

Abstract
Decoys As the Reference State (DARS) is a simple and natural approach to the construction of structure-based intermolecular potentials. The idea is generating a large set of docked conformations with good shape complementarity but without accounting for atom types, and using the frequency of interactions extracted from these decoys as the reference state. In principle, the resulting potential is ideal for finding near-native conformations among structures obtained by docking, and can be combined with other energy terms to be used directly in docking calculations. We investigated the performance of various DARS versions for docking enzyme-inhibitor, antigen-antibody, and other type of complexes. For enzyme-inhibitor pairs, DARS provides both excellent discrimination and docking results, even with very small decoy sets. For antigen-antibody complexes, DARS is slightly better than a number of interaction potentials tested, but results are worse than for enzyme-inhibitor complexes. With a few exceptions, the DARS docking results are also good for the other complexes, despite poor discrimination, and we show that the latter is not a correct test for docking accuracy. The analysis of interactions in antigen-antibody pairs reveals that, in constructing pairwise potentials for such complexes, one should account for the asymmetry of hydrophobic patches on the two sides of the interface. Similar asymmetry does occur in the few other complexes with poor DARS docking results.
INTRODUCTION
Structure-based pairwise potentials (also called knowledge-based or statistical potentials) have emerged as powerful tools for finding near-native conformations in sets of structures generated by search algorithms in macromolecular modeling, and have substantially contributed to improving the accuracy in protein structure prediction (1–8). Such potentials have also been used with success in the discrimination stage of protein-protein docking (9–15). More recently, it was shown that it is even better to use pairwise potentials as part of the scoring function directly in the docking, since one can substantially increase the number of near-native structures found (16,17).
Within the framework of the inverse Boltzmann approach, a statistical potential between two atoms of types I and J, respectively, is defined as
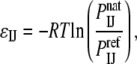 |
where 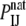 is the probability of contact between the two atoms in the native structure, and
is the probability of contact between the two atoms in the native structure, and 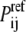 is the probability of the same contact in an appropriate reference state (1). The probability
is the probability of the same contact in an appropriate reference state (1). The probability 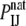 is based on the number
is based on the number 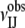 of interactions between atoms of types I and J observed in a protein complex database, usually by calculating the frequency, i.e.,
of interactions between atoms of types I and J observed in a protein complex database, usually by calculating the frequency, i.e.,
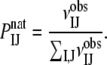 |
However, defining a reference state is more difficult. The general assumption is that the atom-type related properties determining the distribution of pairwise interactions should be removed as much as possible, while retaining all the other properties of the complexes (4). Since experiments do not provide us with such random protein complexes, additional assumptions have to be made, and this is the point where the various structure-based potentials start to differ (4,8,12).
Decoys As the Reference State, or DARS, is a simple and natural approach to the construction of structure-based intermolecular potentials (16). To obtain protein complex structures without atom-type specific interactions for the reference state, we generate a large decoy set of docked conformations based only on shape complementarity as the scoring function, and observe the frequency of interactions in these decoys. Most structure-based potentials have been derived from folded protein structures (2,3,5,7), and generating a meaningful set of random structures would be difficult. However, rigid body protein-protein docking searches only in six dimensions, and it is completely feasible to obtain large sets of docked conformations that do not depend on specific atomic interactions but otherwise look like protein-protein complexes, providing a close to ideal reference state.
The idea of using computationally generated putative decoy ligands for the training of scoring functions has been introduced earlier with applications to virtual screening of small molecular compounds. Smith et al. (18) selected a number of noise molecules, in addition to known ligands, and generated low scoring orientations for both sets of compounds. The parameters of an empirical scoring function for the virtual screening were selected to optimize the average ranking of the known ligand crystal structure for each target within its noise dataset. The method was further developed by Pham and Jain (19,20), who used a library of decoy structures to estimate the value of repulsive terms such as protein-ligand interpenetration instead of relying only upon positive data (protein-ligand complexes of known affinity), again for optimizing screening accuracy. Although DARS also employs negative training (19), we focus on docking rather than screening accuracy. In fact, the decoys are used to obtain a reference state and energy parameters for the optimal selection of correct (i.e., near-native) poses of ligands that, in this case, are also proteins.
As will be discussed, for docking we must employ the DARS potential in combination with other energy terms (e.g., van der Waals and electrostatics). Therefore, we also test various versions of the potential for discrimination accuracy (i.e., the ability of finding near-native conformations in large sets of docked structures). The advantages of the discrimination are that DARS can be used on its own, and the different versions of the potential can be quickly evaluated. However, in this article the discrimination tests are primarily used as surrogates for determining docking accuracy. In fact, our results emphasize that generally it is better to use a high accuracy potential as part of the docking function, rather than generating conformations first and then ranking them based on the potential. Therefore, after selecting the DARS version that provides the best discrimination for a particular class of complexes, we always combine it with the other energy terms and test the resulting function for docking.
It is important to note that the problem of protein-protein docking substantially differs from that of docking small ligands to proteins. In protein-small molecule interactions, the binding pocket of the target is generally known, and due to the restricted nature of the problem and the small size of the ligand, the flexibility of the latter usually can be taken into account. In contrast, in protein-protein docking information on the interaction site is rarely available, and in most cases it is necessary to explore all possible interactions, generating and evaluating billions of putative conformations of the complex. Due to this enormous search space, protein-protein docking generally starts with rigid body search, frequently using simplified protein models and simplified energy functions. The use of rigid protein models requires tolerating some levels of overlaps, and since the energy functions are approximate, the structures that are close to the native conformation do not necessarily have the lowest energies. Thus, to avoid losing potentially useful conformations it is necessary to retain a large number (usually 2000–20,000) of low-energy docked structures for further processing. Thus, the initial docking yields a long list of candidate structures rather than a small number of models, and obtaining meaningful results requires some form of postprocessing, which includes the refinement of the docked conformations, usually accounting for some level of flexibility (21).
Over the last few years we have developed a multistage docking method that performs rigid body docking, retains a number of low energy conformations, clusters them using pairwise RMSD as the distance measure, and then ranks the clusters according to their size, i.e., identifying conformations that have many neighbors within a given clustering radius (11,22). The method is based on the observation that, in the free energy landscapes of partially solvated receptor-ligand complexes, the free energy attractor at the binding site generally has the greatest breadth among all local minima. It was shown that the optimal clustering radius is ∼10 Å—in agreement with the maximum distance two proteins effectively interact in solution (22). Since the native state is identified by clustering, the goal of the rigid body docking is to generate a substantial number of near-native structures or hits within 10 Å RMSD from the native state. Although 10 Å RMSD may appear to be very broad, one has to keep in mind that the prime aim is finding the region of interest in the conformational space, and the structures in this region will be further refined by methods that account for the flexibility of side chains and possibly for the flexibility of some backbone regions.
The goals of this article are the testing and the optimization of DARS potentials for the rigid body docking of enzyme-inhibitor, antigen-antibody, and other type of complexes. The tests will be performed on the complexes of the well-known protein docking benchmark set which, with a few exceptions, includes unbound structures of protein pairs (23). Selecting various reference sets and varying the number of decoys, we have derived and tested many versions of the DARS potential. It is important that we target medium-range potentials that, combined with other energy terms, can produce conformations within 10 Å RMSD from the native state, rather than trying to maximize the fraction of higher accuracy (say, three Å RMSD) structures. The reason is that some side-chain conformations generally differ between bound and unbound states, and due to steric clashes the rigid docking can yield structures with fundamentally correct interactions but with close to 10 Å RMSD. Although the clashes can be easily removed during refinement with flexible side chains, an overly sensitive potential with preference for lower RMSD structures in the rigid body docking could eliminate these conformations. Therefore we try to make the potential relatively flat in the 0–10 Å RMSD range, e.g., by restricting consideration to simple contact potentials rather than developing potentially more sensitive ones with distance-dependent interaction coefficients.
