

Abstract
We report the first pre-steady-state kinetic studies of DNA replication in the absence of hydrogen bonds. We have used nonpolar nucleotide analogues that mimic the shape of a Watson-Crick base pair in order to investigate the kinetic consequences of a lack of hydrogen bonds in the polymerase reaction catalyzed by the Klenow fragment of DNA Polymerase I from Escherichia coli. With a thymine isostere lacking hydrogen bonding ability in the nascent pair, the efficiency (kpol/Kd) of the polymerase reaction is decreased by 30-fold, affecting ground state (Kd) and transition state (kpol) approximately equally. When both thymine and adenine analogues in the nascent pair lack hydrogen bonding ability, the efficiency of the polymerase reaction is decreased by about 1000-fold, with most the decrease attributable to the transition state. Reactions using nonpolar analogues at the primer terminal base pair demonstrated the requirement for a hydrogen bond between the polymerase and the minor groove of the primer-terminal base. The R668A mutation of Klenow fragment abolished this requirement, identifying R668 as the probable hydrogen bond donor. Detailed examination of the kinetic data suggested that Klenow fragment has an extremely low tolerance of even minor deviations of the analogue base pairs from ideal Watson-Crick geometry. Consistent with this idea, some analogue pairings were better tolerated by Klenow fragment mutants having more spacious active sites. By contrast, the Y-family polymerase Dbh was much less sensitive to changes in base pair dimensions, and more dependent on hydrogen bonding between base-paired partners.
INTRODUCTION
The “classical” DNA polymerases from families A (homologous to bacterial DNA polymerase I), B (homologous to eukaryotic DNA polymerase α), and X (homologous to eukaryotic DNA polymerase β) copy DNA with an accuracy far exceeding that predicted solely from the energetics of Watson-Crick hydrogen bonding (1). Structural studies of representatives of all three families, in ternary complexes with a DNA template-primer and a correctly-paired incoming nucleotide (2-7), reveal two features that may explain this impressive fidelity enhancement. First, the active site conforms very closely to the shape of a correct nascent base pair, favoring Watson-Crick geometry over the distorted geometry of nascent mispairs. Secondly, the DNA binding site on the polymerase contains several side chains which form hydrogen bonds to the minor groove of the DNA template primer. The minor groove hydrogen bond acceptors, N3 of purines and O2 of pyrimidines, are positioned almost identically in all four correct pairings but are absent or positioned differently in mispairs (8). Thus, the minor groove interactions provide a mechanism for detecting mispairs at the polymerase active site and within the newly synthesized DNA.
The close steric matching of the polymerase active site to the shape of a Watson-Crick base pair raised the possibility that steric complementarity might be at least as important in recognition of correct base pairs as the hydrogen bonds between the base-paired partners (reviewed in ref. (9). In support of this idea Kool and colleagues showed that adenine could be incorporated opposite the nonpolar thymine analogue F1 with efficiency and fidelity that approached those of a natural hydrogen-bonded base pair. In addition, non-polar nucleotide analogues F and Z (Figure 1A), isosteric with T and A, respectively, could be incorporated opposite each other by Klenow fragment with a reasonable degree of efficiency and selectivity (10). However, the ability to synthesize a DNA base pair in the absence of hydrogen bonding is not a universal trait in the polymerase superfamily; Klenow fragment and its homologues in the A-family are the least affected by the absence of hydrogen bonds in the nascent base pair, while DNA polymerase α is less efficient, and DNA polymerase β and some Y-family polymerases give very poor incorporation of the unnatural base pair (11-13).
Figure 1.

DNA and nucleotide substrates used in this study. A. The natural nucleosides, T and A, and their non-polar isosteres, F, Z and Q. B. Oligonucleotide sequences designed for the investigation of the synthesis and extension of base pairs that lack Watson-Crick hydrogen bonds. The synthesis of these materials has been described in detail previously (10, 14, 17, 18).
Subsequent studies introduced another A isostere, Q (Figure 1A), which differs from Z in the presence of the minor-groove hydrogen bond acceptor (14, 15). This analogue allowed the separation of the effects of Watson-Crick hydrogen bonding and those of minor groove hydrogen bonding. By comparing the effects of Z and Q at a series of positions in a DNA substrate, the requirement for minor groove hydrogen-bonding interactions in a variety of polymerases was determined. For example, Klenow fragment and other A-family polymerases were shown to require a hydrogen bond to the minor groove only at the primer terminus, a conclusion that was independently supported by an alternative approach using the analogue 3-deazaguanine, which selectively removes the minor-groove hydrogen bond acceptor (16). By contrast, experiments with the Z and Q analogues suggested that DNA polymerase β requires hydrogen bonds to the minor groove face of both bases in the nascent base pair, as well as to the primer terminal base (15).
The previous studies involved steady-state kinetic measurements or qualitative gel comparisons. To investigate further the structural features responsible for the recognition of correctly-paired bases by a DNA polymerase, we have used the non-hydrogen-bonding analogues F, Z and Q in single-turnover kinetic studies of nucleotide incorporation by Klenow fragment. We have investigated the kinetic consequences of the absence of hydrogen bonds at either the nascent base pair or the primer-terminal base pair. These studies extend the earlier published data by providing kinetic parameters that can be directly related to ground state or transition state interactions at the DNA polymerase active site. We have also examined the effect of these same analogues on the Y-family polymerase Dbh (DinB homologue) from Sulfolobus acidocaldarius, whose active site differs in some important respects from that of Klenow fragment.
EXPERIMENTAL PROCEDURES
Materials
Oligonucleotides containing analogues F, Z, and Q were synthesized and purified as described (10, 14, 17). Nucleoside triphosphate analogues dFTP, dZTP, and dQTP were prepared following published procedures (10, 18). Wild-type and mutant Klenow fragment derivatives were purified to homogeneity by our standard procedure (19). All Klenow fragment derivatives used in this study contained the D424A mutation that eliminates 3′-5′ exonuclease activity (20).
