

Abstract
Communication between cells is crucial for proper functioning of multicellular organisms. The recently discovered membranous tubes, named tunneling nanotubes, that directly bridge neighboring cells may offer a very specific and effective way of intercellular communication. Our experiments on RT4 and T24 urothelial cell lines show that nanotubes that bridge neighboring cells can be divided into two types. The nanotubes of type I are shorter and more dynamic than those of type II, and they contain actin filaments. They are formed when cells explore their surroundings to make contact with another cell. The nanotubes of type II are longer and more stable than type I, and they have cytokeratin filaments. They are formed when two already connected cells start to move apart. On the nanotubes of both types, small vesicles were found as an integral part of the nanotubes (that is, dilatations of the nanotubes). The dilatations of type II nanotubes do not move along the nanotubes, whereas the nanotubes of type I frequently have dilatations (gondolas) that move along the nanotubes in both directions. A possible model of formation and mechanical stability of nanotubes that bridge two neighboring cells is discussed.
INTRODUCTION
Cell-to-cell communication requires the distribution of signal molecules between donor and acceptor cells. The best-known but most lavish mechanism of intercellular communication depends on secretion of molecules in the extracellular space where they find their targets by diffusion (1). Another acknowledged model of transport of signaling molecules is by communication junctions, such as gap junctions (2), where transport is limited to transfer of small molecules over very short distances between tightly attached cells.
Recently, a new mechanism of cell-to-cell communication was proposed when thin tubular connections between membrane-enclosing compartments were discovered. Basic research was first performed on liposomes on which membranous tubes of thickness less than a micrometer are commonly formed, especially if a mechanical or a chemical disturbance is introduced into the liposome system (3–5). Such lipid bilayer nanotubes may connect two or more liposomes (6). It was observed that a dilatation of the tube forming a gondola may exist and travel along the tube (Fig. 1) (7). Based on this discovery of nanotubes and gondolas in artificial systems (4–6) and the discovery of intratubular particle transport between two liposomes (6), it was suggested that similar mechanisms may also take place in cells (7). In cells, nanotubes and gondolas (forming an integral part of the nanotube) may constitute a transport system within and between cells (5,7). Transport to the target point would be much more selective if the motion of the vesicles were directed by nanotubes. Such nanotube-directed transport might have an important role in the selectivity of specific pathways in cellular systems where the transport vesicles move specifically from one membrane to another (7).
FIGURE 1.