Our results confirm that, for enzyme-inhibitor pairs, DARS provides both excellent discrimination and docking, and the performance remains good even when using very small decoy sets for calculating the reference probabilities. Although DARS is much less accurate for antigen-antibody than for enzyme-inhibitor complexes, it is slightly better than a number of frequently used interaction potentials. Finally, for most other types of complexes, DARS provides strong docking results, substantially better than the ones by the competing potentials, despite its relatively poor performance in the discrimination tests. However, as already mentioned, our primary goal is improving medium-range rigid body docking accuracy, and discrimination is simply a surrogate to test DARS without the additional energy terms.
The performance of DARS for enzyme-inhibitor complexes confirms that the approach can provide an excellent reference state. However, the interpretation of the results for antigen-antibody and other types of complexes is more difficult and poses several questions. First, we explore why pairwise potentials yield worse discrimination and docking results for antigen-antibody than for enzyme-inhibitor complexes. In particular, we argue that for improving potentials for antigen-antibody complexes it is necessary to account for the asymmetry of interactions due to the imperfect complementarity of the hydrophobic patches in the interface. Second, in view of the good docking accuracy but weak discrimination by DARS for most other complexes, we show that discrimination quality is not a valid predictor of docking performance if the component proteins have limited shape complementarity.
METHODS
Developing a DARS potential requires the selection of atom types, defining when two atoms are in contact (i.e., selecting a distance cutoff value), selecting a training set of native protein complex structures, and choosing another set of complexes to generate decoys for the reference state. A benchmark set of complexes is also needed for testing the potential.
Here we use the 18 atom types as introduced for the atomic contact potential (ACP) (24), an atom-level extension of the Miyazawa-Jernigan potential (5), but note that optimizing atom type selection may improve performance (e.g., (25,26)). Atoms i of the receptor (usually the larger protein) and j of the ligand (usually the smaller protein) are considered to interact if their distance rij is <6 Å. For training, we use the nonredundant database of native protein-protein complexes collected by Glaser et al. (27) from the Protein Data Bank (PDB). The original set includes 621 protein interfaces from 492 PDB entries. The nonredundant character of this database was assured by excluding proteins with >30% sequence identity to any other member. We use the protein-protein benchmark set (23) for testing the various potentials. The complexes in the benchmark set were removed from the training set, resulting in 583 interfaces from 466 protein entries. The benchmark set was partitioned into enzyme-inhibitor, antigen-antibody, and other type subsets. As well known (21), these three types of complexes substantially differ from each other in terms of the interface properties, and hence will be treated separately in all tests.
In addition to exploring various DARS parameterizations, we study the performance of three closely related atom-level interaction potentials, the first two based on the same training set of protein complex structures but involving the use of different reference states. In the mole fraction potential (MFP), the reference probability 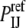 is defined in terms of mole fractions by
is defined in terms of mole fractions by
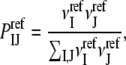 |
where  and
and 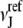 are the numbers of atoms of types I and J, respectively, occurring in a surface layer of each component protein. Thus, the number of contacts between atoms of types I and J is assumed to be proportional to the concentrations of these atoms. This reference state has been used for constructing a variety of interaction potentials (e.g., (3,12,14)). In the uniform reference state (URS) potential
are the numbers of atoms of types I and J, respectively, occurring in a surface layer of each component protein. Thus, the number of contacts between atoms of types I and J is assumed to be proportional to the concentrations of these atoms. This reference state has been used for constructing a variety of interaction potentials (e.g., (3,12,14)). In the uniform reference state (URS) potential 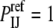 for all I and J, i.e., we assume that all contacts are equally likely. Finally, the atomic contact potential (ACP) (24) was used in our earlier work on protein-protein docking (11).
for all I and J, i.e., we assume that all contacts are equally likely. Finally, the atomic contact potential (ACP) (24) was used in our earlier work on protein-protein docking (11).
Generating decoys for the reference state
As described, the very essence of the DARS method is selecting a set of complexes (the reference set), and for each complex generating a number of docked structures using only shape complementarity as the scoring function. These decoys are then used for calculating the reference probabilities by
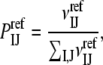 |
where 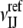 is the number of contacts between atoms of types I and J in the decoys. This involves selecting both a reference set and the number of decoys for each complex. We have tested several options, resulting in a large variety of DARS potentials.
is the number of contacts between atoms of types I and J in the decoys. This involves selecting both a reference set and the number of decoys for each complex. We have tested several options, resulting in a large variety of DARS potentials.
Since we generate up to 20,000 decoys for each complex in the reference set, the size of the latter should be moderate. The main question here is how independent the reference set should be from the training and benchmark sets. One extreme is selecting the benchmark set (23) itself as the reference set. An interesting choice is using the enzyme-inhibitor complexes in the benchmark set as the reference set, since this means a complete overlap when testing DARS on enzyme-inhibitor pairs, but provides complete independence in tests involving antibody-antigen and other types of complexes. Another strategy is selecting proteins that are certainly not homologous to any complex in the benchmark and training sets, and we choose the first 20 targets of the critical assessment of protein interactions (CAPRI) protein docking experiment (28). These targets are novel structures that have been solved after the publication of both benchmark and training sets, which eliminates the possibility of any overlap.
To generate decoys we applied the rigid body docking program PIPER (16) to each complex in the reference set. PIPER is based on the fast Fourier transform correlation approach, and it performs exhaustive evaluation of simplified energy functions in discretized 6D space of mutual orientations of the protein partners (16). In this case, only shape complementarity (with a combination of repulsive and attractive Van der Waals terms) is used for scoring. The 20,000 best scoring docked complexes are kept as the decoy set for calculating the reference probabilities. However, to investigate how the size of the decoy set affects the performance of the potential, the number of decoys for each complex was reduced from 20,000 to 500 and to 1, generating different DARS potentials. If interactions for some atom pair do not occur due to the reduced number of decoys, a large positive interaction potential is assigned.
Discrimination tests
As mentioned, our goal is finding the best DARS potential which, in combination with other energy terms (e.g., van der Waals and electrostatics), will yield sufficiently large sets of near-native docked structures for most of the complexes studied. However, it is easier to evaluate the different versions of DARS first for discrimination accuracy (i.e., the ability of finding near-native conformations in large sets of docked structures). For such discrimination tests we dock the unbound component proteins of the complexes in the benchmark set (23) using only shape complementarity as the scoring function, and retain the best 20,000 structures for each complex. Although this step is the same as generating decoys for the reference proteins, the goal is very different. For each docked structure, we calculate the pairwise energy  where Nr and Nl denote the numbers of atoms in the receptor and the ligand, respectively. For atoms ai and aj of types I and J, respectively, ɛij = ɛIJ if ai and aj are within the cutoff distance D, and ɛij = 0 otherwise. We use the Epair values to rank the 20,000 decoys and select the 2000 structures (1000 for enzyme-inhibitor complexes) with the lowest energies. The quality of these structures is measured in terms of the Cα RMSD between ligand positions in the docked and the experimentally determined structures, calculated after superimposing the receptors and considering only ligand atoms that are within 10 Å from the receptor. This measure, also used in the CAPRI docking experiment (28), will be referred to as binding site RMSD or simply RMSD. A conformation is considered near-native (also called a hit) if its RMSD is <10 Å. As noted, although the 10 Å threshold may appear to be large, the RMSD of such structures can generally be reduced by refinement methods that account for side-chain flexibility (29,30).
