

Abstract
JNK is a stress-activated protein kinase that modulates pathways implicated in a variety of disease states. JNK-interacting protein-1 (JIP1) is a scaffolding protein that enhances JNK signaling by creating a proximity effect between JNK and upstream kinases. A minimal peptide region derived from JIP1 is able to inhibit JNK activity both in vitro and in cell. We report here a series of small molecules JIP1 mimics that function as substrate competitive inhibitors of JNK. One such compound, BI-78D3, dose-dependently inhibits the phosphorylation of JNK substrates both in vitro and in cell. In animal studies, BI-78D3 not only blocks JNK dependent Con A-induced liver damage but also restores insulin sensitivity in mouse models of type 2 diabetes. Our findings open the way for the development of protein kinase inhibitors targeting substrate specific docking sites, rather than the highly conserved ATP binding sites. In view of its favorable inhibition profile, selectivity, and ability to function in the cellular milieu and in vivo, BI-78D3 represents not only a JNK inhibitor, but also a promising stepping stone toward the development of an innovative class of therapeutics.
Keywords: diabetes, drug discovery, JIP1, kinase, NMR
JNKs are serine threonine protein kinases and members of the MAPK family (1–3). JNKs can be expressed as 10 different isoforms by mRNA alternative splicing of three highly related genes, JNK1, JNK2, and JNK3 (4). Although JNK1 and JNK2 are ubiquitous, JNK3 is principally present in the brain, cardiac muscle, and testis (4, 5). JNK activation by extracellular stimuli, such as stress or cytokines, leads to the phosphorylation of several transcription factors, and cellular substrates implicated in cell survival and proliferation, insulin receptor signaling, and mRNA stabilization (6–9). Because these pathways are related to the pathogenesis of several diseases, including diabetes, cancer, atherosclerosis, stroke, and Alzheimer's and Parkinson's diseases, JNKs represent valuable targets in the development of new therapies (10).
JNKs bind to scaffold proteins and substrates containing a D-domain, consensus sequence of which is R/KXXXXLXL (11, 12). JNK-interacting protein-1 (JIP1) is a scaffolding protein that enhances JNK signaling by creating a proximity effect between JNK and upstream kinases (13). The JNK-JIP1 interaction is mediated by a specific, high affinity D-domain on JIP1, the critical features of which were elucidated by Barr and colleagues (14). Overexpression of either the D-domain of JIP1 or the full-length protein potently inhibits JNK signaling in the cell (15). The minimal region of JIP1, consisting of a single D-domain, has been identified as a JNK inhibitor (14, 16). A peptide corresponding to the D-domain of JIP1 (amino acids 153–163; pepJIP1), inhibits JNK activity in vitro toward recombinant c-Jun, Elk, and ATF2 and displays remarkable selectivity with little inhibition of the closely related Erk and p38 MAPKs (17).
The mechanism of JNK1 inhibition by pepJIP1 is mainly because of the competition of pepJIP1 with the D-domains of substrates or upstream kinases (15, 18). Recent in vivo data, generated in studies focusing on a cell-permeable JIP1 peptide consisting in pepJIP1 fused with the cell permeable HIV-TAT peptide, shows that its administration in both genetically and dietary mice models of insulin resistance and type 2 diabetes restores normoglycemia without causing hypoglycemia in lean mice (19). However, the efficacy of TAT mediated drug delivery is still shrouded by controversy (ref. 20 and references therein). This combined with poor cell permeability, peptide instability, and short half-life in vivo have served to hinder the development of peptide-based inhibitors. We report here a series of pepJIP1 mimics that function as JNK inhibitors both in vitro and in cell and exhibiting in vivo efficacy. Our findings represent a significant advance toward the development of small substrate-competitive inhibitors of JNK and possibly other MAPK family members.