Movement of a small phospholipid prolate traveling vesicle (white arrow) along a thin phospholipid tube (black arrow) attached to a spherical liposome (adapted from the study by Iglič et al. (7)). In the final stage, the vesicle was fused with the membrane of liposome.
After discovery of nanotubes in liposome systems, the first indication that nanotubular structures might also be present in cellular systems came from experiments with manipulated erythrocytes. It was observed that small vesicles released from the erythrocytes moved synchronously with the parent cell and that these vesicles were connected to the cell by thin nanotubes (7).
Recently, thin membranous tubes, so-named tunneling nanotubes (TNTs) (8), that bridge distances up to 120 μm have been discovered in immune cells (8), THP-1 monocytes (9), in cultures of DU145 human prostate cancer cells (10), endothelial progenitor cells, rat cardiac myocytes (11), and astrocytes (12). It has been proposed that TNTs represent a new mode of cell-to-cell communication and that they might enable direct transport of molecules and even organelles between cells (8,11–13).
Until these recent discoveries, the nanotubes that bridge neighboring cells (that is, bridging nanotubes) were mostly found in cells that were weakly connected to each other or they would actively migrate and search for bacteria or attachment to eukaryotic cells. The results of this study confirm the previous results of Rustom et al. (8), which demonstrate that bridging nanotubes also exist in cells with a limited ability of movement and strong intercellular connections as in the case of epithelial cells. Furthermore, this study shows that there are two types of bridging nanotubes of different stability, cytoskeletal contents, and function. Nanotubes may also connect intracellular membranous compartments such as Golgi stacks (3,14) and may be a part of the subjacent membrane pool that forms an infrastructure of the cell.
In this study, phase contrast, fluorescence, and electron microscopy were used to study formation and stability of nanotubes that bridge two neighboring urinary bladder epithelial cells. The characterization of different types of bridging nanotubes with respect to their biochemical characteristics and the nature of the process of their formation is presented. Theoretical models that may explain how such nanotubes are created and stabilized are suggested.
MATERIALS AND METHODS
Urothelial cell cultures
Urothelial cell lines RT4 and T24 were cultured in a 1:1 mixture of advanced Dulbecco's modified Eagle's medium (Gibco, Invitrogen, Carlsbad, CA) and Ham's F-12 (Sigma-Aldrich, St. Louis, MO) supplemented with 10% fetal calf serum (FCS), 5 μg/mL insulin, 5 μg/mL transferrin, 100 mg/mL hydrocortisone, and 5 ng/mL selenite (all purchased from Gibco, Invitrogen), as well as 1800 U/mL cristacyclin (Pliva, Zagreb, Croatia) and 0.222 mg/mL streptomycinesulfate (Fatol Arzneilmittel, Schiffweiler, Germany). Cells were incubated at 37°C in a humidified incubator in an atmosphere of 5% CO2.
For actin-green fluorescent protein (actin-GFP) tracking experiments, T24 cells were cultured in Roswell Park Memorial Institute (RPMI) medium 1640 supplemented with 10% FCS (both purchased from Gibco, Invitrogen) and grown at 37°C in a humidified incubator with 5% CO2. Cell culture media, FCS, antibiotics, G418 sulfate (GeneticinÒ), and phosphate-buffered saline (PBS) were all purchased from PAA Laboratories, Pasching, Austria.
A day before the experiments, cells were seeded onto sterile glass coverslips (Brand, Wertheim, Germany) at ∼70–80% confluency and incubated overnight at 37°C.
Antibodies
For labeling with cytokeratin 7, mouse antihuman monoclonal antibody (clone OV-TL 12/30; Dako, Dakocytomation, Glostrup, Denmark) was used at a dilution of 1:20. For desmoplakin labeling, rabbit polyclonal antibodies (AbCam, Cambridge, UK) were used at a dilution of 1:100. Goat antimouse antibodies conjugated with tertamethylrhodamine isothiocyanate (TRITC) (1:100) and goat antirabbit antibodies conjugated with fluorescein isothiocyanate (FITC) (1:100) were used as secondary antibodies.
Cytochalasin D treatment
For treatment with cytochalasin D from Zygosporium mansonii (Sigma-Aldrich), T24 cells were seeded onto glass coverslips at ∼80% confluency and incubated overnight at 37°C. Cells were incubated in 0.14 μM cytochalasin D at 37°C in growth medium. After 30 min, a time-lapse sequence was recorded at room temperature. To check if the actin filaments were depolymerized, cells were labeled with 16.7 μg/mL phalloidin-TRITC (Sigma-Aldrich) in 20% methanol in PBS.
Transfection of T24 cells with actin-GFP
A total of 70% confluent cells were harvested and transfected with 10 μg plasmid DNA (pActin-GFP-N1 construct) using a Gene Pulser electroporation unit (X-cell electroporator; Bio-Rad Laboratories, Hercules, CA) with the following electroporation conditions: 240 V, 950 μF, unlimited resistance, 4 mm gap cuvettes (Bio-Rad), and RPMI1640 (Gibco, Invitrogen) medium without FCS as the electroporation buffer. Cells were plated onto 100 mm culture dishes and grown for 48 h. The medium was removed and replaced with fresh medium supplemented with 400 μg/ml of G418 sulfate (Geneticin; United States Biological, Swampscott, MA). Growth medium was replaced every three days. After 15 to 20 days, individual neomycin-resistant colonies were selected for propagation and analysis. Cells were grown on glass coverslips 24 h before measurements to low confluency to produce TNTs. All experiments were performed in Dulbecco's PBS (PAA Laboratories) at 25°C.
The pActin-GFP-N1 construct was kindly provided by Dr. Hannes Stockinger of the Medical University Vienna.
Labeling with lipophilic stain
To label the plasma membrane, a lipophilic stain (Vybrant DiI; Molecular Probes, Eugene, OR) was used. A solution of membrane marker Vybrant DiI (prepared according to the manufacturer's protocol) was added to T24 cells for 35 min at 37°C. After washing in fresh culture medium for 3 × 10 min, the cells were resuspended and placed on a growing culture of unlabeled T24 cells. After 24 h, the cells were fixed with 2% paraformaldehyde (Merck, Darmstadt, Germany) warmed to 37°C at room temperature for 30 min. After washing three times with PBS for five min, the coverslips with cells were embedded with antibleaching medium (Vectashield; Vector Laboratories, Peterborough, UK).
Immunofluorescence labeling and microscopy
Cells were washed with PBS and fixed for 30 min in 2% paraformaldehyde (Merck). All solutions were warmed to 37°C. After washing with PBS, the coverslips were incubated in ice-cold 0.25% Triton X-100 in PBS for 6 min, washed three times in PBS, and incubated in 0.33 M sucrose in 0.2 M cacodylate buffer for 30 min. Samples were blocked in 2% bovine serum albumin (Sigma-Aldrich) in 0.2% NaN3 in PBS for 30 min at room temperature. Coverslips with cells were incubated in primary antibodies for 1 h at 37°C or overnight at 4°C, then washed in PBS for 10 min, and incubated in secondary antibodies for 30 min at 37°C. Actin labeling in 16.7 μg/mL phalloidin (phalloidin-FITC; Sigma-Aldrich) in 20% methanol (Carlo Erba, Milan, Italy) in PBS for 30 min was carried out after secondary antibody incubation and 10 min washing in PBS. Coverslips were then decanted and embedded in Vectashield with 4′,6-diamidino-2-phenylindole (Vectashield/DAPI; Vector Laboratories), mounted on objective glass, and analyzed in a fluorescence microscope (Eclipse T300; Nikon, Tokyo, Japan) and in an Axio-Imager Z1 microscope (Carl Zeiss MicroImaging, Göttingen, Germany).
Actin-GFP tracking experiments
Fluorescence microscopy was carried out on a setup described previously (15,16). In brief, samples were mounted on a modified epifluorescence microscope (Axiovert 200; Carl Zeiss ) equipped with a Plan-Apochromat objective lens (100×, 1.4 NA; Carl Zeiss). Using the 488 nm line of a Sapphire 488-200 laser (Coherent, Santa Clara, CA), images were recorded on an EMCCD camera (iXon E2V TECH CCD97, 16 μm pixel size; Andor Technology, Belfast, UK). Samples were illuminated at 8 kW/cm2 (see Fig. 5 B) or 15 kW/cm2 (Fig. 5 A) with an illumination time of till = 2 ms or 1 ms. The movie was recorded at a time delay of 22 ms between two consecutive images.
FIGURE 5.

