenomenon in this study. We have used other models such as the F2 or the G2A model (Zeng et al. 2005) and found they would neither eliminate the marker interactions caused by QTL dominance effect nor make the mapping procedure less complicated. The consequence of this phenomenon is that QTL mapping focusing on estimation of marker effects may lead to erroneous conclusions about QTL locations and effects.
model, i.e., Equation 3, to illustrate the phenomenon in this study. We have used other models such as the F2 or the G2A model (Zeng et al. 2005) and found they would neither eliminate the marker interactions caused by QTL dominance effect nor make the mapping procedure less complicated. The consequence of this phenomenon is that QTL mapping focusing on estimation of marker effects may lead to erroneous conclusions about QTL locations and effects.
Comparison of ICIM with CIM:
In Figure 1, each simulated QTL was assigned to a confidence interval of 15 cM centered at the true QTL location, and the power for the confidence interval was estimated. QTL identified in other intervals were viewed as false positives. In the confidence interval, if multiple peaks occurred, only the highest one was counted. In other chromosome regions, all peaks higher than the LOD threshold of 3.0 were counted, regardless of the distance between the significant peaks (Li et al. 2007). Under population size 200, both ICIM and CIM resulted in high powers (i.e., >0.60) for QTL3–QTL6 (Figure 1, A, C, and E). QTL1 and QTL2 explain the least genetic variation (Table 2) and their detection powers were relatively low. The difference in powers between ICIM and CIM is minor, except for QTL1 and QTL2 in models I and II and QTL1–QTL3 in model III (Figure 1, A and E). The distribution of QTL has effects on their detection powers (Figure 1, C and E).
Figure 1.—

Power analysis of CIM and ICIM from 100 simulations. (A) QTL distribution model I and population size 200; (B) QTL distribution model I and population size 500; (C) QTL distribution model II and population size 200; (D) QTL distribution model II and population size 500; (E) QTL distribution model III and population size 200; (F) QTL distribution model III and population size 500. The confidence interval for each predefined QTL was set at 15 cM, and the LOD threshold was 3.0. The last bar in each section represents the false discovery rate (FDR).
As expected, the increase in population size resulted in the increased detection power for both methods (Figure 1A vs. 1B, Figure 1C vs. 1D, and Figure 1E vs. 1F). Under population size 500, both CIM and ICIM had powers close to 1 in detecting all QTL (Figure 1, B, D, and F). The false discovery rate (FDR) is defined as the proportion of false positives to the total number of significant discoveries (Benjamini and Hochberg 1995). The FDR of ICIM was always lower than that of CIM (Figure 1). The increase in population size not only improved the detection power of ICIM, but also reduced its FDR. For CIM, the increases in population size improved its detection power, but did not reduce its FDR. As stated earlier, false positives were counted without considering a confidence interval; that is to say, any significant peaks that were not within the QTL confidence intervals were viewed as false positives, which resulted in a large number of false positives for both methods. In the other aspect, this may indicate that a higher LOD threshold should be applied when using CIM or ICIM.
In Figure 2, power was calculated for every marker interval on the genome, which allows monitoring QTL locations if not located in the predefined intervals. It can be clearly seen that false positives were around the true QTL positions and were less likely to be located in chromosomal regions far from the predefined QTL or in chromosomes where no QTL were located (Figure 2). There is an obvious tendency for significant peaks identified by ICIM for QTL distribution models II and III to be closer to the true QTL locations (Figure 2, C–F), indicating that ICIM is more capable of dissecting linked QTL.
Figure 2.—

Power analysis of every marker interval. (A) QTL distribution model I and population size 200; (B) QTL distribution model I and population size 500; (C) QTL distribution model II and population size 200; (D) QTL distribution model II and population size 500; (E) QTL distribution model III and population size 200; (F) QTL distribution model III and population size 500. The LOD threshold was set at 3.0. Powers were present for all marker intervals in eight chromosomes in A and B, but for marker intervals on the first four chromosomes in C–F.