where Nr and Nl denote the numbers of atoms in the receptor and the ligand, respectively. For atoms ai and aj of types I and J, respectively, ɛij = ɛIJ if ai and aj are within the cutoff distance D, and ɛij = 0 otherwise. We use the Epair values to rank the 20,000 decoys and select the 2000 structures (1000 for enzyme-inhibitor complexes) with the lowest energies. The quality of these structures is measured in terms of the Cα RMSD between ligand positions in the docked and the experimentally determined structures, calculated after superimposing the receptors and considering only ligand atoms that are within 10 Å from the receptor. This measure, also used in the CAPRI docking experiment (28), will be referred to as binding site RMSD or simply RMSD. A conformation is considered near-native (also called a hit) if its RMSD is <10 Å. As noted, although the 10 Å threshold may appear to be large, the RMSD of such structures can generally be reduced by refinement methods that account for side-chain flexibility (29,30).
For enzyme-inhibitor and antibody-antigen complexes in the benchmark set, the best scoring 20,000 structures generated by PIPER were retained for the discrimination test. However, this produced too few near-native structures for other complexes, and hence we used PIPER to generate 70,000 structures and selected the 20,000 with the lowest RMSDs. Since the number of near-native structures among the 20,000 is known, the quality of discrimination for each complex can be described in terms of the receiver operating characteristic (ROC) curve by providing the area under the curve (AUC) value (31). To describe the discrimination quality by each method applied to a set of protein complexes we present both the median and cumulative distribution of the ROC AUC values. The cumulative distributions for the different methods will be compared using Kolmogorov-Smirnov tests.
Docking tests
In these tests we dock the unbound component proteins of the benchmark set (23) using our PIPER program with a combined energy function, and count the number of near-native conformations among the best scoring 2000 (1000 for the enzyme-inhibitor case) structures. The energy function includes terms representing shape complementarity, electrostatic, and desolvation contributions, the latter described by the pairwise potential to be tested,
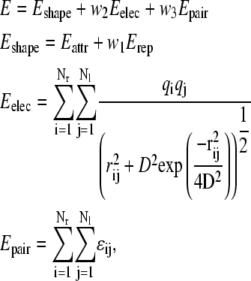 |
where Nr and Nl denote the numbers of atoms in the receptor and the ligand, respectively. The shape complementarity term Eshape is a stepwise implementation of the van der Waals energy, with Eattr and Erep representing its attractive and repulsive components, respectively. Eelec is the Coulombic electrostatic energy, and Epair denotes the pairwise potential defined in the previous section. The implementation of these energy terms on grids has been described previously (16).
The pairwise potential Epair we consider in the docking tests is either DARS or the atomic contact potential (ACP (24)). To assess how the individual energy contributions affect the results, we perform docking calculations with scoring functions that include only the shape complementarity term Eshape, the electrostatic term Eelec, the combination of the two (Eshape + w2Eelec), the combination of the shape complementarity term with the pairwise potential (Eshape + w3Epair), and finally all three terms (Eshape + w2 Eelec + w3 Epair). The performances of the highest scoring DARS and ACP potentials were compared. The w3 coefficients in the energy expression are optimally selected for the particular potential, and hence are different for DARS and ACP.
Since the number of near-native structures over the entire (discretized) conformational space is not determined, no ROC AUC values can be defined for the docking test. However, for any particular set of complexes we can directly compare the numbers of near-native structures provided by two different methods by using the Wilcoxon matched-pair signed-rank test (32), which can be considered as a nonparametric alternative to the paired t-test. The same test can also be used for comparing the number of near-native conformations among the best-scoring 1000 structures from the discrimination test and the number of near-native conformations in the best-scoring 1000 structures obtained directly by docking, where the latter is based on the use of a scoring function that combines DARS with other energy terms.
RESULTS
Enzyme-inhibitor complexes
Table 1 shows discrimination results for 22 enzyme-inhibitor complexes, including the total number of hits among the 20,000 structures generated by PIPER and the number of hits retained in the 1000 best scoring structures selected using DARS, MFPs, URS potentials, and ACPs. The reference probabilities for DARS were obtained using 20,000 decoys for each complex in the CAPRI set. The mole fractions for MFP were extracted from the training set (27). Fig. 1 shows the cumulative distributions of the ROC AUC values for the four methods. Based on the Kolmogorov-Smirnov test, DARS is significantly better (p < 10−5) than any of the three other methods. URS is somewhat better than MFP (p < 0.05), and both URS and MFP are better than ACP (p < 0.01). Since DARS, MFP, and URS are based on the same training set (27) but use different reference states, this result emphasizes the importance of the latter. Table 1 also shows a hydrophobicity score, to be described in the Discussion.
TABLE 1.
Discrimination results for enzyme-inhibitor complexes
| Number of hits in 20,000 decoys | Number of hits in top 1000 decoys selected by
|
Hydrophobicity score | ||||
|---|---|---|---|---|---|---|
| Complex | DARS | MFP* | URS† | ACP‡ | ||
| 1ACB | 261 | 214 | 0 | 100 | 5 | −106.901 |
| 1AVW | 59 | 48 | 49 | 0 | 0 | −114.456 |
| 1BRC | 3384 | 401 | 439 | 198 | 320 | −43.752 |
| 1BRS | 1330 | 270 | 0 | 208 | 50 | −41.276 |
| 1CGI | 943 | 364 | 32 | 127 | 105 | −168.231 |
| 1CHO | 273 | 250 | 1 | 28 | 39 | −51.798 |
| 1CSE | 523 | 86 | 0 | 21 | 7 | −32.692 |
| 1DFJ | 522 | 116 | 40 | 14 | 2 | −42.782 |
| 1FSS | 2 | 2 | 0 | 1 | 0 | −104.730 |
| 1MAH | 3 | 3 | 0 | 3 | 0 | −118.081 |
| 1PPE | 3145 | 838 | 152 | 480 | 284 | −108.031 |
| 1STF | 160 | 102 | 0 | 37 | 0 | −116.980 |
| 1TAB | 871 | 76 | 131 | 147 | 89 | −29.751 |
| 1TGS | 2180 | 595 | 96 | 385 | 448 | −80.820 |
| 1UDI | 37 | 37 | 17 | 11 | 0 | −116.158 |
| 1UGH | 63 | 63 | 46 | 21 | 0 | −133.291 |
| 2KAI | 104 | 0 | 39 | 3 | 2 | −6.454 |
| 2PTC | 1070 | 154 | 84 | 11 | 20 | −57.769 |
| 2SIC | 223 | 97 | 0 | 28 | 18 | −106.294 |
| 2SNI | 112 | 35 | 9 | 1 | 0 | −107.537 |
| 2TEC | 432 | 95 | 0 | 15 | 58 | −49.554 |
| 4HTC | 390 | 261 | 0 | 6 | 8 | −287.830 |
Mole fraction potential.
Uniform reference state potential.
Atomic contact potential.
FIGURE 1.

Cumulative distributions of the ROC AUC values for the discrimination of near-native structures of enzyme-inhibitor complexes.