Kinetic measurements - Klenow fragment
Single-turnover measurements of nucleotide incorporation by Klenow fragment and its mutant derivatives were carried out at room temperature (20 - 22 °C), using a rapid quench-flow instrument (KinTek Corp., Model RQF-3) for fast reactions, and manual quenching when the reaction was sufficiently slow. In either case, the enzyme-DNA solution contained 40 nM of one of the DNA duplexes listed in Figure 1B, 5′-labeled on the primer strand, and 2 μM Klenow fragment in 50 mM Tris-HCl (pH 7.5), 10 mM MgCl2. The reaction was initiated by mixing with an equal volume of an appropriate dNTP in the same Tris-MgCl2 solution. Reactions were quenched at the desired time intervals using an excess of EDTA, and were fractionated on denaturing polyacrylamide-urea gels. The data were processed as described previously (21). Using the oligonucleotide duplex with a terminal (primer)F-Z(template) base pair, we showed that the rate was maximal for wild-type Klenow fragment concentrations of 0.1 μM or greater. Therefore our standard conditions described above should be adequate to ensure that all DNA in the mixture was enzyme-bound, giving pseudo-first-order kinetics for the incorporation reaction.
Kinetic measurements - Dbh
Single-turnover measurements of nucleotide incorporation by Dbh were carried out at room temperature analogously to the method described above for Klenow fragment. The reaction contained 100 nM duplex DNA, 2.5 μM Dbh, and the desired concentration of dNTP, in a buffer containing 5 mM dithiothreitol, 10 mM Hepes, pH 8.5, and 10 mM MgCl2. When using high concentrations of dNTP (> 1 mM), additional MgCl2 equimolar with the dNTP was included.
RESULTS
Non-hydrogen-bonding analogues at the nascent base pair
Using the oligonucleotides listed in Figure 1B, we first measured single-turnover kinetics for incorporation reactions of Klenow fragment in which the nascent base pair completely lacks hydrogen bonds (dZTP incorporation opposite a template F and vice versa), or where one partner of the base pair cannot make hydrogen bonds (A-F pairing) (Figure 2 and Table 1). Compared with the natural A-T pairings, the completely non-polar Z-F pairings resulted in ≈ 7-fold decrease in affinity for the incoming nucleotide [Kd(dNTP)] and ≈ 100-fold reduction in reaction rate (kpol). Overall this gave ≈ 103-fold decrease in the efficiency (kpol/Kd) for the reaction. The results were very similar regardless of which of the hydrophobic partners was the template and which the incoming nucleotide. When only one base in the nascent base pair lacked hydrogen bonding groups (A-F pairing), the results obtained depended on the orientation of the base pair. Incorporation of dFTP opposite template A showed very similar kinetics to incorporation of dFTP opposite Z. However, incorporation of dATP opposite F had a much more favorable kpol than F-dZTP incorporation, although the Kd values were similar.
Figure 2.

Single-turnover kinetics of the formation, by Klenow fragment, of a base-pair that lacks hydrogen-bonds. Examples of the rates measured for dFTP incorporation opposite template Z, and dZTP opposite template F are plotted against nucleotide concentration and fitted to a hyperbolic equation, to give the Kd and kpol values reported in Table 1.
Table 1.
Effect of non-hydrogen-bonding base analogues on Klenow fragment polymerase kinetics
| Enzyme | Reaction | Kd(dNTP) (μM) | kpol (s-1) | kpol/Kd (M-1s-1) |
|---|---|---|---|---|
| Analogues at insertion site: | ||||
| WT | Template-A + dTTP | 11 | 180 | 1.6 × 107 |
| WT | Template-A + dFTPa | 28 | 0.52 | 1.8 × 104 |
| WT | Template-Z + dFTP | 68 | 0.95 | 1.4 × 104 |
| WT | Template-T + dATP | 12 | 250 | 2.1 × 107 |
| WT | Template-F + dATP | 91 | 62 | 6.8 × 105 |
| WT | Template-F + dZTP | 88 | 2.1 | 2.4 × 104 |
| Klenow fragment mutants in insertion reaction: | ||||
| E710A | Template-A + dTTPa | 17 | 11 | 6.2 × 105 |
| E710A | Template-Z + dFTPa | 13 | 0.0067 | 5.1 × 102 |
| E710A | Template-T + dATPa | 6.4 | 5.6 | 8.7 × 105 |
| E710A | Template-F + dZTPa | 19 | 0.25 | 1.4 × 104 |
| Y766A | Template-A + dTTPa | 36 | 20 | 5.5 × 105 |
| Y766A | Template-Z + dFTPa | 26 | 0.027 | 1.0 × 103 |
| Y766A | Template-T + dATPa | 65 | 24 | 3.7 × 105 |
| Y766A | Template-F + dZTP | 6.2 | 0.053 | 8.6 × 103 |
| F762A | Template-T + dATPb | 1.6 × 104 | ||
| F762A | Template-F + dZTPb | 3.2 × 102 | ||
| Analogues at primer terminus: | ||||
| WT | (primer)A-T(template) + dGTP | 3.0 | 190 | 6.3 × 107 |
| WT | (primer)Z-F(template) + dGTP | 29 | 0.0056 | 1.9 × 102 |
| WT | (primer)Q-F(template) + dGTP | 16 | 0.49 | 3.1 × 104 |
| WT | (primer)T-A(template) + dGTP | 4.5 | 63 | 1.4 × 107 |
| WT | (primer)F-Z(template) + dGTP | 140 | 0.16 | 1.1 × 103 |
| WT | (primer)F-Q(template) + dGTP | 81 | 0.18 | 2.2 × 103 |
| R668A | (primer)A-T(template) + dGTP | 40 | 2.6 | 6.6 × 104 |
| R668A | (primer)Z-F(template) + dGTP | 37 | 0.012 | 3.2 × 102 |
| R668A | (primer)Q-F(template) + dGTP | 29 | 0.008 | 2.8 × 102 |
Single measurements. All other determinations were the average of two or more experiments with good agreement.
Because of the high Kd(dNTP) of the F762A mutant protein, it was not possible to use a high enough concentration of dZTP to saturate the reaction, and therefore individual kpol and Kd values could not be obtained. The efficiency (kpol/Kd) was determined from the slope of the plot of rate vs. dNTP concentration, at low dNTP concentrations.
Mutations in the binding pocket for the nascent base pair
The side chains of E710, F762 and Y766 are predicted to contribute to the snug fit of the polymerase binding pocket around the nascent base pair (Figure 3A). We therefore examined the effect of mutation of each of these side chains to alanine, which would be expected to decrease the complementary fit of the surface of the fingers subdomain against a Watson-Crick base pair. When the Z-dFTP pairing was compared with A-dTTP, the mutants showed the same ≈ 103-fold decrease in incorporation efficiency as was previously noted with wild-type Klenow fragment (Table 1). With the opposite orientation, F-dZTP compared with T-dATP, the mutant proteins discriminated less against the non-polar analogues than did wild-type Klenow fragment.
Figure 3.