Estimated QTL location and effects from QTL distribution model II are shown in Table 4. Unbiased estimations of QTL locations and additive effects were observed for ICIM and CIM under the two population sizes. The dominance effects estimated by ICIM were less biased than those estimated by CIM, indicating the advantage of using model (10) in ICIM. Taking population size 500 as an example, the dominance effects estimated by ICIM were 0.05, 0.98, 0.82, −0.87, 1.38, and −1.38, corresponding to the true effects 0, 1, 1, −1, 1.5, and −1.5, respectively. However, the effects estimated by CIM were 0.39, 1.01, 0.61, −0.64, 0.94, and −0.90, respectively. Considering the higher detection power, lower FDR, and less biased estimation of dominance effect, we can conclude that ICIM built on the inclusive linear model (10) is a better method for mapping QTL with additive and dominance in F2 populations. The LOD score from ICIM was always higher than that from CIM (Table 4), indicating the residual variation is better controlled in ICIM.
TABLE 4.
Estimated QTL location and additive and dominance effects from 100 simulations for QTL distribution model II
| Population size | Estimation | Method | QTL1 | QTL2 | QTL3 | QTL4 | QTL5 | QTL6 |
|---|---|---|---|---|---|---|---|---|
| 200 | LOD score | CIM | 6.20 | 5.66 | 7.76 | 7.41 | 8.45 | 8.71 |
| (2.14) | (2.26) | (3.80) | (3.82) | (3.55) | (4.07) | |||
| ICIM | 8.90 | 5.92 | 12.72 | 11.06 | 15.17 | 15.71 | ||
| (3.18) | (2.52) | (5.64) | (4.75) | (6.25) | (5.96) | |||
| Position (cM) | CIM | 25.95 | 54.42 | 25.01 | 54.99 | 24.42 | 55.34 | |
| (3.49) | (3.62) | (3.45) | (3.32) | (3.25) | (3.44) | |||
| ICIM | 25.95 | 54.60 | 24.69 | 55.15 | 24.70 | 55.03 | ||
| (3.92) | (3.42) | (3.39) | (3.71) | (3.31) | (3.05) | |||
| Additive effect | CIM | 1.02 | 0.37 | 1.21 | 1.18 | 1.12 | 1.17 | |
| (0.20) | (0.43) | (0.30) | (0.31) | (0.30) | (0.35) | |||
| ICIM | 0.96 | 0.05 | 1.03 | 0.94 | 0.93 | 0.97 | ||
| (0.18) | (0.18) | (0.37) | (0.33) | (0.40) | (0.42) | |||
| Dominance effect | CIM | 0.44 | 1.20 | 0.65 | −0.67 | 0.99 | −0.96 | |
| (0.31) | (0.25) | (0.34) | (0.26) | (0.30) | (0.32) | |||
| ICIM | 0.05 | 1.13 | 0.72 | −0.73 | 1.28 | −1.29 | ||
| (0.33) | (0.20) | (0.41) | (0.39) | (0.41) | (0.42) | |||
| 500 | LOD score | CIM | 13.19 | 8.64 | 15.11 | 13.93 | 19.63 | 18.71 |
| (4.35) | (3.97) | (7.12) | (6.72) | (8.16) | (7.78) | |||
| ICIM | 18.74 | 9.59 | 26.09 | 25.81 | 33.90 | 34.46 | ||
| (5.85) | (3.95) | (7.13) | (7.12) | (9.42) | (7.54) | |||
| Position (cM) | CIM | 25.31 | 54.30 | 24.81 | 55.31 | 25.08 | 54.67 | |
| (2.53) | (2.75) | (2.73) | (2.28) | (3.14) | (3.02) | |||
| ICIM | 24.69 | 54.67 | 24.66 | 55.24 | 25.10 | 54.80 | ||
| (2.63) | (2.54) | (2.20) | (2.42) | (1.32) | (1.56) | |||
| Additive effect | CIM | 0.96 | 0.24 | 1.12 | 1.07 | 1.13 | 1.13 | |
| (0.15) | (0.32) | (0.24) | (0.23) | (0.29) | (0.26) | |||
| ICIM | 0.96 | 0.04 | 0.99 | 0.98 | 0.95 | 0.98 | ||
| (0.16) | (0.10) | (0.16) | (0.20) | (0.20) | (0.19) | |||
| Dominance effect | CIM | 0.39 | 1.01 | 0.61 | −0.64 | 0.94 | −0.90 | |
| (0.17) | (0.18) | (0.19) | (0.20) | (0.24) | (0.21) | |||
| ICIM | 0.05 | 0.98 | 0.82 | −0.87 | 1.38 | −1.38 | ||
| (0.14) | (0.21) | (0.32) | (0.26) | (0.33) | (0.25) |
The numbers in parentheses are the standard errors.
