

Abstract
We investigate the relaxation dynamics of melanin model constituents including monomers, dimers, and tetramers, upon excitation, using state-of-the-art, time-dependent, density functional theory calculations. The results explain the ability of these molecules to transform photon energy into thermal energy in a remarkably short timescale of ∼100 fs. We find that after electronic excitation by light absorption, ultrafast energy conversion takes place through two novel mechanisms: proton transfer on a timescale of 110 fs and state mixing upon oligomerization on a timescale of <50 fs. These results are in good agreement with available experiments and help elucidate melanin's role in photoprotection against ultraviolet radiation.
INTRODUCTION
Melanin, which is widely present in living organisms including animals, plants, and microorganisms, is believed to be essential for defense against exposure to UV radiation (1,2). This function is achieved through a unique set of optical properties including featureless broadband absorbance (3); extremely low scattering (<6%) and emission (< 0.1%) (4); ultrafast relaxation dynamics (5,6); and the ability to transfer x-ray energy into chemical energy (7), analogous to photosynthesis. Approximately 90% of UV energy absorbed by eumelanin, the dominant subclass of melanin, dissipates as heat on a timescale of <1 ns (5). Ultrafast nonradiative photochemical processes with a characteristic timescale of 150 fs has first been revealed by surface hopping simulations of the primary event in rhodopsin retinal upon visible light illumination (8). Recently, Olsen et al. observed convergent proton transfer in monomers of eumelanin under UV excitation (9), through a process that breaks the mirror-image symmetry between absorption and emission spectra. The microscopic mechanism for this process, and the effects of polymerization on this and on other relaxation pathways available to eumelanin, are not known.
Microscopic understanding of the processes related to melanin's photoprotective role is hindered by the lack of an established molecular structure (10). Eumelanin comprises 5,6-dihydroxyindole (DHI), 5,6-dihydroxy-indole-2-carboxylic acid (DHICA), and their derivatives. The reduced forms of DHI, indolequinone (IQ) and its tautomers quinone-methide (MQ) and quinone-imine (NQ), are all essential components in the Raper-Mason scheme of melanin synthesis (11) and most likely are present in the ultimate melanin structure. MQ and NQ, although marginally present in isolated form (13% and 0.1% compared to IQ), may have a dominant presence in oligomers after polymerization and metal binding (12). In this work we use state-of-the-art electronic structure calculations to investigate nonradiative relaxation processes in eumelanin model constituents including its indole building blocks (IQ, MQ, NQ) as monomers and in oligomers like dimers and tetramers, the latter structures based on a recently proposed porphyrinlike model for eumelanin (13,14). While not intending to extensively investigate all possible melanin constituent species, our results provide useful insights into the specific molecular mechanisms behind melanin's photoprotective properties.
COMPUTATIONAL METHODS
We simulate the nonadiabatic evolution of both electrons and ions in real time under excitation using time-dependent density functional theory (TDDFT) (15) in an implementation based on the SIESTA code (16). The time-dependent Kohn-Sham equations of electrons and the Newtonian motion of ions are solved simultaneously, using numerical atomic orbitals (17) as bases. The electron density is updated self-consistently during the time propagation of Kohn-Sham single-electron wavefunctions. The initial velocity of ions is assigned according to the equilibrium Boltzmann-Maxwell distribution at a given temperature. The real-time implementation of TDDFT is distinct from the usual linear-response form formulated in frequency domain (18), enabling us to capture the nonlinear effects under a strong field and the associated electron dynamics. The use of local bases greatly enhances the efficiency of the real-time TDDFT simulation, reducing the computer time and memory requirement by 1–2 orders of magnitude while maintaining high accuracy from first-principles. It could also lead to linear-scaling performance of excited state simulation, allowing studies on large biological systems and nanomaterials to be possible. Real-time propagation methods similar to ours but formulated in plane-wave bases are used to monitor excited state trajectories of small molecules, obtaining results beyond linear response, with accuracy similar to the CASPT2 method in quantum chemistry (19) and in general agreement with experiment (20). Similar approaches with real-space grid or Gaussian basis are used to investigate harmonic generation (21) and atomic collision (22). The details of this method and the implementation are published elsewhere (23).
Our method implies that our simulations of excited states follow Ehrenfest dynamics, where the quantum states of coupled electron-ion motion are reduced to the classical ionic trajectories. It represents a mean-field approach of the mixed quantum-classical system, with forces on the ions averaged over many adiabatic electronic states induced by the ionic motion. It works well for situations where a single path dominates in the reaction dynamics, for the initial stages of excited states before significant surface crossings take place, or for cases where the states are similar and the state-averaged behavior is of interest when many electron levels are involved as in condensed phases. Advantages of Ehrenfest dynamics include its independence on electron representations used and its efficiency with propagation schemes (24). The approach has limitations when the excited states involve multiple paths, especially when state-specific ionic trajectories are desired. Alternative strategies other than Ehrenfest dynamics for mixed quantum-classical systems include surface hopping methods (25) (which explicitly include electron transitions) and the decay-of-mixing method (26). However, the dependence on diabatic or adiabatic representations and the occurrence of frustrated hops are problematic and could lead to significant errors in surface hopping (24). The mixed quantum-classical methods are accurate in many cases (24). Improvements to include some quantum effects of nuclei are possible in literature such as ab initio multiple spawning methods (27). In this study, Ehrenfest dynamics is suitable because we are interested in the early stages of excited states of model melanin constituents and the mechanisms that lead to branching or nonradiative decay.
