

Abstract
Identifying loci under natural selection from genomic surveys is of great interest in different research areas. Commonly used methods to separate neutral effects from adaptive effects are based on locus-specific population differentiation coefficients to identify outliers. Here we extend such an approach to estimate directly the probability that each locus is subject to selection using a Bayesian method. We also extend it to allow the use of dominant markers like AFLPs. It has been shown that this model is robust to complex demographic scenarios for neutral genetic differentiation. Here we show that the inclusion of isolated populations that underwent a strong bottleneck can lead to a high rate of false positives. Nevertheless, we demonstrate that it is possible to avoid them by carefully choosing the populations that should be included in the analysis. We analyze two previously published data sets: a human data set of codominant markers and a Littorina saxatilis data set of dominant markers. We also perform a detailed sensitivity study to compare the power of the method using amplified fragment length polymorphism (AFLP), SNP, and microsatellite markers. The method has been implemented in a new software available at our website (http://www-leca.ujf-grenoble.fr/logiciels.htm).
ONE of the main challenges of modern biology is to dissect and understand the molecular basis for naturally occurring genetic variation. Recent advances in the fields of computational biology and molecular biology techniques have led to the emerging field of “population genomics,” whose main objective is to characterize the parts of the genome subject to natural selection. This new discipline has important applications in many domains such as medical genetics and the improvement of agricultural crops and breeds. Additionally, ignoring the effect of natural selection in evolutionary studies can lead to wrong estimates of the demographic history of species. Therefore, separating the effect of neutral drift and adaptive genetic differentiation is a necessary preliminary step in most analyses of genomewide data sets, and this distinction can also help us to understand speciation processes.
A wide variety of methods have been developed to identify regions of the genome that have been subject to natural selection (see Nielsen et al. 2005, for a review). Among them, we can distinguish those based on comparative data (taken from different species) that can detect old signatures of selection and those using population genomics data that allow the detection of more recent ones. This latter family of methods has became very popular in the last decade and has been applied to many nonmodel species (see Wilding et al. 2001, for example).
Many of the existing methods for detecting recent selection from population genomics data are based on an idea first introduced by Lewontin and Krakauer (1973) (see, for example, Bowcock et al. 1991; Beaumont and Nichols 1996; Vitalis et al. 2001; Beaumont and Balding 2004). The basic rationale is that loci influenced by directional (also called adaptive or positive) selection will show a larger genetic differentiation than neutral loci, and loci that have been subject to balancing (also called negative or purifying) selection will show a lower genetic differentiation. Thus, the methods generally consist of identifying loci that present FST coefficients that are “significantly” different from those expected under the neutral theory (they are called outlier loci).
Lewontin and Krakauer's (1973) method has raised many criticisms (see Beaumont 2005, for more details about them) and finally fell out of use. More recently, Bowcock et al. (1991) and Beaumont and Balding (2004) showed that problems of a purely statistical nature can be easily solved. In particular, the problem related to the correlation of allele frequencies among demes can be overcome by adopting a Bayesian approach that implements the multinomial-Dirichlet likelihood, which arises in a wide variety of neutral population genetic models (see Balding 2003). One of the scenarios covered consists of an island model (Wright 1931) in which subpopulation allele frequencies are correlated through a common migrant gene pool from which they differ in varying degrees. The difference in allele frequency between this common gene pool and each subpopulation is measured by a subpopulation-specific FST. Therefore, this formulation can consider more realistic ecological scenarios where the effective size and the immigration rate may differ among subpopulations. Additionally, a previous study (Foll and Gaggiotti 2006) has shown that statistical methods based on this approach are robust to deviations from the underlying demographic model.
As opposed to the multinomial-Dirichlet approach, most existing methods to detect outlier loci are based on simpler demographic models. More precisely, Beaumont and Nichols (1996) used a model with an infinite number of islands, all of which have equal sizes and exchange migrants at the same rate. A violation of this model can lead to a high false-positive rate and, in particular, it requires the consideration of a large number of subpopulations (Flint et al. 1999). The mutation rate, the mutation model, and the demographic history may also have a large effect on the distribution of FST, especially if heterozygosities are high (see the manual of the FDist2 program implementing this method). More recently, Vitalis et al. (2001) proposed an alternative model to obtain the expected distribution of FST under the neutral scenario. For this purpose, they use coalescence simulations of pairs of haploid populations that do not exchange migrants and are of constant size, having diverged from an ancestral population that may have experienced a bottleneck before splitting. This method has the disadvantage of considering a model consisting of a single pair of populations and, therefore, the authors recommend to identify a locus as an outlier only if it is identified as such in all or most of the pairwise comparisons that include a particular population (Vitalis et al. 2001). The problem with this approach is that the pairs of populations analyzed are not independent and it is impossible to define rigorous P-values in this case. For this reason, this method is suitable to detect only extreme cases of natural selection. Both of the methods discussed above share an additional drawback: the expected distribution of FST is obtained by simulating a large number of neutral data sets using as input parameters the estimates obtained from the real data set. The problem is that, ideally, input parameters should be based only on neutral markers, and, therefore, the presence of selected loci in the data set can lead to biases. Ad hoc procedures can be used to reduce this problem but they are fairly subjective.
Instead of focusing on FST's as a means of detecting outliers, it is also possible to consider the heterozygosity or some other measure of genetic diversity. Schlotterer (2002) and Kauer et al. (2003) proposed such an approach that is suitable for microsatellites markers because it assumes a strict stepwise mutation model (SMM). They follow Vitalis et al. (2001) and consider statistics based on pairs of populations. More precisely, they proposed to use the ratio R of the genetic diversity θ = 4Neμ of two populations, where Ne is the effective population size and μ is the mutation rate. θ is estimated either from the variance V in repeat number of microsatellites or from the expected heterozygosity H, leading respectively to the so-called ln RV and ln RH statistics. Instead of using coalescence simulations to generate the expected distribution under neutrality, Schlotterer (2002) uses the empirical distribution of ln RV and ln RH statistics and identifies as outliers those loci that fall outside the 95% confidence interval, under the assumption that the distribution is normal. The drawback of this method is that the use of an empirical distribution necessarily leads to a high false-positive rate. In addition, this approach is also primarily designed for pairs of populations and is suitable to detect only extreme cases of natural selection.
As mentioned before, the more recent method proposed by Beaumont and Balding (2004) is based on the multinomial-Dirichlet likelihood and considers that FST values integrate effects that are specific to each population and to each locus. Thus, a locus is deemed to be under selection if the equal-tailed 100(1 – P)% posterior interval for its locus-specific effect excludes zero. Although this method avoids all the above-described problems it still has the drawback of not providing a rigorous way of testing the hypothesis that a locus is subject to selection. To address this problem, Riebler et al. (2008) extended the method by introducing the use of a Bernoulli-distributed auxiliary variable, δi, to indicate whether or not a locus is subject to selection. Then, they classify a locus i as being under selection if the posterior probability P(δi = 1 | data) is larger than a threshold value that is set by means of a simulation study. Although this may represent an improvement over the original method, it still requires the use of a simulation study. The authors propose to use a cutoff value of 0.17 on the basis of their simulations but it is unlikely that this value will be appropriate for all data sets.
Here we propose a different and more rigorous approach to develop a test for selection. More specifically, we directly estimate the posterior probability of a given locus being under the effect of selection by defining two alternative models, one that includes the effect of selection and another that excludes it; we then estimate their respective posterior probabilities using a reversible-jump MCMC approach. Additionally, we address a common limitation of all the methods described above (except that of Beaumont and Nichols 1996) that can be used only with codominant markers. More specifically, we generalize the method of Beaumont and Balding (2004) by making it applicable to dominant markers like amplified fragment length polymorphisms (AFLPs) and perform a detailed sensitivity study to compare the power of the method using AFLP, SNP, and microsatellite markers.
Another issue that has not been considered in the past is the extent to which the demographic history of species and differences in mutation rates among loci can bias the detection of selection using genome scans. To address this issue we present the results of a simulation study that considers the effect of spatial expansions such as that undergone by humans. Additionally, we study the effect of not discriminating between di-, tri-, and tetranucleotide microsatellites when carrying out genome scans. Finally, we illustrate how the method can be applied to study particular cases by analyzing published data sets on humans and periwinkles.
METHODS
Bayesian model for estimation of locus–population-specific FST coefficients:
The model for genetic differentiation used is based on ideas first introduced by Balding and Nichols (1995) and that Beaumont and Balding (2004) later used to detect loci under natural selection. Strictly speaking, the approach applies to an island model (Wright 1931) but it has also been used to describe a fission model (Falush et al. 2003). For the sake of simplicity we describe the details of our approach using the terminology of this latter model. We consider a collection of J subpopulations that evolved in isolation after splitting from an ancestral population. The derived subpopulations may have been subject to different amounts of genetic drift and, therefore, their allele frequencies will show different degrees of differentiation from the ancestral allele frequency. We consider a set of I loci and let Ki be the number of alleles at the ith locus. The extent of differentiation at locus i between subpopulation j and the ancestral population is measured by 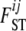 and is the result of its demographic history. Let pi = {pik} denote the allele frequencies of the ancestral population at locus i, where pik is the frequency of the allele k at locus
and is the result of its demographic history. Let pi = {pik} denote the allele frequencies of the ancestral population at locus i, where pik is the frequency of the allele k at locus 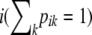 . We use p = {pi} to denote the entire set of allele frequencies of the ancestral population and
. We use p = {pi} to denote the entire set of allele frequencies of the ancestral population and 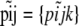 to denote the current allele frequencies at locus i for subpopulation j. Under these assumptions, the allele frequencies at locus i in subpopulation j follow a Dirichlet distribution with parameters
to denote the current allele frequencies at locus i for subpopulation j. Under these assumptions, the allele frequencies at locus i in subpopulation j follow a Dirichlet distribution with parameters 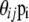 ,
,
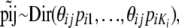 |
(1) |
where 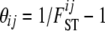 . The parameters
. The parameters 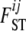 are very closely related to Wright's (1951) FST parameter and are interpreted as measures of the shared ancestry within each of the subpopulations (see Balding 2003, for a more detailed explanation). The full prior distribution can be obtained by multiplying across loci and populations:
are very closely related to Wright's (1951) FST parameter and are interpreted as measures of the shared ancestry within each of the subpopulations (see Balding 2003, for a more detailed explanation). The full prior distribution can be obtained by multiplying across loci and populations:
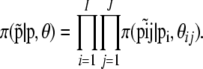 |
(2) |
Hierarchical model for locus- and population-specific effects:
The amount of data available to estimate all locus–population-specific FST coefficients is reduced and this leads to inaccurate estimates, especially for loci with a small number of different alleles. As an alternative, Balding et al. (1996) proposed to decompose locus–population-specific FST coefficients into a population-specific component, βj, shared by all loci and a locus-specific component, αi, shared by all populations. We use the model proposed by Beaumont and Balding (2004) that is based on the following equation:
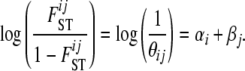 |
(3) |
The advantage of this formulation is that instead of estimating I · J 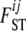 coefficients, we have to estimate only the I parameters αi and the J parameters βj. In the case of absence of natural selection, all αi coefficients are excluded, and the above model is equivalent to the one used by Foll and Gaggiotti (2006), where the term βj is replaced by a generalized linear model. Note that with this formulation,
coefficients, we have to estimate only the I parameters αi and the J parameters βj. In the case of absence of natural selection, all αi coefficients are excluded, and the above model is equivalent to the one used by Foll and Gaggiotti (2006), where the term βj is replaced by a generalized linear model. Note that with this formulation, 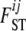 and equivalently θij are no longer model parameters that need to be estimated because they are replaced by αi and βj parameters. In what follows we use θij for the sake of simplicity but note that it can be replaced directly by θij = exp(– (αi + βj)).