Exchange of actin-GFP via a bridging nanotube between two T24 cells. Stable, actin-GFP transfected T24 cells were frequently found to be interconnected by TNTs. Occasionally, connections were observed between a high expressing cell and a cell devoid of actin-GFP (cell borders are indicated by a dashed line). (A) The spreading of actin GFP into the second cell is clearly visible as a cone of fluorescence growing into a GFP-actin negative cell. (B) Two nanotubes indicated by arrows connect the shown nontransfected cell with an actin-GFP positive cell outside the imaged area (the imaging pinhole is indicated by a full line). Multiple diffraction-limited spots could be observed at high mobility, indicating the presence of free actin-GFP molecules in the nontransfected cell (see Movie S3).
Phase-contrast time-lapse microscopy
Subconfluent cultures were observed with a water immersion phase-contrast objective lens (63×) on an Axio-Imager Z1 microscope (Carl Zeiss). Exposures were taken every 3 to 10 s for 20 to 190 cycles at a resolution of 13 M pixels with a time-lapse module of the Axiovision 4.5 SP1 software program (Carl Zeiss).
Scanning electron microscopy
Cells were fixed in 2.5% glutaraldehyde and 2% parafomladehyde (Merck) in cacodylate buffer for 2 h, washed overnight in 0.33 M sucrose in cacodylate buffer, postfixed with 1% OsO4 (Serva Electrophoresis, Heidelberg, Germany) in cacodylate buffer for 1 h, dehydrated in a graded series of acetone, and critical-point dried with a Baltek CPD 030 apparatus (Baltek, Northvale, NJ). Samples were sputtered with gold using a sputter coater (SCD 040; (Balzers Union, Balzers, Lichtenstein) and observed with a scanning electron microscope (JSM 840A; JEOL, Tokyo, Japan).
Transmission electron microscopy
Cells were fixed in 2.5% glutaraldehyde and 2% parafomladehyde (Merck) in cacodylate buffer for 2 h, washed overnight in 0.33 M sucrose in 0.1 M cacodylate buffer, dehydrated in a graded series of ethanol (Carlo Erba), embedded in 1:1 mixture of glycid ether 100 (Serva, Heidelberg, Germany) and propyleneoxide overnight and for 2 h in glycid ether 100. The glycid ether 100 was left to polymerize in a thermostat for five days (one day each at 35°C, 45°C, 60°C, 70°C, and 80°C). Cells embedded in glycid ether 100 were separated from a cover glass in liquid nitrogen. Ultrathin sections were stained in 1.64 M Pb(NO3)2 for 10 min, washed in double-distilled water, and subsequently stained in uranyl acetate. Ultrathin sections were examined with a transmission electron microscope (CM 100; Phillips, Eindhoven, The Netherlands).
EXPERIMENTAL RESULTS
Based on the results, we divided the observed bridging nanotubes into two different types with respect to their formation, stability, and cytoskeletal content.
As type I, we classified membrane tubes that contain actin filaments and begin growing as filopodia but can grow ≤30 μm in length (Fig. 2). This type of tubular protrusion usually appears as bunches of several tubes that dynamically seek connections with neighboring cells (Supplementary Material, Movie S1). Protrusions remain stable even after disintegration of the actin filaments with cytochalasin D (Fig. 3; Movie S2). After reaching an appropriate neighboring cell, the protrusions connect to the target cell (Fig. 2 A) and remain connected for several tens of minutes (average of 20 min). The tube becomes attached to the plasma membrane of the target cell by an anchoring type of intercellular junctions (Fig. 4) From the results of experiments with actin-GFP (Fig. 5), we observed a cytosolic rather than only a membrane connection between the two cells interconnected by type I nanotubes. We found spreading of actin-GFP via a tunneling nanotube from one T24 cell with highly expressed actin-GFP to the other cell that was devoid of actin-GFP (Fig. 5 A). The spreading of actin-GFP into the second cell is clearly visible as a cone of fluorescence at the connection point. To further confirm the connectivity of the two cells, we screened multiple nontransfected cells that were connected to actin-GFP overexpressing cells via type I nanotubes. Frequently, we observed diffraction-limited signals in the nontransfected cell, which moved with very high mobility, too fast to be traceable at the time-delay of 22 ms; the signal amplitudes concur with the brightness of a single or a few GFP molecules (Fig. 5 B; Movie S3). We interpret the signals as free actin-GFP monomers or small associates that have dissociated from the actin filaments upon tube formation. We never observed such signals in nontransfected, unconnected cells.
FIGURE 2.

Type I nanotubes. A is a phase contrast image of live T24 cells, whereas B is a fluorescence micrograph showing actin labeling of the same cells as in A after 15 min of paraformaldehyde fixation. Cell C1 is approaching the cells C2 and C3 (see Movie S1). The white arrows in A and B indicate short and dynamic membrane protrusion with which the approaching cell explores its surroundings. The black arrow in A points at protrusions that have already connected to the target cell. In all these multiple tubular connections, actin filaments are present (arrows in B). Bridging nanotubes of type I can be more than 20 μm in length and occasionally bifurcations are seen (arrow in C).
FIGURE 3.

The stable membrane protrusions after cytochalasin D treatment of T24 cells can be seen by time-lapse phase-contrast microscopy. After incubation in cytochalasin D for 30 min, a time-lapse sequence with Axio-Imager Z1 microscope (Carl Zeiss) was recorded (see Movie S2). The white arrows point to the tip of two nanotubes that move passively. Times indicated in A through F are the times passed from the beginning of the time-lapse sequence.
FIGURE 4.

A transmission electron micrograph showing an anchoring type of intercellular junction (arrow) connecting a nanotubule to the protrusion of a neighboring cell.
The exchange of lipid components of the plasma membrane between the cells connected by nanotubes of type I seems to be almost completely stopped at the junction between the nanotube and the neighboring target cell (Fig. 6). The fluorescent lipid marker DiI did not pass the junction point even after 24 h of cocultivation of labeled T24 cells and nonlabeled cells (Fig. 6). Only some small spots of DiI were found on nonlabeled cells, which could also indicate vesiculation of the labeled cells and the fusion (or adhesion) of the released vesicles with the membranes of the nonlabeled cells.
FIGURE 6.

Urothelial cells T24 labeled with lipophilic stain DiI were cocultured with unlabeled T24 cells. The nanotubes (arrow) of stained cells (red) became protruded and attached to unstained cells (green) in 3 h. However, even after 24 h, the DiI stain did not spread to the connected cells.
Type II bridging nanotubes have cytokeratin filaments and are mainly located more apically on the cell (Fig. 7). These nanotubes start growing as cells move apart (Fig. 8). At the very beginning of the tube formation, some actin is still present at the entry point of the tubes. As the tube elongates, the actin gradually disappears and only cytokeratin filaments remain. After the dissociating cells reach a distance of 30 to 40 μm, only one such cytokeratin containing nanotube remains as a link between the two cells. These longer tubes, which can be up to several 100 μm in length, can connect dissociating cells for more than 2 h (Figs. 7 and 8).
FIGURE 7.