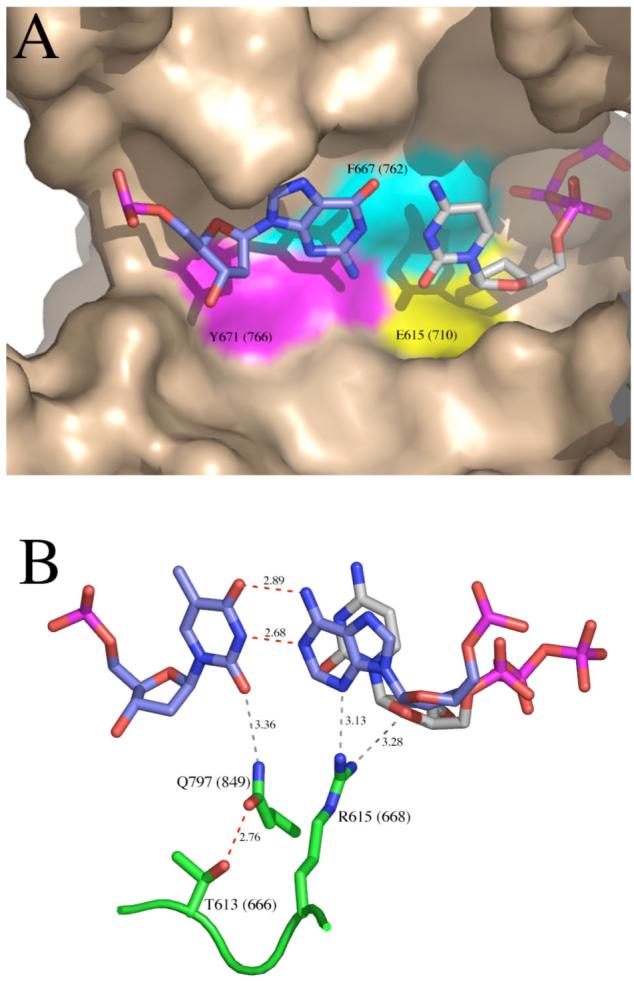
Details of the polymerase active site from ternary complex crystal structures of A-family polymerases, homologous to Klenow fragment. A. Binding pocket for the nascent base pair, illustrated using the ternary complex of Klentaq, PDB file 3KTQ (3). The templating base is colored predominantly in blue, and the incoming dNTP predominantly in gray. On the surface representation of the protein, the colored areas show the side chains homologous to E710 (yellow), F762 (cyan) and Y766 (magenta) of Klenow fragment. The remaining wall of the binding pocket is provided by the terminal base pair (not shown) which would cover the nascent base pair in the view shown here. B. Contacts around the minor groove face of the primer-terminal base pair, illustrated using the ternary complex of Bst DNA polymerase, PDB file 1LV5 (4). The terminal base pair is colored predominantly in blue, and the incoming dNTP predominantly in gray. Protein side chains discussed in the text are shown in green; T613, R615 and Q797 of Bst DNA polymerase are homologous to T666, R668 and Q849 of Klenow fragment. Hydrogen bonds shown in red are predicted to be strong and those in gray weaker. These figures were constructed using PyMOL (DeLano Scientific).
Non-hydrogen-bonding analogues at the primer terminus
We examined the effect on the kinetics of dNTP addition of replacing a normal T-A primer-terminal base pair with a pairing that lacks hydrogen bonds. Oligonucleotide substrates containing an F-Z pair in either orientation at the primer terminus were compared with control substrates having a T-A pair oriented similarly with respect to which strand had the purine-sized base (Figure 1B). When F or T was on the primer strand, the non-polar primer terminus resulted in a decrease of 104-fold in the efficiency of addition of dGTP opposite the following template C, comprised of a 30-fold decrease in dNTP binding affinity and a 400-fold decrease in reaction rate (Table 1). In the opposite orientation (A or Z on the primer strand) the non-polar base pair was even more detrimental to subsequent dNTP addition, resulting in a 3 × 105-fold decrease in efficiency compared with the corresponding natural base pair. Most of this decrease (3 × 104-fold) was attributable to changes in kpol. With the natural T-A pairing at the primer terminus, the orientation with the purine on the primer strand was the more favorable, by about 4-fold (in kpol/Kd).
The F-Z pairing not only eliminates hydrogen bonds between the bases, but also eliminates hydrogen bonds between the polymerase active site and the minor groove side of the base pair. To evaluate the relative contributions of these two classes of hydrogen bonds, we compared the effect of Z in the primer-terminal base pair with the analog Q, which restores the minor groove hydrogen bond to the protein. Restoring the hydrogen bond on the template side caused only a 2-fold increase in the efficiency of dGTP addition, whereas the hydrogen bond on the primer side was associated with a 100-fold improvement in efficiency, almost entirely due to an increase in kpol (Table 1).
Based on crystal structures of A-family DNA polymerases (2-4), R668 of Klenow fragment is predicted to make the hydrogen bond to the minor groove side of the base on the primer side of the terminal base pair (Figure 3B). Consistent with this prediction, the R668A mutant of Klenow fragment did not discriminate between Z and Q at the primer terminus (Table 1). In both cases the kinetic parameters were similar to those obtained with wild-type Klenow fragment and a (primer)Z-F(template) terminal base pair (Figure 4).
Figure 4.

Kinetic demonstration of the requirement for a hydrogen bond from R668 to the minor groove of the primer terminus. Q at the primer terminus, which is capable of hydrogen bonding, is extended much more readily by wild-type Klenow fragment than Z, which has no hydrogen-bonding groups. (Note the change in scale on the vertical axis.) The R668A mutation removes the proposed hydrogen bond donor on the protein and essentially abolishes the distinction between primer-terminal Q and Z.
Comparison of Klenow fragment and the Y-family polymerase Dbh
We examined the incorporation of dATP, dQTP and dZTP opposite F by the Y-family polymerase Dbh. Structural studies suggest that the Y-family lesion-bypass polymerases have a more open active site that makes fewer contacts with the nascent base pair (22, 23). This is reflected in extremely weak binding to the incoming dNTP (KD ≈ 1 mM), which precluded measurement of the separate Kd and kpol values for incorporation of the analogues (24). Based on the efficiency of the reaction (kpol/Kd), all of the incorporations opposite template F were ≈ 1000-fold less favorable than the correct T-dATP incorporation (Table 2). Because the efficiencies of incorporation of dZTP and dQTP were very similar, the data argue against any requirement for a hydrogen bond to the minor groove side of the incoming dNTP.