We use Troullier-Martins pseudopotentials (28), the adiabatic local density approximation (29) for the exchange-correlation functional and a basis of double-ζ polarized orbitals (13 atomic orbitals for C, N, and O and five orbitals for H) (30). With a timestep of 0.02419 fs, the total energy is well conserved to within 4 × 10−5 eV/fs. We emphasize that in the systems we consider here the excited states are all valence excitation states, where self-interaction errors for occupied and unoccupied states are of the same order of magnitude and cancel each other. This cancellation does not occur for situations that involve charge-transfer excited states, in which case TDDFT simulations using standard local approximations of the exchange-correlation functional suffer from significant self-interaction errors and the calculated excitation energies offer no improvement over the energy difference of the corresponding ground-state Kohn-Sham orbitals (31). This is not the case here. The computational scheme we employ successfully reproduces experimental parameters of small molecules in excited states and the ground state. For example, for the CO excited state, we calculate a bond length of 1.24 Å and vibrational frequency of 1597 cm−1, which compare well with the experimentally determined values (32) of 1.24 Å and 1518 cm−1 (the ground-state calculated values are 1.15 Å and 2245 cm−1 and the experimental ones 1.13 Å and 2170 cm−1).
RESULTS AND DISCUSSION
Optical properties of indole monomers
We begin with a discussion on the optical properties of indole monomers. Fig. 1 shows the calculated optical absorption spectra of IQ, MQ, and NQ. The reference molecule IQ has intense absorption bands at 1.69 and 3.57 eV. The first two peaks for MQ at 1.14 and 3.58 eV correspond to excitations from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO), and from the third orbital below the HOMO to the LUMO, respectively. For convenience, in the rest of the article we will number the occupied states as 1, 2, 3… counting from the HOMO toward states of lower energy and the unoccupied ones as 1*, 2*, 3*… counting from the LUMO toward states of higher energy. The two absorption bands are significantly red-shifted in NQ, located at 0.94 and 2.73 eV, respectively, and their intensity is attenuated. From these absorption spectra, we expect the transformation of MQ to NQ to decrease the quantum yield and energy of emitted photons, leading to deactivation of the excited MQ, which is more populated in the ground state (12). Indeed, our simulations, which we discuss next, show that during the excitation, MQ quickly relaxes into the NQ structure by transferring an H atom from the O1 to the O2 site.
FIGURE 1.

The absorption spectra of quinone-methide (MQ, blue solid line) and quinone-imine (NQ, red dashed line). The spectrum of indole quinone (IQ, gray dotted line) is included for comparison. Structures are shown with standard atom labeling (green, C; blue, N; red, and O; white, H).
Proton transfer mechanism upon excitation
Fig. 2 a shows the variation of OH distances 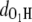 and
and 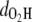 in the MQ monomer after electronic excitation during the TDDFT simulation at 300 K. At time t = 0, an electron at state 1 (the HOMO) is promoted to state 1* (the LUMO). The value
in the MQ monomer after electronic excitation during the TDDFT simulation at 300 K. At time t = 0, an electron at state 1 (the HOMO) is promoted to state 1* (the LUMO). The value 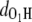 gradually decreases while
gradually decreases while 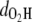 is almost constant until t = 104.79 fs, when the two curves cross, indicating a proton transfer process from O1 to O2. The energy separating state 1 from state 1* changes from 1.5 eV at t = 0 to 0.3–0.5 eV at ∼t = 105 fs. The off-diagonal element Eij, with i and j being the corresponding state indexes of the Hamiltonian matrix, was monitored during the simulation in Fig. 3 a. A large Eij value suggests high tendency of band mixing and high recombination probability between states i and j; it also indicates the failure of the nonadiabatic simulation when it becomes comparable to the typical separation between states, because of the inability to calculate the Hellmann-Feynman forces on the atoms. We found that the off-diagonal element between the excited electron and the hole remains small (<0.2 eV) until t = 114 fs, when H transfer occurs. It increases to 0.4 eV, close to the HOMO-LUMO separation at this time, indicating high tendency of electron-hole recombination (Fig. 3 a).
is almost constant until t = 104.79 fs, when the two curves cross, indicating a proton transfer process from O1 to O2. The energy separating state 1 from state 1* changes from 1.5 eV at t = 0 to 0.3–0.5 eV at ∼t = 105 fs. The off-diagonal element Eij, with i and j being the corresponding state indexes of the Hamiltonian matrix, was monitored during the simulation in Fig. 3 a. A large Eij value suggests high tendency of band mixing and high recombination probability between states i and j; it also indicates the failure of the nonadiabatic simulation when it becomes comparable to the typical separation between states, because of the inability to calculate the Hellmann-Feynman forces on the atoms. We found that the off-diagonal element between the excited electron and the hole remains small (<0.2 eV) until t = 114 fs, when H transfer occurs. It increases to 0.4 eV, close to the HOMO-LUMO separation at this time, indicating high tendency of electron-hole recombination (Fig. 3 a).