Results and Discussion
Structural data (17) (Fig. 1A) coupled with the known inhibitory properties of pepJIP1 suggests that the JIP interaction site of JNK may be a druggable target. Hence, we screened small-molecule libraries for the ability to disrupt the interaction of pepJIP1 with JNK1 by using the Dissociation Enhanced Lanthanide Fluoro-Immuno Assay (DELFIA) platform. DELFIA is a heterogeneous assay whereby a biotin-linked pepJIP1 is adsorbed onto a streptavidin-coated plate followed by incubation with GST-JNK1. Detection of the pepJIP1/GST-JNK1 complex is facilitated by a highly fluorescent anti-GST Eu-antibody conjugate (Perkin–Elmer). To carry out the screening campaign against JNK, a collection of 30,000 compounds (≈16,000 compounds, Maybridge Corporation/Fisher Scientific; 14,000 compounds, ASDI Biosciences) was prepared in mixtures of 20 and screened by using the DELFIA assay. After identifying mixtures that gave greater than or equal to 50% inhibition at 12.5 μM, we carried out dose-response measurements to filter out eventual false positives. We then performed deconvolution of mixtures and retested the individual compounds to identify actual hits.
Fig. 1.

In vitro characterization of pepJIP1 and BI-78D3. (A) Surface representation of JNK1 in complex with pepJIP1 (RPKRPTTLNLF) and the ATP mimic SP600125 (PDB-ID 1UKI). Surface generated with MOLCAD (28) and color coded according to cavity depth (blue, shallow; yellow, deep). (B) Chemical structure of BI-78D3. (C) Dose dependent displacement of biotinylated pepJIP1 from GST-JNK1. (D) Kinase inhibition assay for BI-78D3.
Hits from the DELFIA assay were further tested for their ability to inhibit JNK1 phosphorylation of ATF2 in the Time-Resolved Fluorescence Resonance Energy Transfer (TR-FRET) basedLanthaScreenTM kinase assay (Invitrogen). The compounds able to inhibit the kinase activity of JNK1 were further screened for effects on cell viability and morphology by propidium iodide staining to eliminate overly cytotoxic compounds. These efforts lead to the identification of 4-(2,3-dihydrobenzol[b][1,4]dioxin-6-yl)-5-(5-nitrothiazol-2-ylthio)-4H-1,2,4-triazol-3-ol (BI-78D3; Fig. 1B). BI-78D3 is able to compete with pepJIP1 for JNK1 binding (IC50 = 500 nM; Fig. 1C), and inhibit the JNK kinase activity (IC50 = 280 nM), both with a Hill slope of approximately 1. Using the same in vitro LanthaScreenTM kinase assay and the same ATF2 substrate, BI-78D3 was found to be 100-fold less active vs. p38α, a member of the MAPK family with high structural similarity to JNK, and completely inactive against mTOR and PI3-kinase (α-isoform), both unrelated protein kinases. Furthermore, Lineweaver–Burk analysis clearly indicates that BI-78D3 is competitive with ATF2 for binding to JNK1 with an apparent K(i) value of 200 nM. In addition, the fact that BI-78D3 does not inhibit the phosphorylation of a short peptide substrate lacking a D-domain (data not shown) further confirms that BI-78D3 is substrate competitive.
To further characterize the binding properties of BI-78D3 we performed computational modeling studies supported by experimental NMR (Fig. 2A) and structure activity relationship (SAR) data (Table 1). The docked structure of BI-78D3 onto the binding surface of JNK1 has many features in common with the recently solved x-ray structure of the ternary complex including pepJIP1, JNK1 and the ATP mimic SP600125 (17) (Fig. 1A). For example, our studies predict that the benzodioxan moiety of BI-78D3 occupies a region corresponding to the highly conserved leucines of pepJIP1 (14). In fact, the removal of the benzodioxan moiety results in a twofold reduction in activity in both the DELFIA and kinase assay (BI-83C9; Table 1). In addition, an extensive network of hydrogen bonding interactions is predicted between the triazole, nitro group, and thiazole of BI-78D3 with JNK1 R127, an interaction that has been demonstrated as critical for pepJIP1 binding to JNK1 (17). Whereas substitution of a triazole with a tetrazole seems to be tolerated (cf. BI-83C9 and BI-83C7; Table 1) modifications to the nitro or thiazole moieties result in a marked reduction in inhibitory activity (cf. BI-83C8, BI-83B3, and BI-83C11; Table 1).
Fig. 2.