Estimated QTL locations and effects from large simulated F2 populations:
To further illustrate the outcomes from ICIM, we conducted QTL mapping on the first simulated F2 populations with 500 individuals from the three QTL distribution models (Figures 3, A–C, and 4, A–F). The genotypic values of the two parents and their F1 hybrid were 15, 5, and 16, respectively, for the three QTL models. Phenotypic values in F2 for the three QTL distribution models show continuous distributions (Figure 3, A–C) that are similar to typical quantitative traits. There is no clear classification of the phenotype, and it is impossible to deduce the number of QTL without the assistance of molecular markers.
Figure 3.—

Phenotypic distributions of the three simulated and one actual F2 populations. (A) QTL distribution model I and population size 500; (B) QTL distribution model II and population size 500; (C) QTL distribution model III and population size 500; (D) rice F2 population derived from PA64s and Nipponbare and population size 180.
Figure 4.—

Mapping results from ICIM for the three simulated and one actual F2 populations. (A and B) The first simulated F2 population from QTL distribution model I and population size 500; (C and D) the first simulated F2 population from QTL distribution model II and population size 500; (E and F) the first simulated F2 population from QTL distribution model III and population size 500; (G and H) rice F2 population derived from PA64s and Nipponbare, population size 180. The scanning step was 1 cM.
QTL mapping by ICIM found the difference in genetic mechanism for the three seemingly similar phenotypic distributions in Figure 3, A–C. For QTL distribution model I, six clear peaks on the first six chromosomes can be seen along the one-dimensional LOD profile, indicating six unlinked QTL (Figure 4A). The chromosomes or chromosomal regions not harboring QTL have LOD scores close to 0 (Figure 4A). The six peaks were close to the true QTL position, and the effects at those positions are shown in Table 5. The estimated positions were at 28, 53, 24, 57, 26, and 55 cM, corresponding to the true positions 25, 55, 25, 55, 25, and 55 cM on the first six chromosomes. Along with scanning, the additive and dominance effects (Figure 4B) and variation explained by QTL at the testing positions can also be estimated. The estimated effects at peak positions were close to the true effects in Table 3, although some discrepancies were observed.
TABLE 5.
