

Abstract
Why does hormone replacement therapy (HRT) with estrogens plus progestins increase the risk of breast cancer? First, experimental estrogen receptor-positive (ER+) and progesterone receptor-positive (PR+) human breast cancers contain a rare subpopulation of ER−, PR− cancer stem cells. Especially in small, nascent ER+, PR+ tumor colonies, progestins, but not estrogens, reactivate cells with ER−, PR− stem-like properties. Second, there is a reservoir of occult, undetected, preinvasive breast cancer in some women who are candidates for HRT. We propose that women who develop breast cancer while on estrogens plus progestins harbor undiagnosed nascent disease before the start of therapy. The progestin component, in a nonproliferative step, reactivates receptor-negative cancer stem cells within such germinal, perhaps even dormant tumors. After reacquiring receptors, these tumor cells are expanded by the mitogenic properties of estrogens. We argue that screening methods need to be improved to detect small, preexisting malignancies prior to the start of HRT. Women harboring such disease should be excluded from regimens that include systemic progestins.
Progestins and Breast Cancer Stem Cells
Gene expression profiling separates breast cancers into two major subtypes: estrogen receptor-positive (ER+), progesterone receptor-positive (PR+), cytokeratin 18-positive (CK18+) luminal disease and ER−, PR−, CK5+ basal disease (1). Approximately 80% of breast cancers fall into the luminal ER+, PR+, CK18+ category. These tumors have a better prognosis and more varied treatment options than their basal ER−, PR−, CK5+ counterparts. The origins of luminal vs. basal breast cancers remain unclear.
The cancer stem cell theory proposes that tumors originate from mitotically quiescent stem cells that are capable of self-renewal, while at the same time spawning proliferative, committed progenitor cells that differentiate and expand into the clinically significant tumor mass (2). In the human breast, normal stem cells have been defined by expression of epithelial-specific antigen (ESA) and α6 integrin (CD49f) by markers that include Musashi, the Polycomb group repressor Bmi-1, and CK5/6 or by label-retention and side-population properties (reviewed in Ref. 3). Human breast cancer stem cells are currently characterized by CD44+, CD24−/low, ESA+ expression (4). The concept of breast cancer stem cells remains controversial. Some deny their existence; others question whether they arise from normal breast epithelial cells or from reprogrammed malignant cells. If breast cancers arise from normal stem cells, then a broad consensus, currently elusive, is needed to define their location and properties. Some data support the notion that the normal human breast contains ER−, PR− stem cells that initiate both basal ER−, PR− cancers and more differentiated ER+, PR+ progenitors of luminal cancers. Other data indicate that ER+, PR+ cancers arise from distinct ER+, PR+ stem cells (5,6,7).
Given the different breast cancer subtypes, we were surprised to find that in xenografts of luminal ER+, PR+, CK18+ human breast cancers, progestins up-regulated expression of the basal-like CK5+ marker in a subpopulation of cells (8). They did so without altering growth. Follow-up studies (9) showed that in solid ER+, PR+ experimental tumors, the rare CK5+ cells up-regulated by progestins are ER−, PR−, and CD44+, CD24−/low, a pattern characteristic of tumorigenic breast cancer stem cells (3). Additional details were observed with three-dimensional clonogenic assays of ER+, PR+, CD44+ cells (9). They showed that young, germinal colonies of these cells (i.e. fewer than 10 cells total) contained a subpopulation of ER−, PR−, CK5+ cells, whereas ER+, PR+ cells were rare (Fig. 1). Quantitation of data from many such colonies showed that the ER−, PR−, CK5+ cell number did not expand as colonies enlarged to about 100 cells. That is, there were no more than two or three ER−, PR−, CK5+ putative colony-initiating stem cells, whether a colony was composed of 10 or 100 cells. However, as colonies expanded, there was a linear, non-hormone-dependent increase in the ER+, PR+, CK5− population, so that in larger more evolved colonies, more than 95% of cells exhibited the ER+, PR+, CK5− differentiated signature, with 1–2% of cells retaining the ER−, PR−, CK5+ stem-like signature (9). These properties define breast cancer stem cells as ER−, PR−, CD44+, CK5+, with the capacity to spawn the more differentiated, majority ER+, PR+, CK5− population. Importantly, rare CK5+ stem cells can always be found among the CK5− cells of the more differentiated colonies. This pathway is outlined in Fig. 2. The same sporadic basal-like ER−, PR−, CK5+ subpopulation can be found in ER+, PR+, CK5− luminal breast cancers of patients (9).