and equivalently θij are no longer model parameters that need to be estimated because they are replaced by αi and βj parameters. In what follows we use θij for the sake of simplicity but note that it can be replaced directly by θij = exp(– (αi + βj)).
Beaumont and Balding (2004) originally proposed to include a locus–population parameter γij in their formulation. However, they noted that the posterior probability for this parameter was very similar to the prior used. This indicates that there is not enough information to estimate it and, therefore, we chose here to exclude the γij's from the model.
Estimating the probability that a locus is influenced by selection:
To infer which loci are influenced by selection we focus on the posterior distribution of αi: a positive value suggests that the locus i is subject to directional selection, whereas a negative value suggests balancing selection. However, before deciding on the type of selection we need to decide whether or not there is selection at all. In their original formulation, Beaumont and Balding (2004) focused on the posterior distribution of αi and from this they identified the locus influenced by selection using an approximate method (see below). Here we present a rigorous way of estimating the posterior probability of a given locus being under the effect of selection. Equation 3 can lead to two alternative models, one that includes both effects and another one that does not include the effect of selection. Thus, we use a reversible-jump MCMC algorithm (Green 1995) to estimate the posterior probability of each one of these models. At each iteration of the MCMC algorithm, we propose to remove αi from the model if it is currently present or to add it if it is not included; this is done separately for each locus i. For example, if we propose to add αi to the vector 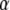 of locus effects, we draw a proposed value from a distribution q. Then we accept to add this locus in the model with probability min(1, A), where
of locus effects, we draw a proposed value from a distribution q. Then we accept to add this locus in the model with probability min(1, A), where
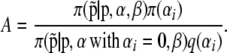 |
Because we only have two models and we choose them uniformly, the ratio of prior model probability simplifies to one. The Jacobian is one because of the canonical jump function used. To consider the reverse move, we simply accept the move deleting αi with probability min(1, 1/A). The proposal distribution q is a normal distribution with its mean and variance pilot tuned to improve convergence (see below).
With this method, we have posterior estimates of the probability that a locus is subject to selection: P(αi included) corresponds to P(αi ≠ 0). This probability is estimated directly from the output of the MCMC by simply by counting the number of times αi is included in the model.
Estimating allele frequencies:
Beaumont and Balding's (2004) original formulation considered codominant markers; here we extend it to dominant ones. Note that we have to estimate the allele frequency of each subpopulation and that of the ancestral population because they are unknowns. Thus, we present two different formulations depending of the type of marker used: codominant (like microsatellites or SNPs) and dominant (like RFLPs or AFLPs).
Codominant markers:
The data consist of allele counts obtained from samples of size nij. We use aijk to denote the number of alleles k observed at locus i in the sample from subpopulation j. Thus, 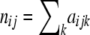 . The full data set can be presented as a matrix
. The full data set can be presented as a matrix 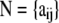 , where
, where 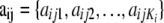 is the allele count at locus i for subpopulation j. The observed allele frequencies,
is the allele count at locus i for subpopulation j. The observed allele frequencies, 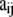 , can be considered as sampled from the true alleles frequencies
, can be considered as sampled from the true alleles frequencies 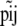 and, therefore, can be described by the multinomial distribution (Holsinger 1999):
and, therefore, can be described by the multinomial distribution (Holsinger 1999):
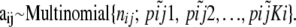 |
(4) |
In principle, we could use as likelihood the multinomial distribution (Equation 4) and consider Equation 1 as a Bayesian prior. However, in our case, we can calculate exactly the marginal distribution of 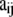 because the Dirichlet distribution is the conjugate prior of the multinomial. This allows us to eliminate the nuisance parameters
because the Dirichlet distribution is the conjugate prior of the multinomial. This allows us to eliminate the nuisance parameters 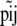 that are not of immediate interest but are needed by the model. Thus, we obtain the multinomial-Dirichlet distribution:
that are not of immediate interest but are needed by the model. Thus, we obtain the multinomial-Dirichlet distribution:
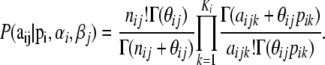 |
The likelihood is obtained by multiplying across all loci and populations:
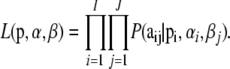 |
(5) |
Since the allele frequencies in the ancestral population are unknown, we have to estimate them by introducing a noninformative Dirichlet prior, 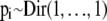 , into our Bayesian model.
, into our Bayesian model.
Dominant markers:
Estimating allele frequencies from dominant markers is more difficult because of the inability to distinguish heterozygous individuals from those that are homozygous for the dominant allele. Nevertheless, they have became very popular in the last decade, mostly due to the development of the AFLP marker, an inexpensive and easy way of obtaining large number of genetic markers from a wide variety of organisms (Bensch and Akesson 2005; Meudt and Clarke 2007). For each individual the information is “band presence” or “band absence,” which can be viewed as a phenotype. One possible solution is to suppose Hardy–Weinberg equilibrium to estimate allele frequencies but this imposes the strong hypothesis of no inbreeding. Holsinger et al. (2002) first proposed a general method that includes the estimation of the inbreeding coefficient FIS.
In the context of dominant markers, the data N consist of the sample counts of observed phenotypes instead of allele counts. Let n[A1],ij and n[A2],ij be the observed number of phenotypes [A1] and [A2] at locus i for population j. The full data set is presented as a matrix 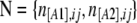 and the sample size at locus i for population j is nij = n[A1],ij + n[A2],ij. We can consider that the number of phenotypes n[A1],ij follows a binomial distribution with parameters g[A1],ij and nij, where g[A1],ij is the unknown [A1] phenotype frequency at locus i in population j:
and the sample size at locus i for population j is nij = n[A1],ij + n[A2],ij. We can consider that the number of phenotypes n[A1],ij follows a binomial distribution with parameters g[A1],ij and nij, where g[A1],ij is the unknown [A1] phenotype frequency at locus i in population j:
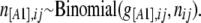 |
(6) |
Note that the binomial distribution is a particular case of the multinomial distribution with only two alleles, and the Dirichlet distribution of Equation 1 reduces to a beta distribution. The beta distribution is the conjugate prior of the binomial distribution, but contrary to the case of codominant markers with the multinomial-Dirichlet distribution, we cannot calculate exactly the marginal distribution. This is due to the fact that, in the case of dominant markers, the parameters of the binomial distribution are the phenotype frequencies instead of the allele frequencies. If we assume independence we can multiply across loci and populations to obtain the likelihood function
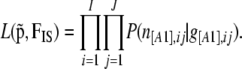 |
The phenotype frequency g[A1],ij can be linked to the corresponding frequency pij of allele A1 and the inbreeding coefficient 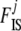 of population j using the following equations:
of population j using the following equations:
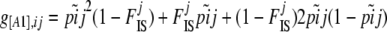 |
(7) |
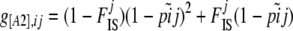 |
(8) |
 |
(9) |
However, Foll et al. (2008) show that estimates obtained from this model are strongly influenced by the ascertainment bias of AFLPs. They proposed an alternative approximate Bayesian computation (ABC) approach that gives unbiased estimates of population-specific FST and FIS coefficients. This solution leads to more uncertainty on posterior distributions, which precludes the estimation of locus-specific αi's. Additionally, the ABC algorithm cannot be used to estimate the posterior probability of each hypothesis of the form αi = 0. Because here values of FIS are not of immediate interest, we propose an intermediate solution: we do not estimate FIS coefficients but we incorporate the full uncertainty on FIS by letting it move freely between 0 and 1 during the MCMC process. This approach has also been proposed for the software Hickory, implementing the method of Holsinger et al. (2002), and is described in the online manual (Holsinger and Lewis 2002). Of course if some other source of information suggests that inbreeding can be bounded within a narrower interval it is possible to restrict it to reduce uncertainty on parameter estimates. We use the prior on ancestral allele frequencies proposed by Foll et al. (2008): 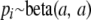 . The parameter a describes the shape of allele frequencies in the ancestral population (Wright 1931) and is estimated using a log-normal positive prior:
. The parameter a describes the shape of allele frequencies in the ancestral population (Wright 1931) and is estimated using a log-normal positive prior:  .
.