Estimated QTL location, additive effect, dominance effect, and variation from ICIM in three simulated and one actual F2 populations
| QTL | Flanking markers with positions (cM) in parentheses | Position (cM) | LOD score | Estimated additive effect | Estimated dominance effect | PVE (%) |
|---|---|---|---|---|---|---|
| QTL distribution model I | ||||||
| QTL1 | M3 (20), M4 (30) | 28 | 16.52 | 0.88 | −0.11 | 6.67 |
| QTL2 | M21 (50), M22 (60) | 53 | 7.67 | 0.03 | 0.85 | 3.27 |
| QTL3 | M33 (20), M34 (30) | 24 | 25.11 | 0.86 | 1.08 | 11.28 |
| QTL4 | M51 (50), M52 (60) | 57 | 35.46 | 0.74 | −1.58 | 16.43 |
| QTL5 | M63 (20), M64 (30) | 26 | 37.12 | 1.05 | 1.38 | 16.74 |
| QTL6 | M81 (50), M82 (60) | 55 | 28.44 | 0.84 | −1.22 | 13.16 |
| QTL distribution model II | ||||||
| QTL1 | M3 (20), M4 (30) | 21 | 9.67 | 0.66 | −0.16 | 2.82 |
| QTL2 | M6 (50), M7 (60) | 54 | 23.53 | 0.06 | 1.60 | 8.43 |
| QTL3 | M18 (20), M19 (30) | 26 | 40.49 | 1.18 | 1.30 | 14.35 |
| QTL4 | M21 (50), M22 (60) | 55 | 24.00 | 0.80 | −1.02 | 8.03 |
| QTL5 | M33 (20), M34 (30) | 24 | 35.93 | 1.02 | 1.35 | 12.82 |
| QTL6 | M36 (50), M37 (60) | 55 | 52.76 | 1.13 | −1.85 | 20.07 |
| QTL distribution model III | ||||||
| QTL1 | M3 (20), M4 (30) | 25 | 11.33 | 0.76 | 0.04 | 4.04 |
| QTL4 | M6 (50), M7 (60) | 53 | 34.54 | 1.22 | −0.85 | 13.59 |
| QTL5 | M18 (20), M19 (30) | 23 | 31.26 | 0.69 | 1.70 | 13.38 |
| QTL2 | M21 (50), M22 (60) | 56 | 11.67 | 0.09 | 1.17 | 4.90 |
| QTL3 | M33 (20), M34 (30) | 25 | 28.89 | 1.10 | 0.87 | 11.84 |
| QTL6 | M36 (50), M37 (60) | 55 | 26.72 | 0.91 | −1.26 | 11.04 |
| Plant height (cm) in rice | ||||||
| qPH1-1 | RM246 (94), RP2 (110) | 103 | 3.32 | 0.18 | −5.32 | 5.28 |
| qPH1-2 | RP82 (164), RP3 (188) | 181 | 14.87 | −9.25 | 1.97 | 29.99 |
| qPH3-1 | RM523 (17), RM251 (57) | 29 | 5.24 | 2.66 | 6.20 | 11.07 |
| qPH3-2 | RP242 (58), RM520 (72) | 67 | 4.96 | −3.90 | 2.89 | 7.80 |
| qPH4 | RM349 (46), RP68 (56) | 49 | 3.68 | −3.05 | −3.10 | 5.56 |
| qPH7-1 | RM82 (0), RM180 (23) | 3 | 3.98 | 0.11 | 5.58 | 5.50 |
| qPH7-2 | RM180 (23), RM119 (75) | 55 | 3.26 | −3.36 | 5.12 | 9.20 |
| qPH7-3 | RM118 (75), RM346 (132) | 95 | 3.30 | −3.95 | 5.72 | 11.75 |
PVE, percentage of variance explained.
For QTL distribution model II, six clear peaks, two each on the first three chromosomes, can be seen on the LOD profile (Figure 4C). The last five chromosomes do not have any QTL and have LOD scores close to 0. The estimated positions were at 21, 54, 26, 55, 24, and 55 cM, corresponding to the true positions 25, 55, 25, 55, 25, and 55 cM on the first three chromosomes. Some bias in estimated effects was observed (Table 5), especially the dominance effect of QTL2. Similar results from Figure 4, E and F, can be observed for QTL distribution model III.
QTL affecting plant height in rice:
The plant height of rice variety PA64s, a carrier of one major dwarfing gene, is 74.4 cm, while that of Nipponbare is 98.3 cm (Figure 3D). The distribution of plant height in their F2 populations is similar to those in Figure 3, A–C. There are a total of 24,660 (i.e., 180 × 137) marker points in the F2 population, 5131 belonging to the PA64s marker type, 6175 to the Nipponbare marker type, and 11,114 to the F1 marker type. A total of 2240 marker points were missing, representing 9.08% of total marker points. Segregation distortions were observed for a few markers as well. LOD scores, along with estimated additive and dominance effects along the rice genome, are shown in Figure 4, G and H. Obviously, the LOD profile in Figure 4G is more complicated than those in Figure 4, A, C, and E, indicating the genetic model with real data may be more complicated than those used in simulation. The other reasons for the rugged LOD profile may be the large amount of missing data and segregation distortions.
