

Abstract
Previous studies have shown that low pHo accelerates depolarization-induced inactivation and decreases the macroscopic conductance by reducing channel availability. To test the hypothesis that outer pore constriction underlies the decreased conductance at low pHo, external Ba2+ was used to examine the accessibility of the channel pore at rest under neutral and acidic conditions. At pHo 7.4, Ba2+ block of closed channels follows a monoexponential time course and involves a low-affinity superficial site (KD ≅ 1 mM, −80 mV, 0 mM 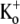 ) and a high-affinity site (KD ≅ 4 μM) deeper in the pore. Depolarization promotes Ba2+ dissociation and an analytical model incorporating state-dependent changes of Ba2+ affinity is presented that replicates the frequency dependence of the time course and the extent of block. Open-channel block by Ba2+ is weak. At pHo 5.5, both the access to and exit from the deep site is inhibited. These results are consistent with a low-pHo-induced conformational change in the outer pore that prevents Ba2+ binding at rest or unbinding during depolarization. If a pore constriction is involved, similar to that proposed to occur during P/C-type inactivation, this would imply that closed-state inactivation in Kv1.5 occurs under acidic conditions.
) and a high-affinity site (KD ≅ 4 μM) deeper in the pore. Depolarization promotes Ba2+ dissociation and an analytical model incorporating state-dependent changes of Ba2+ affinity is presented that replicates the frequency dependence of the time course and the extent of block. Open-channel block by Ba2+ is weak. At pHo 5.5, both the access to and exit from the deep site is inhibited. These results are consistent with a low-pHo-induced conformational change in the outer pore that prevents Ba2+ binding at rest or unbinding during depolarization. If a pore constriction is involved, similar to that proposed to occur during P/C-type inactivation, this would imply that closed-state inactivation in Kv1.5 occurs under acidic conditions.
INTRODUCTION
Kv1.5 is a Shaker-related, voltage-gated potassium (Kv) channel that, in the human heart, underlies the ultrarapid delayed rectifier current (IKur) that contributes to the repolarization of the atrial action potential (1,2). The inhibitory effects of low extracellular pH on Kv1.5 and Shaker channels, as well as the related mammalian Kv1.3 and Kv1.4 channels, have been studied by our group (3–6) and others (7–14).
Decreases in pHo result in an inhibition of macroscopic Kv1.5 currents that is independent of the depolarizing shift of the conductance-voltage relationship caused by the charge-screening effect of the elevated [H+]o (3,8,9,12). It has been suggested that this inhibition is due to an enhancement of slow inactivation; indeed, in addition to decreasing the peak current, low pHo causes an acceleration of slow inactivation in Kv1.5 and in the fast-inactivation-removed mutants of Kv1.4 and Shaker (ShIR) channels (3,7,8,10,11,15). Slow inactivation is thought to proceed in two phases after channel activation. The first phase, known as the pore, or P-type, phase, involves constriction of the outer pore and loss of K+ conductance, and is followed, with continued depolarization, by the C-type phase, where the S4 transmembrane segments are stabilized in the “open” conformation through interactions with the pore (for a recent review, see Kurata and Fedida (16)). Here, we refer to these processes collectively as “P/C-type” inactivation. Consistent with H+-mediated enhancement of P/C-type inactivation, maneuvers that impede inactivation, such as raising external [K+] or mutating a specific outer pore residue (Kv1.5 R487, ShIR T449, or Kv1.4 K532) to a valine or tyrosine residue, also antagonize the inhibition of Kv1.5 current by low pHo (3,10,11).
It is important to note, however, that an enhancement of the rate of depolarization-induced P/C-type inactivation and accumulation of channels in the inactivated state does not fully account for the decrease in peak macroscopic current observed at low pHo. We have shown previously that the inhibition of Kv1.5 current at pH 5.9 does not show use dependence, as would be expected if it were caused by cumulative inactivation (3). In addition, analyses of single-channel currents show that low pHo mainly blocks current by increasing the probability that closed Kv1.5 channels enter an unavailable mode (5). Fluorescence measurements from ShIR channels have also provided evidence that protons induce inactivation of channels from closed, resting states (6). These results therefore suggest that external acidification blocks Kv1.5 current by enhancing open- as well as closed-state inactivation.
Whether the 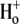 -induced closed-state inactivation process involves the same conformational changes as P/C-type inactivation, however, remains unclear. In this article, we have used externally applied Ba2+ as a molecular probe to examine possible changes in the accessibility of the outer pore of Kv1.5 under acidic conditions. A Ba2+ ion has essentially the same radius as a K+ ion and is able to enter the pore of several classes of K+ channels from either side of the membrane (13,17–22). Regardless of the side of the channel to which it is applied, Ba2+ binding is thought to occur within the selectivity filter (19,20,23) and, perhaps because of its greater charge, Ba2+ tends to have a long mean dwell time, resulting in a block of K+ current. In ShIR channels, Ba2+ has been shown to bind to two sequential sites within the pore: a low-affinity superficial site that gives rise to a near-instantaneous component of block, and a deep site with a higher affinity for Ba2+ that is associated with a comparatively much slower block onset and offset (20).