FIGURE 2.

Time evolution of MQ excited state. (a) The OiH (i = 1,2) distances and (b) electronic state energies (diagonal Hamiltonian matrix elements) as a function of time after excitation. State number and state character (bond type) are indicated in panel b.
FIGURE 3.

Time evolution of (a) the off-diagonal Hamiltonian matrix element, (b) adiabatic and diabatic energy gap, and (c) transition probability (calculated from Landau-Zener formula, see text) between states 1 and 1* upon excitation in MQ.
Indeed, we have calculated the transition probability between state 1 and 1* along the trajectory based on the Landau-Zener theory, which is originally formulated to study the nonadiabatic transition at the linear crossing of two electronic energy levels i and j (33,34). Only the two states are included in the analysis because only the off-diagonal elements between them are significant. The Landau-Zener decay probability is 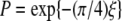 (34,35), where ξ is the Massey parameter
(34,35), where ξ is the Massey parameter 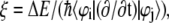 with ΔE being the energy difference between two adiabatic states, φi and φi, calculated by diagonalizing the matrix
with ΔE being the energy difference between two adiabatic states, φi and φi, calculated by diagonalizing the matrix 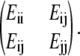 The adiabatic and diabatic energy gaps between state 1 and 1* are also shown in Fig. 3 b, with the decay probability shown in Fig. 3 c. Generally the diabatic energy gap shows negligible deviation from the adiabatic one except for 40 fs < t < 60 fs, and t > 100 fs. The decay probability is <5% for t < 100 fs with two small peaks at 17 fs and 95 fs, but increases to ∼30% at t = 107 fs, indicating a fairly large transition probability from state 1* to 1. If this does occur, the molecule returns to the ground state of NQ either by emitting photons of low energy (0.5 eV), evidenced by a small but finite oscillation strength ∼0.002, or through radiationless internal conversion. This process may continue as NQ transforms into MQ (thermodynamically the more stable form) in the ground state, releasing 0.2 eV of energy as heat.
The adiabatic and diabatic energy gaps between state 1 and 1* are also shown in Fig. 3 b, with the decay probability shown in Fig. 3 c. Generally the diabatic energy gap shows negligible deviation from the adiabatic one except for 40 fs < t < 60 fs, and t > 100 fs. The decay probability is <5% for t < 100 fs with two small peaks at 17 fs and 95 fs, but increases to ∼30% at t = 107 fs, indicating a fairly large transition probability from state 1* to 1. If this does occur, the molecule returns to the ground state of NQ either by emitting photons of low energy (0.5 eV), evidenced by a small but finite oscillation strength ∼0.002, or through radiationless internal conversion. This process may continue as NQ transforms into MQ (thermodynamically the more stable form) in the ground state, releasing 0.2 eV of energy as heat.
This proton transfer is an activated process. The potential energy was monitored in Fig. 4, where a barrier of <0.2 eV is identified. Indeed, our simulations reveal that the process takes place when the initial temperature is T0 = 300 K, but does not occur at T0 = 0 K, as shown by the values of 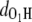 and
and 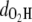 in Fig. 2 a. During H transfer, the electron density of the HOMO shows a concerted transition of π-bond patterns from MQ to NQ, which involves double-bond breaking and reforming (Fig. 4). Please check the Supplementary Material for the real time evolution of wavefunctions (see Movie S1). The transition can have momentary setbacks, and the molecule returns to its initial state with a broken C7-C8 bond, if the H is not successfully transferred (as at the time at ∼t = 103 fs). When it does successfully transfer (at ∼t = 114 fs), the bonding in the molecule evolves into a new pattern and the potential energy is lowered by ∼0.1 eV (t = 118 fs). Therefore we have directly observed π-bond breaking processes in real-time through concerted electron-proton dynamics simulation. As shown in Fig. 4 (inset), the proton transfer process is strongly coupled with electron redistribution around the neighboring O sites, indicating it is actually an H atom transfer. This is in general agreement with previous photochemistry studies, which reveal that excited-state intramolecular proton transfer is mostly H atom transfer with intrinsic electron-proton couplings (36,37). For convenience, we do not make an explicit difference between proton transfer and H atom transfer in this work, collectively referred to as H transfer.
in Fig. 2 a. During H transfer, the electron density of the HOMO shows a concerted transition of π-bond patterns from MQ to NQ, which involves double-bond breaking and reforming (Fig. 4). Please check the Supplementary Material for the real time evolution of wavefunctions (see Movie S1). The transition can have momentary setbacks, and the molecule returns to its initial state with a broken C7-C8 bond, if the H is not successfully transferred (as at the time at ∼t = 103 fs). When it does successfully transfer (at ∼t = 114 fs), the bonding in the molecule evolves into a new pattern and the potential energy is lowered by ∼0.1 eV (t = 118 fs). Therefore we have directly observed π-bond breaking processes in real-time through concerted electron-proton dynamics simulation. As shown in Fig. 4 (inset), the proton transfer process is strongly coupled with electron redistribution around the neighboring O sites, indicating it is actually an H atom transfer. This is in general agreement with previous photochemistry studies, which reveal that excited-state intramolecular proton transfer is mostly H atom transfer with intrinsic electron-proton couplings (36,37). For convenience, we do not make an explicit difference between proton transfer and H atom transfer in this work, collectively referred to as H transfer.