Docking studies and NMR analysis. (A) Chemical structure and predicted binding mode of SP600125 derived indazole-TEMPO compound. (B) 1D 1H-NMR T1ρ spectra of BI-78D3 (500 μM) in the presence of 5 μM JNK-2 (blue) or 5 μM JNK-2 and 200 μM ATP-TEMPO (red) at 100 ms. NMR resonance assignments for the hydrogen nuclei of the small molecule are reported. Signal reductions are as follows: H1 46%, H2 56%, H3/H5 65%, and H4 72%. (C) Docked structure of BI-78D3 into the x-ray structure of JNK1 (PDB ID 1UKI). Predicted hydrogen bonding interactions between the compound and the residue Arg-127 (displayed) are highlighted with yellow dashed lines. Computational docking studies were performed with GOLD 2.1 (The Cambridge Crystallographic Data Centre) (28, 29) and analyzed with Sybyl. Molecular models were generated with CONCORD (29) and energy minimized with Sybyl.
Table 1.
In vitro activity data for BI-78D3 and related compounds
| Molecule | ID | MW | DELFIA assay data | LanthaScreenTM kinase assay data |
|---|---|---|---|---|
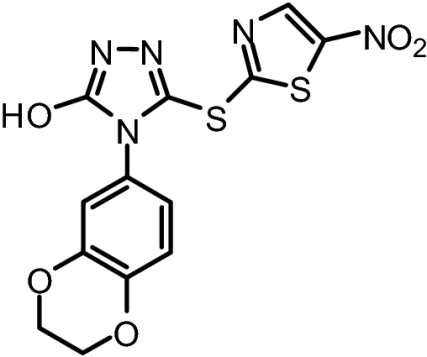 |
BI-78D3* | 379.37 | IC50 = 500 nM | IC50 = 280 nM |
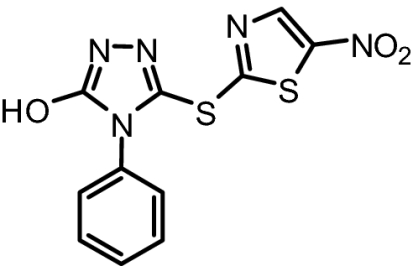 |
BI-83C9 | 321.33 | IC50 = 237 nM | IC50 = 3.1 μM |
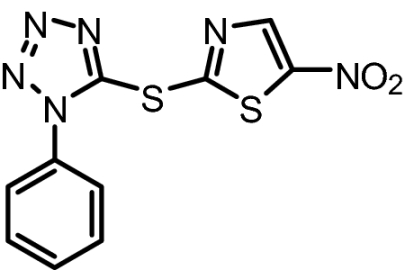 |
BI-83C7 | 306.32 | IC50 = 183 nM | IC50 = 3.3 μM |
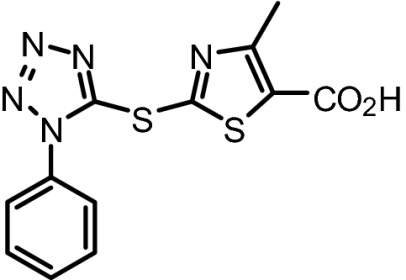 |
BI-83C8 | 319.36 | IC50 = 7.5 μM | No Inhibition at 100 μM |
 |
BI-83B3 | 268.33 | No Displacement at 25 μM | No Inhibition at 100 μM |
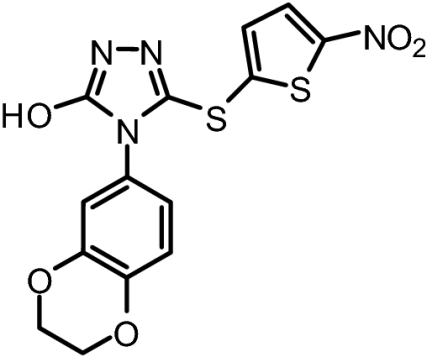 |
BI-83C11 | 378.38 | No Displacement at 25 μM | No Inhibition at 100 μM |
The contribution of JNK1 R127 (JNK2 amino acid numbering is the same) was confirmed in isothermal titration calorimetry (ITC) experiments where the free energy of binding (ΔG) of BI-78D3 to WT JNK2 was compared with that of an R127A mutant thereof. The twofold reduction in the Kd (Fig. 3) was consistent with those data generated for pepJIP1 (17) thus confirming the role of R127 in the binding of BI-78D3. Likewise the docked structure of BI-78D3 onto the binding surface of JNK1 points to a possible role of C162 in BI-78D3 binding. The ITC data shown in Fig. 3 shows a significant decrease in the Kd of BI-78D3 for the C162S JNK2 mutant as compared with the wild-type construct. This finding raises the distinct possibility of a specific involvement of C167 of JNK2 in binding BI-78D3.