Implementation:
For codominant markers, the full Bayesian model represented by the directed acyclic graph (DAG) in Figure 1A is given by
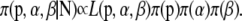 |
(10) |
Figure 1.—

DAG of the models given in Equation 10 (A) and Equation 11 (B). The square node denotes known quantity (i.e., data) and circles represent parameters to be estimated. Lines between nodes represent direct stochastic relationships within the model. The variables within each node correspond to the different model parameters discussed in the text. N is the genetic data, that is, allele-frequency counts for codominant markers or phenotype-frequency counts for dominant data. 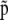 and p are, respectively, the allele frequencies in each local population and in the ancestral population.
and p are, respectively, the allele frequencies in each local population and in the ancestral population. 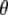 is the vector of the genetic differentiation coefficient for each local population.
is the vector of the genetic differentiation coefficient for each local population. 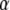 and
and 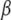 are, respectively, the vectors of locus- and population-specific effects of the genetic differentiation. The vector
are, respectively, the vectors of locus- and population-specific effects of the genetic differentiation. The vector 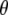 is represented within a dashed circle because it is not actually a parameter of the model: it can be calculated directly from Equation 3, but we represent it for a better understanding of the diagram.
is represented within a dashed circle because it is not actually a parameter of the model: it can be calculated directly from Equation 3, but we represent it for a better understanding of the diagram.  is the vector of inbreeding coefficients and a is the hyperprior determining the shape of the ancestral allele frequencies.
is the vector of inbreeding coefficients and a is the hyperprior determining the shape of the ancestral allele frequencies.
For dominant markers, the full Bayesian model represented by the DAG in Figure 1B is given by
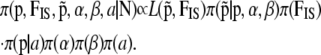 |
(11) |
Following Beaumont and Balding (2004), for the population effects βj, we used a Gaussian prior with mean −2 and standard deviation 1.8; for the locus effects, αi, we used a Gaussian prior with a zero mean and a standard deviation of 1. As explained above, 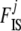 's are not estimated during the MCMC algorithm but are used to incorporate the uncertainty on inbreeding in the model with dominant markers.
's are not estimated during the MCMC algorithm but are used to incorporate the uncertainty on inbreeding in the model with dominant markers.
The estimation of model parameters is carried out using a combination of MCMC and reversible-jump (RJ)MCMC (Green 1995) techniques that are described in the supplemental information. We evaluated the convergence of the method using the diagnostic tests implemented in the R BOA package (Smith 2005). The tests indicated that a burn-in of 10,000 iterations was enough to attain convergence and it has been implemented as part of the pilot-tuning process (see below). We used a sample size of 10,000 and a thinning interval of 50 as suggested by an autocorrelation analysis. With these parameter values, the total length of the chain was 500,000 iterations. The method has been implemented in a software written in C++. We provide a command line version for Linux and a version with a friendly user graphical interface for Microsoft Windows.
Proposal distributions have to be adjusted to have acceptance rates between 0.25 and 0.45. If we propose values in a very wide interval, most moves will be rejected because they will correspond to areas of low posterior probability. On the other hand, if we propose values very close to the current one, the move will be almost always accepted but the chain will take a long time to explore all the parameter space. These values are automatically tuned on the basis of short pilot runs: we run 2000 iterations, and for each parameter the proposal is adjusted to reduce or increase the acceptance rate. We make 10 such pilot runs before starting the sampling, which also play the role of a burn-in period. At the same time, we can choose the proposal distribution q for the reversible jump. Brooks et al. (2003) showed that the best choice is to take 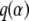 to be the full conditional distribution of
to be the full conditional distribution of 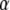 in the saturated model. Because we do not know this distribution, we use the pilot run to get rough estimates of the mean mi and the variance vi for all αi under the saturated model (in which all parameters αi are included). Then we propose a new value for αi from
in the saturated model. Because we do not know this distribution, we use the pilot run to get rough estimates of the mean mi and the variance vi for all αi under the saturated model (in which all parameters αi are included). Then we propose a new value for αi from 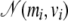 that is generally close to the full conditional distribution.
that is generally close to the full conditional distribution.
SIMULATION STUDY
We investigated the performance of our method under different scenarios using a simulation study and also compared its performance with that of Beaumont and Balding's (2004) approach. This latter approach has already been shown to perform better in various scenarios than the previous approaches based on the same idea (Beaumont and Balding 2004).
Our first simulation approach uses the same statistical model assumed by our method (the inference model) and allows us to study the effect of three critical parameters of the model in the identification of selection: the sample sizes, the number of populations, and the level of genetic differentiation. We also use this simulation scheme to compare the power of three different types of markers: AFLPs, SNPs, and microsatellites. The first marker is a dominant marker while the two others are codominant.
We also used a second simulation approach to investigate the effect of departures from the demographic model assumed by our method. For this purpose we generated neutral marker data sets under a population expansion model that assumes a stepping-stone colonization process (SPLATCHE, Currat et al. 2004). This allows us to investigate if the confounding effect of selection and demographic history can lead the method to identify as selected loci that are in fact neutral (false-positive detection of selection).
Basic simulation design:
Our initial simulation scheme assumes 1000 loci of which 100 are under directional selection and 100 are under balancing selection. Later on we also considered cases where only 100 loci are influenced by selection (50 balancing and 50 directional). We introduced selection using α = 2 and α = −2 for directional and balancing selection, respectively. To have an idea of the strength of natural selection implied by these values, it is necessary to consider the extent to which a value of FST for a neutral marker (background FST) is increased when a value of 2 is used for αi in Equation 3. Figure 2 shows the effect of αi when the background FST is 0.1. Additionally, Figure 2 in Beaumont and Balding (2004) shows FST values for three different selection coefficients (s = 0.02, 0.05, 0.1) and for the same background FST. From these figures it is possible to obtain Table 1, which relates the selection coefficient with the αi-values. Table 1 shows that an αi = 2 is equivalent to an s slightly >0.05.
Figure 2.—

Influence of the α-coefficient on FST for a background 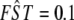 . On the basis of Equation 3 we calculate the FST coefficient that a locus under selection with a given αi would have. For this we first obtain the value of the population-specific effect for a chosen background FST from
. On the basis of Equation 3 we calculate the FST coefficient that a locus under selection with a given αi would have. For this we first obtain the value of the population-specific effect for a chosen background FST from 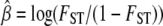 and then obtain the corresponding value under selection using
and then obtain the corresponding value under selection using 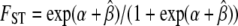 . For example, if an initially neutral marker exhibiting an
. For example, if an initially neutral marker exhibiting an 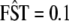 is subject to selection with α = 2, then we expect that its FST will increase to 0.45 once a new equilibrium is reached. Dashed lines connect the FST values given in Table 1 with the corresponding αi-values.
is subject to selection with α = 2, then we expect that its FST will increase to 0.45 once a new equilibrium is reached. Dashed lines connect the FST values given in Table 1 with the corresponding αi-values.
TABLE 1.
Equivalence between αi and selection coefficient
| s | FST range | Mean | α |
|---|---|---|---|
| 0.02 | [0.1; 0.4] | 0.25 | 1.12 |
| 0.05 | [0.2; 0.6] | 0.4 | 1.8 |
| 0.1 | [0.5; 0.8] | 0.65 | 2.8 |
The relationship between the selection coefficients, s, and locus-specific effects αi for a background FST of 0.1 is shown. The second and third columns show the FST's of the loci under selection and were obtained from Figure 2 in Beaumont and Balding (2004). The fourth column shows the α-values for a given FST under selection and is obtained from Figure 2.
To investigate the performance of the method under different scenarios, we considered a default set of values for parameters that were common to both codominant and dominant markers and then changed values of one parameter at a time. This procedure led to 10 different data sets that are described in Table 2. The default values were six populations, a sample size of 30 individuals per population, and an FST coefficient of 0.10.
TABLE 2.
Simulation parameters
| Name | Populations | FST | Sample size |
|---|---|---|---|
| Pop-2 | 2 | 0.15 | 30 |
| Pop-6a | 6 | 0.15 | 30 |
| Pop-10 | 10 | 0.15 | 30 |
| Pop-20 | 20 | 0.15 | 30 |
| Fst-0.05 | 6 | 0.05 | 30 |
| Fst-0.10 | 6 | 0.1 | 30 |
| Fst-0.15a | 6 | 0.15 | 30 |
| Fst-0.25 | 6 | 0.25 | 30 |
| Size-15 | 6 | 0.15 | 15 |
| Size-30a | 6 | 0.15 | 30 |
| Size-50 | 6 | 0.15 | 50 |
| Size-100 | 6 | 0.15 | 100 |
Parameters used in data simulated under the inference model discussed in the text are shown.
These three parameters are in fact the same data set used as a reference. In all other data sets, we modified parameters one by one from this reference.
In the particular case of AFLP markers we also need to consider the effect of inbreeding so we used a default value of 0.5 and two additional values (0 and 1; corresponding data sets are called Fis-0, Fis-0.5, and Fis-1 in Table 3) leading to a total of 12 different data sets. Additionally, we included in the simulation the ascertainment bias process observed for AFLP markers and described by Foll et al. (2008). The bias we imposed ensures that at least 2% of the total number of individuals have a band and that at most 2% do not have a band. We used default parameters that we modified one by one to obtain the 12 data sets.
TABLE 3.