-induced closed-state inactivation process involves the same conformational changes as P/C-type inactivation, however, remains unclear. In this article, we have used externally applied Ba2+ as a molecular probe to examine possible changes in the accessibility of the outer pore of Kv1.5 under acidic conditions. A Ba2+ ion has essentially the same radius as a K+ ion and is able to enter the pore of several classes of K+ channels from either side of the membrane (13,17–22). Regardless of the side of the channel to which it is applied, Ba2+ binding is thought to occur within the selectivity filter (19,20,23) and, perhaps because of its greater charge, Ba2+ tends to have a long mean dwell time, resulting in a block of K+ current. In ShIR channels, Ba2+ has been shown to bind to two sequential sites within the pore: a low-affinity superficial site that gives rise to a near-instantaneous component of block, and a deep site with a higher affinity for Ba2+ that is associated with a comparatively much slower block onset and offset (20).
Here, we first characterize the block of Kv1.5 by externally applied Ba2+ and show that for the block of resting channels there are two sequential binding sites, and that the affinity of the deep binding site is much higher than reported for ShIR. Again in contrast to ShIR, there is only weak fast block of open channels by Ba2+ in Kv1.5. We then assess whether Ba2+ can either access or exit the deep binding site at pHo 5.5 and find that external acidification can prevent either the loading or unloading of the deep pore site. These results suggest that the decrease of channel availability caused by external acidification is associated with a constriction of the outer pore mouth and adds support to the hypothesis that a P-type inactivation process occurs at rest at low pHo.
METHODS
Cell preparation
Wild-type hKv1.5 currents were recorded from channels stably expressed in a human embryonic kidney cell line (HEK293, American Type Culture Collection, Rockville, MD), as reported previously (3). Cells were passaged using trypsin-EDTA and maintained in minimum essential medium supplemented with 10% fetal bovine serum, 1% penicillin-streptomycin, and 0.5 mg ml−1 geneticin at 37°C in an atmosphere of 5% CO2 in air. All tissue culture supplies were obtained from Invitrogen (Burlington, ON, Canada).
Recording solutions
Unless otherwise stated, the standard, nominally K+-free bath solution contained (in mM): 143.5 NaCl, 2 CaCl2, 1 MgCl2, 5 glucose, and 10 HEPES, and was titrated at room temperature to pH 7.4 with NaOH. In experiments where 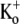 was present, KCl was substituted for NaCl. Ba2+-containing solutions were prepared by isoosmotic substitution of NaCl with BaCl2. The standard low-pH bath solution was prepared using 10 mM MES instead of HEPES and titrated to pH 5.5 with NaOH. The patch pipette solution contained (in mM): 130 KCl, 4.75 CaCl2 (pCa2+ = 7.3), 1.38 MgCl2, 10 EGTA, and 10 HEPES, and was adjusted to pH 7.4 with KOH. Chemicals were obtained from the Sigma Aldrich Chemical (Mississauga, ON, Canada).
was present, KCl was substituted for NaCl. Ba2+-containing solutions were prepared by isoosmotic substitution of NaCl with BaCl2. The standard low-pH bath solution was prepared using 10 mM MES instead of HEPES and titrated to pH 5.5 with NaOH. The patch pipette solution contained (in mM): 130 KCl, 4.75 CaCl2 (pCa2+ = 7.3), 1.38 MgCl2, 10 EGTA, and 10 HEPES, and was adjusted to pH 7.4 with KOH. Chemicals were obtained from the Sigma Aldrich Chemical (Mississauga, ON, Canada).
In most experiments, a section of glass coverslip to which cells had attached was placed in a saline-filled recording chamber (volume 0.5 ml) and perfused with control solution (2 ml min−1). We found no difference between experiments using continuous perfusion and those where control and test responses were recorded without perfusion (not shown).
Fast perfusion
For experiments requiring rapid solution exchange, a Warner Instruments (Hamden, CT) SF-77B Perfusion Fast Step system was used. Perfusion pipettes were made using borosilicate theta glass tubing (2 mm outer diameter; Warner Instruments) that was pulled in one step to an outer diameter at the tip of ∼300 μm and then cut into two parts using a ceramic wafer (for details, see Jonas (24)). Deactivated fused silica tubing (0.18 mm inner diameter, Agilent Technologies, Mississauga, ON, Canada) was then inserted into the back ends of the pipette barrels, advanced as far as possible toward the tip and fixed in place by back-filling with epoxy glue. The free ends of the silica tubing were then connected to the solution reservoirs via polyethylene tubing and the manifolds provided with the Fast Step system. While the bath was being constantly perfused with control solution, the assembled perfusion pipette was positioned as close as possible to the cell of choice, so that the cell was exposed to solution outflow from only one barrel. Rapid switching of the pipette position (200 μm movement) exposed the cell to the test solution that flowed continuously from the second barrel. Using a method described by Panyi and Deutsch (25), the time course of solution exchange, estimated from the decay in current amplitude at 0 mV when the [K+] of the perfusion solution was switched from 0.5 to 140 mM K+ (not shown), was ∼150 ms.