Fig. 3.

Binding affinity data. The dissociation constants for BI-78D3 as determined by isothermal titration calorimetry are reported for JNK2 (A), JNK2 (B) R127A, and JNK2 C162S (C).
To confirm the predicted binding mode of BI-78D3, we used a recently developed chemical probe (21) that, by anchoring in the ATP binding site, brings a paramagnetic spin label into proximity of the JIP binding site of JNK (Fig. 2A). The paramagnetic spin label used is an ATP-mimic linked to 2,2,6,6-tetramethylpiperidine 1-oxyl (indazole-TEMPO; Fig. 2A). If a test compound binds in close proximity to indazole-TEMPO, its NMR signal is going to be affected by the unpaired electron. The effect, manifested in a reduction in signal intensity in the 1D 1H-NMR spectrum of the test compound, is distance dependent thus allowing a rough estimate of the binding mode of the test compound with respect to indazole-TEMPO. As shown in Fig. 2C, the signals of BI-78D3 are attenuated when in presence of indazole-TEMPO. This attenuation is strong evidence of binding to JNK in the JIP binding site. Moreover, the signals of the aliphatic protons on the benzodioxan moiety are most affected whereas the signal of the proton on the thiazole ring is least affected (Fig. 2C). These data clearly indicate that the benzodioxan moiety is closest to ATP-TEMPO, thus further confirming the orientation of BI-78D3 predicted by in silico docking (Fig. 2B).
In an attempt to profile the properties of BI-78D3 in the context of a complex cellular milieu, we used the cell-based LanthaScreenTM kinase assay (22). In this assay BI-78D3 is able to inhibit TNF-α stimulated phosphorylation of c-Jun in cell (EC50 = 12.4 μM; Fig. 4A). It should be noted that the cell-based system used makes use of a GFP-c-Jun stable expression system. As a result, the levels of GFP-c-Jun in these cells are higher than endogenous levels. This could have an inflationary effect on the EC50 values obtained with this assay when testing substrate competitive compounds. Nonetheless, this finding establishes that BI-78D3 is able to function in a cellular context. Given that TNF-α stimulates both JNK and p38 and that ATF2 is a substrate for both kinases, one would predict that a JNK selective compound such as BI-78D3 would not affect the ability of p38 to phosphorylate ATF2. Indeed, ATF2 phosphorylation was not affected in the cell-based LanthaScreenTM kinase assay further demonstrating the selectivity of BI-78D3.
Fig. 4.

Bioevaluation studies with BI-78D3. (A) TR-FRET analysis of c-Jun phosphorylation upon TNF-alpha stimulation of HeLa cells in the presence of increasing BI-78D3. (B) BI-78D3 effect on serum alanine-aminotransferase levels after 7.5 h of exposure to ConA compared with DMSO control. Results shown are averages ± SD (n = 7). (C) Effect of BI-78D3 (25 mg/kg) on insulin resistance in 11-week-old BKS.Cg-+Leprdb/+Leprdb/OlaHsd db/db mice (Harlan Sprague–Dawley). Closed diamonds, vehicle control; closed squares, 25 mg/kg BI-78D3. Data shown as means ± SD (n = 5). *, P < 0.0003; **, P < 0.0001.