AFLP simulation results
| True: | Balancing selection
|
Neutral
|
Directional selection
|
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Classified: | Bal. | Neut. | Direc. | Bal. | Neut. | Direc. | Bal. | Neut. | Direc. | |
| Pop-2 | 0 (0) | 100 (100) | 0 (0) | 0 (0) | 794 (788) | 6 (12) | 0 (0) | 70 (59) | 30 (41) | |
| Pop-6a | 37 (60) | 63 (40) | 0 (0) | 20 (61) | 758 (702) | 22 (37) | 0 (0) | 29 (28) | 71 (72) | |
| Pop-10 | 72 (81) | 28 (19) | 0 (0) | 21 (45) | 755 (721) | 24 (34) | 0 (0) | 21 (19) | 79 (81) | |
| Pop-20 | 95 (97) | 5 (3) | 0 (0) | 26 (50) | 749 (709) | 25 (41) | 0 (0) | 14 (10) | 86 (90) | |
| Fst-0.05 | 1 (14) | 99 (86) | 0 (0) | 0 (19) | 784 (752) | 16 (29) | 0 (0) | 33 (22) | 67 (78) | |
| Fst-0.10 | 27 (49) | 73 (51) | 0 (0) | 17 (46) | 761 (712) | 22 (42) | 0 (0) | 26 (22) | 74 (78) | |
| Fst-0.15a | 37 (60) | 63 (40) | 0 (0) | 20 (61) | 758 (702) | 22 (37) | 0 (0) | 29 (28) | 71 (72) | |
| Fst-0.25 | 51 (67) | 49 (33) | 0 (0) | 23 (47) | 763 (724) | 14 (29) | 0 (0) | 30 (29) | 70 (71) | |
| Size-15 | 15 (35) | 85 (65) | 0 (0) | 8 (33) | 776 (736) | 16 (31) | 0 (1) | 38 (32) | 62 (67) | |
| Size-30a | 37 (60) | 63 (40) | 0 (0) | 20 (61) | 758 (702) | 22 (37) | 0 (0) | 29 (28) | 71 (72) | |
| Size-50 | 47 (65) | 53 (35) | 0 (0) | 38 (65) | 734 (684) | 28 (51) | 0 (0) | 21 (17) | 79 (83) | |
| Size-100 | 61 (73) | 39 (27) | 0 (0) | 40 (66) | 732 (688) | 28 (46) | 0 (0) | 19 (18) | 81 (82) | |
| Fis-0 | 39 (59) | 61 (41) | 0 (0) | 23 (50) | 760 (722) | 17 (28) | 0 (0) | 29 (24) | 71 (76) | |
| Fis-0.5a | 37 (60) | 63 (40) | 0 (0) | 20 (61) | 758 (702) | 22 (37) | 0 (0) | 29 (28) | 71 (72) | |
| Fis-1 | 37 (57) | 63 (43) | 0 (0) | 25 (60) | 755 (703) | 20 (37) | 0 (0) | 25 (21) | 75 (79) | |
Numbers of AFLP loci simulated under (“true”) balancing selection, neutrality, and directional selection that were classified in each category using a reversible-jump cutoff of 0.79 (0.69) that give false-positive rates of 5% (10%) are shown. Bal., balancing selection; Neut., neutrality; Direc., directional selection.
These data sets represent the same reference data set that is replicated in the table to make comparisons between results.
Allele frequencies in the ancestral population for both AFLPs and SNPs were generated from a U-shaped beta distribution with both parameters equal to 0.7. As Wright (1931) showed, at equilibrium, this implies 4Nμ = 0.7, where N is the effective population size and μ is the mutation rate. In the case of microsatellites, we could simply use the noninformative Dirichlet prior with all parameters equal to 1 assumed by our inference model for multiallelic markers. However, Pritchard and Feldman (1996) showed that the stepwise mutation model describes better the mutation process of microsatellites, and Graham et al. (2000) found that the Dirichlet distribution is not appropriate in that case. In particular, simulating ancestral allele frequencies from their prior distribution would lead to a higher variability than what is generally observed in real data sets, and this would artificially increase the power of our method. To take into account this finding and at the same time evaluate the influence of a violation of the underlying infinite-alleles model assumption, we follow the approach of Lockwood et al. (2001) to simulate allele frequencies similar to those observed in real microsatellites. They considered a maximum of seven different alleles and fixed the vector of allele frequencies in the ancestral population at each locus to (0.05, 0.1, 0.2, 0.3, 0.2, 0.1, 0.05). Although Moran (1975) showed that no equilibrium distribution can be obtained under a stepwise mutation model, this provides a practical way to simulate realistic microsatellite data sets. To allow variability in the ancestral population, we simulate the vector of allele frequencies at each locus from a Dirichlet distribution with parameters (10, 20, 40, 60, 40, 20, 10).
For some of the scenarios considered for AFLPs and SNPs, we observed a true-positive rate of 1 for microsatellite data sets. Thus, we decided to enlarge the range of parameter values considered for this marker and instead of presenting results for data sets where all 200 loci under selection were correctly identified we simulated additional data sets corresponding to samples of lower quality. More specifically, we added simulations with four populations, FST = 0.01, FST = 0.03, 10 individuals in each population, |α| = 0.5, |α| = 1.0, and |α| = 1.5 (respectively called Pop-4, Fst-0.01, Fst-0.03, size-10, alpha-0.5, alpha-1.0, and alpha-1.5 in Table 5).
TABLE 5.
Microsatellite simulation results
| True: | Balancing selection
|
Neutral
|
Directional selection
|
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Classified: | Bal. | Neut. | Direc. | Bal. | Neut. | Direc. | Bal. | Neut. | Direc. | |
| Pop-2 | 75 (47) | 25 (53) | 0 (0) | 23 (4) | 765 (792) | 12 (4) | 0 (0) | 3 (12) | 97 (88) | |
| Pop-4 | 100 (98) | 0 (2) | 0 (0) | 20 (7) | 764 (788) | 16 (5) | 0 (0) | 0 (0) | 100 (100) | |
| Fst-0.01 | 27 (1) | 73 (99) | 0 (0) | 21 (1) | 763 (793) | 16 (6) | 0 (0) | 0 (1) | 100 (99) | |
| Fst-0.03 | 90 (67) | 10 (33) | 0 (0) | 35 (12) | 744 (782) | 21 (6) | 0 (0) | 0 (0) | 100 (100) | |
| Fst-0.05 | 100 (100) | 0 (0) | 0 (0) | 17 (9) | 761 (786) | 22 (5) | 0 (0) | 0 (0) | 100 (100) | |
| Size-10 | 97 (88) | 3 (12) | 0 (0) | 17 (2) | 761 (792) | 22 (6) | 0 (0) | 0 (0) | 100 (100) | |
| Size-15 | 100 (100) | 0 (0) | 0 (0) | 20 (6) | 766 (791) | 14 (3) | 0 (0) | 0 (0) | 100 (100) | |
| Alpha-0.5 | 31 (13) | 69 (87) | 0 (0) | 25 (4) | 755 (792) | 20 (4) | 1 (0) | 49 (69) | 50 (31) | |
| Alpha-1.0 | 92 (79) | 8 (21) | 0 (0) | 15 (3) | 762 (791) | 23 (6) | 0 (0) | 7 (14) | 93 (86) | |
| Alpha-1.5 | 100 (97) | 0 (3) | 0 (0) | 22 (4) | 761 (792) | 17 (4) | 0 (0) | 0 (2) | 100 (98) | |
Numbers of microsatellites loci simulated under (“true”) balancing selection, neutrality, and directional selection that were classified in each category using a reversible-jump cutoff of 0.85 (0.98) are shown. The 0.85 cutoff gives the same false-positive rate of 5% used for AFLP and SNP data sets. The 0.98 cutoff gives a false-positive rate of only 1%. Bal., balancing selection; Neut., neutrality; Direc., directional selection.
To decide whether or not a locus i is influenced by selection, we need to choose a cutoff value for the posterior probability P(αi ≠ 0). All loci for which this posterior probability is larger than the cutoff value are considered as outliers. Ideally, one should choose a high value such as 0.95 or 0.99; however, if the purpose is to compare the performance of different types of markers, one needs to chose cutoff values that depend on the quality of the data set considered. For example, retaining only loci with a posterior probability >0.99 using microsatellites with a large sample size and many populations will lead to both a very low false-positive rate and a very high true-positive rate. By contrast, using the same cutoff value with the much less informative dominant markers will not allow us to detect many markers that are indeed under selection. For this reason, a pragmatic way to compare results between different kinds of data sets is to choose cutoff values that give the same false-positive rate and to compare the rate of true positives. With a fixed false-positive rate, the true-positive rate is directly related to the positive predictive value (PPV, or precision rate) defined as the proportion of markers detected as being under selection that are correctly classified. Here we choose to present results using a cutoff value that leads to a global false-positive rate of 5% in each of the three sets of simulations (it corresponds to posterior probabilities of 0.79, 0.86, and 0.85 for AFLPs, SNPs, and microsatellites, respectively). For AFLPs and SNPs we also show results using a false-positive rate of 10% that increases the true-positive rate (it corresponds, respectively, to posterior probabilities of 0.69 for AFLPs and 0.79 for SNPs). For microsatellites, because the true-positive rate is already very high with 5% of false positives, we also show results with a false-positive rate of 1% (it corresponds to a posterior probability of 0.98).
Spatial population expansion model:
Currat et al. (2006) showed that a spatial population expansion can lead to false-positive detection of selection when using a simple comparison of haplotype frequencies. Thus, we use SPLATCHE (Currat et al. 2004) to evaluate the performance of our method (and also other genome-scan methods based on FST) when the true evolutionary model differs radically from the inference model we used. More specifically, SPLATCHE simulates a population expansion from a single origin in a two-dimensional habitat (strict two-dimensional stepping-stone model) and generates genetic samples for geographic locations chosen by the user. Here, we are interested in estimating the proportion of neutral loci that could be identified as being under the effect of selection by our method if the population underwent a recent spatial expansion.
We used the human example as a template and simulated the population expansion with the origin in East Africa. We used a growth rate of 0.3, a carrying capacity of 100 for all demes, and a migration rate of 0.2. With these settings, the whole world is colonized after ∼4000 generations. Because we apply below our method to the Human Genome Diversity Project–Centre d'Etude du Polymorphisme Humain (HGDP–CEPH) data set (Cann et al. 2002), we “sampled” populations at the same locations using the same sample sizes as in this database. To study the effect of sampling design, we considered five sampling scenarios. The first one included all 53 populations while the second considered only 26 because it excluded populations with sample sizes <20 to minimize sampling error. The two last scenarios explored the effect of including populations that underwent a severe bottleneck, which can lead to false-positive detection of selection (Currat et al. 2006). Thus, we considered a third scenario that excludes the 4 Amerindian populations, all issued from the same bottleneck, leaving us with a data set of 22 populations, and a fourth scenario that also excludes 3 isolated insular populations (Orcadian, Sardinian, and Papuan), leaving us with only 19 populations. Finally, we considered a fifth extreme case where we “collected” samples for 36 populations chosen uniformly on the map (cf. Figure 3 in Foll and Gaggiotti 2006) with many isolated insular populations (Greenland, Australia, New Zealand, etc.) Genetic data were generated with SPLATCHE, using a stepwise mutation model (SMM). We simulated 1000 independent loci with a fixed mutation rate of 7.0 × 10−4 under the full sampling scenario containing 53 populations and then used this data set to obtain the three others by removing some of the populations. For the last scenario we generated an independent data set with the same mutation rate and number of loci.