Figure 1.

MPA reactivates stem-like properties in human breast cancer cells. Cells isolated from human ER+, PR+, xenografted, T47D cell-derived solid tumors were FACS sorted for CD44+ cells, plated into minimal medium in eight-well plates, and allowed to form colonies for 3–14 d. Cells were either vehicle treated (control) or treated with 10 nm of the progestin MPA for 24 h before fixation and immunostaining for CK5 (green) and PR (red) and counterstained with DAPI (blue). Because the cells express PR constitutively, no estrogen was required. Two young colonies (3 d, 10–15 cells), one control and one MPA-treated, are shown. Cells are either CK5+ (stem-like) or ER+, PR+ (more differentiated) but not both (ER are not shown). Arrow points to a rare PR+, CK5+, transient intermediate cell (see Fig. 2), usually found only in progestin-treated colonies. Scale bars, 10 μm. [Reproduced with permission from K. B. Horwitz et al.: Proc Natl Acad Sci USA 105:5774 (9). ©The National Academy of Sciences.]
Figure 2.
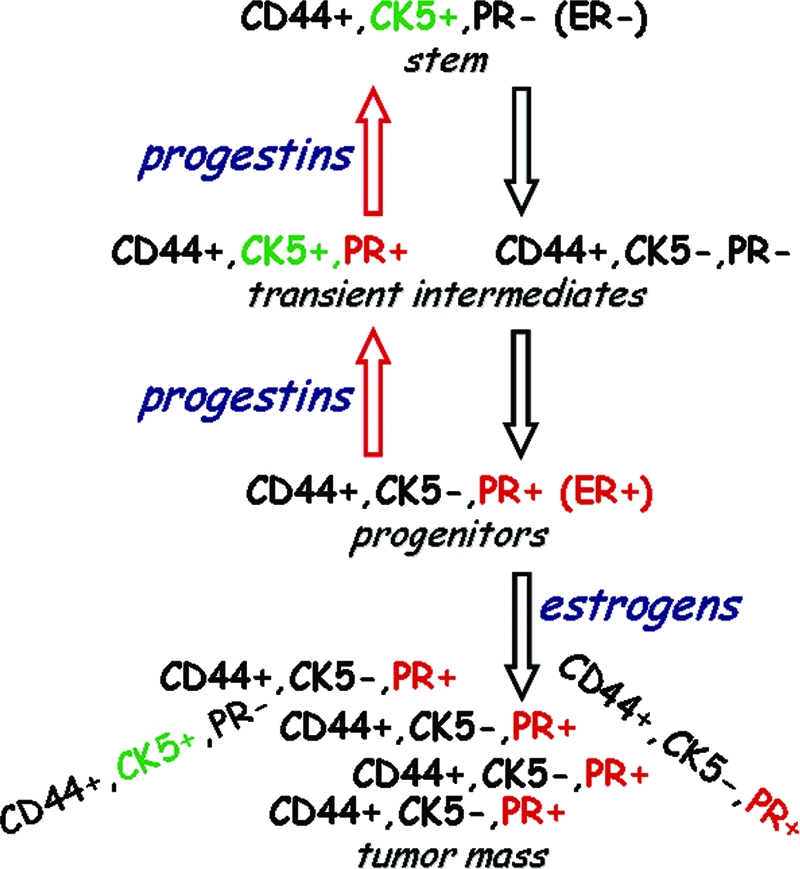
Proposed differentiation pathway of ER+, PR+ breast cancers from ER−, PR− stem cells, and the reversal induced by progestins. Data in support of this pathway are reported in Refs. 3, 8, and 9. All cells are CD44+, CD24−/low (not shown). Tumors are also ER+, but PR, the targets of progestins, are mainly shown for simplicity. Without stimulating proliferation, both progesterone and MPA reactivate CK5 in ER+, PR+ cells within 24 h especially in young, germinal tumors. Restoration of steroid receptors to CK5+ stem-like cells does not require hormones. Mitogenic effects of estrogens occur in later stages of these pathways after cells have acquired ER and PR.