Electrophysiological procedures
Whole-cell currents were recorded at room temperature (20–25°C) using an EPC-8 patch-clamp amplifier and Pulse + Pulsefit software (HEKA Electronik, Lambrecht/Pfalz, Germany), connected through an ITC-18 digital interface (Instrutech, Port Washington, NY). Patch electrodes were made from thin-walled borosilicate glass (World Precision Instruments, Sarasota, FL) and had resistances of 1.0–2.5 MΩ measured in the bath with standard internal and external solutions. Capacitance and series resistance were compensated (typically 80%) using the circuitry of the amplifier. Leak subtraction was achieved using the online P/N protocol in Pulse, for which the holding potential was −100 mV and the scaling factor was −0.25. Current signals filtered at 3 kHz (−3 dB, 8 pole Bessel filter) were digitized (18 bit) at a sampling interval of 100 μs. Voltages were corrected for liquid junction potentials. Cells were held at −80 mV and a test potential of +50 mV was standard. Test pulses were 1.5 s long in experiments examining open-channel Ba2+ dissociation. In experiments involving repetitive stimulation at different frequencies, the pulse duration was 10 ms and the current amplitude was measured at the end of the voltage pulse. When applicable, current amplitudes were normalized to control measurements made at the beginning of an experiment. Data from cells showing <85% recovery to control levels were discarded.
Data analysis
Data are presented as mean ± SE, except for the values derived from non-linear least-squares fitting routines, which are expressed as mean ± SD (Igor, Wavemetrics, Portland, OR); n represents the number of cells. Due to the sigmoidal nature of current activation under control (0 mM Ba2+) conditions, the time course of activation was estimated by fitting the portion of the trace between 50 – 100% of the peak current to a single exponential function (using a 1.5 s pulse; Fig. 6). After Ba2+ exposure, the entire rising phase of the first current trace obtained upon the return to control conditions could be fit to an exponential function. The time courses of currents obtained using short pulses (10 ms) were not quantified (e.g., traces shown in Fig. 1).
FIGURE 6.

Ba2+ unbinding from the open state is voltage-dependent and inhibited by increasing the [K+]o. (A) Schematic of voltage-pulse and Ba2+ application protocol. A 1.5 s pulse to +50 mV was applied under 0.5 mM [K+]o, pHo 7.4 conditions. The cell was then exposed to 5 mM Ba2+ for 2 min, followed by a 4-min wash in control solution, after which the test pulse was repeated. (B) After Ba2+ exposure (gray trace), the current activation was much slower and well fitted by a single exponential (τact,app = 28.14 ms; compare to τact = 1.25 ms for the control response (black trace). (Inset) Current activation on an expanded timescale. These results are representative of those obtained from 11 cells. (C) Normalized currents after a 2-min exposure to 5 mM Ba2+ (with 0.5 mM 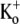 ) recorded at two different test-pulse potentials. The activation time course at both voltages was well fitted by a single exponential. At +50 mV, τact,app was 22.56 ± 1.25 ms (n = 11), which was significantly faster (p < 0.001) than at +25 mV (τact,app = 65.59 ± 7.33 ms; n = 3). Each trace was normalized with respect to its own peak current. (D) Plot of the Ba2+ off rate (1/τact,app) derived from experiments such as those described in C, obtained between 0 and +100 mV in 25-mV increments. A fit of the data to the Woodhull model (see Methods) gave an estimate for δBa,off of 0.51 ± 0.04. Data points are from a total of 21 cells; 3–11 cells/point. (E) After Ba2+ loading (in 0.5 mM
) recorded at two different test-pulse potentials. The activation time course at both voltages was well fitted by a single exponential. At +50 mV, τact,app was 22.56 ± 1.25 ms (n = 11), which was significantly faster (p < 0.001) than at +25 mV (τact,app = 65.59 ± 7.33 ms; n = 3). Each trace was normalized with respect to its own peak current. (D) Plot of the Ba2+ off rate (1/τact,app) derived from experiments such as those described in C, obtained between 0 and +100 mV in 25-mV increments. A fit of the data to the Woodhull model (see Methods) gave an estimate for δBa,off of 0.51 ± 0.04. Data points are from a total of 21 cells; 3–11 cells/point. (E) After Ba2+ loading (in 0.5 mM 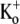 ), the time course of the current onset in 0.5 mM or 10 mM
), the time course of the current onset in 0.5 mM or 10 mM 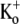 was examined. The current activated significantly faster with 0.5 mM
was examined. The current activated significantly faster with 0.5 mM 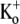 (τact,app = 22.56 ± 1.25 ms; n = 11) than with 10 mM
(τact,app = 22.56 ± 1.25 ms; n = 11) than with 10 mM 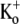 (τact,app = 145.40 ± 6.15 ms; n = 5; p < 0.001). Traces were normalized as in C. (F) Plot of the Ba2+ off rate under varying [K+]o at a test-pulse potential of +50 mV. The best fit of the data to the Hill equation (see Methods) gave a KD for the
(τact,app = 145.40 ± 6.15 ms; n = 5; p < 0.001). Traces were normalized as in C. (F) Plot of the Ba2+ off rate under varying [K+]o at a test-pulse potential of +50 mV. The best fit of the data to the Hill equation (see Methods) gave a KD for the 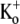 inhibition of Ba2+ unbinding of 1.00 ± 0.08 mM. Data points are from a total of 31 cells, 5–11 cells/point.