In urothelial cell line T24, a long tubular structure connects cells of the two cell clusters C1 and C2 (A). B is a magnified region of the area in the black frame in A. Such long, singular tubes of type II contain thin cytokeratin filaments (arrow in C). In C, cytokeratin 7 is labeled in red, actin in green, and the nucleus with DAPI in blue.
FIGURE 8.

Two separating T24 cells (C1 and C2) having actin (A) and cytokeratin (B) filaments present in the forming protrusions. Membranes of the two cells detach at certain sites, forming tail-like protrusions between the membranes. The membranes gradually separate as the cells move apart, pulling and dividing their cytoskeletal content. Note that both actin (A) and cytokeratin (B) filaments are still present in growing tubular connections.
On many nanotubes that connect neighboring urothelial cells, vesicular dilatations were found (Fig. 9). Vesicular dilatations can be seen in both types of bridging nanotubes. The dilatations on type II nanotubes are larger, usually placed in the middle of the tube, and do not move along the tube (Movie S4). In contrast, type I nanotubes frequently have vesicular dilatations that move along the tubes in both directions, as seen in Fig. 10, B–E, and Movie S5. Such vesicular dilatations (gondolas) move for 5 to 15 μm in a certain direction with an average speed of 40 nm per second. They sometimes appear in the middle of the nanotube and travel along the nanotube until they fuse with the cell body (Fig. 11).
FIGURE 9.

Membrane nanotubes with gondolas (arrows) observed between cells in the human urothelial cell line RT4 (A) and T24 (B and C) by scanning electron microscopy under physiological conditions. Note that the gondolas are an integral part of the tubes.
FIGURE 10.

Movement of small vesicles along membrane bridging nanotubes connecting two locations (white arrows in A) on the membrane surface of cells in the human urothelial cell line RT4 observed by phase contrast microscopy in cell culture under physiological conditions. Black arrows point to two carrier vesicles (gondolas) that moved in opposite directions (B–E).
FIGURE 11.

Fusion of a gondola (arrows) with a cell body is seen after a time-lapse sequence showing directional movement of the gondola along a nanotube. The time sequence in seconds is indicated on the upper left side of each micrograph.
DISCUSSION
Models of the formation and stability of nanotubes in liposomes and cellular systems
Formation of tubular membrane bilayer structures (nanotubes) is a common phenomenon in both artificial membrane and cellular systems (3,5–7,15–19). Usually these nanotubes are very thin structures. Sometimes vesicles, which seem to be freely diffusing in solution, are attached to the parent cell by nanotubes. This finding was observed for erythrocytes that moved synchronously with some small, released vesicles nearby (20). Once pulled out from the liposome membrane (see, for example, the study by Roux et al. (21)), the nanotubular membrane protrusions in liposomes or the nanotubular connections between two liposomes can also be mechanically stable without any permanent external (pulling) stabilization force (5,7). This observation was theoretically explained with weak average orientational ordering and direct interactions between oriented lipids in highly curved tubular membrane regions (5,22).
Recently, thin membranous bridging tubes, including TNTs, which are the most significant (8), have been discovered in several cell lines (for review, see the study by Gerdes et al. (19)). On the basis of our experiments on epithelial cells and experiments by other authors (8,11,13), we divided the nanotubes that bridge neighboring cells into two different types with respect to their formation, stability and cytoskeletal content.
Type I nanotubes and tubular protrusions that form this type of bridging nanotubes contain actin filaments, which give them dynamic properties. Using these tubular protrusions, cells can explore their surroundings (also see the study by Faix and Rottner (23)). Growth and movement of such tubes depend on actin polymerization and actin-dependent motor proteins. Once formed, the bridging nanotubes of type I remain stable even if actin filaments are disintegrated by cytochalasin D (Fig. 3; Movie S2).
We suggest that nanotubular membrane protrusions and bridging nanotubes are also mechanically stabilized by energetically favorable clustering of anisotropic (flexible) membrane nanodomains (that is, small domains composed of lipids and proteins) in nanotubes (12,24,25) (Figs. 12 and 13).
FIGURE 12.

Schematic figure of three different kinds of intrinsic shapes of flexible membrane nanodomains: partly cylindrical, flat, and saddle-like. The intrinsic shape of the nanodomain can be characterized by two intrinsic (spontaneous) principal curvatures C1m and C2m. When C1m = C2m, the nanodomain is isotropic, whereas if C1m ≠ C2m, the nanodomain is anisotropic (12). Bending deformation and rotation of the nanodomain allow the nanodomain to adapt its shape and orientation to the actual membrane curvature, which in turn is influenced by the nanodomain. The nanodomains with C1m > 0 and C2m 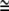 0 favor cylindrical geometry of the membrane. Nanodomains with C1m = C2m = 0 prefer flat membrane shape, whereas nanodomans with C1m > 0 and C2m < 0 favor saddle-like membrane geometry (as, for example, in the membrane neck connecting the daughter vesicle to the parent membrane).
0 favor cylindrical geometry of the membrane. Nanodomains with C1m = C2m = 0 prefer flat membrane shape, whereas nanodomans with C1m > 0 and C2m < 0 favor saddle-like membrane geometry (as, for example, in the membrane neck connecting the daughter vesicle to the parent membrane).
FIGURE 13.