Of interest was the effect of progestins, but not estrogens, on this pathway (9). Treatment of colonies for 24 h with progesterone, or the synthetic progestin commonly used for hormone replacement therapies (HRT), medroxyprogesterone acetate (MPA; Depo Provera), led to an increase in the ER−, PR−, CK5+ stem cell-like subpopulation, from 1–2% to more than 20%. In cells with constitutive PR, this step did not require estrogens, and did not involve proliferation. In small colonies, almost 100% of cells acquired the ER−, PR−, CK5+ stem-like phenotype (Figs. 1 and 2). Additionally, progestin treatment unmasked a unique, ER+, PR+, CK5+ transient intermediate between the more differentiated ER+, PR+, CK5− cells that are the likely targets of progestins and the revertant ER−, PR−, CK5+ stem-like cells (Fig. 1, arrow). Together, these data fit a model in which tumor cells retrogress from a differentiated state to a more stem-like, primitive state in response to progestins.
Tumorigenicity assays of ER+, PR+ human breast cancer cells grown into tumors in mammary glands of ovariectomized mice supplemented with estradiol showed similar effects of progestins. Nascent, 30-d-old tumors contained rare CK5+ cells and only sporadic ER+ and PR+ cells. Strong CK18 expression confirmed the luminal subtype of all the cells. In older, 60-d tumors, CK5+ cells remained rare, but the majority of cells had acquired an ER+, PR+ status. Twenty-four hours of MPA or progesterone led to extensive CK5 reactivation, which was much more pronounced in the younger, nascent tumors (9).
In summary, with regard to the role of progestins on putative breast cancer stem cells, 1) ER+, PR+, CK5− luminal breast cancers contain a small tumorigenic ER−, PR−, CK5+ stem cell-like subpopulation. 2) Progestins via PR act on the more abundant ER+, PR+ CK5− differentiated cells to reactivate ER−, PR−, CK5+ cells with stem-like features. Estrogens are not required for this unless they are needed to induce PR. 3) Small, nascent tumors or cell clusters are more sensitive to stem cell reactivation by progestins than large, mature tumors. Of note, this model critically separates the functions of progestins and estrogens in breast cancers. We propose that what progestins do is restore cancer stem cell-like properties (CD44+, CK5+, PR−, ER−) to some ER+, PR+ breast cancer cells. This happens in 24 h or less and does not require proliferation. Up-regulation by progestins of cells with stem-like properties sets the stage for their subsequent differentiation into transient intermediates/progenitors and restoration of ER and PR in a step that is not hormone regulated, followed by resumption of growth under the control of estrogens. Our hypothesis thus relegates to progestins the function of stem cell reactivation without concomitant proliferation, while leaving to estrogens their well-known mitogenic/proliferative functions (see Fig. 2).
Progestins, occult breast cancers, and HRT
The combined estrogen plus progestin (E+P) arm of the Women’s Health Initiative menopausal HRT trial was stopped prematurely in 2002, because of failure to demonstrate an overall health benefit and because of an increased risk of invasive, ER+ breast cancer, compared with the estrogen-only arm (10). The data have been the subject of considerable controversy since then due to concerns about the statistical analytic methods employed, the high proportion of elderly women begun on hormones several years after menopause, hormone restriction to conjugated equine estrogens plus MPA, and because the results contravened prevailing views that progestins should be protective. Nevertheless, subsequent studies have tended to support the notion that E+P increases the risk of developing clinically relevant breast cancers (11,12,13) or stimulates growth of tumor microdeposits in breast cancer survivors (14).