The link between ConA-induced liver failure, TNF receptor signaling, and JNK function has been established by studies employing JNK1−/− and JNK2−/− mice (23, 24). Likewise, the link between the JNK pathway and type 2 diabetes has been established (10, 25–27). Thus, in an attempt to further our bio-analysis of the JNK-inhibitory properties of BI-78D3, we tested its efficacy in blocking ConA-induced liver failure as measured by the release of the liver enzyme alanine-aminotranferase (ALT) to the circulation (Fig. 4B). Furthermore, we monitored the ability of BI-78D3 to restore insulin sensitivity in a mouse model of type 2 diabetes. For this analysis, insulin insensitive mice from Harlan (Harlan Sprague–Dawley) were injected only once with 25 mg/kg BI-78D3, 30 min before insulin injection. The effect of insulin on blood glucose levels was then measured. BI-78D3 resulted in a statistically significant reduction in blood glucose levels as compared with the vehicle control (Fig. 4C). Thus, the ability of BI-78D3 to abrogate ConA-induced liver damage and restore insulin sensitivity is consistent with its proposed function as an effective JNK inhibitor. Liquid chromatography/mass spectrometry bio-availability analysis demonstrates that BI-78D3 has favorable microsome and plasma stability (T1/2 = 54 min).
In conclusion, we report here a pepJIP1 mimic, BI-78D3 that functions as a substrate competitive inhibitor of JNK. In view of its favorable inhibition profile, selectivity, and ability to function in the cellular milieu and in vivo, BI-78D3 is not only a JNK inhibitor, but also a promising stepping stone toward the development of an innovative class of therapeutics. Further and more extensive SAR studies, including in vivo pharmacokinetics of most promising compounds are ongoing to validate the proposed compound series for continued drug development. Finally, our data should open the way for the development of protein kinase inhibitors targeting substrate specific docking sites, rather than the highly conserved ATP binding sites.
Materials and Methods
DELFIA Assay (Dissociation Enhanced Lanthanide Fluoro-Immuno Assay).
To each well of 96-well streptavidin-coated plates (Perkin-Elmer), 100 μl of a 100 ng/ml solution of biotin-labeled pepJIP1 (Biotin-lc-KRPKRPTTLNLF, where lc indicates a hydrocarbon chain of six methylene groups) was added. After 1-h incubation and elimination of unbound biotin-pepJIP1 by three washing steps, 87 μl of Eu-labeled anti-GST antibody solution (300 ng/ml; 1.9 nM), 2.5 μl DMSO solution containing test compound, and 10-μl solution of GST-JNK2 for a final protein concentration of 10 nM was added. After 1-h incubation at 0°C, each well was washed five times to eliminate unbound protein and the Eu-antibody if displaced by a test compound. Subsequently, 200 μl of enhancement solution (Perkin-Elmer) was added to each well and fluorescence measured after 10-min incubation (excitation wavelength, 340 nm; emission wavelength, 615 nm). Controls include unlabeled peptide and blanks receiving no compounds. Protein and peptide solutions were prepared in DELFIA buffer (Perkin-Elmer).
In Vitro Kinase Assay.
LanthaScreenTM assay platform from Invitrogen was used. The time-resolved fluorescence resonance energy transfer assay (TR-FRET) was performed in 384 well plates. Each well received JNK1 (100 ng/ml), ATF2 (200 nM), and ATP (1 μM) in 50 mM Hepes, 10 mM MgCl2, 1 mM EGTA, 0.01% Brij-35, pH 7.5, and test compounds. The kinase reaction was performed at room temperature for 1 h. After this time, the terbium labeled antibody and EDTA were added into each well. After an additional hour incubation, the signal was measured at 520/495 nm emission ratio on a BMG Pherastar fluorescence plate reader.
NMR Spectroscopy.
NMR spectra were acquired on a 600-MHz Bruker Avance spectrometer equipped with a Bruker TXI probe. All 1D 1H experiments carried out with samples containing unlabeled His-tagged JNK-2 at a concentration of 5 μM. 1D T1ρ experiments were measured with a spin-lock time of 100 (or 50) ms, a recycle delay of 1.5 sec, and water suppression based on the Watergate sequence.
Cell-Based Assays for c-Jun and ATF2 Phosphorylation.