The outlier behavior of a locus can be due to selection but also to a mutation rate that differs from that of most of the other loci. Thus, we investigated the effect of mutation rates on the performance of the method using the scenario with 19 populations (which excludes small samples and populations that underwent a severe bottleneck). Here, we also tried to be as close as possible to the real HGDP–CEPH data set; thus we used different mutation rates for di-, tri-, and tetranucleotidic microsatellites. More precisely, we used the values estimated by Zhivotovsky et al. (2003) from this same data set: 1.52 × 10−3, 7.0 × 10−4, and 6.4 × 10−4 for di-, tri-, and tetranucleotides, respectively. We simulated 1000 markers of each type and conducted three separate analyses and also one analysis containing the 3000 markers at the same time. We also investigated the influence of variability in mutation rates within each class of microsatellite following Xu et al. (2005), who used 5252 dinucleotide markers from the Genome Database and showed that the distribution of mutation rates can be approximated by a gamma distribution with a shape parameter of 1.3327. The scale parameter was chosen to obtain the mean mutation rates μ given above and we generated a data set with 1000 loci.
Results:
Comparison among markers:
The detailed results obtained for AFLPs, SNPs, and microsatellites are presented in Tables 3, 4, and 5, respectively. Table 7 presents a summary for comparing the power to detect selection among markers. The first interesting observation is the very similar results obtained for AFLPs and SNPs, which indicates that they have similar power to detect selection. Moreover, the fact of being dominant does not seem to be a big handicap for AFLPs. The precision rate is slightly higher for both balancing and directional selection with SNPs. With microsatellites, the results are much better than with the two other biallelic markers. Note that in Table 7 the results for microsatellites are presented for poor-quality data sets and are still much better than those of AFLPs or SNPs. Our study shows that the polymorphism of microsatellites is a very strong advantage for the detection of selection. For example, with only two populations, we did not identify any loci under balancing selection for SNPs, but we obtained a true-positive rate of 75% with microsatellites. We were also able to detect weak effects of selection (|α| = 0.5) with microsatellites whereas no loci were detected at this level with SNPs (results not shown). Beaumont and Balding (2004) concluded from simulations of biallelic codominant markers that the method could not distinguish loci under balancing selection even when the selection coefficient is 20 times the migration rate. The results we obtained here show that microsatellites can be used to detect balancing selection, especially with data sets containing a large number of populations. Of course, the advantage of microsatellites over SNPs may disappear if one can group SNPs that are in complete linkage disequilibrium and treat them as haplotypes (see discussion).
TABLE 4.
SNP simulation results
| True: | Balancing selection
|
Neutral
|
Directional selection
|
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Classified: | Bal. | Neut. | Direc. | Bal. | Neut. | Direc. | Bal. | Neut. | Direc. | |
| Pop-2 | 0 (0) | 100 (98) | 0 (2) | 0 (0) | 799 (743) | 1 (57) | 0 (0) | 86 (62) | 14 (38) | |
| Pop-6a | 58 (73) | 42 (26) | 0 (1) | 34 (44) | 751 (714) | 15 (42) | 0 (0) | 39 (29) | 61 (71) | |
| Pop-10 | 82 (85) | 18 (13) | 0 (2) | 31 (44) | 747 (719) | 22 (37) | 0 (0) | 18 (11) | 82 (89) | |
| Pop-20 | 97 (98) | 3 (2) | 0 (0) | 25 (40) | 756 (728) | 19 (32) | 0 (0) | 8 (7) | 92 (93) | |
| Fst-0.05 | 10 (25) | 90 (74) | 0 (1) | 10 (34) | 772 (728) | 18 (38) | 0 (0) | 27 (18) | 73 (82) | |
| Fst-0.10 | 33 (49) | 67 (50) | 0 (1) | 22 (39) | 763 (720) | 15 (41) | 0 (0) | 38 (28) | 62 (72) | |
| Fst-0.15a | 58 (73) | 42 (26) | 0 (1) | 34 (44) | 751 (714) | 15 (42) | 0 (0) | 39 (29) | 61 (71) | |
| Fst-0.25 | 62 (76) | 38 (23) | 0 (1) | 34 (51) | 756 (701) | 10 (48) | 0 (0) | 32 (17) | 68 (83) | |
| Size-15 | 32 (52) | 68 (47) | 0 (1) | 14 (33) | 772 (729) | 14 (38) | 0 (0) | 32 (25) | 68 (75) | |
| Size-30a | 58 (73) | 42 (26) | 0 (1) | 34 (44) | 751 (714) | 15 (42) | 0 (0) | 39 (29) | 61 (71) | |
| Size-50 | 61 (78) | 39 (22) | 0 (0) | 31 (51) | 741 (710) | 28 (39) | 0 (0) | 22 (17) | 78 (83) | |
| Size-100 | 61 (76) | 39 (24) | 0 (0) | 30 (52) | 745 (711) | 25 (37) | 0 (0) | 25 (22) | 75 (78) | |
Numbers of SNP loci simulated under (“true”) balancing selection, neutrality, and directional selection that were classified in each category using a reversible-jump cutoff of 0.86 (0.79) that give false-positive rates of 5% (10%) are shown. Bal., balancing selection; Neut., neutrality; Direc., directional selection.
These data sets represent the same reference data set that is replicated in the table to make comparisons between results.
TABLE 7.
Simulation results summary
| Marker: | AFLPs
|
SNPs
|
Microsatellites
|
||||
|---|---|---|---|---|---|---|---|
| Method used: | IC (%) | RJ (%) | IC (%) | RJ (%) | IC (%) | RJ (%) | |
| Directional selection | |||||||
| False positive | 3.2 | 2.5 | 2.3 | 2.1 | 2.5 | 2.3 | |
| True positive | 70.8 | 70.4 | 65.6 | 67.3 | 94.1 | 94.0 | |
| PPV | 73.5 | 78.0 | 78.5 | 80.1 | 82.4 | 83.7 | |
| Balancing selection | |||||||
| False positive | 1.9 | 2.5 | 2.8 | 2.9 | 2.5 | 2.7 | |
| True positive | 33.7 | 40.2 | 45.8 | 49.6 | 78.7 | 81.2 | |
| PPV | 69.4 | 66.7 | 67.1 | 68.2 | 79.8 | 79.1 | |
| Total | |||||||
| False positive | 5.0 | 5.0 | 5.0 | 5.0 | 5.0 | 5.0 | |
| True positive | 52.3 | 55.3 | 55.7 | 58.5 | 86.4 | 87.6 | |
| PPV | 72.2 | 73.5 | 73.3 | 74.6 | 81.2 | 81.5 | |
A summary of the results for AFLP, SNP, and microsatellite data sets in Tables 3–5 is shown. The PPV is defined as the proportion of markers detected as being under selection that are correctly classified. Results are presented using cutoff values that lead to a 5% total false-positive rate for both the reversible-jump (RJ) method introduced here and the informal criterion (IC) originally proposed by Beaumont and Balding (2004).
Influence of data set characteristics:
The number of populations is a key parameter for the identification of selection, especially for balancing selection. For directional selection, we observed that for all the data sets 6 populations are enough to have a good true-positive rate. However, for balancing selection, we need 10 populations with AFLPs and SNPs to reach a comparable result. Microsatellites, on the other hand, perform fairly well even with only 2 populations.
The level of genetic differentiation also plays an important role for the detection of balancing selection. Weak genetic differentiation (FST ≤ 0.05) makes it almost impossible to detect balancing selection with AFLP or SNP data. On the other hand, with microsatellites, even a small amount of genetic differentiation FST = 0.01 allows us to detect balancing selection. Here we did not note a negative influence of high genetic differentiation on the detection of directional selection but we conducted further simulations (not presented here) with only two populations, and, in that case, having a high genetic differentiation (0.25) leads to low power to detect directional selection.
The sample size is also important for the detection of balancing selection. The lower false-positive rate observed in cases of small sample size is due to the lack of power of the method to detect any loci under selection (being true or false positives). For directional selection, increasing the sample size is less valuable; it is possible to obtain a correct true-positive rate with only 15 individuals per population for AFLPs or SNPs and with only 10 individuals per population for microsatellites. Note that this result is valid only because we used six populations and FST = 0.15; but, for example, if we had only two populations and a higher genetic differentiation, it would be necessary to have larger sample sizes.
In terms of the effect of inbreeding on the power to detect selection using dominant markers such as AFLPs, the results are very similar for all the FIS values considered (Table 3), which suggests that inbreeding is not an issue for the application of our method.
It is possible that the false-positive rate is influenced by the proportion of selected loci in the genome. Thus, we carried out additional simulations with a smaller proportion (10%) of loci under selection for the default scenario with six populations for AFLPs and SNPs and four populations for microsatellites (see Table 2). The false-positive and false-negative rates are similar to those observed when 20% of the loci are under selection (see Table 6). This is another advantage of the model proposed by Beaumont and Balding (2004) that we use here over previous approaches. As explained in the Introduction, this is due to the fact that this model does not require us to input parameters estimated on neutral markers that could be biased by the presence of a high number of selected loci (Beaumont and Nichols 1996; Vitalis et al. 2001).
TABLE 6.
Simulation results when 10% of the loci are subjected to selection
| True: | Balancing selection
|
Neutral
|
Directional selection
|
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Classified: | Bal. | Neut. | Direc. | Bal. | Neut. | Direc. | Bal. | Neut. | Direc. | |
| AFPLs 10% | 18 (31) | 32 (19) | 0 (0) | 11 (49) | 861 (802) | 28 (49) | 0 (0) | 17 (15) | 33 (35) | |
| AFLPs 20% | 37 (60) | 63 (40) | 0 (0) | 20 (61) | 758 (702) | 22 (37) | 0 (0) | 29 (28) | 71 (72) | |
| SNPs 10% | 28 (38) | 22 (12) | 0 (0) | 26 (48) | 856 (911) | 18 (41) | 0 (0) | 19 (11) | 31 (39) | |
| SNPs 20% | 58 (73) | 42 (26) | 0 (1) | 34 (44) | 751 (714) | 15 (42) | 0 (0) | 39 (29) | 61 (71) | |
| SSRs 10% | 50 (50) | 0 (0) | 0 (0) | 22 (5) | 866 (891) | 12 (4) | 0 (0) | 0 (0) | 50 (50) | |
| SSRs 20% | 100 (98) | 0 (2) | 0 (0) | 20 (7) | 764 (788) | 16 (5) | 0 (0) | 0 (0) | 100 (100) | |
Numbers of AFLP, SNP, and microsatellite loci simulated under (“true”) balancing selection, neutrality, and directional selection that were classified in each category using reversible-jump cutoff values given in Tables 3–5 are shown. The results for the simulations with 20% of selected loci in the genome are also shown. Bal., balancing selection; Neut., neutrality; Direc., directional selection.