FIGURE 4.

Concerted electron-proton motion. Total energy (dashed blue line) and potential energy (solid black line) are plotted as a function of time. Snapshots of the HOMO electron density (at contour level 0.02 e/Å3) and corresponding bonding diagrams show the evolution of π-bond patterns during proton transfer.
It was found from previously studies that the reaction coordinate in intramolecular H transfer involves mainly heavy-atom bond alternation instead of proton movement (37), consistent with our trajectories where forces on the transferring proton are minimal. More important, conical intersections involving two (37,38) or three states (39) are critical for deactivation dynamics and responsible for the sharp decrease in fluorescence quantum yield. Although the capability of TDDFT to describe such conical intersections depends case by case and is to be explored (40), we expect similar conical intersections between excited states and the ground state to exist in MQ, as sampled in some excited-state trajectories. To confirm the above picture, we performed simulations on multiple trajectories. The ground-state ab initio molecular dynamics was run for 1200 fs after equilibrating the system at 300 K for 500 fs. Twelve ground-state configurations at every 100 fs during the MD trajectory were collected for excited state simulations. The result shows that all these trajectories involve H transfer within 120 fs after photoexcitation, three of which have H transfer within 20 fs. The Landau-Zener transition probability at approximately the time of H transfer ranges from 10% to 98%. The large variation is due to great thermal fluctuations in the MQ molecule because of its small size. The excitation at 3.58 eV shows similar behavior, namely, proton transfer occurs at ∼110 fs after excitation at 300 K.
The above H transfer mechanism explains the observed broken mirror-image symmetry in the absorption and emission spectra of the melanin DHICA monomers. DHICA solutions exhibit two absorption peaks at 3.8 and 4.2 eV in the UV region while displaying a single emission peak at 3.0 eV (9). From our TDDFT molecular dynamics simulation, this is the result of molecules in the methide form transforming into the imine form after excitation, so that both forms absorb photons of different energy while only those in the imine form emit light, in agreement with the mechanism suggested by Olsen et al. (9). This type of photoinduced H transfer has been discussed intensively in relation to DNA basepairs as a cause of gene mutation (41–43). Recent ultrafast measurements (44,45) consistently report a biexponential decay behavior with two decay constants of ∼0.2 ps and 1.1 ps, corresponding to a double-proton transfer process. Although still under debate, the lifetime for the first H transfer (∼130 fs) is very close to what we find here for excited-state H transfer in melanin (110 fs), if a stepwise proton transfer mechanism is assumed (45).
We note that in real experiments in vivo and in vitro, melanin is surrounded by solvent molecules that might serve as hydrogen reservoirs and thus be involved in the excited H transfer processes. The barrier for proton transfer, dependent on the pKa difference between the donor and acceptor groups, is sensitive to the presence of solvents; it can be significantly changed if the solvent effect is missing (46). That is not the case in this study because the transferring H is closely bonded to the two neighboring -C-O- sites throughout the process and unlikely to be much perturbed by solvents. This is evidenced by the small barrier ∼0.1–0.2 eV identified. We expect that solvent-mediated H transfer might take place at a similar or slightly longer timescale. Full treatment of solvent molecules in first-principles is very challenging given the current capabilities of time-dependent ab initio calculations, but solvent molecules can be explicitly included in the hybrid QM/MM method to fully account for the dynamical effects of the solvation structure and the large system size (47). While the integration of the current method with QM/MM approaches is interesting and subject to future work, our present results on model structures in vacuum represent a simplified description of the variety of real processes, upon which a more complete picture incorporating solvent effects may be built.
State-mixing in the dimer due to polymerization
A more important question is how polymerization of the melanin monomers affects H transfer and other relaxation processes. We therefore chose a dimer structure, composed by an MQ and an IQ unit covalently bonded at the 2 and 7′ sites and compared its relaxation dynamics with those of the monomers. Dimers coupled at 2–7′ sites are the most stable (48) and dominant during melanin synthesis (49). We found that the H transfer process also occurs in the MQ-IQ dimer for low-energy excitations (0.9 eV) at 300 K, albeit with a shorter timescale for electron-hole recombination. More interestingly, we found that excited states in MQ-IQ are more likely to experience ultrafast nonradiative relaxation through the mixture of the hole and the occupied states before H transfer can take place. This is demonstrated by electron dynamics at the excitation energy of 3.48 eV in Fig. 5, where an electron is promoted from state 8 to 1*. For comparison, excitation dynamics at 3.58 eV in the MQ monomer is also shown. In both cases the initial temperature was set to 0 K for simplicity. MQ has a long lifetime (>190 fs) when electron-hole recombination or band mixture is not active, while MQ-IQ experiences ultrafast electronic relaxation processes. At t = 42.55 fs, the off-diagonal matrix element between state 8 (the hole) and 2 becomes large (>0.4 eV). As a result, the two bands start to mix and their energy approaches each other, clearly shown by a significant decrease of the state 2 energy after this time (Fig. 5 b). The mixing is more obvious for states 3 and 9 (the latter being almost degenerate with state 8 after excitation), which start to mix at the same time. At t = 0 the two bands are separated by 2.14 eV with very different bonding patterns in the MQ constituent: C3-C9 and C4-C5 bonds for state 3 and N1-C2 and C9-C4 bonds for state 9 (shown in Fig. 5 c). However, as they start to mix, the two bands develop into almost the same character at t = 48.38 fs with bonding at N1-C2 and C3-C9-C4-C5 sites. The Eij value at this time becomes even larger (0.7 eV) than their energy separation of 0.5 eV. As the Hellmann-Feynman theorem fails at this point, the simulation was stopped. We expect states 2 and 8 (the hole) a few femtoseconds later to exhibit the same mixing behavior as states 3 and 9 and thus quench the excited state without any (or with very low) emission. The nonzero transition probability starts to develop. During this process, direct recombination of the excited electron and the hole would not happen, due to negligible interaction: the Eij value is <0.08 eV and the transition probability is zero. We find that energy dissipation occurs mainly through excitation of vibrational modes at 1346 cm−1 (C-C and C-N stretch) and 673 cm−1 (C-H out-of-plane bending) and a small contribution at 3363 cm−1 (O-H stretch). The main cause of the nonradiative relaxation process is the presence of extra states (9, 3, and 2) introduced by polymerization, which greatly increases the possibility of band mixing. Simulations at an initial temperature of 300 K show the same dynamics, indicating that this electronic relaxation process is not affected by temperature.