Table 2.
Effect of non-hydrogen-bonding base analogues on reactions catalyzed by Dbh polymerase
| Reaction | Rate (s-1) | kpol/Kd (M-1s-1) |
|---|---|---|
| Analogues at insertion sitea: | ||
| Template-T + dATP | 1.2 × 103 | |
| Template-F + dATP | 0.72 | |
| Template-F + dZTP | 0.46 | |
| Template-F + dQTP | 0.62 | |
| Analogues at primer terminusb: | ||
| (primer)T-A(template) + dGTP | 0.10 | |
| (primer)F-Z(template) + dGTP | 1.3 × 10-4 | |
| (primer)F-Q(template) + dGTP | 2.9 × 10-4 | |
| (primer)A-T(template) + dGTP | 1.4 × 10-2 | |
| (primer)Z-F(template) + dGTP | 2.9 × 10-5 | |
| (primer)Q-F(template) + dGTP | 2.3 × 10-4 | |
Because of the high Kd(dNTP) of Dbh, it was not possible to use a high enough dNTP concentration to saturate the reaction, and therefore individual kpol and Kd values could not be obtained. Even with T-dATP, the reaction rate did not plateau at concentrations up to 4 mM dATP, and we estimate a Kd in excess of 1.5 mM. The efficiency (kpol/Kd) was determined from the slope of the plot of rate vs. dNTP concentration, at low dNTP concentrations.
Rates were compared at 2 mM dGTP. In every case, the rate at 2 mM dGTP was 54 to 77% of that at 10 mM, suggesting no substantial variation in dGTP affinity in these reactions.
The lack of a minor groove hydrogen bond acceptor on the primer terminal base had a smaller effect on Dbh than on Klenow fragment (Table 2). Comparing (primer)Q-F with (primer)Z-F, the presence of the hydrogen bond acceptor was associated with an increase of only ≈ 8-fold in reaction rate. On the template strand, substitution of Q for Z (F-Q vs. F-Z) caused ≈ 2-fold increase in the rate of nucleotide addition by Dbh, similar in magnitude to the effect seen with Klenow fragment. The reaction rates for nucleotide addition by Dbh to all four of these analogue-containing terminal base pairs were similar except for (primer)Z-F, which was 5- to 10-fold slower than the other three.
DISCUSSION
Incorporation of non-hydrogen-bonding analogues
The formation of hydrogen bonded natural base pairs reflects a delicate free energy balance. When, for example, A pairs with T in an aqueous environment, energy input is required for desolvation of the hydrogen bonding face of the two bases and then energy is recovered due to the formation of hydrogen bonds. By contrast, when the isosteric nonpolar analogues Z and F are paired opposite one another, there is no requirement for desolvation of polar groups, but also no gain in free energy due to hydrogen bonding. Also important are steric effects; although the analogue F is nearly perfect in its size/shape mimicry of thymine, analogues Z and Q possess a C-H in place of N1 of adenine, which makes them slightly larger than their natural counterpart though still able to assume a conformation close to Watson-Crick geometry (25). Base stacking is another important contributor to the energetics of base pairing, but is unlikely to be substantially altered when a natural base is replaced by an isosteric analogue (9). If anything, the hydrophobic analogues would be expected to improve base stacking (26). It is in the context of all these energetic factors that we should consider the single-turnover data for synthesis of pairings involving F, Z, or Q by Klenow fragment.
Our data show that incorporation of dZTP opposite F, or dFTP opposite Z, is ≈ 1000-fold less efficient than synthesis of a normal Watson-Crick base pair. The decrease in efficiency derives primarily from the transition state for nucleotide insertion (kpol, ≈ 100-fold decrease), whereas changes in ground state dNTP binding are more modest (Kd, ≈ 7-fold increase). Currently we do not know whether phosphoryl transfer or the preceding noncovalent step is rate-limiting in the incorporation of the nonpolar analogues by Klenow fragment (see ref. (27); thus we can only conclude that the rate of at least one of these steps is substantially decreased by the use of the non-polar analogues. The decrease in efficiency reported here is about 10-fold greater than was estimated from steady-state measurements in an earlier publication (10).
Although less efficient than synthesis of a Watson-Crick base pair, synthesis of the Z-F pairing by Klenow fragment is remarkably robust compared with, for example, incorporation of the natural bases in a mispair. The highest Klenow fragment misincorporation efficiencies, for T-dGTP or A-dATP, are ≈ 104-fold lower than for a correct base pair, and the lowest, for pyrimidine-pyrimidine pairings are typically 106-fold lower (28, 29). Therefore synthesis of a base pair that mimics the shape of a correct base pair but lacks hydrogen bonds is 10- to 1000-fold more favorable than misincorporation. Nevertheless, the 1000-fold lower efficiency that we observe indicates that the Z-F pairing is at some disadvantage relative to A-T. Very recent experiments in which the size of the non-polar analogues was varied systematically suggest that Klenow fragment can be highly discriminating against small changes in base pair size (30); thus it seems most likely that the Z-F pair is at a kinetic disadvantage largely because of the increased size of Z, described above.
The idea that the slightly larger size of the Z-F pair compared with A-T might be particularly problematic in nucleotide incorporation catalyzed by Klenow fragment is consistent with the considerably higher efficiency seen for incorporation of dATP opposite analogue F, where the base pair size is closer to that of the natural A-T base pair. Comparing the kinetic parameters for F-dATP and F-dZTP incorporation, it can be seen that F-dATP has a much more favorable kpol than F-dZTP, though the dNTP binding constants are very similar (Table 1). A plausible interpretation is that the lack of hydrogen bonds primarily affects the ground state dNTP binding, accounting for the ≈ 7-fold change in Kd(dNTP) for both F-dATP and F-dZTP incorporation, whereas the effects of inappropriate base pair size are felt in the transition state. This interpretation is undoubtedly oversimplified because F-dATP and F-dZTP are not strictly comparable in that A must be desolvated in order to participate in a base pair, whereas the same is not true for Z. It is also puzzling that A-dFTP is much less efficient than F-dATP incorporation, the kinetic parameters for A-dFTP being very similar to those of the completely nonpolar F-dZTP pairing.