The cell based kinase assays for c-Jun and ATF2 phosphorylation carried out by using the LanthaScreenTM c-Jun (1–79) HeLa and LanthaScreenTM ATF2 (19–106) A549 cell lines (Invitrogen) which stably express GFP-c-Jun 1–79 and GFP-ATF2 19–106, respectively. Phosphorylation was determined by measuring the time resolved FRET (TR-FRET) between a terbium labeled phospho-specific antibody and the GFP-fusion protein (22). The cells were plated in white tissue culture treated 384 well plates at a density of 10,000 cell per well in 32 μl assay medium (Opti-MEM®, supplemented with 1% charcoal/dextran-treated FBS, 100 U/ml penicillin and 100 μg/ml streptomycin, 0.1 mM nonessential amino acids, 1 mM sodium pyruvate, 25 mM Hepes, pH 7.3, and lacking phenol red). After overnight incubation, cells were pretreated for 60 min with compound (indicated concentration) followed by 30 min of stimulation with 2 ng/ml of TNF-α that stimulates both JNK and p38. The medium was then removed by aspiration and the cells were lysed by adding 20 μl of lysis buffer (20 mM Tris·HCl, pH 7.6, 5 mM EDTA, 1% Nonidet P-40 substitute, 5 mM NaF, 150 mM NaCl, and 1:100 protease and phosphatase inhibitor mix, SIGMA P8340 and P2850, respectively). The lysis buffer included 2 nM of the terbium-labeled anti-pc-Jun (pSer73) or anti-pATF2 (pThr71) detection antibodies (Invitrogen). After allowing the assay to equilibrate for 1 h at room temperature, TR-FRET emission ratios were determined on a BMG Pherastar fluorescence plate reader (excitation at 340 nm, emission 520 nm and 490 nm; 100 μs lag time, 200 μs integration time, emission ratio = Em520/Em 490).
Molecular Modeling.
Computational docking studies were performed with GOLD 2.1 (The Cambridge Crystallographic Data Centre (28, 29) and analyzed with Sybyl (Tripos). Molecular surfaces were generated with MOLCAD (30). The x-ray coordinates of JNK1/pepJIP1/SP600125 (PDB-ID 1UKI) were used to dock the compounds. Molecular models were generated with CONCORD (31) and energy minimized with Sybyl. Ten solutions were generated and subsequently ranked according to Chemscore. The top five solutions converged to a similar pose that was used to represent the docked geometry of BI-78D3. Peptide and SP600125 poses reported in Fig. 1 of the manuscript correspond to those obtained directly from the x-ray coordinates.
Isothermal Titration Calorimetry.
Titrations were done by using a VP-ITC calorimeter from Microcal. Wild-type, R127A or C167S JNK2 were used at concentrations between 50 and 100 μM in 20 mM sodium phosphate buffer (pH 7.4) and 0.5% DMSO. BI-78D3 and JIP peptide were used at concentrations 10–15× of the protein in the same buffer. Titrations were carried out at 25°C and repeated between two and four times. Data were analyzed by using Microcal Origin software provided by the ITC manufacturer (Microcal).
Liver Injury Model.
ConA (Sigma) and BI-78D3 was injected i.v. at 10 mg/kg into 6 to 8 weeks old male BL/6 mice. For partial hepatectomy, mice were anesthetized with isofluorane and subjected to midventral laparotomy followed by removal of the left lateral and median lobes. Animals were killed, blood was collected by cardiac puncture, and livers were surgically removed. Serum was separated and analyzed for alanine-aminotranferase levels (Sigma).
Insulin Tolerance Test.
Eleven-week-old male BKS.Cg-+Leprdb/+Leprdb/OlaHsd db/db mice from Harlan (Harlan Sprague–Dawley, Inc.) were randomized based on blood glucose levels acclimated three days before drug dosing. Blood glucose was read by using a hand-held glucose meter (OneTouch Ultra, LifeScanafter tail snipping. Mice were fasted 6 h before i.p. (i.p.) administration of 25 mg/kg BI-78D3. Thirty minutes after test article administration, Bovine Insulin (Sigma, I-0516 at 0.75 mg/kg) was administered via i.p. injection. Blood samples were taken at designated time points and blood glucose levels were measured as described. Food was returned three hours after test article administration.
Chemistry.
All anhydrous solvents were commercially obtained and stored in Sure-seal bottles under nitrogen. All other reagents and solvents were purchased as the highest grade available and used without further purification. TLC (TLC) analysis of reaction mixtures was performed by using Merck silica gel 60 F254 TLC plates, and visualized by using UV light. 1H NMR data were collected by using a 300-MHz Varian instrument and recorded in deuteriochloroform (CDCl3) or dimethyl sulfoxide- d6 (DMSO-d6). Chemical shifts (δ) are reported in ppm referenced to 1H (Me4Si at 0.00). Mass spectral data were acquired on a Shimadzu LCMS-2010EV for low resolution, and on an Agilent ESI-TOF for high resolution and low resolution. Purity of compounds was determined by using a Waters HPLC. Purity of compounds was obtained in a HPLC Breeze from Waters Co. by using an Atlantis T3 3 μm 4.6 × 150 mm reverse-phase column.