inhibition of Ba2+ unbinding of 1.00 ± 0.08 mM. Data points are from a total of 31 cells, 5–11 cells/point.
FIGURE 1.

Ba2+ blocks Kv1.5 in a concentration- and frequency-dependent manner. (A) With 0 mM 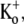 pHo 7.4 solution, 10-ms test pulses to +50 mV were applied at 1 Hz without (uppermost trace) and with 5 mM Ba2+. Shown are 65 consecutive traces from the time of Ba2+ application, which began immediately after the control trace. (Inset) The trace at the steady-state level of block was scaled 1.87× to match the peak amplitude of the control current. There was a small effect of Ba2+ on the onset of the K+ current. (B (i)) Trace 1 was recorded using a 10-ms pulse to +50 mV after a 1-min exposure to 1 mM Ba2+ in 0 mM
pHo 7.4 solution, 10-ms test pulses to +50 mV were applied at 1 Hz without (uppermost trace) and with 5 mM Ba2+. Shown are 65 consecutive traces from the time of Ba2+ application, which began immediately after the control trace. (Inset) The trace at the steady-state level of block was scaled 1.87× to match the peak amplitude of the control current. There was a small effect of Ba2+ on the onset of the K+ current. (B (i)) Trace 1 was recorded using a 10-ms pulse to +50 mV after a 1-min exposure to 1 mM Ba2+ in 0 mM 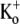 at −80 mV. Traces 2–5 show the current from subsequent pulses applied at 1 Hz in the continued presence of Ba2+. Trace 25 shows the steady-state current in 1 mM Ba2+ with 1 Hz stimulation and Trace 100 is the steady-state current after recovery in 0 mM Ba2+. (B (ii)) Traces 1–5 and 25 are normalized with respect to Trace 100. There was a marked slowing of the rising phase of the current elicited by the first pulse after Ba2+ exposure, but this effect is diminished with subsequent pulses in the continued presence of Ba2+. (C) The time course of the onset and offset of the Ba2+ block. The average current amplitude in the last 1 ms of each pulse was normalized to the Ba2+-free control current. Down and up arrows indicate the time of fast Ba2+ application and withdrawal, respectively. Black circles (•) represent the normalized current amplitudes for the experiment shown in A. Single-exponential fits to the onset of and recovery from Ba2+ block give τblock = 6.84 s and τunblock = 19.21 s, respectively. When the experiment was performed with the same stimulus protocol but with 20 mM Ba2+ (solid triangles), block onset was slightly faster (τblock = 4.97 s) and the normalized steady-state current was smaller (0.37 vs. 0.50 for 5 mM Ba2+), but the time course of current recovery was the same. If the [Ba2+] was kept constant at 5 mM but the pulse frequency decreased to 0.2 Hz (open diamonds), the normalized steady-state level was also reduced (steady-state normalized current = 0.27). Although the onset of the block was not affected by the decrease in stimulation frequency, recovery with a 0.2-Hz pulse frequency was much slower (τunblock = 63.20 s) than that seen at 1 Hz. (D) The blocking rate (1/τblock) is a nonlinear function of the [Ba2+]. τblock values were assessed in 0 mM
at −80 mV. Traces 2–5 show the current from subsequent pulses applied at 1 Hz in the continued presence of Ba2+. Trace 25 shows the steady-state current in 1 mM Ba2+ with 1 Hz stimulation and Trace 100 is the steady-state current after recovery in 0 mM Ba2+. (B (ii)) Traces 1–5 and 25 are normalized with respect to Trace 100. There was a marked slowing of the rising phase of the current elicited by the first pulse after Ba2+ exposure, but this effect is diminished with subsequent pulses in the continued presence of Ba2+. (C) The time course of the onset and offset of the Ba2+ block. The average current amplitude in the last 1 ms of each pulse was normalized to the Ba2+-free control current. Down and up arrows indicate the time of fast Ba2+ application and withdrawal, respectively. Black circles (•) represent the normalized current amplitudes for the experiment shown in A. Single-exponential fits to the onset of and recovery from Ba2+ block give τblock = 6.84 s and τunblock = 19.21 s, respectively. When the experiment was performed with the same stimulus protocol but with 20 mM Ba2+ (solid triangles), block onset was slightly faster (τblock = 4.97 