Schematic illustration of stabilization of type I nanotubular membrane protrusions by accumulation of anisotropic membrane nanodomains in the tubular region. Growing actin filaments push the membrane outward (A). The protrusion is additionally stabilized by accumulated anisotropic nanodomains with C1m > 0 and C2m 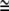 0 (see Fig. 12) that favor anisotropic cylindrical geometry of the membrane (12,30). Possible candidates for such anisotropic membrane nanodomains might be prominin-containing nanodomains (24,25,28). The cylindrical-shaped anisotropic membrane domains, once assembled in the membrane region of a nanotubular membrane protrusion, keep the protrusion mechanically stable even if the cytoskeletal components (actin filaments) are disintegrated by cytochalasin D (B).
0 (see Fig. 12) that favor anisotropic cylindrical geometry of the membrane (12,30). Possible candidates for such anisotropic membrane nanodomains might be prominin-containing nanodomains (24,25,28). The cylindrical-shaped anisotropic membrane domains, once assembled in the membrane region of a nanotubular membrane protrusion, keep the protrusion mechanically stable even if the cytoskeletal components (actin filaments) are disintegrated by cytochalasin D (B).
Much experimental and theoretical evidence indicate the existence of membrane micro- and nanodomains (12,24–32 and the references therein). To consider the biological membrane as only a mixture of different types of individual molecules with different intrinsic shapes without explicitly taking into account the possibility of their self-assembly into mixed, energetically favorable membrane micro- and/or nanodomains (which may be composed of many different types of molecules, Fig. 12) would be an overestimate of the role of the individual molecular intrinsic shape in the mechanics of biological membranes and would neglect the role of direct interactions between the molecules that compose the membrane. For example, membrane lipids, which comprise an impressively large number of molecular species with different intrinsic shapes (30,33), may self-assemble into various micro- and nanodomains with an average intrinsic shape (spontaneous curvature) of the domain that can be different from the intrinsic shapes of the lipids constituting the domain (30,34). A proper theoretical description of the mechanics of biological membranes should therefore also take into account the possibility that membrane molecules may form small, flexible micro- and nanodomains with different intrinsic shapes (Fig. 12).
Because of the very large number of different types of molecules constituting the biological membrane (33), however, it would be an extremely difficult task to simultaneously consider in the theoretical model the various intrinsic shapes of all the membrane molecules and the different kinds of direct interactions between them (which my lead to energetically favorable formation of different membrane micro- and nanodomains). Consequently, in the model of the membrane used in this work, we introduce the concept of a flexible membrane nanodomain, which is defined as a small complex of membrane molecules (lipids, proteins) (Fig. 12), for the sake of simplicity. Such flexible membrane nanodomains can be then considered as membrane building blocks (24,30).
In this model, we assume that membrane nanodomains (Fig. 12), as a result of their structure and local interactions, energetically prefer a local geometry that is described by the two intrinsic principal curvatures (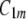 and
and 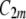 ). The intrinsic principal curvatures (spontaneous curvatures)
). The intrinsic principal curvatures (spontaneous curvatures)  and
and 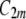 are, in general, different (
are, in general, different (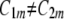 ) (Fig. 12). If they are identical (
) (Fig. 12). If they are identical (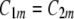 ), the nanodomain is isotropic. If
), the nanodomain is isotropic. If 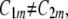 the nanodomain is anisotropic. The location and orientation of the anisotropic nanodomain are important for its energy. An anisotropic nanodomain (Fig. 12) will therefore prefer to accumulate in the membrane region with the principal curvatures C1 and C2, close to the values of its intrinsic principal curvatures C1m and C2m (14), and, on average, spend more time in the orientation that is energetically more favorable than any other orientation (24,30). A coupling between the membrane shape (that is, curvature) and the nonhomogeneous lateral distribution of membrane nanodomains have been predicted (14).
the nanodomain is anisotropic. The location and orientation of the anisotropic nanodomain are important for its energy. An anisotropic nanodomain (Fig. 12) will therefore prefer to accumulate in the membrane region with the principal curvatures C1 and C2, close to the values of its intrinsic principal curvatures C1m and C2m (14), and, on average, spend more time in the orientation that is energetically more favorable than any other orientation (24,30). A coupling between the membrane shape (that is, curvature) and the nonhomogeneous lateral distribution of membrane nanodomains have been predicted (14).
The curvature-mediated accumulation of interacting membrane nanodomains having C1m > 0 and 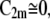 which prefer the cylindrical shape of the membrane (Figs. 12 and 13), in bridging nanotubes might thus create a phase separation with respect to the surrounding microenvironment (12,24,25). The self-assembly of interacting nanodomains, which prefer the cylindrical membrane shape in larger tubular domains (12,24–28), may thus explain the tubular budding of the membrane (Fig. 13) even if there are no actin fibers generating a pulling or pushing force (12,26,29,30). Accordingly, it was recently observed that curvature-driven self-assembly of interacting (anisotropic) membrane nanodomains is sufficient to promote growth and stability of membrane nanotubes in erythrocytes (30), primary murine astrocytes (12), other cells (25,30), and even in model membranes (29).
which prefer the cylindrical shape of the membrane (Figs. 12 and 13), in bridging nanotubes might thus create a phase separation with respect to the surrounding microenvironment (12,24,25). The self-assembly of interacting nanodomains, which prefer the cylindrical membrane shape in larger tubular domains (12,24–28), may thus explain the tubular budding of the membrane (Fig. 13) even if there are no actin fibers generating a pulling or pushing force (12,26,29,30). Accordingly, it was recently observed that curvature-driven self-assembly of interacting (anisotropic) membrane nanodomains is sufficient to promote growth and stability of membrane nanotubes in erythrocytes (30), primary murine astrocytes (12), other cells (25,30), and even in model membranes (29).