Acknowledgments.
In vivo experiments related to the insulin tolerance were performed by Explora Biolabs. Small amounts of compound BI78D3 for research purposing are available on request. This work was supported by National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases branch, Grants R21DK073274 and R24DK080263. Financial support was also provided by Syndexa Pharmaceuticals.
Footnotes
Conflict of interest statement: The series of compounds discovered were licensed to Syndexa Pharmaceuticals. M.P. and M.K. are shareholders and consultants for Syndexa Pharmaceuticals.
This article is a PNAS Direct Submission.
References
- 1.Derijard B, et al. JNK1: A protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell. 1994;76:1025–1037. doi: 10.1016/0092-8674(94)90380-8. [DOI] [PubMed] [Google Scholar]
- 2.Kallunki T, et al. JNK2 contains a specificity-determining region responsible for efficient c-Jun binding and phosphorylation. Genes Dev. 1994;8:2996–3007. doi: 10.1101/gad.8.24.2996. [DOI] [PubMed] [Google Scholar]
- 3.Kyriakis JM, Avruch J. pp54 microtubule-associated protein 2 kinase. A novel serine/threonine protein kinase regulated by phosphorylation and stimulated by poly-L-lysine. J Biol Chem. 1990;265:17355–17363. [PubMed] [Google Scholar]
- 4.Gupta S, et al. Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J. 1996;15:2760–2770. [PMC free article] [PubMed] [Google Scholar]
- 5.Martin JH, Mohit AA, Miller CA. Developmental expression in the mouse nervous system of the p493F12 SAP kinase. Brain Res Mol Brain Res. 1996;35:47–57. doi: 10.1016/0169-328x(95)00181-q. [DOI] [PubMed] [Google Scholar]
- 6.Ip YT, Davis RJ. Signal transduction by the c-Jun N-terminal kinase (JNK)–from inflammation to development. Curr Opin Cell Biol. 1998;10:205–219. doi: 10.1016/s0955-0674(98)80143-9. [DOI] [PubMed] [Google Scholar]
- 7.Leppa S, Bohmann D. Diverse functions of JNK signaling and c-Jun in stress response and apoptosis. Oncogene. 1999;18:6158–6162. doi: 10.1038/sj.onc.1203173. [DOI] [PubMed] [Google Scholar]
- 8.Minden A, Karin M. Regulation and function of the JNK subgroup of MAP kinases. Biochim Biophys Acta. 1997;1333:F85–104. doi: 10.1016/s0304-419x(97)00018-8. [DOI] [PubMed] [Google Scholar]
- 9.Whitmarsh AJ, Davis RJ. Transcription factor AP-1 regulation by mitogen-activated protein kinase signal transduction pathways. J Mol Med. 1996;74:589–607. doi: 10.1007/s001090050063. [DOI] [PubMed] [Google Scholar]
- 10.Manning AM, Davis RJ. Targeting JNK for therapeutic benefit: From junk to gold? Nat Rev Drug Discov. 2003;2:554–565. doi: 10.1038/nrd1132. [DOI] [PubMed] [Google Scholar]
- 11.Kallunki T, Deng T, Hibi M, Karin M. c-Jun can recruit JNK to phosphorylate dimerization partners via specific docking interactions. Cell. 1996;87:929–939. doi: 10.1016/s0092-8674(00)81999-6. [DOI] [PubMed] [Google Scholar]
- 12.Yang SH, Whitmarsh AJ, Davis RJ, Sharrocks AD. Differential targeting of MAP kinases to the ETS-domain transcription factor Elk-1. EMBO J. 1998;17:1740–1749. doi: 10.1093/emboj/17.6.1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Whitmarsh AJ, et al. A mammalian scaffold complex that selectively mediates MAP kinase activation. Science. 1998;281:1671–1674. doi: 10.1126/science.281.5383.1671. [DOI] [PubMed] [Google Scholar]
- 14.Barr RK, Kendrick TS, Bogoyevitch MA. Identification of the critical features of a small peptide inhibitor of JNK activity. J Biol Chem. 2002;277:10987–10997. doi: 10.1074/jbc.M107565200. [DOI] [PubMed] [Google Scholar]