Comparison with Beaumont and Balding's (2004) method:
Instead of using an approach to estimate the probability that αi is different from 0, such as the one we propose here, Beaumont and Balding (2004) adopted a simple informal criterion for identifying values of αi that are “significant.” More precisely, they define αi to be “significant at level P” if its equal-tailed 100(1 – P)% posterior interval excludes zero. For example, if P = 5%, then αi is significantly positive if its 2.5% quantile is positive and is significantly negative if its 97.5% quantile is negative. In the former case we would conclude that the locus is subjected to directional selection while in the latter we would conclude that it is subjected to balancing selection. We use the three series of data sets presented above to compare the two different ways of detecting selection. We applied the informal criterion on all these simulated data sets using the same false-positive rate of 5% (it corresponds, respectively, to a cutoff value of the informal criterion of 0.95, 0.96, and 0.98 for AFLPs, SNPs, and microsatellites). A summary of the results is presented in Table 7. The global PPVs are sightly higher for the reversible jump than for the informal criterion in seven of the nine cases. The results are very similar between the two approaches for microsatellites and the new method seems to be particularly useful for AFLPs and SNPs.
Spatial population expansion model:
The results of the simulations show that sampling design can affect the ability of our method to detect outliers. Indeed, even though we used a neutral mutation model to generate the synthetic data, we identified loci that had a posterior probability >0.99 of being outliers. More precisely, we observed 3.5, 3.0, 2.2, and 1.7% of false positives for scenarios based on the HGDP–CEPH data set with 53, 26, 22, and 19 populations, respectively. Additionally, the scenario with uniform sampling led to a false-positive rate of 10.6%.
Including markers with different mutation rates can also affect the performance of our method. The analysis that included all 3000 loci without distinguishing between di-, tri-, or tetranucleotides led to a false-positive rate of 2.2%. On the other hand, carrying out separate analyses for each type of microsatellite and then pooling the results led to a false-positive rate of 1.6%. Additionally, the simulations allowing for variable mutation rates within each class of microsatellite led to 4.5% false positives when carrying out a separate analysis for each type and to 5.6% when analyzing all 3000 markers simultaneously.
Our results are in accordance with those of Currat et al. (2006), showing that severe population bottlenecks during a geographic expansion can lead to false-positive detection of selection. However, this problem can be avoided by excluding isolated populations from the analysis. In the case of humans, this is done by considering only the 19 continental populations of Africa, Europe, and Asia. It is worth emphasizing that a uniform sampling design that includes many isolated populations is likely to lead to a high false-positive rate and, therefore, should be avoided. Finally, we showed that including in the same analysis loci with different mutation rates can also increase the false-positive rate. For microsatellites, performing separate analyses on di-, tri-, and tetranucleotides solves this problem, but biases due to variable mutation rate within each class of microsatellite are difficult to avoid.
APPLICATION
Humans:
We use the HGDP–CEPH Human Genome Diversity Cell Line Panel presented by Cann et al. (2002) to identify regions of the human genome that may be influenced by selection. The last version of this data set consists of 1056 individuals from 53 subpopulations, which were scored for 835 microsatellites. On the basis of the results of our realistic simulation study, we chose to use the same 19 continental populations from Africa, Europe, and Asia to minimize the false-positive rate. We kept only microsatellites that were strictly di-, tri-, and tetranucleotidic, which led us to select 106 dinucleotides, 127 trinucleotides, and 327 tetranucleotides, leading to a total of 560 markers. To further minimize the detection of false positives we adopted the best strategy identified by our simulations and conducted separated analyses for each of the three types of markers and grouped the results. We used the same cutoff value of 0.99 as in the simulated data set. We found 131 loci under selection: 86 were detected as being under directional selection and 45 under balancing selection. This represents 23% of the studied loci and is much higher than the false-positive rate estimated from the simulation study that considered similar demographic and sampling scenarios (4.5%). These results suggest that a high number of loci have been subject not only to directional (15%) but also to balancing selection (8%) in the course of human evolution.
We identified the microsatellite loci that are located within a gene whose position is well defined, using the NCBI UniSTS database (http://www.ncbi.nlm.nih.gov/sites/entrez?db=unists). We found eight microsatellites close to known genes under directional selection of which two were located on the X chromosome and 10 known genes under balancing selection, all located on autosomes (Table 8). We then used the Online Mendelian Inheritance in Man (OMIM) database (ftp.ncbi.nih.gov/repository/OMIM/morbidmap) to establish the putative function of the 18 genes identified using NCBI and established that 15 genes (8 under balancing and 7 under directional selection) are referenced as implicated in a genetic disease. These results are in accordance with those of Clark et al. (2003) who showed that the genes under selection are overrepresented in this database.
TABLE 8.
Genes under natural selection
| CEPH index | Chromosome | α | FST | OMIM | Gene |
|---|---|---|---|---|---|
| 602 | 3 | −1.45 | 0.00997 | No | ZPLD1 |
| 570 | 12 | −1.42 | 0.00882 | Yes | TMEM16B |
| 471 | 12 | −1.09 | 0.0117 | Yes | PRMT8 |
| 604 | 6 | −1.02 | 0.015 | Yes | EPHA7 |
| 674 | 10 | −0.981 | 0.0157 | Yes | OIT3 |
| 234 | 16 | −0.96 | 0.0213 | No | RPL3L |
| 600 | 5 | −0.955 | 0.013 | Yes | PDE4D |
| 477 | 3 | −0.919 | 0.0134 | Yes | GPD1L |
| 433 | 7 | −0.905 | 0.0136 | Yes | PRKAG2 |
| 532 | 7 | −0.694 | 0.02 | Yes | PLXNA4 |
| 495 | 1 | 0.523 | 0.0633 | Yes | C8B |
| 337 | X | 0.589 | 0.0551 | Yes | IL1RAPL1 |
| 15 | 4 | 0.738 | 0.0771 | Yes | SLEB3 |
| 739 | 17 | 0.865 | 0.0721 | Yes | RAB37 |
| 351 | 2 | 0.945 | 0.0766 | No | ARMC9 |
| 22 | 1 | 1.07 | 0.103 | Yes | LYST |
| 265 | 6 | 1.08 | 0.136 | Yes | PHACTR1 |
| 26 | X | 1.28 | 0.125 | Yes | EBM |
Genes identified as under balancing (α < 0) and directional (α > 0) selection with the corresponding posterior estimate of FST are shown. Highest absolute values of α suggest a stronger effect of selection. For each gene, we present the chromosome location and the corresponding marker number in the HGDP–CEPH database. We also indicate whether the genes are present in the Online Mendelian Inheritance in Man (OMIM) database.
Littorina saxatilis:
To present an application to AFLPs, we reanalyzed the Littorina saxatilis data set of Wilding et al. (2001), studied also by Grahame et al. (2006). The data consist of 290 polymorphic AFLP loci, surveyed in four different rocky shores in Britain: Thornwick Bay, Flamborough (TH); Filey Brigg (FY); Old Peak (OP); and Robin Hood's Bay (RB). In this region L. saxatilis is found as two morphological forms (“H” and “M”) that show good evidence of partial reproductive isolation. One set of individuals of each morphological form was sampled in each shore, with the exception of the RB shore where two sets of M were sampled. Each of the eight resulting samples is composed of 43–51 individuals.
In each shore two hypotheses can explain the observed divergence between the two morphological forms (Grahame et al. 2006): an allopatric divergence followed by a secondary contact or a primary parapatric divergence (Wilding et al. 2001). In both cases populations are likely to be exchanging genes only in the region of contact, and using the eight populations in a single analysis would lead to a violation of the demographic model assumed by our inference method. This is also supported by the neighbor-joining tree constructed by Wilding et al. (2001) from the loci they identified as neutral: populations were clustered by site (they also constructed a tree using all loci, which led to a grouping of populations by morphotypes H and M).
Wilding et al. (2001) used a modified version of the Fdist model (Beaumont and Nichols 1996) to detect selection from dominant markers. They analyzed three data sets, corresponding to the three shores where both morphotypes were sampled, each one containing two populations. One potential problem of the Beaumont and Nichols (1996) method is the necessity to estimate Nm from the data set to perform simulations with this target value. However, the estimation of Nm assumes neutrality and is overestimated in the presence of directional selection. To avoid this problem, they used an iterative procedure whereby the mean FST calculated from the full data set is used as input of a first Fdist run, and then it is iteratively modified as outlier loci are removed. After four such steps, Wilding et al. (2001) retained only loci that were lying above the 0.99 quantile in all three H–M comparisons and identified 15 loci under selection.
We made the same three analyses of each two-population data set using our method. The Bayesian model we used takes explicitly into account the loci under selection in the estimation of FST coefficients in Equation 3 and, therefore, does not suffer from the problem mentioned above. Beaumont and Balding (2004) compared the critical P-values between the Bayesian method and Fdist by matching the false-positive rate of 6800 neutral loci. They showed that a level of 1% for Fdist is equivalent to a level of 10% for the Bayesian model. Here, the sensitivity study above indicates that a 10% level for the informal criterion used by Beaumont and Balding (2004) is equivalent to a cutoff value of 0.7 for the posterior probability estimated by our reversible-jump version of the method. We identified 13 loci with a probability >0.7 and they all belong to the list of 15 loci identified by Wilding et al. (2001). The two missing loci are named “A37” and “F11” by Wilding et al. (2001) and, according to our method, both are identified as outlier in only two of the data sets. More precisely, the A37 locus has a posterior probability of only 0.53 in the Filey data set, and the F11 locus has a posterior probability of 0.65 in the Old Peak data set. These loci are at the lower tail of the allele-frequency distribution estimated by Wilding et al. (2001) in two of the three data sets considered. If we were to use a cutoff value of 0.65 instead of 0.7 we would include the F11 locus in the list of selected loci but also an additional marker not found by Wilding et al. (2001).