FIGURE 5.

Evolution of the energy of electronic states after excitation at 350 nm in (a) the MQ monomer and (b) the MQ-IQ dimer. Dashed lines indicate states between which the excitation takes place. (c) Electron charge density (at contour level of 0.01 e/Å3) in the MQ-IQ dimer for states 9 and 3 (dot-dashed lines in b) at two different times.
Ultrafast relaxation in tetramers
As the polymerization of eumelanin's constituent monomers proceeds further, the nonradiative relaxation through state-mixing could take place even faster. However, the study is hindered by the lack of a realistic model for the secondary structure of eumelanin at the atomic level. Two qualitatively different models, the cross-linked heteropolymer model (50) and the stacked oligomer model (51), have been proposed to account for the observed melanin properties, but neither of them has been firmly established from experimental observations. There is, however, increasingly more evidence in support of the finite size of melanin constituent particles or protomolecules, presumably composed of small melanin oligomers (10). Recently, a new, detailed structural model for eumelanin protomolecules was proposed based on a tetramer structure consisting of four indole monomers in arrangements that contain an inner porphyrin ring (13). This model provides a natural explanation of the finite size of eumelanin protomolecules, x-ray diffraction data, and metal-binding properties (14). Most importantly, the broadband absorbance of eumelanin is reproduced by superposition of the spectra of all likely tetramer structures in agreement with the chemical disorder concept (52).
Motivated by the promising character of this tetramer model for melanin protomolecules, we study its relaxation dynamics. We will use the most stable tetramer structure composed of MQ-IQ-MQ-IQ as an example. This molecule can be thought of as linking two stable MQ-IQ dimers shown in Fig. 5, through the C2-C7′ sites (Fig. 6, inset). TDDFT calculations for this structure predict two major peaks in the adsorption spectrum shown in Fig. 6 a, at 3.48 eV (356 nm) and 2.58 eV (480 nm), a shoulder at 2 eV, and a broad peak at ∼0.5–1.5 eV. The 3.48-eV absorption comprises mainly excitation from state 2 to state 6*. The simulation of electron dynamics after this excitation is shown in Fig. 6 b, where the energy of electronic states is plotted as a function of time. The energy of state 6* starts to decrease abruptly at t = 24 fs after excitation, an indication that this state starts to mix with other states. Analysis on the off-diagonal Hamiltonian elements shows that it sequentially crosses with state 5*, 4*, 2*, 2*, and 1*, and becomes lower in energy than all unoccupied states at t = 50 fs, consistent with Kasha's rule. Occupied states around state 2 (the hole) also mix strongly, because they are even closer in energy with each other, and probably mix with the lowest unoccupied state, now state 6*. The ground state is thus restored without emission, and the photon energy is transferred into thermal energy. Overall this process is quite similar to that in the MQ-IQ dimer in Fig. 5, but the state-mixing takes place ∼10-fs earlier in the tetramer case. Simulations of excitation at 2.58 eV show energy dissipation on the same timescale. This supports the idea that the larger the extent of oligomerization, the faster the nonradiative relaxation is, after UV illumination. Therefore, after completion of oligomerization, the photoprotective function through state-mixing is fully developed due to the presence of dense and sensitively entangled states. Ultrafast processes with a timescale <50 fs have been reported in femtosecond pump-probe spectroscopy of eumelanin (6), which is consistent with our results.
FIGURE 6.

Optical properties of a tetramer protomolecule. (a) Absorption spectrum of the MQ-IQ-MQ-IQ protomolecule (inset) and (b) evolution of the energy of electronic states after excitation at ∼350 nm. Dashed lines indicate states between which the excitation takes place. The Fermi energy level is set to zero.