- 15.Dickens M, et al. A cytoplasmic inhibitor of the JNK signal transduction pathway. Science. 1997;277:693–696. doi: 10.1126/science.277.5326.693. [DOI] [PubMed] [Google Scholar]
- 16.Bonny C, et al. Cell-permeable peptide inhibitors of JNK: Novel blockers of beta-cell death. Diabetes. 2001;50:77–82. doi: 10.2337/diabetes.50.1.77. [DOI] [PubMed] [Google Scholar]
- 17.Heo YS, et al. Structural basis for the selective inhibition of JNK1 by the scaffolding protein JIP1 and SP600125. EMBO J. 2004;23:2185–2195. doi: 10.1038/sj.emboj.7600212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ho DT, Bardwell AJ, Abdollahi M, Bardwell L. A docking site in MKK4 mediates high affinity binding to JNK MAPKs and competes with similar docking sites in JNK substrates. J Biol Chem. 2003;278:32662–32672. doi: 10.1074/jbc.M304229200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaneto H, et al. Possible novel therapy for diabetes with cell-permeable JNK-inhibitory peptide. Nat Med. 2004;10:1128–1132. doi: 10.1038/nm1111. [DOI] [PubMed] [Google Scholar]
- 20.Nori A, Kopecek J. Intracellular targeting of polymer-bound drugs for cancer chemotherapy. Adv Drug Deliv Rev. 2005;57:609–636. doi: 10.1016/j.addr.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 21.Vazquez J, et al. Development of paramagnetic probes for molecular recognition studies in protein kinases. J Med Chem. 2008;51:3460–3465. doi: 10.1021/jm800068w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Robers MB, et al. High-throughput cellular assays for regulated posttranslational modifications. Anal Biochem. 2008;372:189–197. doi: 10.1016/j.ab.2007.09.012. [DOI] [PubMed] [Google Scholar]
- 23.Maeda S, et al. IKKbeta is required for prevention of apoptosis mediated by cell-bound but not by circulating TNFalpha. Immunity. 2003;19:725–737. doi: 10.1016/s1074-7613(03)00301-7. [DOI] [PubMed] [Google Scholar]
- 24.Wang Y, et al. Tumor necrosis factor-induced toxic liver injury results from JNK2-dependent activation of caspase-8 and the mitochondrial death pathway. J Biol Chem. 2006;281:15258–15267. doi: 10.1074/jbc.M512953200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bennett BL, Satoh Y, Lewis AJ. JNK: A new therapeutic target for diabetes. Curr Opin Pharmacol. 2003;3:420–425. doi: 10.1016/s1471-4892(03)00068-7. [DOI] [PubMed] [Google Scholar]
- 26.Kaneto H, et al. Oxidative stress and the JNK pathway as a potential therapeutic target for diabetes. Drug News Perspect. 2004;17:447–453. doi: 10.1358/dnp.2004.17.7.863704. [DOI] [PubMed] [Google Scholar]
- 27.Kaneto H, et al. Oxidative stress and the JNK pathway are involved in the development of type 1 and type 2 diabetes. Curr Mol Med. 2007;7:674–686. doi: 10.2174/156652407782564408. [DOI] [PubMed] [Google Scholar]
- 28.Eldridge MD, et al. Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J Comput Aided Mol Des. 1997;11:425–445. doi: 10.1023/a:1007996124545. [DOI] [PubMed] [Google Scholar]
- 29.Jones G, et al. Development and validation of a genetic algorithm for flexible docking. J Mol Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 30.Teschner M, et al. Texture mapping: A new tool for molecular graphics. J Mol Graphics. 1994;12:98–105. doi: 10.1016/0263-7855(94)80074-x. [DOI] [PubMed] [Google Scholar]
- 31.Pearlman R. S. CONCORD. 1998 Distributed by Tripos (St. Louis) [Google Scholar]