Table 2 compares the overall discriminatory performance of the different potentials, including the various parameterizations of DARS, in terms of the median ROC AUC values. The table starts with the worst performers, i.e., the ACP, MFP using CAPRI for calculating the mole fractions, MFP with mole fractions derived from the entire training set (27), and the URS potential. The remainder of the table shows the performance of DARS potentials with different reference sets and/or different numbers of decoys generated for each complex. According to the last five rows, DARS performs reasonably well for enzyme-inhibitor complexes regardless of the specific choice of the reference set as long as the latter comprises of a wide range of complexes such as the 20 CAPRI targets. In the latter case, the quality of discrimination is completely independent of the number of decoys, and DARS performs very well even with a single decoy for each complex.
TABLE 2.
Overall discrimination for enzyme-inhibitor complexes by various potentials
| Potential | Reference set | Number of decoys | ROC† |
|---|---|---|---|
| ACP | — | — | 0.464 |
| MFP | CAPRI | — | 0.585 |
| MFP | Training | — | 0.645 |
| URS | — | — | 0.718 |
| DARS (clustered) | CAPRI | 20000 | 0.775 |
| DARS | Benchmark (E-I)* | 20000 | 0.811 |
| DARS | Benchmark | 20000 | 0.843 |
| DARS | CAPRI | 1 | 0.854 |
| DARS | CAPRI | 500 | 0.853 |
| DARS | CAPRI | 20000 | 0.854 |
Enzyme-inhibitor subset of the benchmark set.
Median ROC AUC.
Table 3 shows docking results for the enzyme-inhibitor subset of the benchmark set obtained using different combinations of energy function terms, including the best scoring DARS potential for the enzyme-inhibitor set as shown in Tables 1 and 2. With the exception of 1TAB, where it has a slight negative effect, adding DARS to shape complementarity and electrostatics greatly increases the number of hits generated. The combination of shape complementarity and DARS terms performs better than the combination of shape complementarity and electrostatics, and the best scoring function to capture hits includes all three terms. The only complex where the combined potential does not generate any near-native structures is 2KAI. The most likely origin of this problem is the steric clash of side chains when the unbound proteins are superimposed over their structures in the complex. Table 3 also shows the docking results obtained with the ACP in place of DARS in the scoring function. Based on the Wilcoxon matched-pair signed-rank test (32), the combined potential with DARS is significantly better (p < 0.0003). In fact, apart from the complexes 1TAB and 2KAI, adding ACP to shape complementarity and electrostatic terms is substantially less favorable than adding DARS.
TABLE 3.
Number of hits in the top 1000 docked structures for enzyme-inhibitor complexes
| DARS
|
ACP
|
||||||
|---|---|---|---|---|---|---|---|
| Complex | V* | E† | VE‡ | VP¶ | VEP§ | VP¶ | VEP§ |
| 1ACB | 14 | 0 | 51 | 346 | 436 | 15 | 36 |
| 1AVW | 0 | 0 | 0 | 42 | 75 | 0 | 0 |
| 1BRC | 286 | 178 | 465 | 389 | 566 | 304 | 432 |
| 1BRS | 34 | 0 | 111 | 133 | 164 | 18 | 99 |
| 1CGI | 78 | 0 | 102 | 376 | 327 | 78 | 100 |
| 1CHO | 43 | 0 | 85 | 73 | 127 | 49 | 99 |
| 1CSE | 0 | 0 | 0 | 39 | 52 | 0 | 0 |
| 1DFJ | 67 | 237 | 237 | 136 | 382 | 74 | 245 |
| 1FSS | 0 | 23 | 0 | 1 | 30 | 0 | 0 |
| 1MAH | 0 | 1 | 0 | 63 | 107 | 0 | 0 |
| 1PPE | 417 | 0 | 414 | 847 | 899 | 411 | 411 |
| 1STF | 33 | 0 | 41 | 153 | 168 | 29 | 37 |
| 1TAB | 199 | 0 | 249 | 133 | 120 | 195 | 257 |
| 1TGS | 300 | 0 | 303 | 506 | 478 | 317 | 325 |
| 1UDI | 0 | 14 | 10 | 206 | 353 | 0 | 10 |
| 1UGH | 11 | 0 | 22 | 279 | 352 | 10 | 18 |
| 2KAI | 2 | 0 | 3 | 0 | 0 | 3 | 3 |
| 2PTC | 79 | 0 | 103 | 220 | 243 | 62 | 82 |
| 2SIC | 28 | 0 | 40 | 332 | 291 | 32 | 39 |
| 2SNI | 7 | 0 | 7 | 59 | 65 | 5 | 7 |
| 2TEC | 5 | 0 | 17 | 159 | 197 | 4 | 19 |
| 4HTC | 74 | 0 | 26 | 252 | 201 | 56 | 19 |
Eshape.
Eelec.
Eshape + w2 Eelec.
Eshape + w3Epair..
Eshape + w2Eelec + w3Epair.
Antigen-antibody complexes
For antigen-antibody complexes, both discrimination and docking are generally more difficult than for enzyme-inhibitor complexes, and hence we retain the 2000 (rather than the 1000) best scoring conformations. As shown in Table 4, discrimination by DARS substantially varies among the complexes. According to the cumulative distributions of the ROC AUC values (Fig. 2), the discrimination results for DARS and MFP do not significantly differ (p ≈ 0.2). Both DARS and MFP are significantly better than URS or ACP (p < 0.05) by the Kolmogorov-Smirnov test, but the differences are not significant (p ≈ 0.2) by the Wilcoxon test. As shown in Table 5, discrimination results are weakest for ACP and URS. In terms of the median ROC AUC values, MFP with mole fractions based on the CAPRI set is slightly better than the best DARS. However, as discussed, the difference is not significant. The docking results for antigen-antibody pairs are generally also worse than for enzyme-inhibitor complexes (Table 6). Shape complementarity alone captures very few hits. Adding DARS to Evdw improves the result to a certain degree. The combination of shape complementarity, electrostatics, and DARS provides the best performance, better than the energy function that includes ACP (p < 0.01 based on the Wilcoxon test).
TABLE 4.
Discrimination results for antigen-antibody complexes
| Number of hits in 20,000 decoys | Number of hits in top 2000 decoys selected by
|
Hydrophobicity score | ||||
|---|---|---|---|---|---|---|
| Complex | DARS | MFP* | URS† | ACP‡ | ||
| 1AHW | 194 | 3 | 13 | 0 | 1 | −17.579 |
| 1BQL | 140 | 0 | 49 | 0 | 0 | −54.895 |
| 1BVK | 206 | 81 | 4 | 2 | 3 | −36.090 |
| 1DQJ | 111 | 15 | 75 | 0 | 9 | −7.955 |
| 1EO8 | 41 | 40 | 40 | 0 | 0 | −142.676 |
| 1FBI | 141 | 0 | 0 | 0 | 0 | −33.439 |
| 1IAI | 136 | 72 | 63 | 9 | 7 | −151.531 |
| 1JHL | 243 | 9 | 1 | 5 | 0 | −43.759 |
| 1MEL | 464 | 407 | 72 | 218 | 134 | −150.946 |
| 1MLC | 48 | 4 | 35 | 48 | 48 | −16.247 |
| 1NCA | 147 | 1 | 1 | 0 | 3 | −61.264 |
| 1NMB | 0 | 0 | 0 | 0 | 0 | −29.297 |
| 1QFU | 247 | 206 | 1 | 7 | 3 | −142.245 |
| 1WEJ | 947 | 208 | 226 | 3 | 3 | −29.742 |
| 2JEL | 327 | 134 | 134 | 6 | 0 | −64.268 |
| 2VIR | 120 | 39 | 25 | 5 | 12 | −90.544 |
Mole fraction potential.