The results shown in Fig. 6, which indicate that lipids are probably not transferred in greater amounts between two neighboring cells connected by type I nanotubes, support the hypothesis of energetically favorable self-assembly of specific anisotropic membrane nanodomains into larger domain-forming nanotubes (24,25,30). We propose that the anisotropic membrane nanodomains that comprise the membrane of bridging nanotubes energetically prefer highly curved cylindrical geometry (C1 > 0 and C2 = 0) over flat geometry and so do not spread into the nearly flat (that is, noncylindrical) membrane of the target cell. It is therefore not a surprise that strongly anisotropic dimeric fluorescent markers such as DiI (which energetically prefer the cylindrical geometry of the membrane (30,35)) are also building blocks of the above-mentioned anisotropic nanodomains. These markers stabilize the nanotubular geometry and do not cross the junction between the type I nanotube and the membrane of the target cell in greater amounts (as shown in Fig. 6).
The increased concentration of Lubrol rafts (24,25,27) and Gb3 glycolipid-binding β-subunits (29) in tubular membrane protrusions (inward or outward) supports the above-suggested mechanism for the mechanical stability of bridging nanotubes. Lubrol rafts containing the protein prominin are considered to be a special type of membrane raft that is distinct from the cholesterol-sphingolipid (Triton-resistant) rafts that occur in the planar parts of the membrane (25,28). It has been suggested that, due to their specific molecular shape (28), prominin molecules form small, anisotropic protein-lipid nanodomains (having C1m > 0 and C2m 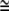 0) that associate into larger, two-dimensional aggregates (Lubrol rafts) upon their curvature-induced accumulation in tubular membrane protrusions (24,25) (Fig. 13). The predicted lateral phase separation of anisotropic prominin nanodomains (24,30), that is, their accumulation in membrane nanotubes, is possible only if the anisotropy of prominin nanodomains is large enough (that is, C1m > 0 at C2m
0) that associate into larger, two-dimensional aggregates (Lubrol rafts) upon their curvature-induced accumulation in tubular membrane protrusions (24,25) (Fig. 13). The predicted lateral phase separation of anisotropic prominin nanodomains (24,30), that is, their accumulation in membrane nanotubes, is possible only if the anisotropy of prominin nanodomains is large enough (that is, C1m > 0 at C2m 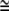 0) and if the radius of the nanotube is small enough (24). The nearest-neighbor direct interactions between the prominin nanodomains also promote their accumulation in membrane nanotubes. Our theoretical model thus provides an explanation (24,25) for the observed curvature-induced enrichment of Lubrol raft markers in tubular membrane protrusions (28).
0) and if the radius of the nanotube is small enough (24). The nearest-neighbor direct interactions between the prominin nanodomains also promote their accumulation in membrane nanotubes. Our theoretical model thus provides an explanation (24,25) for the observed curvature-induced enrichment of Lubrol raft markers in tubular membrane protrusions (28).
Type II nanotubes are longer and more stable than type I nanotubes. They also have cytokeratin filaments and are formed when two cells that are already connected start to move apart. The actin filaments disappear from these nanotubes as they elongate, and only cytokeratin filaments remain. Usually only a single nanotube of type II is present between two cells. The length of such bridging nanotubes can be >100 μm. Along the whole length of such bridging nanotube, cytokeratin filaments are always preserved. Cytokeratins provide these nanotubes with stronger mechanical properties that prevents tearing caused by cell migration or external influences (36,37).
It can be speculated that bridging nanotubes may also be formed between cell organelles that are considered to be separated. Therefore, studies (14,38) have suggested that Golgi apparatus cisterns might also be interconnected by continuous membranous structures, thus enabling selective and polarized vesicle trafficking from one Golgi stack to another.
Possible origins of gondola formations and their movement along bridging nanotubes
The observed distensions of the nanotubes (gondolas) moving along the bridging nanotubes of type I (Figs. 10 and 11) may be formed in different ways. In some cases, the formation of gondolas corresponding to transient excited states may be induced by a sudden tension (caused, for example, by diverging cells) in the membrane nanotubes at specific sites where the local membrane constituents of the nanotubes enable and favor the formation of such dilatations. The tension-induced dilatation of the nanotubes may appear anywhere along the nanotube and then travel as a wave along the bridging nanotube in the direction that is energetically favorable. The tension might be the most probable origin of gondolas that suddenly appear in the middle of the nanotube. These tension-induced dilatation of the nanotubes, like any other excited states of the membrane, are relaxed after a certain time. It was previously reported that slight undulations are relaxed in seconds, whereas sphere-like blobs are relaxed in minutes (39).
The distension of the nanotubes also may be formed because of a small organelle inside the nanotubes, if the diameter of the organelle is greater than the inner diameter of the nanotube (19). The organelles inside the nanotubes my be actively transported by different actomyosin-dependent mechanisms (8,13,19).
The observed vesicular dilatations of the nanotubes moving along the bridging nanotubes of type I (Figs. 10 and 11) show a striking similarity to the dilatations of phospholipid nanotubes that move along these nanotubes (Fig. 1). Therefore, it is also possible that the initiation of gondola formation (Fig. 14 A) may be based on physical mechanisms similar to those governing the formation of free membrane daughter vesicles, which are created in the processes of budding. In contrast to the latter process, however, the connection that gondolas have to the parent membrane, from which they originate, is not disrupted when the gondola is detached from the parent cell (Fig. 14 B).
FIGURE 14.