Effect of active site “tightness”
We hypothesized that mutations that increase the volume of the binding pocket for the nascent base pair, thus giving a less snug fit around the templating base and the incoming dNTP, might make the incorporation reaction more dependent on hydrogen bonding between the base pairing partners, as was observed for the Y-family DNA polymerases κ and η (12, 13). We tested this hypothesis using Klenow fragment mutants with alanine substitutions in three important active-site residues: E710, F762 and Y766. From the ternary complex crystal structures of other A-family polymerases (2-4), E710 should be positioned beneath the sugar of the incoming dNTP, F762 should contact the base and sugar of the dNTP, and Y766 should contribute to the floor of the binding pocket, primarily on the template side (Figure 3A). The results with alanine substitutions at these positions were the opposite of our expectation: rather than an increased dependence on hydrogen bonding (relative to wild-type Klenow fragment), the mutants showed the same or lower discrimination against the F-Z pairing. Moreover, there were some interesting effects of the orientation of the base pair. When dFTP was incorporated opposite Z, the overall (kpol/Kd) discrimination compared with the correct pairing was similar to that observed with wild-type Klenow fragment; however, with the mutant proteins, almost all the discrimination was seen in the transition state (kpol). We suggest that the less constrained active site in these mutant polymerases makes the initial binding less stringent, and this, combined with the absence of hydrogen bonds between the bases, allows deviations of the nascent base pair from the normal Watson-Crick geometry required for optimal catalysis. With the opposite orientation, dZTP opposite F, the mutant proteins discriminated about 10-fold less than wild-type against incorporation of the nonpolar analogue, suggesting that these mutations may be particularly helpful in accommodating the slightly oversized dZTP in the nucleotide binding site. Remarkably, E710A and Y766A were only slightly less efficient than wild-type when incorporating dZTP opposite F (Table 1). The results with the Y766A mutant protein were particularly interesting in that they suggested that the mutation allowed a more favorable binding of dZTP (10-fold decrease in Kd) but that this was associated with mispositioning that compromised the geometry required for catalysis (400-fold decrease in kpol). We were unable to carry out a similar analysis for the F762A mutant because the extremely weak dNTP binding of this protein made it impractical to measure the individual Kd and kpol values on account of the high dZTP concentrations that would have been required.
The suggestion of mispositioning in the absence of the Y766 side chain is intriguing in light of the mutator phenotypes of the Y766A and Y766S Klenow fragment mutants (28, 31, 32). In an earlier kinetic study of misinsertion by the Y766S mutant polymerase, the mispaired dNTP was bound more strongly by the mutant protein than by wild-type Klenow fragment in seven of the twelve possible misinsertion reactions (28). Together with the results of the present study, this underscores the importance of Y766 in controlling the geometry of the nascent base pair, probably via positioning of the templating base.
Minor groove hydrogen bonds at the primer terminus
Morales and Kool (15) presented evidence, in Klenow fragment and its homologues, for a functionally important hydrogen-bond to the minor groove face of the primer-terminal base, but not to its template partner. Our data with the hydrophobic F, Q, and Z analogues at the terminal base pair confirm these observations and additionally provide insights into the effects on ground state and transition state interactions that were not obtainable from the earlier steady-state data. The key comparisons involve DNA substrates that are identical aside from the presence of Q or Z (paired opposite F) at a particular position, corresponding to the presence or absence, respectively, of the minor groove hydrogen bond acceptor. With wild-type Klenow fragment, replacement of Z by Q at the primer terminal base resulted in a 100-fold improvement in the efficiency of incorporation of the following nucleotide. This improvement (almost all in kpol) provides evidence for a functionally important hydrogen bond to the minor groove of the primer terminus, which may facilitate catalysis by maintaining the correct geometry at the primer terminus. By contrast, the same substitution on the template side of the terminal base pair had essentially no effect, implying that the hydrogen bond (to the homologue of Gln849) seen in A-family polymerase structures does not influence the reaction parameters we have measured.
Detailed examination of the data obtained with wild-type Klenow fragment and hydrophobic analogues at the primer terminus reveals some interesting asymmetries. For example, incorporation of the next correct nucleotide (dGTP) was 6-fold less efficient onto a primer terminal Z, paired with template F, than onto the opposite orientation, primer F paired with Z. This is in spite of the fact that, with the natural bases, a primer-terminal purine (A) gave 5-fold more efficient incorporation than a primer-terminal pyrimidine (T), probably because of better stacking of the incoming dGTP on an adjacent purine. These data suggest that the slightly larger Z is better tolerated on the template side of the terminal base pair. All three analogues, F, Z and Q, at the primer terminus destabilized binding of the incoming dGTP, but the effect was greatest with F, again consistent with better stacking of dGTP onto the purine analogues. Transition state interactions show the opposite trend; thus, kpol was ≈ 30-fold lower with primer-terminal Z than with primer-terminal F, whereas ground-state binding (Kd) was about 5-fold more favorable. These opposing effects on kpol and Kd are highly suggestive of mispositioning of the substrate, a plausible scenario being that stacking of dGTP on the purine analogue, Z, enhances binding but also pulls the nucleotide away from the optimal position for catalysis. Conversely, a primer terminal F, contributing less to binding, might be less likely to distort the active site geometry.
The R668A mutation eliminated the kinetic differences between DNA substrates having primer terminal Z and Q, indicating that R668 most likely contributes the hydrogen bond to the minor groove of the primer terminal base (Figure 3B). This result was expected from the ternary complex crystal structures of three Klenow fragment homologues (2-4), and from biochemical studies by Spratt, who reported an analogous effect of the R668A mutation when comparing dG and 3-deazaguanine at the primer terminal base, an alternative strategy for probing the requirement for a minor groove hydrogen bond to N3 of purines (16). Our experiments using the Z and Q analogues, and the 3-deazaguanine studies of Spratt, demonstrate the lack of a functionally important hydrogen bond to the template side of the terminal base pair, in spite of the presence of an invariant glutamine side chain, equivalent to Q849 of Klenow fragment, in the appropriate location in all three ternary complex structures of A-family polymerases (2-4). Examination of these three structures indicates that the active site glutamine, equivalent to Q849, forms only a weak hydrogen bond to the DNA minor groove, but forms a shorter hydrogen bond to an invariant threonine, two residues N-terminal to the homologue of R668 (Figure 3B). Perhaps the more important function of Q849 is in positioning the β-turn containing the invariant arginine that interacts with the primer-terminal base. Consistent with its presence in all A-family polymerase sequences, mutational data indicate an important role for Q849 of Klenow fragment. Substitution of this side chain caused substantial decreases in reaction rate and binding affinity for DNA and dNTP, and was associated with an antimutator phenotype (32-34).