Uniform reference state potential.
Atomic contact potential.
FIGURE 2.

Cumulative distributions of the ROC AUC values for the discrimination of near-native structures of antigen-antibody complexes.
TABLE 5.
Overall discrimination for antigen-antibody complexes by various potentials
| Potential | Reference set | Number of decoys | ROC† |
|---|---|---|---|
| URS | — | — | 0.453 |
| ACP | — | — | 0.376 |
| MFP | CAPRI | — | 0.716 |
| MFP | Training | — | 0.619 |
| DARS | CAPRI | 20000 | 0.610 |
| DARS (clustered) | CAPRI | 20000 | 0.599 |
| DARS | Benchmark | 20000 | 0.675 |
| DARS | Benchmark (E-I)* | 20000 | 0.656 |
Enzyme-inhibitor subset of the benchmark set.
Median ROC AUC.
TABLE 6.
Number of hits in the top 2000 docked structures for antigen-antibody complexes
| DARS
|
ACP
|
||||||
|---|---|---|---|---|---|---|---|
| Complex | V* | E† | VE‡ | VP¶ | VEP§ | VP¶ | VEP§ |
| 1AHW | 5 | 251 | 70 | 44 | 103 | 3 | 34 |
| 1BQL | 0 | 0 | 0 | 0 | 0 | 2 | 0 |
| 1BVK | 15 | 0 | 16 | 65 | 60 | 54 | 56 |
| 1DQJ | 0 | 0 | 0 | 15 | 14 | 2 | 0 |
| 1EO8 | 0 | 0 | 0 | 26 | 30 | 0 | 0 |
| 1FBI | 3 | 0 | 11 | 0 | 0 | 0 | 3 |
| 1IAI | 10 | 0 | 8 | 135 | 135 | 45 | 46 |
| 1JHL | 28 | 0 | 27 | 12 | 11 | 17 | 14 |
| 1MEL | 30 | 0 | 14 | 260 | 241 | 74 | 72 |
| 1MLC | 0 | 0 | 1 | 3 | 20 | 5 | 19 |
| 1NCA | 5 | 0 | 12 | 0 | 0 | 4 | 8 |
| 1NMB | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 1QFU | 16 | 0 | 23 | 99 | 111 | 0 | 0 |
| 1WEJ | 118 | 5 | 226 | 79 | 241 | 105 | 167 |
| 2JEL | 10 | 0 | 35 | 56 | 83 | 9 | 20 |
| 2VIR | 10 | 0 | 11 | 16 | 17 | 11 | 11 |
Eshape.
Eelec.
Eshape + w2 Eelec.
Eshape + w3Epair.
Eshape + w2Eelec + w3Epair.
Other complexes
We recall that, for the other complexes, the 20,000 PIPER-generated structures with the best shape complementarity do not provide enough hits, and hence we selected the 20,000 structures with the lowest RMSD values to obtain a more meaningful decoy set for the discrimination test. Although we retain the best scoring 2000 structures, there are very few hits in Table 7, which was derived using the enzyme-inhibitor subset of the benchmark set as the reference. As shown in Fig. 3, DARS provides worse discrimination than the other three potentials, and the differences are significant by both the Kolmogorov-Smirnov and Wilcoxon tests (p < 0.01). The best discrimination is achieved by ACP, which is significantly better than the other three by the Kolmogorov-Smirnov test (p < 0.01), but not better than MFP or URS by the Wilcoxon test. Discrimination by DARS is poor using any reference set, and the other potentials perform better than DARS (Table 8). However, as shown in Table 9, in a complete reversal the docking results are significantly better using DARS than using ACP (p < 0.05 by the Wilcoxon test), with DARS producing a substantial number of hits for seven of the 10 complexes.
TABLE 7.
Discrimination results for other type of complexes
| Number of hits in 20,000 decoys | Number of hits in top 2000 decoys selected by
|
Hydrophobicity score | ||||
|---|---|---|---|---|---|---|
| Complex | DARS | MFP* | URS† | ACP‡ | ||
| 1AVZ | 117 | 25 | 20 | 60 | 30 | −62.695 |
| 1L0Y | 104 | 7 | 15 | 47 | 33 | −7.092 |
| 1A0O | 86 | 0 | 41 | 7 | 18 | −80.814 |
| 1ATN | 17 | 0 | 1 | 7 | 8 | −124.488 |
| 1GLA | 4 | 0 | 0 | 3 | 0 | −105.213 |
| 1IGC | 51 | 1 | 1 | 25 | 14 | −45.924 |
| 1SPB | 362 | 34 | 38 | 76 | 84 | −87.611 |
| 2BTF | 200 | 20 | 53 | 39 | 35 | −67.276 |
| 1WQ1 | 366 | 7 | 62 | 121 | 106 | −102.925 |
| 2PCC | 79 | 1 | 4 | 20 | 18 | −28.573 |
Mole fraction potential.
Uniform reference state potential.
Atomic contact potential.
FIGURE 3.

Cumulative distributions of the ROC AUC values for the discrimination of near-native structures of other complexes.
TABLE 8.
Overall discrimination for other type of complexes by various potentials
| Potential | Reference set | Number of decoys | ROC† |
|---|---|---|---|
| DARS | CAPRI | 20,000 | 0.364 |
| DARS | Benchmark | 20,000 | 0.362 |
| DARS | Benchmark (E-I)* | 20,000 | 0.399 |
| DARS (clustered) | CAPRI | 20,000 | 0.361 |
| MFP | Training | — | 0.611 |
| MFP | CAPRI | — | 0.608 |
| ACP | — | — | 0.666 |
| URS | — | — | 0.723 |
Enzyme-inhibitor subset of the benchmark set.
Median ROC AUC.
TABLE 9.
Number of hits in the top 2000 docked structures for other type of complexes
| DARS
|
ACP
|
||||||
|---|---|---|---|---|---|---|---|
| Complex | V* | E† | VE‡ | VP¶ | VEP§ | VP¶ | VEP§ |
| 1AVZ | 0 | 5 | 1 | 0 | 0 | 0 | 0 |
| 1L0Y | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 1A0O | 0 | 873 | 12 | 0 | 198 | 0 | 30 |
| 1ATN | 0 | 0 | 0 | 105 | 196 | 0 | 0 |
| 1GLA | 0 | 0 | 8 | 323 | 434 | 0 | 8 |
| 1IGC | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 1SPB | 105 | 0 | 107 | 440 | 487 | 100 | 96 |
| 2BTF | 0 | 0 | 7 | 21 | 71 | 0 | 0 |
| 1WQ1 | 248 | 27 | 436 | 192 | 429 | 291 | 430 |
| 2PCC | 0 | 35 | 50 | 0 | 166 | 0 | 44 |
Eshape.
Eelec.
Eshape + w2 Eelec.
Eshape + w3Epair.
Eshape + w2Eelec + w3Epair.
DISCUSSION
Enzyme-inhibitor complexes
Developing DARS potentials we compare the frequency of contacts between two specific atom types in the x-ray structures of protein complexes to the frequency of contacts in the decoys that are devoid of specific interactions. Since in discrimination tests the goal is finding complex conformations close to the native among the many structures that all have good shape complementarity, this scoring scheme is very natural, as it rewards the occurrence in the interface of the atom pairs that are frequently seen to interact in the native complexes. Thus, we expect excellent results both in discrimination and docking, and the results for enzyme-inhibitor complexes shown in Tables 1–3 fully support this expectation. DARS performs much better than the other potentials considered here (MFP, URS, and ACP).