s) and the normalized steady-state current was smaller (0.37 vs. 0.50 for 5 mM Ba2+), but the time course of current recovery was the same. If the [Ba2+] was kept constant at 5 mM but the pulse frequency decreased to 0.2 Hz (open diamonds), the normalized steady-state level was also reduced (steady-state normalized current = 0.27). Although the onset of the block was not affected by the decrease in stimulation frequency, recovery with a 0.2-Hz pulse frequency was much slower (τunblock = 63.20 s) than that seen at 1 Hz. (D) The blocking rate (1/τblock) is a nonlinear function of the [Ba2+]. τblock values were assessed in 0 mM 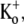 with 10-ms pulses to +50 mV applied at 1 Hz. The solid line represents the best fit of the data to a sequential two-site binding model (Eq. 1; see text for fit parameters). Data points are from a total of 46 cells, 3–8 cells/point. (E) The unblocking rate is linearly related to the pulse frequency. Values for τunblock were assessed with 0 mM
with 10-ms pulses to +50 mV applied at 1 Hz. The solid line represents the best fit of the data to a sequential two-site binding model (Eq. 1; see text for fit parameters). Data points are from a total of 46 cells, 3–8 cells/point. (E) The unblocking rate is linearly related to the pulse frequency. Values for τunblock were assessed with 0 mM 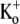 and 20 mM
and 20 mM 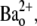 using 10-ms pulses to +50 mV applied at 1, 0.7, 0.2, 0.066, or 0.033 Hz. The extrapolated off rate without pulsing (0 Hz) is 0.001 ± 0.002 s−1. Data points are from a total of 53 cells, 5–24 cells/point.
using 10-ms pulses to +50 mV applied at 1, 0.7, 0.2, 0.066, or 0.033 Hz. The extrapolated off rate without pulsing (0 Hz) is 0.001 ± 0.002 s−1. Data points are from a total of 53 cells, 5–24 cells/point.
The analysis of the relationship between the blocking rate (1/τblock) and [Ba2+]o (Fig. 1 C) was based on a sequential two-site binding model:
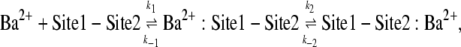 |
(Scheme 1) |
where Sites 1 and 2 are, respectively, the superficial and deep sites, k1 and k2 are the respective binding rates at Sites 1 and 2, and k−1 and k−2 are the respective rates of unbinding from Sites 1 and 2. This same model has been used previously to describe the concentration dependence of the slow block of ShIR channels by Ba2+ (20). At the superficial site, the binding rate is proportional to [Ba2+]. The deeper site is not directly exposed to the external bulk solution, or to the internal bulk solution by virtue of the closed activation gate and, as a consequence, k2 is proportional to the probability that Site 1 is occupied. We make the assumption that k−2 is independent of the occupancy of Site 1; this arose from the assumption in the original model that a coulombic interaction precludes simultaneous occupation of the two sites by Ba2+. Even if this constraint is removed, there is little change in the outcome of the model, because k−2, derived independently from the time dependence of the current recovery in Ba2+-free medium (Fig. 1 E), is very small relative to k2 at Ba2+ concentrations that produce a significant degree of block. Indeed, making the off rate a function of the probability that the superficial site is not occupied (i.e., k−2 × 1/(1 + [Ba2+]o/KBa,s)) has minimal effect on the fit parameters (not shown). Thus, the data in Fig. 1 C were fit to the equation
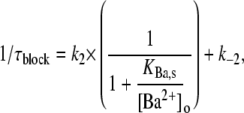 |
(1) |
where k2 and k−2 are, respectively, the binding and unbinding rates at the deep site, KBa,s is the apparent equilibrium dissociation constant of the superficial site, and 1/(1 + KBa,s/[Ba2+]o) is the probability that Ba2+ is bound to the superficial site.
Primarily to allow a comparison with the results of others, the [Ba2+]o dependence of the steady-state residual current at a given pulse frequency was also quantified by fitting the data to the Hill equation:
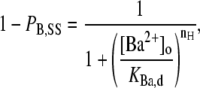 |
(2) |
where PB,SS is the proportion of macroscopic current blocked at the steady state, KBa,d is the apparent equilibrium dissociation constant of the deep Ba2+ binding site and nH is the Hill coefficient reflecting the number of Ba2+ ions binding per channel.