Schematic illustration of nanotubule-directed transport of small carrier vesicles (gondolas) transporting granular content and membrane particles.
From observations in pure lipid systems (Fig. 1), it is clear that, for the existence of a vesicle that is a distended integral part of the nanotube membrane (Fig. 14), it is not always necessary that the diameter of the enclosed material (as, for example, organelles) is greater than the inner diameter of the nanotube (19). Transported material (multiple small particles moving synchronously within the distension) may be enclosed within a gondola or may be a part of the gondola membrane (Fig. 14).
Once the gondola is formed, its movement along the nanotube (Fig. 14 C) requires no additional bending energy. Nevertheless, some process is needed to provide energy for the gondola to travel along the nanotube. It is possible that the gondola movement is driven by the difference in chemical potential between the molecules packed inside the gondola and the molecules in the interior of the target cell. The movement could also be caused by the difference in chemical potential between the molecules composing the membrane of the gondola and the molecules in the membrane of the target cell.
The final event of the transport is the fusion of the gondola with the target membrane (7). In this process, molecules of the gondola's membrane that originate from the parent, nearly flat, membrane redistribute again in an almost flat target membrane (Fig. 14 D). This redistribution may be energetically favorable and so also part of a driving mechanism to facilitate fusion of the gondola with the membrane. Before fusion of the gondola with the target cell membrane, no neck formation is needed (Fig. 14 C), because the neck is already part of the nanotube connecting the gondola to the membrane of the target cell. This observation is contrary to the case of a free transport vesicle. One can therefore conclude that the transport of material in gondolas (or the transport of molecules composing the membrane of gondolas) may be more energetically advantageous than free vesicle transport.
CONCLUSION
In conclusion, our results indicate that at least two different kinds of bridging nanotubes exist in cellular systems. Nanotubes of type I are shorter and more dynamic than type II nanotubes. They also contain actin filaments and are formed when cells explore their surroundings to make contact with another cell. Nanotubes of type II are longer and more stable than type I nanotubes. They also have cytokeratin filaments and are formed when two cells that are already connected start to move apart. Both types of bridging nanotubes may form vesicular dilatations. Although dilatations on type II nanotubes do not demonstrate any dynamics, those on type I nanotubes can move along the nanotubes to fuse with the target membrane; therefore, we consider them to be gondolas.
Because the bridging nanotubes differ in their structural components, they probably also differ in their functions. Some of the observed bridging nanotubes are certainly TNTs (8). The cloud of cytosolic nonpolymerized (free) actin-GFP molecules in a cell originally devoid of actin-GFP (Fig. 5) clearly shows the transport of free actin-GFP molecules through the observed bridging nanotubes connecting two neighboring cells. Nevertheless, further investigations are required to explain in detail the type of material, signals, or information that could be exchanged via the observed bridging nanotubes and nanotube-directed gondolas, and the form and type of cytoskeletal components that are involved in the possible nanotube-mediated communications between neighboring cells.
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Supplementary Material
Acknowledgments
The authors thank B. Likar and B. Babnik for help with preparation of the figures and movies. G.J.S., J.W., and S.W. were funded by the Austrian Science Fund (FWF project Y250-B10) and the GEN-AU project of the Austrian Federal Ministry for Science and Research.
Editor: Alberto Diaspro.
References
- 1.Linder, M. E., and A. G. Gilman. 1992. G-proteins. Sci. Am. 267:36–43. [DOI] [PubMed] [Google Scholar]
- 2.Kumar, N. M., and N. B. Gilula. 1996. The gap junction communication channel. Cell. 84:381–388. [DOI] [PubMed] [Google Scholar]
- 3.Mathivet, L., S. Cribier, and P. F. Devaux. 1996. Shape change and physical properties of giant phospholipids vesicles prepared in the presence of an AC electric field. Biophys. J. 70:1112–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kralj-Iglič, V., G. Gomiscek, J. Majhenc, V. Arrigler, and S. Svetina. 2001. Myelin-like protrusions of giant phospholipid vesicles prepared by electroformation. Colloids Surf. A. 181:315–318. [Google Scholar]
- 5.Kralj-Iglič, V., A. Iglič, G. Gomišček, V. Arrigler, and H. Hägerstrand. 2002. Microtubes and nanotubes of phospholipid bilayer vesicles. J. Phys. Math. Gen. 35:1533–1549. [Google Scholar]
- 6.Karlsson, A., R. Karlsson, M. Karlsson, A. S. Cans, A. Strömberg, F. Ryttsén, and O. Orwar. 2001. Networks of nanotubes and containers. Nature. 409:150–152. [DOI] [PubMed] [Google Scholar]
- 7.Iglič, A., H. Hägerstrand, M. Bobrowska-Hägerstrand, V. Arrigler, and V. Kralj-Iglič. 2003. Possible role of phospholipid nanotubes in directed transport of membrane vesicles. Phys. Lett. 310:493–497. [Google Scholar]
- 8.Rustom, A., R. Saffrich, I. Marković, P. Walther, and H. H. Gerdes. 2004. Nanotubular highways for intercellular organelle transport. Science. 303:1007–1010. [DOI] [PubMed] [Google Scholar]
- 9.Watkins, S. C., and R. D. Salter. 2005. Functional connectivity between immune cells mediated by tunneling nanotubules. Immunity. 23:309–318. [DOI] [PubMed] [Google Scholar]
- 10.Vidulescu, C., S. Clejan, and K. C. O'Connor. 2004. Vesicle traffic through intercellular bridges in DU 145 human prostate cancer cells. J. Cell. Mol. Med. 8:388–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koyanagi, M., R. P. Brandes, J. Haendeler, A. M. Zeiher, and S. Dimmeler. 2005. Cell-to-cell connection of endothelial progenitor cells with cardiac myocytes by nanotubes: a novel mechanism for cell fate changes? Circ. Res. 96:1039–1041. [DOI] [PubMed] [Google Scholar]