Two important factors are likely to contribute to this success. First, docking enzyme-inhibitor complexes is relatively easy (21). The affinity is generally high, with ΔG values ranging from −17.5 kcal/mol to −13.0 kcal/mol. The convex-concave interface has good geometric complementarity, and it is largely desolvated, with crystallographic water molecules visible only around the perimeter (33,34). It will be important for our discussion that there is generally very good complementarity of hydrophobic patches on the two sides of the interface, favorably contributing to the binding free energy. The second factor is that the current training set is very good for developing potentials for enzyme-inhibitor complexes. In fact, of the 621 interfaces, 404 are from homodimers that, similarly to enzyme-inhibitor complexes, have excellent pairing of shapes and hydrophobic patches on the two sides of the interface. In addition, the set also includes a number of enzyme-inhibitor pairs.
Due to these favorable conditions, testing DARS on enzyme-inhibitor complexes provides the best opportunity to explore the fundamental properties of the approach. First, results show that DARS performs reasonably well regardless of the specific choice of the reference set as long as the latter includes a wide range of complexes. Second, the performance remains excellent even when using very small decoy sets for calculating the reference probabilities. As shown in Table 2, the 20 complexes in the CAPRI set provide an adequate number of contacts for most atom pairs even with a single decoy structure for each complex. We think that this independence of the number of decoys is due to the clustering of the ligand positions at a few locations on the receptor surface (35). Therefore, increasing the number of decoys yields almost no new interactions. This observation also explains why clustering of the decoys with a given clustering radius and selecting a single representative from each cluster performs worse than its unclustered counterpart (Table 2). In fact, since only a few clusters are well populated, with this strategy we force a number of outliers into the decoy set with the same weights as the representatives of the meaningful clusters.
As already mentioned, the primary goal of developing DARS potentials is to improve docking results, but the different DARS versions and other methods are also compared in substantially simpler discrimination tests. In these tests, a large number (20,000) structures are generated by shape complementarity alone, and ranked by one of the pairwise potentials. The number of near-native conformations among the best scoring 1000 structures can be compared to the number of near-native conformations among the top 1000 structures generated by docking using Eshape + w3 Epair. For enzyme-inhibitor complexes discrimination and docking perform equally well, with no significant difference (p > 0.1 by the Wilcoxon test). However, adding electrostatics to the docking potential, i.e., using Eshape + w2 Eelec + w3 Epair makes the docking results significantly better (p < 0.001) than the discrimination results.
Antigen-antibody complexes
It is well known that docking antigen-Fab complexes is more challenging than docking enzyme-inhibitor complexes (21). The complexes are generally weaker, with ΔG values ranging from −13.0 kcal/mol to −6.5 kcal/mol. Since the interfaces are close to planar, shape complementarity provides limited information for docking. The interface is generally less hydrophobic than in enzyme-inhibitor complexes (33,34). According to Tables 4–6, both discrimination and docking results are relatively poor. We note that not only DARS but all four pairwise potentials (DARS, MFP, URS, and ACP) have difficulties with antigen-antibody complexes.
Since we assume that more accurate DARS potentials can be developed specifically for antigen-antibody complexes, it is important to explore the problems that reduce performance. As shown in Table 5, the selection of the reference set had no major impact on the results. A potentially more important factor is the training set. As noted, the current training set is biased toward homodimers and enzyme-inhibitor complexes that have excellent pairing of hydrophobic patches on the two sides of the interface. Therefore, interactions between hydrophobic atoms contribute very favorably to the energy function (16). It is easy to show that such a potential works for some but not for all antigen-antibody complexes. In fact, one can raise antibodies against virtually any region of an antigen surface, with some of the epitopes being fairly polar. For example, Fig. 4, a and b, shows complexes of lysozyme with the variable domain of Fab fragments from two different antibodies (PDB codes 1BQL and 1MLC, respectively). In both figures the Fab fragment is shown as the white solid model, with teal patches representing the regions with maximum hydrophobicity. The lysozyme is shown as a brown cartoon, with light brown patches as regions with maximum hydrophobicity. The CDRs are oriented upward, the teal hydrophobic patch sitting among the hypervariable loops, directly interacting with the lysozyme. However, the two most hydrophobic regions of the lysozyme do not directly interact with the CDRs in either of the antibodies. We understand that the interface on the lysozyme must exhibit some level of hydrophobicity, or otherwise binding would become highly unfavorable. However, according to Fig. 4, a and b, these interface regions are far from being the most hydrophobic ones on the lysozyme.
FIGURE 4.

Patches of maximum hydrophobicity in antigen-antibody complexes. (a) Hyhel-5 Fab antibody fragment in complex with chicken lysozyme (PDB code 1BQL). (b) Monoclonal antibody Fab D44.1 in complex with chicken lysozyme (PDB code 1MLC). In both panels the antibody fragment is shown as the white solid model, with teal patches representing the regions with maximum hydrophobicity. The lysozyme is shown as a brown cartoon, with light brown patches as regions of maximum hydrophobicity. In both figures the antibody CDRs are oriented upward, showing that the CDR regions include strongly hydrophobic patches, but these do not interact with regions of maximum hydrophobicity on the lysozyme.
To demonstrate the importance of hydrophobic interactions, we calculated the pairwise hydrophobicity score  where the sum is restricted to hydrophobic atoms on the two sides of the interface, i.e., interaction energies among all other atoms types are set to zero. As shown in Table 4, the Ehyd values vary substantially for antigen-antibody complexes, and a favorable (large negative) Ehyd generally implies good discrimination. The correlation coefficient between the Ehyd values in Table 4 and the corresponding enrichment factors is r = −0.88. This correlation is highly significant (p < 0.0001), confirming that good complementarity of hydrophobic patches on the two sides of the interface is required for successful discrimination, but this occurs only in some of the antigen-antibody complexes.
where the sum is restricted to hydrophobic atoms on the two sides of the interface, i.e., interaction energies among all other atoms types are set to zero. As shown in Table 4, the Ehyd values vary substantially for antigen-antibody complexes, and a favorable (large negative) Ehyd generally implies good discrimination. The correlation coefficient between the Ehyd values in Table 4 and the corresponding enrichment factors is r = −0.88. This correlation is highly significant (p < 0.0001), confirming that good complementarity of hydrophobic patches on the two sides of the interface is required for successful discrimination, but this occurs only in some of the antigen-antibody complexes.
The presence of hydrophobic complementarity in enzyme-inhibitor complexes (see Table 1) and the lack of it in antigen-antibody pairs could simply indicate that the current DARS potential is overtrained on a set that does not properly represent the interactions specific to antigen-antibody complexes. However, training the potential on antigen-antibody structures (36) did not improve discrimination, and hence the results are not shown here. We argue that the accuracy of pairwise potentials for antigen-antibody complexes is reduced by the usual, and so far unquestioned, assumption that ɛIJ = ɛJI, where I and J denote the atom types in the antigen and in the antibody, respectively. Due to this assumption of symmetry, the current pairwise potentials are unable to distinguish between atoms on the antibody and on the antigen. Therefore, any docked conformation that aligns the hydrophobic patches well is considered favorably, resulting in false positives for antibodies that recognize relatively polar epitopes. We currently explore several potential solutions to this problem, including the use of one-side hydrophobicity terms in the scoring function and the construction of asymmetric DARS potentials.