The kinetic parameters (both kpol and Kd) for wild-type Klenow fragment with (primer)Z-F are very similar to those for R668A with either Z-F or Q-F at the primer terminus. The simplest inference is that the kinetic parameters in all three cases reflect the loss of the minor groove hydrogen bond to the terminal base pair and, moreover, that this hydrogen-bonding interaction is the only function provided by R668 when hydrophobic analogues are present at the primer terminus. However, structural and biochemical evidence suggest that R668 uses its bifunctional guanidino side chain to make a second important interaction, to the ring oxygen of the deoxyribose of the incoming nucleotide (2-4, 35). Loss of this interaction could certainly account for the higher Kd(dGTP) in all the reactions catalyzed by R668A, compared with the value for wild-type Klenow fragment with a normal DNA substrate. It is less obvious why the Kd(dGTP) for wild-type Klenow fragment with (primer)Z-F is similar to the values obtained with the R668A protein, even though the interaction of R668 with the dNTP sugar should be possible in the wild-type situation. A reasonable inference is that the non-hydrogen-bonding analogues at the primer terminal base pair compromise the ability of R668 to interact with the incoming dNTP, perhaps by changing the position of the R668 side chain or the dNTP in the polymerase active site so the interaction is not fully formed except in the case of wild type Klenow fragment and an A-T primer terminus. Regardless of the precise details, the 103-fold decrease in kpol/Kd seen when comparing R668A with wild-type Klenow fragment on normal (AT) primer termini must indicate the combined contribution of all the interactions made by R668 in both ground state and transition state. Like Q849, R668 is invariant in A-family DNA polymerases. Substitution of R668 results in compromised kinetic parameters and a mutator phenotype, due to poor discrimination against mispaired primer termini (32, 33, 36).
It is clear from our kinetic data that the absence of the hydrogen bond to the minor groove of the primer terminus is not the only problem associated with hydrophobic analogues at the primer terminus. If it were, the Q-F primer terminus would behave the same as A-T in reactions catalyzed by wild-type Klenow fragment, and R668A would not discriminate between Watson-Crick base pairs and their hydrophobic isosteres. In reality, the differences in reaction efficiency are 2,000- and 200-fold, respectively. Thus Klenow fragment has significant difficulty with non-polar base analogues at the primer terminus even when the minor groove hydrogen bond acceptor is present. It is possible that the slightly larger size of the Z or Q analogues causes distortions in binding of the terminal base pair, and this may mean that the Q substitution only partially restores the minor groove hydrogen bond. This scenario would be consistent with the changes to the positioning of the incoming dNTP in the polymerase active site inferred from individual kpol and Kd values, discussed above.
Y-family DNA polymerase
We expected that the response of the Y-family DNA polymerase, Dbh from Sulfolobus solfataricus, to the non-hydrogen-bonding nucleotide analogues might be different from that of Klenow fragment. Structural studies of the Y-family lesion bypass polymerases indicate a more open active site than in classical DNA polymerases such as Klenow fragment, which could make dNTP incorporation more dependent on hydrogen bonding in the nascent base pair (22, 23, 37, 38). However, replacement of T-dATP by F-dZTP in the nascent base pair caused ≈ 1000-fold decrease in kpol/Kd for Dbh, very similar in magnitude to the effect seen with Klenow fragment. If Dbh were more dependent than Klenow fragment on hydrogen bonding, one might have expected a larger decrease in kpol/Kd. Alternatively, a greater dependence on hydrogen-bonding by Dbh may be compensated by some other favorable factor. For example, as suggested above for the Klenow fragment binding pocket mutations, a less snugly fitting active site could be more permissive for the slightly larger Z-F base pair. Our results for F-dATP incorporation support the latter interpretation. The efficiencies of F-dATP and F-dZTP incorporation by Dbh were very similar, implying that the larger size of the Z-F base pair did not affect the reaction. Moreover, the rather poor efficiency of F-dATP incorporation suggests that the lack of hydrogen bonds assumes greater importance in the Dbh active site than in the Klenow fragment active site. Similar conclusions have been reached in studies of DNA polymerases η and κ (12, 13).
The ternary complex crystal structure of the Dpo4 polymerase, a close homologue of Dbh, shows no potential hydrogen bond donors to the minor groove of the DNA primer-template duplex or the nascent base pair (23). Consistent with the structural data, the similar kinetics of dZTP and dQTP incorporation indicated that Dbh does not require a minor groove hydrogen-bond acceptor on the incoming nucleotide. Analogous studies of Y-family polymerases using 3-deazaguanine have indicated that DNA polymerase κ is not strongly dependent on a minor groove hydrogen bond acceptor on the incoming dNTP, whereas DNA polymerase η is more dependent on this hydrogen bond (13, 39). When Q replaced Z at the primer terminus, Dbh showed some increase in reaction rate but this was less than seen with Klenow fragment (10-fold vs. 100-fold). It should be noted that (primer)Z-F was the least efficiently extended of all the hydrophobic pairings by both polymerases in this study, raising the possibility that there could be some inherent problem with this base pair, regardless of any requirement for minor groove hydrogen bonding groups.
Conclusions
The kinetic parameters determined in this study show that incorporation catalyzed by Klenow fragment can be reasonably efficient in the absence of hydrogen bonds between the template and incoming nucleotide, as illustrated by F-dATP incorporation. The much less favorable kpol for synthesis of the slightly larger Z-F pairing demonstrates the extremely stringent steric matching between the Klenow fragment active site and the nascent base pair in the transition state. Mutations which increase the size of the Klenow fragment binding pocket improve F-dZTP incorporation, presumably by permitting altered binding geometry which impacts ground state and transition state kinetic parameters to different extents in the different mutants. In contrast to Klenow fragment, the more open active site of the Y-family polymerase, Dbh, results in a greater dependence on hydrogen bonding between the base-paired partners and much less effect of small changes in base pair dimensions. When the hydrophobic analogues were present at the primer terminus, the Klenow fragment reaction kinetics revealed the requirement, in the transition state, for a hydrogen bond between the protein and the minor groove of the primer-terminal base. However, even when using the Q analogue to allow this interaction, the reaction rate is substantially lower than that obtained with the natural bases. The changes in individual kinetic parameters due to hydrophobic analogues at the primer terminus are very suggestive of altered positioning relative to the natural substrates. Thus there may be some plasticity of the polymerase active site which allows unnatural substrates to be accommodated, though at the price of a lower reaction rate. This is reminiscent of the structures reported for an A-family polymerase with DNAs containing terminal mispairs, where a wide range of active site distortions was observed (40). Moreover, it may explain the paradoxical observation of relatively efficient polymerase-catalyzed incorporation of hydrophobic analogues that are very poor shape mimics, unlike those used in the present study (41, 42).