Under the LOD threshold of 3.0, eight QTL affecting plant height in the F2 population were identified: two each on chromosomes 1 and 3, one on chromosome 4, and three on chromosome 7 (Table 5). Locus qPH1-2, a major QTL explaining ∼30% of the phenotypic variation, has been detected by other methods (Ye et al. 2005). The PA64s allele at qPH1-2 can reduce plant height by ∼10 cm, and the dominance effect is relatively small.
Few F2 individuals are shorter than PA64s (Figure 3D), indicating most, if not all, reduced-height alleles are harbored by PA64s. However, many F2 individual plants are taller than the taller parent Nipponbare (Figure 3D), which may indicate the presence of overdominance. Five QTL have negative additive effects (Table 5), indicating the reduced-height alleles at these loci are also from PA64s. Overdominance effects were observed for qPH1-1, PH3-1, qPH7-1, qPH7-2, and qPH7-3, which explains the large number of F2 individuals that are taller than Nipponbare. For qPH1-1 and qPH7-1, the additive effects were close to 0, indicating that these loci will be less likely to be detected in other populations, such as recombination inbred lines, where heterozygosity is not present. So it is not unusual that different QTL are detected even when using the populations derived from the same parents.
DISCUSSION
Properties of the proposed inclusive linear model:
In an F2 population, all three genotypes at a locus are represented, which allows the estimation of additive effects and dominance deviations for individual QTL (Paterson et al. 1991). At the same time, the genetic analysis can be very complicated, as more genetic parameters have to be considered simultaneously. In this study, we proposed an inclusive linear model where the included marker variables can completely explain the additive and dominance effects of QTL. Model (10), built on solid genetic and statistical theories, is the theoretical basis for QTL mapping in F2 populations. It has the properties similar to those reported by Zeng (1994) for CIM, which are summarized as follows.
Property 1:
The QTL additive effect causes marker additive effects, while the QTL dominance effect causes marker dominance effects, as well as additive-by-additive and dominance-by-dominance interactions between the two flanking markers. By including two multiplication variables between flanking markers, the additive and dominance effects of one QTL can be completely absorbed. This property comes from Equations 6 and 7.
Property 2:
Assuming the additivity of QTL effects on a phenotypic trait, i.e., model (9), the expectation of the main marker effect in model (10), i.e.,  or
or  , depends only on those QTL located on two intervals where the current marker is involved. The expectation of marker interaction in model (10), i.e.,
, depends only on those QTL located on two intervals where the current marker is involved. The expectation of marker interaction in model (10), i.e.,  or
or  , depends only on the QTL located in the interval flanked by the two markers. This property can be seen from the deriving process of Equation 10. Thereby, under the condition of isolated QTL (Whittaker et al. 1996), the six coefficients of the jth marker interval, i.e.,
, depends only on the QTL located in the interval flanked by the two markers. This property can be seen from the deriving process of Equation 10. Thereby, under the condition of isolated QTL (Whittaker et al. 1996), the six coefficients of the jth marker interval, i.e.,  ,
,  ,
,  ,
,  ,
,  , and
, and  , contain and contain only the effect and location information of the QTL located in the interval.
, contain and contain only the effect and location information of the QTL located in the interval.
Property 3:
Under the condition of isolated QTL, adjusted phenotypic values by Equation 11 retain the effect and location information of the QTL located in the current interval; at the same time, QTL in other intervals and chromosomes have been controlled. Therefore, conditioning on both linked and unlinked markers in the second step of interval mapping reduces the sampling variance of the test statistic by controlling the residual genetic variation, thus increasing the power of QTL mapping.