To model the frequency dependence of the Ba2+ block of Kv1.5, we employed Eqs. 3–7, which were originally applied by Starmer et al. (26) to describe the block of voltage-gated Na+ (NaV) channels by the quaternary lidocaine derivative QX222. Briefly, a bimolecular scheme involving unblocked channels (U), drug (D), and blocked channels (B) is
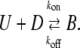 |
(Scheme 2) |
For a given channel conformation (i.e., open or closed), the steady-state level of block is kon/(kon + koff) and the time constant for the block is (kon + koff)−1. However, if the channel conformation affects the binding equilibrium, by virtue of an effect on the accessibility of the site and/or a change of kon and/or koff, and if the lifetime of a particular conformation is short-lived with respect to the blocking time constant, then the proportion of channels blocked can be critically influenced by the frequency and duration of test pulses that cycle channels in and out of different conformations. In the case of QX222, block is enhanced by increasing the pulse frequency, which increases the proportion of time spent in one or more conformations favoring drug binding and decreases the time spent in conformations favoring drug unbinding. As we will show, binding and unbinding of Ba2+ exhibits the opposite relationship: channel opening during test pulses to +50 mV promotes Ba2+ unbinding, whereas the reaction is biased toward binding in a closed channel at −80 mV. Consequently, increasing the frequency of depolarizing test pulses decreases the fractional block by Ba2+. The blockade with repetitive stimulation (see the Appendix of Starmer et al. (26) for details of the derivation) is described by the equation
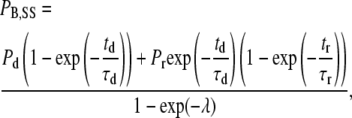 |
(3) |
where PB,SS is the proportion of channels blocked at the steady state, Pd is the proportion of channels blocked at the deep site after a prolonged depolarization, Pr is the proportion of channels blocked at the deep site after a prolonged rest at the holding potential, td is the average amount of time that a channel is open during a depolarizing pulse, τd is the time constant for unblock during a depolarization to the test voltage, tr is the time at the holding potential between test pulses, τr is the time constant for the block at the holding potential, and λ is equal to the unitless quantity (td/τd + tr/τr). With the assumption that Pd is zero (20,21), Eq. 3 simplifies to
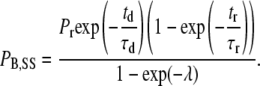 |
(4) |
Since Ba2+ can be trapped in open-inactivated channels (21,27), this equation is valid for prolonged depolarizations only if the value for td is appropriately adjusted; otherwise, the degree of unblock would be overestimated. With the 10-ms test depolarizations used in the quantification of the Ba2+ block, inactivation is not a significant mitigating factor, and the estimate of td is calculated from the open probability estimated from prior single-channel recordings (see Results).
The fractional block at the end of the xth pulse of a train (PB,x) is
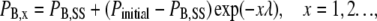 |
(5) |
where Pinitial is the proportion of the current blocked at the point in time when the pulse frequency is changed, λ is as defined for Eq. 3, and x is the pulse number in the stimulus train. In words, Eq. 5 indicates that changing the stimulus frequency causes the proportion of current blocked during a pulse train to relax monoexponentially to a new steady-state level. The relationship between the time constant (τ) for that relaxation and λ is:
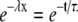 |
(6) |
Given that t, the time elapsed since the change of the pulse frequency, is equal to the product of the pulse number (x) and the cycle length (CL), then the time constant can be calculated by
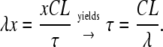 |
(7) |
To describe the [K+]o dependence of the open-channel Ba2+ unbinding rate (see Fig. 6 F), the Ba2+ dissociation rates measured at +50 mV with different [K+]o were fit to the Hill equation,
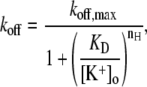 |
(8) |
where koff is the apparent rate of Ba2+ unbinding from open channels at +50 mV (taken as 1/τact,app, where τact,app is the apparent activation time constant (see Fig. 6)), koff,max is the unbinding rate with 0 mM 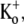 KD is the apparent equilibrium dissociation constant for the
KD is the apparent equilibrium dissociation constant for the 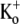 inhibition of Ba2+ unbinding, and nH is the Hill coefficient reflecting the number of K+ ions binding per channel to slow Ba2+ unbinding.
inhibition of Ba2+ unbinding, and nH is the Hill coefficient reflecting the number of K+ ions binding per channel to slow Ba2+ unbinding.
To quantify the voltage dependence of the open-channel Ba2+ dissociation rate (see Fig. 6 D), koff was measured over a range of test potentials (with a fixed [K+]o and [K+]i), plotted against the test potential and fit to the Woodhull model:
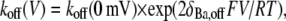 |
(9) |
where koff(V) is the Ba2+ unbinding rate at the test voltage, koff (0 mV) is the unbinding rate at 0 mV, V is the test potential, δBa,off is the electrical distance between the deep Ba2+-binding site and the rate-limiting barrier for exit, and F, R, and T have their usual meanings (28).