We also analyzed these three sets of two populations as a single data set of six populations to investigate the influence of the violation of the demographic model assumed by our method. Using a cutoff value of 0.99, all 13 loci found in the pairwise analyses are identified as outliers, but we also find 4 additional loci. The results of the simulations of the spatial expansion model suggest that these loci could be false positives due to the violation of the demographic model assumed. As was the case for the human data set, these 4 loci have a posterior estimate of α situated at the tail of the distribution of α-values for loci with a posterior probability >0.99. More precisely, the maximum estimated value of α for these 4 additional loci is 1.89, while most of the loci identified as outliers (7 of 13) in the pairwise analyses have a posterior estimate of α greater than this value.
To establish which of the two approaches is the most appropriate one, we modified the simulation scheme presented above to incorporate a different demographic scenario. More precisely, instead of simulating the six populations under an island model, we simulated first three populations from this model (for the three shores) and then, from each one of them, generated allele frequencies for two populations (corresponding to the two different morphotypes). This demographic history mimics the neighbor-joining tree constructed by Wilding et al. (2001) from the loci they identified as neutral. We chose simulation parameters to obtain data sets close to the real one. We simulated 290 such loci and 50 individuals in each population. We used FST = 0.05 between the ancestral population and the three intermediate populations and FST = 0.03 between the intermediate populations and the six populations sampled. The ancestral allele frequencies were simulated from a beta distribution with both parameters equal to 0.5 and we chose FIS = 0.5. We added selection to 20 loci, using α = 2.5.
We performed the same analysis on this data set as with the real one: 17 loci over the 20 loci under selection had a posterior probability >0.7 in all three pairwise analyses. We did not detect any false positives and all 3 false-negative loci were identified as outliers in two of the three analyses. We then carried out an analysis with the six populations as a single data set and identified all 20 loci as selected with a posterior probability >0.99. However, we also identified 4 additional false-positive loci. The maximum estimated value of α for these 4 additional loci is 2.02, while only 11 of the 20 true outlier loci have a posterior estimate of α greater than this value. These results suggest that, under this particular demographic model, it is better to carry out pairwise analyses instead of a single global one. Moreover, it seems that the best strategy is to identify as selected all loci that are outliers in at least two of the three pairwise analyses. Indeed, if we use such an approach, then we retrieve all 20 loci under selection without identifying any false positives. Note that we can obtain the same result even if we raise the cutoff probability to 0.78.
Applying this approach to the periwinkle data set of Wilding et al. (2001), we identify as selected all 15 loci originally found by them and also 6 additional outliers. We obtain the same result even if we raise the cutoff probability to 0.81. Thus, our analyses suggest that a total of 21 loci are influenced by selection in this species.
DISCUSSION
We present an extension of Beaumont and Balding's (2004) method to detect outlier loci that is applicable to both dominant and codominant markers. Additionally, we propose a rigorous way of estimating the posterior probability of a given locus being under the effect of selection. In their original formulation, Beaumont and Balding (2004) focus on the posterior distribution of locus-specific effects, αi, and use an approximate method to determine if a given locus is significantly influenced by selection. On the other hand, the RJMCMC method we implemented is based on the idea that Equation 3 can give rise to two models, a null model M0 that includes only population-specific effects and an alternative model M1 that includes both locus- and population-specific effects. Thus, it is possible to directly estimate the posterior probability of each alternative model and on the basis of them decide which are the loci subject to selection. The main difference between the two methods is that the original one uses a cutoff value based on the false-positive rate that one is willing to accept. For example, if the threshold value is 99%, we expect to have 1% of false positives. However, one is not able to determine what is the probability that a given locus is or is not influenced by selection. In this sense, Beaumont and Balding's (2004) approach uses the same strategy as that of frequentist methods, where the objective is to reject a null hypothesis without being able to estimate what is the probability that this hypothesis is true. Our method, being fully Bayesian, allows us to rigorously estimate both P(αi = 0 | data) [from the posterior probability P(M0 | data)] and P(αi ≠ 0 | data) [from the posterior probability P(M1 | data)]. Once a locus has been identified as being influenced by selection on the basis of P(M1 | data) we can determine if it is under balancing or directional selection using the mode of the posterior distribution P(αi | data); a negative value indicates the former while a positive value indicates the latter.
Recently Riebler et al. (2008) presented an approximate method to identify nonneutral loci that is not based on the posterior distribution of αi. They propose to introduce a Bernoulli-distributed auxiliary variable, δi to indicate whether or not a locus is subjected to selection. Then, they classify a locus i as being under selection if the posterior probability P(δi = 1 | data) is larger than a threshold value that is set by means of a simulation study. The authors propose to use a cutoff value of 0.17 based on simulations of a selective sweep under a Wright–Fisher model. The problem with this approach is that it is unlikely that this cutoff value is generally applicable to all data sets and all demographic scenarios. Additionally, it is clear that P(δi = 1 | data) cannot be interpreted as the probability that the locus is under selection; the most we can say is that it is proportional to this probability. Otherwise, it would imply that we are willing to accept that a locus is nonneutral even if the probability for this to be true is as low as 0.17. Our method, on the other hand, directly estimates this probability and allows us to avoid the use of simulations to choose a cutoff value. Of course, our method would still need simulations to adjust this cutoff value in cases of strong violation of the demographic model assumed, like we did for the human case.
With our approach, it is clear that for good-quality data we should choose a stringent criterion such as P(αi ≠ 0 | data) ≥ 0.99, which leaves very little room for false positives. Of course, as our simulation study suggests, we may want to choose a somewhat lower threshold (e.g., 0.95) for dominant markers to take into account the fact that they are less informative than codominant ones. The full Bayesian estimation of the probability that a locus is influenced by selection provided by our method also allows the consideration of other factors when choosing a cutoff value. For example, if the purpose is to study local adaptation using a model species for which many genetic resources exist, we may be willing to use a not very stringent criterion (e.g., 0.90) because the costs associated with localizing the position of the candidate loci for subsequent sequencing may not be too high. On the other hand, if we are dealing with a nonmodel species, we may want to use a very restrictive criterion [e.g., P(αi ≠ 0 | data) ≥ 0.99 or 0.999] for deciding whether or not the species in question is appropriate for a study of local adaptation, based on the number of candidate loci found in a genome scan.
Our simulation study demonstrates that, as expected, codominant markers are better suited for detecting selection than dominant ones. More precisely, if we were to use the same cutoff value for both AFLPs and SNPs, we would obtain a much lower true-positive rate (proportion of loci that are correctly identified as subject to selection) for the former than for the latter. Of course, the false-positive rate for AFLPs would be lower but simply because we would identify very few (if any) loci as being influenced by selection. For this reason we decided to compare performance among markers by fixing the false-positive rate to 5% by choosing a lower threshold value for AFLPs than for SNPs and microsatellites. Another interesting but also expected result is that multiallelic markers such as microsatellites are much more powerful than biallelic markers such as SNPs (cf. Table 7). Moreover, we note that although Beaumont and Balding (2004) conclude that the power of the method to detect balancing selection is very low, our analyses showed that this is not the case if we use microsatellites or some other multiallelic marker. Thus, the degree of polymorphism is one of the most important factors determining the power to detect outlier loci. In this regard, we note that the simulation results indicate that microsatellites seem more informative than SNPs. However, the latter are much more abundant and many SNPs may be available for a given region, in which case, they could be grouped into haplotypes and be treated in a similar way to microsatellites. Of course, this is possible only in the case of species whose genome is fairly well known, such as humans and other model species.
We also identified three other parameters that are particularly important determinants of power: the sample size, the number of populations, and the level of genetic differentiation. In general, a sample size of 30 individuals seems enough when the study considers six or more populations. In terms of the effect of neutral genetic differentiation, extremely low FST values decrease the power to detect balancing selection. On the other hand, very high values limit the detection of directional selection. Note, however, that these problems are avoided by using multiallelic markers.
Foll et al. (2008) showed that the estimation of FIS from dominant markers is strongly biased by the ascertainment of markers when assuming the island model. However, in our case we are concerned only with the potential bias in the estimation of the locus-specific effect and our simulation study shows that it suffices to incorporate the uncertainty about the inbreeding coefficient to avoid such a bias. In the present formulation we let FIS move freely between 0 and 1. It would be possible to incorporate prior knowledge about the degree of inbreeding by using a narrower interval for the prior distribution of FIS.
A common problem in all genome-scan methods (e.g., Beaumont and Nichols 1996; Vitalis et al. 2001; Schlotterer 2002), including ours, is the assumption of independence among loci. Although this may not be important when considering a limited number of markers, it may have a large effect in the case of genome scans that use millions of SNPs, in which case, many markers will be in linkage disequilibrium. However, it is not clear what type of bias will be observed. For this it would be necessary to carry out a detailed simulation study that considers linked loci, something that falls outside the scope of this article. In any case, it may be possible to minimize potential biases using the strategy mentioned above, namely, the grouping of markers from the same region into haplotypes.
Another common problem in all genome-scan methods is the possibility that the outlier behavior of a locus is due to differences in mutation rates among loci and not to selection; this is particularly the case when using microsatellites. Our simulation study shows that this problem can be avoided by carrying out separate analyses for each type of marker. For example, in the case of microsatellites it is best to do separate analyses for di-, tri-, and tetranucleotides.
We investigated the potential biases that could be introduced if the demographic history of the species under study does not follow the model assumed by our method. For this purpose we generated synthetic data with SPLATCHE (Currat et al. 2004), using the human population expansion example as a template. The results show that including populations that underwent severe bottlenecks can increase the false-positive rate, particularly for directional selection. However, this problem disappears if all isolated populations are excluded from the analysis, in which case the false-positive rate is 4.5% when mutation rates within each class of microsatellite vary. Moreover, it can be as low as 1.6% if mutation rates do not vary within each class of microsatellites (but they do vary among markers).