Finally, we note that the above ultrafast state-mixing as a nonradiative decay pathway is by no means limited to the simple structural models we studied as illustrative examples. According to the chemical disorder model recently proposed (52), there is large structural variability in eumelanin constituents including monomers, dimers, and oligomers, as well as those structures comprising DHICA. Indeed, nonplanar DHICA dimer structures coupled at C4-C7′ (53) and tetramers involving novel couplings at C2-C3′ (54) have also been synthesized recently. The state-mixing processes and the corresponding timescales in the more complicated structures encountered in actual eumelanin would be essentially similar to those of the MQ-IQ dimer and MQ-IQ-MQ-IQ tetramer presented above, since they all have the same structural characteristics, that is, single bonds between monomers and the presence of extra bands due to oligomerization.
CONCLUDING REMARKS
By monitoring the evolution of the excited states of eumelanin constituent building-blocks (monomers and dimers) and model protomolecules (tetramers), we find that eumelanin experiences ultrafast nonradiative excited-state relaxation, which effectively transfers photon energy into heat, thus blocking possible light-induced damage. These relaxation processes take place through H transfer and state mixture, for both of which we have given direct evidence. The H transfer mechanism explains well the observed mirror-image symmetry breaking in the absorption and emission spectra of eumelanin monomers (9). The timescale of 110 fs for the H transfer at 300 K agrees with that measured for the first-proton transfer in a model DNA basepair (45), ∼130 fs. Further, we find that the timescale for state mixing is very sensitive to the extent of polymerization: monomers do not show significant state-mixture, while dimers have state mixing on a 50-fs timescale, and tetramers even shorter, on a scale of 30–40 fs. The overall nonradiative relaxation in the building blocks and protomolecules of eumelanin takes place within a time <50 fs through state mixture, in agreement with the transient adsorption data in femtosecond pump-probe experiments (6).
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Supplementary Material
Acknowledgments
We acknowledge helpful discussions with P. Meredith and J. D. Simon.
This work was partially supported by Department of Energy Computational Materials Science Network grant No. DE-FG02-05ER46226 and the Harvard University Center for the Environment.
Editor: Gregory A. Voth.
References
- 1.Kollias, N., R. M. Sayre, L. Zeise, and M. R. Chedekel. 1991. Photoprotection by melanin. J. Photochem. Photobiol. B. 9:135–160. [DOI] [PubMed] [Google Scholar]
- 2.Seagle, B. L., E. M. Gasyna, W. F. Mieler, and J. R. Norris, Jr. 2006. Photoprotection of human retinal pigment epithelium cells against blue light-induced apoptosis by melanin free radicals from Sepia officinalis. Proc. Natl. Acad. Sci. USA. 103:16644–16648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Meredith, P., and T. Sarna. 2006. The physical and chemical properties of eumelanin. Pigment Cell Res. 19:572–594. [DOI] [PubMed] [Google Scholar]
- 4.Meredith, P., and J. Riesz. 2004. Radiative relaxation quantum yields for synthetic eumelanin. Photochem. Photobiol. 79:211–216. [DOI] [PubMed] [Google Scholar]
- 5.Nofsinger, J. B., S. E. Forest, and J. D. Simon. 1999. Explanation for the disparity among absorption and action spectra of eumelanin. J. Phys. Chem. 103:11428–11432. [Google Scholar]
- 6.Nofsinger, J. B., and J. D. Simon. 2001. Radiative relaxation of Sepia eumelanin is affected by aggregation. Photochem. Photobiol. 74:31–37. [DOI] [PubMed] [Google Scholar]
- 7.Dadachova, E., R. A. Bryan, X. Huang, T. Moadel, A. D. Schweitzer, P. Aisen, J. D. Nosanchuk, and A. Casadevall. 2007. Ionizing radiation changes the electronic properties of melanin and enhances the growth of melanized fungi. PloS ONE. 2:e457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Warshel, A. 1976. Bicycle-pedal model for 1st step in vision process. Nature. 260:679–683. [DOI] [PubMed] [Google Scholar]
- 9.Olsen, S., J. Riesz, I. Mahadevan, A. Coutts, B. J. Powell, R. H. McKenzie, S. C. Smith, and P. Meredith. 2007. Convergent proton transfer photocycles violate mirror-image symmetry in a key melanin monomer. J. Am. Chem. Soc. 129:6672–6673. [DOI] [PubMed] [Google Scholar]
- 10.Bush, W. D., J. Carguilo, F. A. Zucca, A. Albertin, L. Zecca, G. S. Edwards, R. J. Nemanich, and J. D. Simon. 2006. The surface oxidation potential of human neuromelanin reveals a spherical architecture with a pheomelanin core and a eumelanin surface. Proc. Natl. Acad. Sci. USA. 103:14785–14789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ito, S. 2003. A chemist's view of melanogenesis. Pigment Cell Res. 16:230–236. [DOI] [PubMed] [Google Scholar]
- 12.Il'ichev, Y. V., and J. D. Simon. 2003. Building blocks of eumelanin: relative stability and excitation energies of tautomers of 5,6-dihydroxyindole and 5,6-indolequinone. J. Phys. Chem. 107:7162–7171. [Google Scholar]
- 13.Kaxiras, E., A. Tsolakidis, G. Zonios, and S. Meng. 2006. Structural model of eumelanin. Phys. Rev. Lett. 97:218102. [DOI] [PubMed] [Google Scholar]
- 14.Meng, S., and E. Kaxiras. 2008. Theoretical models of eumelanin protomolecules and their optical properties. Biophys. J. 94:2095–2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Runge, E., and E. K. U. Gross. 1984. Density-functional theory for time-dependent systems. Phys. Rev. Lett. 52:997–1000. [Google Scholar]
- 16.Soler, J. M., E. Artacho, J. D. Gale, A. García, J. Junquera, P. Ordejón, and D. Sánchez-Portal. 2002. The SIESTA method for ab initio order-N materials simulation. J. Phys. Condens. Matter. 14:2745–2779. [Google Scholar]
- 17.Junquera, J., O. Paz, D. Sanchez-Portal, and E. Artacho. 2001. Numerical atomic orbitals for linear-scaling calculations. Phys. Rev. B. 64:235111. [Google Scholar]
- 18.Casida, M. E. 1996. Recent Developments and Applications in Density Functional Theory. J. M. Seminario, editor. Elsevier, Amsterdam, The Netherlands.