RESULTS
Ba2+ blocks closed Kv1.5 channels at pH 7.4
Our first objective was to test the hypothesis that Ba2+ is able to access and bind within the pore of Kv1.5 channels and block current. We began by characterizing the effects of external Ba2+ on Kv1.5 currents at pH 7.4 using a stimulus protocol comparable to that used to investigate Ba2+ block of ShIR channels (20). Fig. 1 A illustrates a representative experiment examining the effect of 5 mM Ba2+ on currents evoked by 10-ms pulses to +50 mV applied at a frequency of 1 Hz from a holding potential of −80 mV. Tail current was recorded at −80 mV. Scaling of the steady-state current evoked in 5 mM Ba2+ and superimposition on the control current (Fig. 1 A, inset) revealed that the activation and deactivation kinetics of the steady-state current observed with 1 Hz stimulation were slightly affected by the 5 mM Ba2+ treatment.
For the currents shown in Fig. 1 B (i), 1 mM Ba2+ was applied for 1 min in the absence of pulses before initiating a train of 1-Hz pulses to +50 mV. The current in the first pulse had a small initial amplitude and slowly activating time course, which is consistent with the block of virtually all of the channels at the start of the pulse, followed by a slow unblock during the pulse (21). With continued pulsing in the presence of Ba2+, the degree of block at equilibrium (Trace 25) was nearly identical to that in Fig. 1 A. Trace 100 is the steady-state recovery response obtained in Ba2+-free medium. For Fig. 1 B (ii), select traces were normalized with respect to Trace 100 to better illustrate their relative activation time courses. These data suggest that repetitive pulsing has a substantial effect on the steady-state level of block of Kv1.5 by Ba2+, consistent with the observation made by Armstrong on the Ba2+ block of the squid giant axon delayed rectifier channels (17). A detailed analysis of the frequency dependence of Ba2+ block of Kv1.5 is presented below.
To illustrate the time dependence of the onset and offset of Ba2+ block, the normalized mean current in the last millisecond of each test pulse from the experiment described by Fig. 1 A has been plotted against time in Fig. 1 C (solid circles); the down arrow indicates the time at which the 5 mM Ba2+ fast perfusion began. In five experiments of this type, the relaxation of the normalized test current (Inorm) to a steady-state level of 0.52 ± 0.01 was well fitted by a single exponential with a time constant (τblock) of 7.2 ± 0.6 s. After returning to Ba2+-free medium, marked by the up black arrow, the time course for complete reversal of the Ba2+ block was also monoexponential (τunblock = 18.5 ± 2.6 s). In an experiment with a fourfold higher concentration (20 mM) of Ba2+ (Fig. 1 C, solid triangles) but with the same pulse frequency of 1 Hz, the onset of the block was only slightly faster (τblock = 4.8 ± 0.5 s; n = 9), and the steady-state block (1 − Inorm = 0.61 ± 0.01) was ∼20% greater. Note that after switching from 20 mM Ba2+ back to control solution τunblock was 12.8 ± 2.1 s, which was not significantly different (p > 0.05) from τunblock measured after the 5 mM Ba2+ treatment.
For the third set of responses (Fig. 1 C, open diamonds), a 5-mM concentration of Ba2+ was used but the stimulus frequency was 0.2 Hz. The τblock (9.2 ± 0.3 s; n = 5) was similar to that observed with a 1-Hz pulse frequency using either 5 or 20 mM Ba2+. However, the degree of block (0.74 ± 0.01) was greater and unblock was slower (τunblock = 60.5 ± 2.9 s) than with the 1-Hz test-pulse frequency after channel loading using either 5 or 20 mM Ba2+. Thus, the Ba2+ block of Kv1.5 is similar to that reported for ShIR (20,21) in that the steady-state level of block and the kinetics of block onset are dependent on both the Ba2+ concentration and frequency of test pulses applied, whereas the time course of current recovery from Ba2+ block is sensitive only to the pulse frequency.
Despite these qualitative similarities in the features of the Ba2+ block of Kv1.5 (Fig. 1, A and C) and ShIR currents, the results of Fig. 1 also reveal substantial qualitative and quantitative differences, which is perhaps unexpected given the similarity of their pore structures (Fig. 2). For example, in ShIR with 20 mM Ba2+, the inhibition of current consists of a very rapid phase, attributable to fast block of open channels during the test pulse, as well as a slow phase that relaxes to the steady state with a time constant of ∼100 s (Fig. 2 of (20)). In contrast, but similar, to findings in Kv1.3 (13,14), there is no rapid component of block of Kv1.5 by 20 mM Ba2+ (Fig. 1 C) and the slow block occurs roughly 10 times faster than in ShIR. In light of these differences, in the second part of this article we undertook to analyze the Ba2+ block of Kv1.5 in more detail in the interest of highlighting mechanistic differences in the Ba2+ block of these closely related channels, and to validate the use of Ba2+ as a tool to probe the Kv1.5 pore at low pHo.
FIGURE 2.