As an example of an application of the method with codominant markers, we analyzed the HGDP microsatellite database (Cann et al. 2002) and found that 15% of the markers are under directional selection and 8% are under balancing selection. Eighteen of the outlier loci are located within known genes whose position is well defined. Interestingly, a total of 15 correspond to genes implicated in genetic diseases. None of them are included in the table of the top 50 genes showing evidence for positive selection presented by Nielsen et al. (2005). This difference is likely to be due to the fact that their analysis is based on the comparison of human and chimpanzee genomes and includes only genes that are present in both species.
We also present an example of an application with dominant markers. We analyzed the L. saxatilis data set published by Wilding et al. (2001) and consisting of two samples from each of three shores in which two different morphotypes coexisted. Following Wilding et al. (2001) we first carried out separate analyses for each shore, each including a sample from each morphotype, and chose to identify as selected only the loci that were outliers in all three pairwise analyses. Using this approach we detected only 13 of the 15 loci found by Wilding et al. (2001). We also conducted an analysis with all six populations and identified a total of 17 outlier loci. To decide which of the two approaches was more appropriate we carried out a simulation study based on the demographic history of L. saxatilis and found that the use of the first approach can lead to false negatives while the use of the second one can lead to false positives. Thus, we propose a third strategy consisting of identifying as selected all loci that are outliers in at least two of the three pairwise analyses. Using this approach on the simulated data recovers all the selected loci and does not lead to false positives. In the case of the L. saxatilis data set we identified 21 selected loci, 6 more than Wilding et al. (2001).
The human and the L. saxatilis data sets are two good examples of how to deal with the violation of the demographic model assumed by genome-scan methods based on the multinomial-Dirichlet distribution. For the human data set we showed that excluding populations that underwent a severe bottleneck and identifying as selected only those loci for which P(α ≠ 0 | data) ≥ 0.99 eliminate most of the false positives. For the L. saxatilis data set, taking into account the demographic history by carrying out three pairwise analyses, one for each shore, allows us to avoid all false-positive and false-negative loci at the same time. Thus, some preliminary information about the demographic history of the species under study suffices to come up with an analysis strategy that minimize biases.
A natural extension of our method would be to incorporate the full demographic history into the analysis instead of imposing a simple demographic model. If the demographic history is known, it may be possible to simply incorporate it into the estimation process.
Acknowledgments
We thank Mark Beaumont for the many helpful discussions we had on the subject of this article. The comments of two anonymous reviewers greatly improved an earlier version of the manuscript. Most the computations presented in this article were performed on the cluster HealthPhy (Calcul Intensif, Modélisation, Expérimentation Numérique et Technologique, Grenoble, France). The software implementing the method is available at http://www-leca.ujf-grenoble.fr/logiciels.htm both for Unix and for Windows platforms. This work was supported by the Fond National de la Science (grant ACI-IMPBio-2004-42-PGDA). M.F. holds a Ph.D. studentship from the Ministère de la Recherche.
References
- Balding, D. J., 2003. Likelihood-based inference for genetic correlation coefficients. Theor. Popul. Biol. 63 221–230. [DOI] [PubMed] [Google Scholar]
- Balding, D. J., and R. A. Nichols, 1995. A method for quantifying differentiation between populations at multi-allelic loci and its implications for investigating identity and paternity. Genetica 96 3–12. [DOI] [PubMed] [Google Scholar]
- Balding, D. J., M. Greenhalgh and R. A. Nichols, 1996. Population genetics of STR loci in Caucasians. Int. J. Leg. Med. 108 300–305. [DOI] [PubMed] [Google Scholar]
- Beaumont, M. A., 2005. Adaptation and speciation: What can F-st tell us? Trends Ecol. Evol. 20 435–440. [DOI] [PubMed] [Google Scholar]
- Beaumont, M. A., and D. J. Balding, 2004. Identifying adaptive genetic divergence among populations from genome scans. Mol. Ecol. 13 969–980. [DOI] [PubMed] [Google Scholar]
- Beaumont, M. A., and R. A. Nichols, 1996. Evaluating loci for use in the genetic analysis of population structure. Proc. R. Soc. Lond. Ser. B Biol. Sci. 263 1619–1626. [Google Scholar]
- Bensch, S., and M. Akesson, 2005. Ten years of AFLP in ecology and evolution: Why so few animals? Mol. Ecol. 14 2899–2914. [DOI] [PubMed] [Google Scholar]
- Bowcock, A., J. Kidd, J. Mountain, J. Herbert, L. Carotenuto et al., 1991. Drift, admixture, and selection in human evolution: a study with DNA polymorphisms. Proc. Natl. Acad. Sci. USA 88 839–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks, S. P., P. Giudici and G. O. Roberts, 2003. Efficient construction of reversible jump Markov chain Monte Carlo proposal distributions. J. R. Stat. Soc. Ser. B Stat. Methodol. 65 3–39. [Google Scholar]
- Cann, H. M., C. de Toma, L. Cazes, M. F. Legrand, V. Morel et al., 2002. A human genome diversity cell line panel. Science 296 261–262. [DOI] [PubMed] [Google Scholar]
- Clark, A. G., S. Glanowski, R. Nielsen, P. D. Thomas, A. Kejariwal et al., 2003. Inferring nonneutral evolution from human-chimp-mouse orthologous gene trios. Science 302 1960–1963. [DOI] [PubMed] [Google Scholar]
- Currat, M., N. Ray and L. Excoffier, 2004. Splatche: a program to simulate genetic diversity taking into account environmental heterogeneity. Mol. Ecol. Notes 4 139–142. [Google Scholar]
- Currat, M., L. Excoffier, W. Maddison, S. P. Otto, N. Ray et al., 2006. Comment on “ongoing adaptive evolution of ASPM, a brain size determinant in homo sapiens” and “microcephalin, a gene regulating brain size, continues to evolve adaptively in humans”. Science 313 172. [DOI] [PubMed] [Google Scholar]
- Falush, D., M. Stephens and J. K. Pritchard, 2003. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164 1567–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint, J., J. Bond, D. C. Rees, A. J. Boyce, J. M. Roberts-Thomson et al., 1999. Minisatellite mutational processes reduce fst estimates. Hum. Genet. 6 567–576. [DOI] [PubMed] [Google Scholar]
- Foll, M., and O. Gaggiotti, 2006. Identifying the environmental factors that determine the genetic structure of populations. Genetics 174 875–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foll, M., M. Beaumont and O. Gaggiotti, 2008. An approximate Bayesian computation approach to overcome biases that arise when using AFLP markers to study population structure. Genetics 179 927–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham, J., J. Curran and B. S. Weir, 2000. Conditional genotypic probabilities for microsatellite loci. Genetics 155 1973–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grahame, J. W., C. S. Wilding and R. K. Butlin, 2006. Adaptation to a steep environmental gradient and an associated barrier to gene exchange in Littorina saxatilis. Evolution 60 268–278. [PubMed] [Google Scholar]
- Green, P. J., 1995. Reversible jump Markov chain Monte Carlo computation and Bayesian model determination. Biometrika 82 711–732. [Google Scholar]
- Holsinger, K. E., 1999. Analysis of genetic diversity in geographically structured populations: a Bayesian perspective. Hereditas 130 245–255. [Google Scholar]
- Holsinger, K. E., and P. O. Lewis, 2002. Hickory: A Package for Analysis of Population Genetic Data v1.1. http://darwin.eeb.uconn.edu/hickory/hickory.html.
- Holsinger, K. E., P. O. Lewis and D. K. Dey, 2002. A Bayesian approach to inferring population structure from dominant markers. Mol. Ecol. 11 1157–1164. [DOI] [PubMed] [Google Scholar]
- Kauer, M. O., D. Dieringer and C. Schlotterer, 2003. A microsatellite variability screen for positive selection associated with the “out of Africa” habitat expansion of Drosophila melanogaster. Genetics 165 1137–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewontin, R., and J. Krakauer, 1973. Distribution of gene frequency as a test of theory of selective neutrality of polymorphisms. Genetics 74 175–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockwood, J. R., K. Roeder and B. Devlin, 2001. A Bayesian hierarchical model far allele frequencies. Genet. Epidemiol. 20 17–33. [DOI] [PubMed] [Google Scholar]
- Meudt, H. M., and A. C. Clarke, 2007. Almost forgotten or latest practice? AFLP applications, analyses and advances. Trends Plant Sci. 12 106–117. [DOI] [PubMed] [Google Scholar]
- Moran, P. A. P., 1975. Wandering distributions and electrophoretic profile. Theor. Popul. Biol. 8 318–330. [DOI] [PubMed] [Google Scholar]
- Nielsen, R., C. Bustamante, A. G. Clark, S. Glanowski, T. B. Sackton et al., 2005. A scan for positively selected genes in the genomes of humans and chimpanzees. PLoS Biol. 3 976–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard, J. K., and M. W. Feldman, 1996. Statistics for microsatellite variation based on coalescence. Theor. Popul. Biol. 50 325–344. [DOI] [PubMed] [Google Scholar]
- Riebler, A., L. Held and W. Stephan, 2008. Bayesian variable selection for detecting adaptive genomic differences among populations. Genetics 178 1817–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlotterer, C., 2002. A microsatellite-based multilocus screen for the identification of local selective sweeps. Genetics 160 753–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, B. J., 2005. Bayesian Output Analysis Program (BOA), version 1.1.5. http://www.public-health.uiowa.edu/boa.
- Vitalis, R., K. Dawson and P. Boursot, 2001. Interpretation of variation across marker loci as evidence of selection. Genetics 158 1811–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilding, C. S., R. K. Butlin and J. Grahame, 2001. Differential gene exchange between parapatric morphs of Littorina saxatilis detected using AFLP markers. J. Evol. Biol. 14 611–619. [Google Scholar]
- Wright, S., 1931. Evolution in Mendelian populations. Genetics 16 97–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, S., 1951. The genetic structure of populations. Ann. Eugen. 15 323–354. [DOI] [PubMed] [Google Scholar]
- Xu, H., R. Chakraborty and Y. Fu, 2005. Mutation rate variation at human dinucleotide microsatellites. Genetics 170 305–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhivotovsky, L. A., N. A. Rosenberg and M. W. Feldman, 2003. Features of evolution and expansion of modern humans, inferred from genomewide microsatellite markers. Am. J. Hum. Genet. 72 1171–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]