Marker coefficients are biased in the first step of model selection using stepwise regression:
In the QTL and marker distribution model used in our simulation study, there were a total of 464 variables included in model (10), i.e., x1, x2,· · · , x120, y1, y2,· · · , y120, x1 × x2,· · · , x119 × x120, y1 × y2,· · · , and y119 × y120, where the multiplication of the last marker in a chromosome with the first marker in the next chromosome was excluded. When the largest P-value for entering variables and the smallest P-value for removing variables were set at 0.01 and 0.02, only a few of the six variables were picked up by stepwise regression. For QTL1 in distribution model I, only variable x4 was retained and its coefficient was estimated as 0.841. Without the second step of interval mapping, one could conclude that one additive QTL was located at 30 cM. For QTL1 in distribution models II and III, only variable x3 was retained and its coefficient was estimated as 0.648 and 0.713, respectively. Without the second step of interval mapping, one could conclude that one additive QTL was located at 20 cM. However, the second step of interval mapping found the largest LOD score was achieved at 21 cM for model II and at 25 cM for model III (Table 5), which are closer or the same to the true QTL position. For QTL5 and QTL6, the interaction coefficients were more important (Table 2). Under distribution model II, the four variables for QTL6 retained by stepwise regression were x36, x37, x36 × x37, and y36 × y37. But this does not mean there were two interacting QTL located at 50 and 60 cM on chromosome 3. Rather, the dominance effect of QTL6 caused interactions between the 36th and 37th markers.
Model (10) is a linear regression model, and the choice of variables is a typical model selection issue (Miller 1990). Treating QTL mapping as a model selection problem and the use of model selection criteria to identify the best model have been intensely investigated by many authors (Piepho and Gauch 2001; Broman and Speed 2002; Bogdan et al. 2004; Baierl et al. 2006). A number of statistical methods are available to search through the space of models, and various criteria can be used to select the best model (Miller 1990; Piepho and Gauch 2001). However, there is no general conclusion in statistics as to which model selection method is best (Miller 1990). In the first step of ICIM, we use stepwise regression for model selection. However, we do not exclude the possibility that other model selection methods may achieve similar or even better performance than the stepwise regression used in ICIM. If better model selection methods than stepwise regression are identified, they should be readily used in the first step of ICIM.
The second step of interval mapping is necessary in ICIM:
At first glance, the result of ICIM seems to depend on the identification of an appropriate regression model in the first step. However, the two-step approach we adopted in ICIM has the advantage that the best regression model in the first step does not need to be very close to the true model. Ideally, the second step of interval mapping can correct the imprecision of coefficient estimation in the first step. For all three QTL distribution models, a large bias has been observed between the true marker effects in Table 3 and the estimated marker effects. In addition, some nonrelevant variables were also selected by stepwise regression. Some were close to the markers flanking QTL, but some were not. For example, the marker for x53 in model I was close to the markers flanking QTL4, but the marker for y97 and the markers for x104 × x105 were on the seventh chromosome where no QTL was located. However, all biases were apparently corrected to some extent by the second step of interval mapping (Figure 4, A–F; Table 5), which indicated the necessity of fine tuning using interval mapping in the second step of ICIM.
In ICIM, the inference of QTL is not built on the estimated coefficients in model (10). Actually, model (10) is used to control background genetic variation in the second step of interval mapping. In this sense, the predictability of model (10) for the background genetic effects that can be used to adjust the phenotypic performance in Equation 11 becomes more important. In the regression theory, it is generally agreed that collinearity between regression variables in model (10) can seriously bias the estimation of their effects, but this undesirable bias does not extend to the model's fit (Miller 1990; Harrell 2001). In other words, collinearity does not affect predictions made on the same data set used to estimate the model parameters. This may have explained the advantage of using the two-step strategy in ICIM.