Footnotes
Supported by NIH grants GM-28550 (N.D.F.G.) and GM-072705 (E.T.K.)
- F
- 2,4-difluorotoluene
- Z
- 4-methylbenzimidazole
- Q
- 9-methyl-1 H-imidazo[4,5-b]-pyridine
- Dbh
- DinB homologue DNA polymerase from Sulfolobus acidocaldarius
REFERENCES
- 1.Kunkel TA, Bebenek K. DNA replication fidelity. Annu. Rev. Biochem. 2000;69:497–529. doi: 10.1146/annurev.biochem.69.1.497. [DOI] [PubMed] [Google Scholar]
- 2.Doublié S, Tabor S, Long A, Richardson CC, Ellenberger T. Crystal structure of a bacteriophage T7 DNA replication complex at 2.2 Å resolution. Nature. 1998;391:251–258. doi: 10.1038/34593. [DOI] [PubMed] [Google Scholar]
- 3.Li Y, Korolev S, Waksman G. Crystal structures of open and closed forms of binary and ternary complexes of Thermus aquaticus DNA polymerase I: structural basis for nucleotide incorporation. EMBO J. 1998;17:7514–7525. doi: 10.1093/emboj/17.24.7514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnson SJ, Taylor JS, Beese LS. Processive DNA synthesis observed in a polymerase crystal suggests a mechanism for the prevention of frameshift mutations. Proc. Natl. Acad. Sci. U.S.A. 2003;100:3895–3900. doi: 10.1073/pnas.0630532100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Franklin MC, Wang J, Steitz TA. Structure of the replicating complex of a pol α family DNA polymerase. Cell. 2001;105:657–667. doi: 10.1016/s0092-8674(01)00367-1. [DOI] [PubMed] [Google Scholar]
- 6.Pelletier H, Sawaya MR, Kumar A, Wilson SH, Kraut J. Structures of ternary complexes of rat DNA polymerase β, a DNA template-primer, and ddCTP. Science. 1994;264:1891–1903. [PubMed] [Google Scholar]
- 7.Sawaya MR, Prasad R, Wilson SH, Kraut J, Pelletier H. Crystal structures of human DNA polymerase β complexed with gapped and nicked DNA: evidence for an induced fit mechanism. Biochemistry. 1997;36:11205–11215. doi: 10.1021/bi9703812. [DOI] [PubMed] [Google Scholar]
- 8.Seeman NC, Rosenberg JM, Rich A. Sequence-specific recognition of double-helical nucleic acids by proteins. Proc. Natl. Acad. Sci. USA. 1976;73:804–808. doi: 10.1073/pnas.73.3.804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kool ET. Active site tightness and substrate fit in DNA replication. Annu. Rev. Biochem. 2002;71:191–219. doi: 10.1146/annurev.biochem.71.110601.135453. [DOI] [PubMed] [Google Scholar]
- 10.Morales JC, Kool ET. Efficient replication between non-hydrogen-bonded nucleoside shape analogs. Nature Struct. Biol. 1998;5:950–954. doi: 10.1038/2925. [DOI] [PubMed] [Google Scholar]
- 11.Morales JC, Kool ET. Varied molecular interactions at the active sites of several DNA polymerases: nonpolar isosteres as probes. J. Am. Chem. Soc. 2000;122:1001–1007. doi: 10.1021/ja993464+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Washington MT, Helquist SA, Kool ET, Prakash L, Prakash S. Requirement of Watson-Crick hydrogen bonding for DNA synthesis by yeast DNA polymerase η. Mol. Cell. Biol. 2003;23:5107–5112. doi: 10.1128/MCB.23.14.5107-5112.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wolfle WT, Washington MT, Kool ET, Spratt TE, Helquist SA, Prakash L, Prakash S. Evidence for a Watson-Crick hydrogen bonding requirement in DNA synthesis by human DNA polymerase κ. Mol. Cell Biol. 2005;25:7137–7143. doi: 10.1128/MCB.25.16.7137-7143.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morales JC, Kool ET. Minor groove interactions between polymerase and DNA: more essential to replication than Watson-Crick hydrogen bonds? J. Am. Chem. Soc. 1999;121:2323–2324. doi: 10.1021/ja983502+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morales JC, Kool ET. Functional hydrogen-bonding map of the minor groove binding tracks of six DNA polymerases. Biochemistry. 2000;39:12979–12988. doi: 10.1021/bi001578o. [DOI] [PubMed] [Google Scholar]
- 16.Spratt TE. Identification of hydrogen bonds between Escherichia coli DNA polymerase I (Klenow fragment) and the minor groove of DNA by amino acid substitution of the polymerase and atomic substitution of the DNA. Biochemistry. 2001;40:2647–2652. doi: 10.1021/bi002641c. [DOI] [PubMed] [Google Scholar]
- 17.Moran S, Ren RX-F, Rumney S, Kool ET. Difluorotoluene, a nonpolar isostere of thymine, codes specifically and efficiently for adenine in DNA replication. J. Am. Chem. Soc. 1997;119:2056–2057. doi: 10.1021/ja963718g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moran S, Ren RX-F, Kool ET. A thymidine triphosphate shape analog lacking Watson-Crick pairing ability is replicated with high sequence selectivity. Proc. Natl. Acad. Sci. U S A. 1997;94:10506–10511. doi: 10.1073/pnas.94.20.10506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Joyce CM, Derbyshire V. Purification of E. coli DNA polymerase I and Klenow fragment. Methods Enzymol. 1995;262:3–13. doi: 10.1016/0076-6879(95)62003-6. [DOI] [PubMed] [Google Scholar]
- 20.Derbyshire V, Freemont PS, Sanderson MR, Beese L, Friedman JM, Joyce CM, Steitz TA. Genetic and crystallographic studies of the 3′,5′-exonucleolytic site of DNA polymerase I. Science. 1988;240:199–201. doi: 10.1126/science.2832946. [DOI] [PubMed] [Google Scholar]
- 21.Astatke M, Grindley NDF, Joyce CM. How E. coli DNA polymerase I (Klenow fragment) distinguishes between deoxy- and dideoxynucleotides. J. Mol. Biol. 1998;278:147–165. doi: 10.1006/jmbi.1998.1672. [DOI] [PubMed] [Google Scholar]