Why does addition of progestins to estrogens increase breast cancer risk? Various explanations have been advanced: that progestins cause cancer directly, that they enhance the effect of carcinogens, that progestins are antiproliferative in the uterus but proliferative in the breast, and that effects of progestins are subject to lifestyle factors like diet and alcohol use or to genetic factors, reproductive history, environmental exposure, mammographic features, and the like (reviewed in15). Some of these explanations remain unproven; others may be too complex and confounding for practical clinical decision making.
Is there another explanation? Among seven autopsy series of women not known to have breast cancer during life, the median prevalence of invasive breast cancer was 1.3% (range, 0–1.8%), and the median prevalence of ductal carcinoma in situ was 8.9% (range, 0–14%). Importantly, this occult disease was found only in women over 40 yr of age (16). A separate study in 1987 sought to analyze the radiographic detectability of such occult disease. The authors also concluded that it was restricted to women over 39 yr old but found, discouragingly, that 82% of such tumors would have been mammographically undetectable in life by the methods then in use and were only detectable histologically postmortem (17). Clearly, a substantial reservoir (16) of early breast cancers is undetected in women who are at an age when HRT is often prescribed. Importantly, younger women who are more likely to be using oral contraceptives that include progestins appear not to be susceptible to this problem. In a related issue, women presumed to have recovered from breast cancer may harbor occult nanometastases that are mammographically undetectable and may even escape routine microscopy (18). Use of systemic progestins in such patients should be viewed with caution.
Hypothesis
Based on these studies, and our discovery that progestins, but not estrogens, reactivate stem-like properties in breast cancer cells (9), we propose a simple and potentially testable explanation for the effects of progestins in HRT. Namely, that women who develop breast cancer while on E+P had undiagnosed breast cancer before the start of HRT, and the progestin component reactivated occult, possibly even dormant (17), breast cancer stem cells. Importantly, the experimental data indicate that estrogen alone is incapable of such an effect. Once reactivated, however, mitogens like estrogen can expand the tumor cells. The data (9) also suggest that small, nascent, preinvasive disease, like atypical intraductal hyperplasia, ductal carcinoma in situ, or nanometastases, are at greatest risk of stem cell reactivation by systemic progestins. If our hypothesis is correct, it would suggest that 1) sensitive methods need to be developed to detect occult, possibly dormant, breast cancers; 2) women should be screened for preexisting malignancies by the best available methods before the start of HRT and, if harboring such disease, be excluded from regimens that include systemic progestins; and 3) given current limitations of screening methods, patients should be fully informed about the benefits and risks they face when they are prescribed HRTs that include systemic progestins. Instead, local progestin delivery, as with intrauterine systems, could provide the desired protective effects of these hormones in the uterus, without their possible harmful effects in the breast (19).
Acknowledgments
We thank Lawrence D. Horwitz, M.D., for helpful suggestions and members of the Horwitz lab for data. Presented in part as “The Year in Hormones & Cancer” lecture at ENDO ’08.
Footnotes
Disclosure Statement: The authors have nothing to disclose.
This work was funded by the Susan G. Komen Breast Cancer Foundation (BCTR0402682), the University of Colorado Cancer Center, the National Institutes of Health National Cancer Institute (CA26869), the National Foundation for Cancer Research, the Breast Cancer Research Foundation, and the Avon Foundation.
First Published Online July 22, 2008
Abbreviations: CK18, Cytokeratin 18; E+P, estrogen plus progestin; ER, estrogen receptor; HRT, hormone replacement therapy; MPA, medroxyprogesterone acetate; PR, progesterone receptor.