Acknowledgments
The authors thank Ping Li, Rice Research Institute, Sichuan Agricultural University, for providing the rice F2 population. Software for ICIM, called QTL IciMapping, is available from http://www.isbreeding.net, and the genetics and breeding simulation tool of Quline is available from http://www.uq.edu.au/lcafs/qugene/. This work was supported by the National 863 Programme of China (no. 2006AA10Z1B1), the Natural Science Foundation of China (no. 30771351), the Bill and Melinda Gates Foundation (drought tolerant maize for Africa), and the Generation Challenge Programme of the Consultative Group for International Agricultural Research.
References
- Baierl, A., M. Bogan, F. Frommlet and A. Futschik, 2006. On locating multiple interacting quantitative trait loci in intercross designs. Genetics 173 1693–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball, R. D., 2001. Bayesian methods for quantitative trait locus mapping based on model selection: approximate analysis using the Bayesian information criterion. Genetics 159 1351–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton, N. H., and P. D. Keightley, 2002. Understanding quantitative genetic variation. Nat. Rev. Genet. 3 11–21. [DOI] [PubMed] [Google Scholar]
- Benjamini, Y., and Y. Hochberg, 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 57 289–300. [Google Scholar]
- Bogdan, M., J. K. Ghosh and R. W. Doerge, 2004. Modifying the Schwarz Bayesian information criterion to locate multiple interacting quantitative trait loci. Genetics 167 989–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broman, K. W., and T. P. Speed, 2002. A model selection approach for the identification of quantitative trait loci in experimental crosses. J. R. Stat. Soc. Ser. B 64 641–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempster, A., N. Laird and D. Rubin, 1977. Maximum likelihood from incomplete data via the EM algorithm. J. R. Stat. Soc. Ser. B 39 1–38. [Google Scholar]
- Doerge, R. W., 2002. Mapping and analysis of quantitative trait loci in experimental populations. Nat. Rev. Genet. 3 43–52. [DOI] [PubMed] [Google Scholar]
- Feenstra, B., I. M. Skovgaard and K. W. Broman, 2006. Mapping quantitative trait loci by an extension of the Haley–Knott regression method using estimating equations. Genetics 173 2269–2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haley, C. S., and S. A. Knott, 1992. A simple regression method for mapping quantitative loci in line crosses using flanking markers. Heredity 69 315–324. [DOI] [PubMed] [Google Scholar]
- Haley, C. S., S. A. Knott and J.-M. Elsen, 1994. Mapping quantitative trait loci in crosses between outbred lines using least squares. Genetics 136 1195–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrell, F. E., 2001. Regression Modeling Strategies, With Applications to Linear Models, Logistic Regression, and Survival Analysis. Springer, New York.
- Hua, J., Y. Xing, W. Wu, C. Xu, X. Sun et al., 2003. Single-locus heterotic effects and dominance by dominance interactions can adequately explain the genetic basis of heterosis in an elite rice hybrid. Proc. Natl. Acad. Sci. USA 94 2574–2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia, Z., and S. Xu, 2007. Mapping quantitative trait loci for expression abundance. Genetics 176 611–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, C., and Z. Zeng, 1995. Multiple trait analysis of genetic mapping for quantitative trait loci. Genetics 140 1111–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao, C.-H., Z-B. Zeng and R. D. Teasdale, 1999. Multiple interval mapping for quantitative trait loci. Genetics 152 1203–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott, S. A., and C. S. Haley, 1992. Aspects of maximum likelihood methods for the mapping of quantitative trait loci in line crosses. Genet. Res. 60 139–151. [Google Scholar]
- Lander, E. S., and D. Botstein, 1989. Mapping Mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics 121 185–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H., G. Y. Ye and J. Wang, 2007. A modified algorithm for the improvement of composite interval mapping. Genetics 175 361–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H., J.-M. Ribaut, Z. Li and J. Wang, 2008. Inclusive composite interval mapping (ICIM) for digenic epistasis of quantitative traits in biparental populations. Theor. Appl. Genet. 116 243–260. [DOI] [PubMed] [Google Scholar]
- Lynch, M., and B. Walsh, 1998. Genetics and Analysis of Quantitative Traits. Sinauer Associates, Sunderland, MA.