Other complexes
As shown in Table 9, the scoring function Eshape + w2 Eelec + w3 Epair with DARS as the pairwise potential generates adequate numbers of hits for seven of the 10 other complexes, but no hits for three complexes (PDB codes 1L0Y, 1AVZ, and 1IGC). According to Table 7, these complexes have weak pairwise hydrophobic complementarity. As discussed for antigen-antibody complexes, this generally implies relatively poor discrimination. Hydrophobic complementarity also impacts the quality of docking: the correlation coefficient between Ehyd values and the number of hits obtained by the combined potential for the other complexes is r = −0.67, which is significant at p < 0.02. We note that one of the component proteins in each of the complexes 1L0Y, 1AVZ, and 1IGC is a recognition domain which can bind to a variety of proteins. The interface in each complex has a strong hydrophobic patch on the side of recognition domain, which presumably contributes to the promiscuous binding; however, this patch does not interact with any of the most hydrophobic regions of the partner protein, and this results in poor hydrophobic complementarity.
Based on the experience with enzyme-inhibitor complexes we expect that the quality of discrimination and that of docking are similar. However, for other complexes the docking results are significantly better (p < 0.03 by the Wilcoxon test) than discrimination by DARS. The main difference between the enzyme-inhibitor and the other complexes is that shape complementarity provides information for the first, but almost none for the second. In fact, for the other complexes the 20,000 PIPER-generated structures with the best shape complementarity include so few hits that we had to select the 20,000 structures with the best RMSD values for the discrimination test. Thereby we force a number of low RMSD structures into the test set (Table 7), but apparently these structures include too few of the native contacts, and ranking them by DARS fails to improve discrimination. In contrast, the direct use of the combined potential Eshape + w2Eelec + w3Epair for docking selects near-native structures that are not present in the discrimination test set. Thus, we conclude that the quality of discrimination is not necessarily a valid predictor of docking performance if the component proteins have limited shape complementarity.
Comparison of potential functions
In the Supplementary Material, Table S1, Table S2, and Table S3 list the pairwise interaction coefficients for the DARS, MFP, and URS potentials. The current version of the ACP potential has been implemented as a server (37) that also provides the ACP coefficient matrix. In all four potentials we used the 18 atom types defined by Zhang et al. (24), based on considerations of chemical properties. The principle used in the atom type selection was to group all atoms that behave similarly in interactions. For example, Cβ atoms of all amino acids were grouped as a single class, apart from those of Ser and Thr that have nonnegligible partial charges. Side-chain atoms were also grouped if they behaved similarly, e.g., both Cɛ and Nζ atoms of the Lys side chain belong to the KNζ group. Most hydrophobic side-chain atoms are in the FCζ and LCδ categories. In contrast, the backbone atoms are considered as separate atom types N, CA, C, and O. A detailed description of the 18 atom types is given in the original ACP article (24).
It is far from simple to compare the four 18 × 18 tables of interaction coefficients, and here we restrict consideration to atom types for which the coefficients substantially differ. The interaction energies among hydrophobic side-chain atoms FCζ and LCδ are negative in all four potentials, with somewhat more favorable values for LCδ in DARS than in the other three. There are substantial differences for interactions among backbone atoms. In ACP and URS these are relatively large and attractive, indicating that such interactions frequently occur in the interface; in MFP, the coefficients are similarly large, but repulsive, clearly because of the high mole fractions of backbone atoms. In contrast, in DARS all backbone-backbone interactions are close to zero for non-Gly residues, indicating that such interactions occur with similar frequency in the complexes and in the random docked decoys. The four potentials also substantially differ in the interaction coefficients between charged side-chain atoms. In ACP, all such coefficients are positive. In fact, as described previously (16), ACP does not represent well the electrostatic interactions, and was always used in conjunction with a Coulombic potential. In DARS, most interaction coefficients are reasonable: e.g., DOδ-DOδ is strongly repulsive, whereas DOδ-RNη and DOδ-RNɛ are strongly attractive. RNη-RNη is close to zero, most likely due to the interactions between the hydrophobic parts of the arginine side chains compensating for the unfavorable charge interactions. The only somewhat unexpected observation is that the interactions between Lys side-chain atoms and any other atom are repulsive, indicating that Lys is more frequently seen in the interfaces of docked decoys than in the interfaces of protein complexes. The coefficients for Lys are also positive in the URS potential, indicating that Lys is quite rare in the interface. Both URS and MFP present some coefficients that are more difficult to explain, such as the favorable DOδ-DOδ self-interaction. Since DARS, URS, and MFP are based on the same interaction data, these differences demonstrate the substantial impact of the reference state on the interaction coefficients.
CONCLUSIONS
DARS is a very natural approach to the construction of structure-based intermolecular potentials. The idea is generating a large set of docked conformations with only shape complementarity in the scoring function (i.e., without accounting for any atom-type specific property), and determining the frequency of atom pairs in these decoys for the calculation of interaction probabilities in the reference state. The motivation for this article has been to test and possibly to optimize DARS potentials for docking enzyme-inhibitor, antigen-antibody, and other complex-types of a well-known benchmark set for protein-protein docking (23). Selecting various reference sets and varying the number of decoys we have derived many versions of the potential and tested them both for discrimination (i.e., finding near-native conformations in large sets of docked structures) and for docking (in combination with van der Waals and electrostatics energy terms). Considerations were restricted to the simple case of contact (rather than distance-dependent) type potentials.
Results for enzyme-inhibitor complexes confirm that the DARS approach can provide an excellent reference state, and that the performance does not heavily depend on the selection of complexes used for generating the decoys and on the number of decoys generated. We have also shown that discrimination and docking yield similarly good results. For antigen-antibody complexes all four potentials considered in this article are less accurate than for enzyme-inhibitor complexes. Finally, DARS provides strong docking results for almost all other types of complexes, substantially better than the ones by the competing potentials. However, docking does not yield any near-native structures for a few other complexes. In addition, for all complexes in the other category, the discrimination results are generally much weaker than the docking results, and we have argued that discrimination tests are not appropriate surrogates for docking tests if the component proteins have limited shape complementarity.
Further analysis of our antigen-antibody results reveals that neither discrimination nor docking can be accurate for complexes in which the hydrophobic patches on the two side of the interface do not properly align with each other. Calculating a pairwise hydrophobicity potential we have shown that many antigen-antibody and some of the other complexes are in this category, resulting in poor docking and discrimination results. We suggest that the problem may be avoided by allowing for the asymmetry of the potentials, e.g., in antigen-antibody complexes, considering the hydrophobic atoms to be more favorable for interactions if they are located on the antibody rather than on the antigen. Since accounting for the asymmetry is independent of the reference state, our results are likely to help in the development of improved DARS or other structure-based interaction potentials, especially for antigen-antibody docking.
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Supplementary Material
Acknowledgments
We are grateful to the Boston University Scientific Computing and Visualization Center for the opportunity to use the Blue Gene/L Supercomputer.
This work has been supported by grants No. GM61867 and No. GM64700 from the National Institutes of Health.
Gwo-Yu Chuang, Dima Kozakov, and Ryan Brenke contributed equally to this article.
Editor: Ruth Nussinov.