- 19.Tavernelli, I., U. F. Rohrig, and U. Rothlisberger. 2005. Molecular dynamics in electronically excited states using time-dependent density functional theory. Mol. Phys. 103:963–981. [Google Scholar]
- 20.Tateyama, Y., N. Oyama, T. Ohno, and Y. Miyamoto. 2006. Real-time propagation time-dependent density functional theory study on the ring-opening transformation of the photoexcited crystalline benzene. J. Chem. Phys. 124:124507. [DOI] [PubMed] [Google Scholar]
- 21.Castro, A., M. A. L. Marques, J. A. Alonso, G. F. Bertsch, and A. Rubio. 2004. Excited states dynamics in time-dependent density functional theory—high-field molecular dissociation and harmonic generation. Eur. Phys. J. D. 28:211–218. [Google Scholar]
- 22.Isborn, C. M., X. Li, and J. C. Tully. 2007. Time-dependent density functional theory Ehrenfest dynamics: collisions between atomic oxygen and graphite clusters. J. Chem. Phys. 126:134307. [DOI] [PubMed] [Google Scholar]
- 23.Meng, S., and E. Kaxiras. 2008. Real-time, local basis-set implementation of time-dependent density functional theory for excited state dynamics simulations. J. Chem. Phys. 129:054110. [DOI] [PubMed] [Google Scholar]
- 24.Hack, M. D., and D. G. Truhlar. 2000. Nonadiabatic trajectories at an exhibition. J. Phys. Chem. A. 104:7917–7926. [Google Scholar]
- 25.Tully, J. C. 1990. Molecular-dynamics with electronic-transitions. J. Chem. Phys. 93:1061–1071. [Google Scholar]
- 26.Zhu, C. Y., S. Nangia, A. W. Jasper, and D. G. Truhlar. 2004. Coherent switching with decay of mixing: an improved treatment of electronic coherence for non-Born-Oppenheimer trajectories. J. Chem. Phys. 121:7658–7670. [DOI] [PubMed] [Google Scholar]
- 27.Ben-Nun, M., and T. J. Martinez. 2002. Ab initio quantum molecular dynamics. Adv. Chem. Phys. 121:439–512. [Google Scholar]
- 28.Troullier, N., and J. L. Martins. 1991. Efficient pseudopotentials for plane-wave calculations. Phys. Rev. B. 43:1993–2006. [DOI] [PubMed] [Google Scholar]
- 29.Ceperley, D. M., and B. J. Alder. 1980. Ground-state of the electron-gas by a stochastic method. Phys. Rev. Lett. 45:566–569. [Google Scholar]
- 30.Tsolakidis, A., and E. Kaxiras. 2005. A TDDFT study of the optical response of DNA bases, base pairs, and their tautomers in the gas phase. J. Phys. Chem. A. 109:2373–2380. [DOI] [PubMed] [Google Scholar]
- 31.Dreuw, A., and M. Head-Gordon. 2004. Failure of time-dependent density functional theory for long-range charge-transfer excited states: the zincbacteriochlorin-bacteriochlorin and bacteriochlorophyll-spheroidene complexes. J. Am. Chem. Soc. 126:4007–4016. [DOI] [PubMed] [Google Scholar]
- 32.Linstrom, P. J., and W. G. Mallard, editors. 2005. N. I. S. T. Chemistry WebBook. NIST Standard Reference Database 69. http://webbook.nist.gov.