- Mackay, T. F. C., 2001. Quantitative trait loci in Drosophila. Nat. Rev. Genet. 2 11–20. [DOI] [PubMed] [Google Scholar]
- Martinez, O., and R. N. Curnow, 1992. Estimating the locations and the sizes of the effects of quantitative trait loci using flanking markers. Theor. Appl. Genet. 85 480–488. [DOI] [PubMed] [Google Scholar]
- Miller, A. J., 1990. Subset Selection in Regression (Monographs on Statistics and Applied Probability, Vol. 40). Chapman & Hall, London.
- Paterson, A. H., S. Damon, J. D. Hewitt, D. Zamir, H. D. Rabinowitch et al., 1991. Mendelian factors underlying quantitative traitis in tomato: comparison across species, generations, and environments. Genetics 127 181–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Enciso, M., and L. Varona, 2000. Quantitative trait loci mapping in F2 crosses between outbred lines. Genetics 155 391–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piepho, H.-P., and H. G. Gauch, 2001. Marker pair selection for mapping quantitative trait loci. Genetics 157 433–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satagopan, J. M., B. S. Yandell, M. A. Newton and T. C. Osborn, 1996. A Bayesian approach to detect quantitative trait loci using Markov chain Monte Carlo. Genetics 144 805–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen, S., and G. A. Churchill, 2001. A statistical framework for quantitative trait mapping. Genetics 159 371–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sillanpää, M. J., and J. Corander, 2002. Model choice in gene mapping: what and why. Trends Genet. 18 302–307. [DOI] [PubMed] [Google Scholar]
- Wang, J., M. van Ginkel, D. Podlich, G. Ye, R. Trethowan et al., 2003. Comparison of two breeding strategies by computer simulation. Crop Sci. 43 1764–1773. [Google Scholar]
- Wang, J., M. Van Ginkel, R. Trethowan, G. Ye, I. Delacy et al., 2004. Simulating the effects of dominance and epistasis on selection response in the CIMMYT Wheat Breeding Program using QuCim. Crop Sci. 44 2006–2018. [Google Scholar]
- Wang, S., C. J. Basten and Z-B. Zeng, 2005. Windows QTL Cartographer 2.5. North Carolina State University, Raleigh, NC.
- Whittaker, J. C., R. Thompson and P. M. Visscher, 1996. On the mapping of QTL by regression of phenotype on marker-type. Heredity 77 23–32. [Google Scholar]
- Wright, A. J., and R. P. Mowers, 1994. Multiple regression for molecular-marker, quantitative trait data from large F2 populations. Theor. Appl. Genet. 89 305–312. [DOI] [PubMed] [Google Scholar]
- Ye, S.-P., Q.-J. Zhang, J.-Q. Li, B. Zhao and P. Li, 2005. QTL mapping for yield component traits using (Pei'ai 64s/Nipponbare) F2 population. Acta Agron. Sin. 31 1620–1627 (in Chinese with English abstract). [Google Scholar]
- Ye, S.-P., Q.-J. Zhang, J.-Q. Li, B. Zhao, D.-S. Yin et al., 2007. Mapping of quantitative trait loci for six agronomic traits of rice in Pei'ai 64s/Nipponbare F2 population. Chin. J. Rice Sci. 21 39–43 (in Chinese with English abstract). [Google Scholar]
- Yi, N., V. George and D. B. Allison, 2003. Stochastic search variable selection for identifying multiple quantitative trait loci. Genetics 164 1129–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, S. B., J. X. Li, C. G. Xu, Y. F. Tan, Y. J. Gao et al., 1997. Importance of epistasis as the genetic basis of heterosis in an elite rice hybrid. Proc. Natl. Acad. Sci. USA 94 9226–9231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng, Z-B., 1994. Precision mapping of quantitative trait loci. Genetics 136 1457–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng, Z-B., T. Wang and W. Zou, 2005. Modeling quantitative trait loci and interpretation of models. Genetics 169 1711–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]