References
- 1.Sippl, M. J. 1990. Calculation of conformational ensembles from potentials of mean force. An approach to the knowledge-based prediction of local structures in globular proteins. J. Mol. Biol. 213:859–883. [DOI] [PubMed] [Google Scholar]
- 2.Skolnick, J., L. Jaroszewski, A. Kolinski, and A. Godzik. 1997. Derivation and testing of pair potentials for protein folding, when is the quasichemical approximation correct? Protein Sci. 6:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lu, H., and J. Skolnick. 2001. A distance-dependent atomic knowledge-based potential for improved protein structure selection. Proteins. 44:223–232. [DOI] [PubMed] [Google Scholar]
- 4.Godzik, A. 1996. Knowledge-based potentials for protein folding: what can we learn from known protein structures? Structure. 4:363–366. [DOI] [PubMed] [Google Scholar]
- 5.Miyazawa, S., and R. Jernigan. 1985. Estimation of effective interresidue contact energies from protein crystal structures: quasi-chemical approximation. Macromolecules. 18:534–552. [Google Scholar]
- 6.Miyazawa, S., and R. Jernigan. 1996. Residue-residue potentials with a favorable contact pair term and an unfavorable high packing density term, for simulation and threading. J. Mol. Biol. 256:623–644. [DOI] [PubMed] [Google Scholar]
- 7.Rojnuckarin, A., and S. Subramaniam. 1999. Knowledge-based interaction potentials for proteins. Proteins. 36:54–67. [DOI] [PubMed] [Google Scholar]
- 8.Zhou, H., and Y. Zhou. 2002. Distance-scaled, finite ideal-gas reference state improves structure-derived potentials of mean force for structure selection and stability prediction. Protein Sci. 11:2714–2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Camacho, C., D. Gatchell, S. Kimura, and S. Vajda. 2000. Scoring docked conformations generated by rigid-body protein-protein docking. Proteins. 40:525–537. [DOI] [PubMed] [Google Scholar]
- 10.Li, L., R. Cheng, and Z. Weng. 2003. RDOCK: refinement of rigid-body protein docking predictions. Proteins. 53:693–707. [DOI] [PubMed] [Google Scholar]
- 11.Comeau, S., D. Gatchell, S. Vajda, and C. Camacho. 2004. ClusPro: an automated docking and discrimination method for the prediction of protein complexes. Bioinformatics. 20:45–50. [DOI] [PubMed] [Google Scholar]
- 12.Moont, G., H. Gabb, and M. Sternberg. 1999. Use of pair potentials across protein interfaces in screening predicted docked complexes. Proteins. 35:364–373. [PubMed] [Google Scholar]
- 13.Murphy, J., D. Gatchell, J. Prasad, and S. Vajda. 2003. Combination of scoring functions improves discrimination in protein-protein docking. Proteins. 53:840–854. [DOI] [PubMed] [Google Scholar]
- 14.Lu, H., L. Lu, and J. Skolnick. 2003. Development of unified statistical potentials describing protein-protein interactions. Biophys. J. 84:1895–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu, S., C. Zhang, H. Zhou, and Y. Zhou. 2004. A physical reference state unifies the structure-derived potential of mean force for protein folding and binding. Proteins. 56:93–101. [DOI] [PubMed] [Google Scholar]
- 16.Kozakov, D., R. Brenke, S. R. Comeau, and S. Vajda. 2006. PIPER: an FFT-based protein docking program with pairwise potentials. Proteins. 65:392–406. [DOI] [PubMed] [Google Scholar]
- 17.Mintseris, J., B. Pierce, K. Wiehe, R. Anderson, R. Chen, and Z. Weng. 2007. Integrating statistical pair potentials into protein complex prediction. Proteins. 69:511–520. [DOI] [PubMed] [Google Scholar]
- 18.Smith, R., R. Hubbard, D. Gschwend, A. Leach, and A. Good. 2003. Analysis and optimization of structure-based virtual screening protocols. 3. New methods and old problems in scoring function design. J. Mol. Graph. Model. 22:41–53. [DOI] [PubMed] [Google Scholar]
- 19.Pham, T., and A. Jain. 2008. Customizing scoring functions for docking. J. Comput. Aided Mol. Des. 22:269–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pham, T., and A. Jain. 2006. Parameter estimation for scoring protein-ligand interactions using negative training data. J. Med. Chem. 49:5856–5868. [DOI] [PubMed] [Google Scholar]
- 21.Vajda, S., and C. Camacho. 2004. Protein-protein docking: is the glass half full or half empty? Trends Biotechnol. 22:110–116. [DOI] [PubMed] [Google Scholar]
- 22.Kozakov, D., K. Clodfelter, S. Vajda, and C. Camacho. 2005. Optimal clustering for detecting near-native conformations in protein docking. Biophys. J. 89:867–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen, R., J. Mintseris, J. Janin, and Z. Weng. 2003. A protein-protein docking benchmark. Proteins. 52:88–91. [DOI] [PubMed] [Google Scholar]
- 24.Zhang, C., G. Vasmatzis, J. Cornette, and C. DeLisi. 1997. Determination of atomic desolvation energies from the structures of crystallized proteins. J. Mol. Biol. 267:707–726. [DOI] [PubMed] [Google Scholar]
- 25.Mintseris, J., and Z. Weng. 2004. Optimizing protein representations with information theory. Genome Inform. 15:160–169. [PubMed] [Google Scholar]
- 26.Ruvinsky, A. M., and A. V. Kozintsev. 2005. The key role of atom types, reference states, and interaction cutoff radii in the knowledge-based method: New variational approach. Proteins. 58:845–851. [DOI] [PubMed] [Google Scholar]
- 27.Glaser, F., D. M. Steinberg, I. A. Vakser, and N. Ben-Tal. 2001. Residue frequencies and pairing preferences at protein-protein interfaces. Proteins. 43:89–102. [PubMed] [Google Scholar]
- 28.Janin, J., K. Henrick, J. Moult, L. Ten Eyck, M. Sternberg, S. Vajda, I. Vakser, and S. Wodak. 2003. CAPRI: a critical assessment of predicted interactions. Proteins. 52:2–9. [DOI] [PubMed] [Google Scholar]
- 29.Camacho, C., and S. Vajda. 2002. Protein docking along smooth association pathways. Proc. Natl. Acad. Sci. USA. 23:319–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paschalidis, I. C., Y. Shen, P. Vakili, and S. Vajda. 2007. SDU: a semi-definite programming-based underestimation method for global optimization in molecular docking. IEEE Trans. Automat. Contr. 52:664–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jain, A., and A. Nicholls. 2008. Recommendations for evaluation of computational methods. J. Comput. Aided Mol. Des. 22:133–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bradley, J. 1968. Distribution Free Statistical Tests. Prentice Hall, Englewood Cliffs, NJ.
- 33.Jackson, R. 1999. Comparison of protein-protein interactions in serine protease-inhibitor and antibody-antigen complexes: Implications for the protein docking problem. Protein Sci. 8:603–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.LoConte, L., C. Chothia, and J. Janin. 1999. The atomic structure of protein-protein recognition sites. J. Mol. Biol. 285:2177–2198. [DOI] [PubMed] [Google Scholar]
- 35.O'Toole, N., and I. A. Vakser. 2008. Large-scale characteristics of the energy landscape in protein-protein interactions. Proteins. 71:144–152. [DOI] [PubMed] [Google Scholar]
- 36.Ponomarenko, J. V., and P. E. Bourne. 2007. Antibody-protein interactions: benchmark datasets and prediction tools evaluation. BMC Struct. Biol. 7:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Camacho, C., and C. Zhang. 2005. FastContact: rapid estimate of contact and binding free energies. Bioinformatics. 21:2534–2536. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.