- 12.Gimsa, U., A. Iglič, S. Fiedler, M. Zwanzig, V. Kralj-Iglič, L. Jonas, and J. Gimsa. 2007. Actin is not required for nanotubular protrusions of primary astrocytes grown on metal nano-lawn. Mol. Membr. Biol. 24:243–255. [DOI] [PubMed] [Google Scholar]
- 13.Önfelt, B., S. Nedvetzki, K. Yanagi, and D. M. Davis. 2004. Cutting edge: membrane nanotubes connect immune cells. J. Immunol. 173:1511–1513. [DOI] [PubMed] [Google Scholar]
- 14.Iglič, A., M. Fošnarič, H. Hägerstrand, and V. Kralj-Iglič. 2004. Coupling between vesicle shape and the non-homogeneous lateral distribution of membrane constituents in Golgi bodies. FEBS Lett. 574:9–12. [DOI] [PubMed] [Google Scholar]
- 15.Wieser, S., M. Moertelmaier, E. Fuertbauer, H. Stockinger, and G. J. Schutz. 2007. (Un)confined diffusion of CD59 in the plasma membrane determined by High-resolution single molecule microscopy. Biophys. J. 92:3719–3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wieser, S., G. J. Schütz, M. E. Cooper, and H. Stockinger. 2007. Single molecule diffusion analysis on cellular nanotubes: implications on plasma membrane structure below the diffraction limit. Appl. Phys. Lett. 91:233901–13. [Google Scholar]
- 17.Galkina, S. I., J. G. Molotkovsky, V. Ullrich, and G. F. Sudina. 2005. Scanning electron microscopy study of neutrophil membrane tubulovesicular extensions (cytonemes) and their role in anchoring, aggregation and phagocytosis. The effect of nitric oxide. Exp. Cell Res. 304:620–629. [DOI] [PubMed] [Google Scholar]
- 18.Sun, M., J. S. Graham, B. Hegedüs, F. Marga, Y. Zhang, and G. Forgacs. 2005. Multiple membrane tethers probed by atomic force microscopy. Biophys. J. 89:4320–4329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gerdes, G. G., N. V. Bukoreshtliev, and J. F. V. Barroso. 2007. Tunneling nanotubes: a new route for the exchange of components between animal cells. FEBS Lett. 581:2194–2201. [DOI] [PubMed] [Google Scholar]
- 20.Kralj-Iglič, V., A. Iglič, M. Bobrowska-Hägerstrand, and H. Hägerstrand. 2001. Tethers connecting daughter vesicles and parent red blood cell may be formed due to ordering of anisotropic membrane constituents. Colloids Surf. A. 180:57–64. [Google Scholar]
- 21.Roux, A., G. Cappello, J. Cartaud, J. Prost, B. Goud, and P. Bassereau. 2002. A minimal system allowing tubulation with molecular motors pulling on giant liposomes. Proc. Natl. Acad. Sci. USA. 99:5394–5399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kralj-Iglič, V., B. Babnik, D. R. Gauger, S. May, and A. Iglič. 2006. Quadrupolar ordering of phospholipid molecules in narrow necks of phospholipid vesicles. J. Stat. Phys. 125:727–752. [Google Scholar]
- 23.Faix, J., and K. Rottner. 2006. The making of filopodia. Curr. Opin. Cell Biol. 18:18–25. [DOI] [PubMed] [Google Scholar]
- 24.Iglič, A., H. Hägerstrand, P. Veranič, A. Plemenitaš, and V. Kralj-Iglič. 2006. Curvature induced accumulation of anisotropic membrane components and raft formation in cylindrical membrane protrusions. J. Theor. Biol. 240:368–373. [DOI] [PubMed] [Google Scholar]
- 25.Janich, P., and D. Corbeil. 2007. GM1 and GM3 gangliosides highlight distinct lipid microdomains with the apical domain of epithelial cells. FEBS Lett. 581:1783–1787. [DOI] [PubMed] [Google Scholar]
- 26.Fargeas, C. A., A. V. Fonseca, W. B. Huttner, and D. Corbeil. 2006. Prominin-1 (CD133): from progenitor cells to human diseases. Future Lipidol. 1:213–225. [Google Scholar]
- 27.Corbeil, D., K. Röper, C. A. Fargeas, A. Joester, and W. B. Huttner. 2001. Prominin: a story of cholesterol, plasma membrane protrusions and human pathology. Traffic. 2:82–91. [DOI] [PubMed] [Google Scholar]
- 28.Huttner, W. B., and J. Zimmerberg. 2001. Implications of lipid microdomains for membrane curvature, budding and fission. Curr. Opin. Cell Biol. 13:478–484. [DOI] [PubMed] [Google Scholar]
- 29.Römer, W., L. Berland, V. Chambon, K. Gaus, B. Windschiegl, D. Tenza, M. R. E. Aly, V. Fraisier, J.-C. Florent, D. Perrais, C. Lamaze, G. Raposo, C. Steinem, P. Sens, P. Bassereau, and L. Johannes. 2007. Shiga toxin induces tubular membrane invaginations for its uptake into cells. Nature. 450:670–679. [DOI] [PubMed] [Google Scholar]
- 30.Iglič, A., M. Lokar, B. Babnik, T. Slivnik, P. Veranič, H. Hägerstrand, and V. Kralj-Iglič. 2007. Possible role of flexible red blood cell membrane nanodomains in the growth and stability of membrane nanotubes. Blood Cells Mol. Dis. 39:14–23. [DOI] [PubMed] [Google Scholar]
- 31.Salzer, U., and R. Prohaska. 2003. Segregation of lipid raft proteins during calcium-induced vesiculation of erythrocytes. Blood. 101:3751–3753.12707226 [Google Scholar]
- 32.Al-Nedawi, K., B. Meehan, J. Micallef, V. Lhotak, L. May, A. Guga, and J. Rak. 2007. Intercellular transfer of oncogenic receptor EGFRvIII by microvesicles derived from tumor cells. Nat. Cell Biol. 10:619–624. [DOI] [PubMed] [Google Scholar]
- 33.Roelofsen, B., F. A. Kuypers, J. A. Op den Kamp and L. L. van Deenen. 1989. Influence of phosphatidiylcholine molecular species composition on stability of the erythrocyte membrane. Biochem. Soc. Trans. 17:284–286. [DOI] [PubMed] [Google Scholar]
- 34.Kuypers, F. A., B. Roelofsen, W. Berendsen, J. A. F. Op den Kamp, and L. L. M. van Deenen. 1984. Shape changes in human erythrocytes induced by replacement of the native phosphatidiylcholine with species containing various fatty acids. J. Cell Biol. 99:2260–2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kralj-Iglič, V., A. Iglič, H. Hägerstrand, and P. Peterlin. 2000. Stable tubular microexovesicles of the erythrocyte membrane induced by dimeric detergent. Phys. Rev. E Stat. Phys. Plasmas Fluids Relat. Interdiscip. Topics. 61:4230–4234. [DOI] [PubMed] [Google Scholar]
- 36.Coulombe, P. A., O. Bousquet, L. Ma, S. Yamada, and D. Wirtz. 2000. The ‘ins’ and ‘outs’ of intermediate filament organization. Trends Cell Biol. 10:420–428. [DOI] [PubMed] [Google Scholar]
- 37.Magin, T. M., P. Vijayaraj, and R. E. Leube. 2007. Structural and regulatory functions of keratins. Exp. Cell Res. 313:2021–2032. [DOI] [PubMed] [Google Scholar]
- 38.Beznoussenko, G. V., and A. A. Mironov. 2002. Models of intracellular transport and evolution of the Golgi complex. Anat. Rec. 268:226–238. [DOI] [PubMed] [Google Scholar]
- 39.Bar-Ziv, R., and E. Moses. 1994. Instability and “pearling” states produced in tubular membranes by competition of curvature and tension. Phys. Rev. Lett. 73:1392–1395. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.