- 33.Weiss, R. M., and A. Warshel. 1979. New view of the dynamics of singlet cis-trans photoisomerization. J. Am. Chem. Soc. 101:6131–6133. [Google Scholar]
- 34.Desouter-Lecomte, M., and J. C. Lorquet. 1979. Non-adiabatic interactions in unimolecular decay. 4. Transition probability as a function of the Massey parameter. J. Chem. Phys. 71:4391–4403. [Google Scholar]
- 35.Frutos, L. M., T. Andruniów, F. Santoro, N. Ferré, and M. Olivucci. 2007. Tracking the excited-state time evolution of the visual pigment with multiconfigurational quantum chemistry. Proc. Natl. Acad. Sci. USA. 104:7764–7769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tanner, C., C. Manca, and S. Leutwyler. 2003. Probing the threshold to H atom transfer along a hydrogen-bonded ammonia wire. Science. 302:1736–1739. [DOI] [PubMed] [Google Scholar]
- 37.Coe, J. D., B. G. Levine, and T. J. Martinez. 2007. Ab initio molecular dynamics of excited-state intramolecular proton transfer using multireference perturbation theory. J. Phys. Chem. A. 111:11302–11310. [DOI] [PubMed] [Google Scholar]
- 38.Paterson, M. J., M. A. Robb, L. Blancafort, and A. D. DeBellis. 2005. Mechanism of an exceptional class of photostabilizers: a seam of conical intersection parallel to excited state intramolecular proton transfer (ESIPT) in o-hydroxyphenyl-(1,3,5)-triazine. J. Chem. Phys. A. 109:7527–7537. [DOI] [PubMed] [Google Scholar]
- 39.Coe, J. D., and T. J. Martinez. 2005. Competitive decay at two- and three-state conical intersections in excited-state intramolecular proton transfer. J. Am. Chem. Soc. 127:4560–4561. [DOI] [PubMed] [Google Scholar]
- 40.Levine, B. G., C. Ko, J. Quenneville, and T. J. Martinez. 2006. Conical intersections and double excitations in time-dependent density functional theory. Mol. Phys. 104:1039–1051. [Google Scholar]
- 41.Löwdin, P.-O. 1965. Quantum genetics and the aperiodic solid. Some Aspects on the biological problems of heredity, mutations, ageing and tumours in view of the quantum theory of the DNA molecule. Adv. Quantum Chem. 2:213–361. [Google Scholar]
- 42.Groenhof, G., L. V. Schafer, M. Boggio-Pasqua, M. Goette, H. Grubmüller, and M. A. Robb. 2007. Ultrafast deactivation of an excited cytosine-guanine base pair in DNA. J. Am. Chem. Soc. 129:6812–6819. [DOI] [PubMed] [Google Scholar]
- 43.Hudock, H. R., B. G. Levine, A. L. Thompson, H. Satzger, D. Townsend, N. Gador, S. Ullrich, A. Stolow, and T. J. Martinez. 2007. Ab initio molecular dynamics and time-resolved photoelectron spectroscopy of electronically excited uracil and thymine. J. Phys. Chem. A. 111:8500–8508. [DOI] [PubMed] [Google Scholar]
- 44.Tahara, T., and S. Takeuchi. 2007. The answer to concerted versus step-wise controversy for the double proton transfer mechanism of 7-azaindole dimer in solution. Proc. Natl. Acad. Sci. USA. 104:5285–5290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kwon, O.-H., and A. H. Zewail. 2007. Double proton transfer dynamics of model DNA base pairs in the condensed phase. Proc. Natl. Acad. Sci. USA. 104:8703–8708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kato, M., A. V. Pisliakov, and A. Warshel. 2006. The barrier for proton transport in aquaporins as a challenge for electrostatic models: the role of protein relaxation in mutational calculations. Proteins Struct. Funct. Bioinform. 64:829–844. [DOI] [PubMed] [Google Scholar]
- 47.Warshel, A., and Z. T. Chu. 2001. Nature of the surface crossing process in bacteriorhodopsin: computer simulations of the quantum dynamics of the primary photochemical event. J. Phys. Chem. B. 105:9857–9871. [Google Scholar]
- 48.Stark, K. B., J. M. Gallas, G. W. Zajac, M. Eisner, and J. T. Golab. 2003. Spectroscopic study and simulation from recent structural models for eumelanin. I. Monomer, dimers. J. Phys. Chem. B. 107:3061–3067. [Google Scholar]
- 49.Pezzella, A., L. Panzella, O. Crescenzi, A. Napolitano, S. Navaratman, R. Edge, E. J. Land, V. Barone, and M. d'Ischia. 2006. Short-lived quinonoid species from 5,6-dihydroxyindole dimers en route to eumelanin polymers: integrated chemical, pulse radiolytic, and quantum mechanical investigation. J. Am. Chem. Soc. 128:15490–15498. [DOI] [PubMed] [Google Scholar]
- 50.McGinness, J., P. Corry, and P. Proctor. 1974. Amorphous semiconductor switching in melanins. Science. 183:853–855. [DOI] [PubMed] [Google Scholar]
- 51.Cheng, J., S. C. Moss, M. Eisner, and P. Zschack. 1994. X-ray characterization of melanins— II. Pigment Cell Res. 7:263–273. [DOI] [PubMed] [Google Scholar]
- 52.Tran, M. L., B. J. Powell, and P. Meredith. 2006. Chemical and structural disorder in eumelanins: a possible explanation for broadband absorbance. Biophys. J. 90:743–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Butcher, J. 2007. Spectroscopy of eumelanin monomers and dimers. Masters thesis. University of Queensland, Brisbane, Australia.
- 54.Panzella, L., A. Pezzella, A. Napolitano, and M. d'Ischia. 2007. The first 5,6-dihydroxyindole tetramer by oxidation of 5,5′,6,6′-tetrahydroxy-2,4′-biindolyl and an unexpected issue of positional reactivity en route to eumelanin-related polymers. Org. Lett. 7:1411–1414. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.