References
- Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, Fluge O, Pergamenschikov A, Williams C, Zhu SX, Lønning PE, Børresen-Dale AL, Brown PO, Botstein D 2000 Molecular portraits of human breast tumours. Nature 406:747–752 [DOI] [PubMed] [Google Scholar]
- Reya T, Morrison SJ, Clarke MF, Weissman IL 2001 Stem cells, cancer, and cancer stem cells. Nature 414:105–111 [DOI] [PubMed] [Google Scholar]
- Clarke RB 2006 Ovarian steroids and the human breast: regulation of stem cells and cell proliferation. Maturitas 54:327–334 [DOI] [PubMed] [Google Scholar]
- Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF 2003 Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA 100:3983–3988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke RB, Spence K, Anderson E, Howell A, Okano H, Potten CS 2005 A putative human breast stem cell population is enriched for steroid receptor-positive cells. Dev Biol 277:443–456 [DOI] [PubMed] [Google Scholar]
- Dontu G, Al-Hajj M, Abdallah WM, Clarke MF, Wicha MS 2003 Stem cells in normal breast development and breast cancer. Cell Prolif 36(Suppl 1):59–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dontu G, El-Ashry D, Wicha MS 2004 Breast cancer, stem/progenitor cells and the estrogen receptor. Trends Endocrinol Metab 15:193–197 [DOI] [PubMed] [Google Scholar]
- Sartorius CA, Harvell DM, Shen T, Horwitz KB 2005 Progestins initiate a luminal to myoepithelial switch in estrogen-dependent human breast tumors without altering growth. Cancer Res 65:9779–9788 [DOI] [PubMed] [Google Scholar]
- Horwitz KB, Dye WW, Harrell JC, Kabos P, Sartorius CA 2008 Rare steroid receptor-negative basal-like tumorigenic cells in luminal subtype human breast cancer xenografts. Proc Natl Acad Sci USA 105:5774–5779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, Jackson RD, Beresford SA, Howard BV, Johnson KC, Kotchen JM, Ockene J; Writing Group for the Women’s Health Initiative Investigators 2002 Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results From the Women’s Health Initiative randomized controlled trial. JAMA 288:321–333 [DOI] [PubMed] [Google Scholar]
- Beral V, Million Women Study Collaborators 2003 Breast cancer and hormone replacement therapy in the Million Women Study. Lancet 362:419–427 [DOI] [PubMed] [Google Scholar]
- Fournier A, Berrino F, Riboli E, Avenel V, Clavel-Chapelon F 2005 Breast cancer risk in relation to different types of hormone replacement therapy in the E3N-EPIC cohort. Int J Cancer 114:448–454 [DOI] [PubMed] [Google Scholar]
- Prentice RL, Chlebowski RT, Stefanick ML, Manson JE, Langer RD, Pettinger M, Hendrix SL, Hubbell FA, Kooperberg C, Kuller LH, Lane DS, McTiernan A, O'Sullivan MJ, Rossouw JE, Anderson GL 2008 Estrogen plus progestin therapy and breast cancer in recently postmenopausal women. Am J Epidemiol 167:1207–1216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmberg L, Iversen OE, Rudenstam CM, Hammar M, Kumpulainen E, Jaskiewicz J, Jassem J, Dobaczewska D, Fjosne HE, Peralta O, Arriagado R, Holmqvist M, Maenpa J; on behalf of the HABITS Study Group 2008 Increased risk of recurrence after hormone replacement therapy in breast cancer survivors. J Natl Cancer Institute100:475–482 [DOI] [PubMed] [Google Scholar]
- Santen RJ 2003 Risk of breast cancer with progestins: critical assessment of current data. Steroids 68:953–964 [DOI] [PubMed] [Google Scholar]
- Welch HG, Black WC 1997 Using autopsy series to estimate the disease “reservoir” for ductal carcinoma in situ of the breast: how much more breast cancer can we find? Ann Int Med 127:1023–1028 [DOI] [PubMed] [Google Scholar]
- Pollei SR, Mettler Jr FA, Bartow SA, Moradian G, Moskowitz M 1987 Occult breast cancer: prevalence and radiographic detectability. Radiology 163:459–462 [DOI] [PubMed] [Google Scholar]
- Ross JS, Symmans, WF, Pusztai L, Hortobagyi G 2007 Standardizing slide-based assays in breast cancer: hormone receptors, HER2, and sentinel lymph nodes. Clin Can Res 13:2831–2835 [DOI] [PubMed] [Google Scholar]
- Backman T, Rauramo I, Jaakkola K, Inki P, Vaahtera K, Launonen A, Koskenvuo M 2005 Use of the Levonorgestrel-releasing intrauterine system and breast cancer. Obst Gynecol 106:813–817 [DOI] [PubMed] [Google Scholar]