- 22.Zhou B-L, Pata JD, Steitz TA. Crystal structure of a lesion bypass DNA polymerase catalytic fragment reveals a classical polymerase catalytic domain for the UmuC/DinB enzyme family. Mol. Cell. 2001;8:427–437. doi: 10.1016/s1097-2765(01)00310-0. [DOI] [PubMed] [Google Scholar]
- 23.Ling H, Boudsocq F, Woodgate R, Yang W. Crystal structure of a Y-family DNA polymerase in action: a mechanism for error-prone and lesion-bypass replication. Cell. 2001;107:91–102. doi: 10.1016/s0092-8674(01)00515-3. [DOI] [PubMed] [Google Scholar]
- 24.Potapova O, Grindley NDF, Joyce CM. The mutational specificity of the Dbh lesion bypass polymerase and its implications. J. Biol. Chem. 2002;277:28157–28166. doi: 10.1074/jbc.M202607200. [DOI] [PubMed] [Google Scholar]
- 25.Guckian KM, Krugh TR, Kool ET. Solution structure of a nonpolar, non-hydrogen-bonded base pair surrogate in DNA. J. Am. Chem. Soc. 2000;122:6841–6847. doi: 10.1021/ja994164v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guckian KM, Ren RX-F, Chaudhuri NC, Tahmassebi DC, Kool ET. Factors contributing to aromatic stacking in water: evaluation in the context of DNA. J. Am. Chem. Soc. 2000;122:2213–2227. doi: 10.1021/ja9934854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Joyce CM, Benkovic SJ. DNA polymerase fidelity: kinetics, structure and checkpoints. Biochemistry. 2004;43:14317–14324. doi: 10.1021/bi048422z. [DOI] [PubMed] [Google Scholar]
- 28.Carroll SS, Cowart M, Benkovic SJ. A mutant of DNA polymerase I (Klenow fragment) with reduced fidelity. Biochemistry. 1991;30:804–813. doi: 10.1021/bi00217a034. [DOI] [PubMed] [Google Scholar]
- 29.Minnick DT, Liu L, Grindley NDF, Kunkel TA, Joyce CM. Discrimination against purine-pyrimidine mispairs in the polymerase active site of DNA polymerase I: a structural explanation. Proc. Natl. Acad. Sci. USA. 2002;99:1194–1199. doi: 10.1073/pnas.032457899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim TW, Delaney JC, Essigmann JM, Kool ET. Probing the active site tightness of DNA polymerase in subangstrom increments. Proc. Natl. Acad. Sci. U S A. 2005;102:15803–15808. doi: 10.1073/pnas.0505113102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bell JB, Eckert KA, Joyce CM, Kunkel TA. Base miscoding and strand misalignment errors by mutator polymerases with amino acid substitutions at tyrosine 766 in the O helix of the fingers subdomain. J. Biol. Chem. 1997;272:7345–7351. doi: 10.1074/jbc.272.11.7345. [DOI] [PubMed] [Google Scholar]
- 32.Minnick DT, Bebenek K, Osheroff WP, Turner RM, Jr., Astatke M, Liu L, Kunkel TA, Joyce CM. Side chains that influence fidelity at the polymerase active site of Escherichia coli DNA polymerase I (Klenow fragment) J. Biol. Chem. 1999;274:3067–3075. doi: 10.1074/jbc.274.5.3067. [DOI] [PubMed] [Google Scholar]
- 33.Polesky AH, Steitz TA, Grindley NDF, Joyce CM. Identification of residues critical for the polymerase activity of the Klenow fragment of DNA polymerase I from Escherichia coli. J. Biol. Chem. 1990;265:14579–14591. [PubMed] [Google Scholar]
- 34.Polesky AH, Dahlberg ME, Benkovic SJ, Grindley NDF, Joyce CM. Side chains involved in catalysis of the polymerase reaction of DNA polymerase I from Escherichia coli. J. Biol. Chem. 1992;267:8417–8428. [PubMed] [Google Scholar]
- 35.Meyer AS, Blandino M, Spratt TE. Escherichia coli DNA polymerase I (Klenow Fragment) uses a hydrogen-bonding fork from Arg668 to the primer terminus and incoming deoxynucleotide triphosphate to catalyze DNA replication. J. Biol. Chem. 2004;279:33043–33046. doi: 10.1074/jbc.C400232200. [DOI] [PubMed] [Google Scholar]
- 36.Thompson EHZ, Bailey MF, van der Schans EJC, Joyce CM, Millar DP. Mismatch recognition within the polymerase domain of the Klenow Fragment. Biochemistry. 2002;41:713–722. doi: 10.1021/bi0114271. [DOI] [PubMed] [Google Scholar]
- 37.Silvian LF, Toth EA, Pham P, Goodman MF, Ellenberger T. Crystal structure of a DinB family error-prone DNA polymerase from Sulfolobus solfataricus. Nature Struct. Biol. 2001;8:984–989. doi: 10.1038/nsb1101-984. [DOI] [PubMed] [Google Scholar]
- 38.Trincao J, Johnson RE, Escalante CR, Prakash S, Prakash L, Aggarwal AK. Structure of the catalytic core of S. cerevisiae DNA polymerase η: implications for translesion DNA synthesis. Mol. Cell. 2001;8:417–426. doi: 10.1016/s1097-2765(01)00306-9. [DOI] [PubMed] [Google Scholar]
- 39.Washington MT, Wolfle WT, Spratt TE, Prakash L, Prakash S. Yeast DNA polymerase η makes functional contacts with the DNA minor groove only at the incoming nucleoside triphosphate. Proc. Natl. Acad. Sci. USA. 2003;100:5113–5118. doi: 10.1073/pnas.0837578100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Johnson SJ, Beese LS. Structures of mismatch replication errors observed in a DNA polymerase. Cell. 2004;116:803–816. doi: 10.1016/s0092-8674(04)00252-1. [DOI] [PubMed] [Google Scholar]
- 41.Henry AA, Romesberg FE. Beyond A, C, G and T: augmenting nature’s alphabet. Curr. Opin. Chem. Biol. 2003;7:727–733. doi: 10.1016/j.cbpa.2003.10.011. [DOI] [PubMed] [Google Scholar]
- 42.Chiaramonte M, Moore CL, Kincaid K, Kuchta RD. Facile polymerization of dNTPs bearing unnatural base analogues by DNA polymerase α and Klenow fragment (DNA polymerase I) Biochemistry. 2003;42:10472–10481. doi: 10.1021/bi034763l. [DOI] [PubMed] [Google Scholar]