Sequence alignment of hKv1.5 and Shaker from the C-terminal end of S5 to the end of the selectivity filter. The putative regions forming the turret, pore helix, and selectivity filter are shown.
τblock is not a linear function of [Ba2+]
To develop a kinetic model for the Ba2+ block, we examined the relationship between the blocking rate, taken as the inverse of τblock after Ba2+ application in K+-free bath solution, and the concentration of Ba2+ applied (Fig. 1 D). As reported for ShIR, the blocking rate of Kv1.5 current saturated at a high [Ba2+]o, indicating that a bimolecular blocking reaction was not involved. The data were fit very well to a sequential two-site binding model used to describe the Ba2+ block of closed ShIR channels (see Methods and Hurst et al. (20)). The best fit of the experimental data to Eq. 1, which is based on Scheme 1, is shown in Fig. 1 D (solid line) and gave an estimate of 0.93 ± 0.15 mM for the apparent equilibrium dissociation constant (KBa,s = k−1/k1) of the superficial Ba2+ binding site, 0.16 ± 0.01 s−1 for k2, and 0.0025 ± 0.0019 s−1 for k−2.
An independent estimate of k−2 was obtained from experiments where Kv1.5 channels were first loaded with Ba2+, and after switching back to Ba2+-free medium, τunblock was measured with test pulses to +50 mV applied at varying frequencies. The underpinning assumption of the experimental approach, which is validated below in Kv1.5 and supported by results in ShIR (21), is that because depolarization facilitates the unblocking reaction, and since recovery occurs in Ba2+-free medium, rebinding of Ba2+ is therefore unlikely to occur, and the relationship between τunblock and the pulse frequency allows k−2 to be estimated. Recovery time courses were always well fitted by a single exponential, regardless of the stimulus frequency (e.g., Fig. 1 C), and τunblock was independent of either the extent of the block or the Ba2+ concentration used in the loading phase of the experiment (not shown). As expected for an unblocking reaction that is facilitated by depolarization, the apparent unblocking rate (1/τunblock) is proportional to the stimulus frequency (Fig. 1 E) when the test-pulse duration is fixed and brief. Extrapolation of the fitted line to the y intercept provides an estimate of k−2 in the absence of pulsing, or, in other words, the value for the Ba2+ off rate from the deep binding site at the holding potential of −80 mV and with 0 mM external K+. The best estimate of k−2 derived from Fig. 1 E is 0.001 ± 0.002 s−1 and buttresses the estimate of 0.0025 ± 0.0019 s−1 in Fig. 1 D. However, both estimates have a large standard deviation and their accuracy is therefore uncertain. In comparison, the upper limit for the closed-state off rate in ShIR channels is 0.0132 s−1 (21).
Ba2+ block of Kv1.5 is dependent on stimulation frequency
Although an effect of the test-pulse frequency on the steady-state level of Ba2+ block has been noted with ShIR and other K+ channels (17,21), to our knowledge, this relationship has not been systematically studied. Indeed, an explicit assumption has been made that a brief test pulse would have little effect on the slow component of block (20). Especially in view of the greatly enhanced block observed after a 1-min interpulse interval (Fig. 1 B), a more detailed examination of the frequency dependence of the Ba2+ block was warranted.
To quantify the relationship between the Ba2+ block and the test-pulse frequency, the steady-state level of block was assessed at two different stimulation frequencies over a range of [Ba2+]. Current amplitude was measured at the end of 10-ms test pulses to +50 mV applied once every 30 s (0.033 Hz) or once every 1.43 s (0.7 Hz). Ba2+ was applied by a computer-controlled fast perfusion system and the blocking reaction was allowed to proceed to the steady-state. Steady-state residual current was calculated by normalizing the peak test current at the steady state to the control test currents obtained before Ba2+ application. Fig. 3 shows the concentration-response relationship at 0.033 Hz (open circles) and at 0.7 Hz (solid circles). Separate fits of the two data sets to the Hill equation (Fig. 3, black lines, and Eq. 2) gave estimates for the apparent affinity of the deep Ba2+ binding site, KBa,d, and the Hill coefficient of 20.3 ± 2.6 μM and 1.12, respectively, for the 0.033-Hz pulse frequency, as compared to 361 ± 151 μM and 1.16 for responses evoked at 0.7 Hz. This demonstrates that increasing the pulse frequency from 0.033 Hz to 0.7 Hz causes an 18-fold increase of the midpoint of the concentration-response relationship while having no effect on the Hill coefficient, which had a value consistent with deep block arising from the binding of one Ba2+ ion. Increasing the pulse frequency to 0.7 Hz also caused an upward shift of the foot of the concentration-response relationship. This upward shift reflects the fact that with a saturating concentration of Ba2+ the degree of block with test pulses of a fixed duration (and a fixed td) is proportional to the time spent at rest (tr). In other words, the decrease of tr as the stimulus frequency increases means there is less time for the reestablishment of block at rest and, as a consequence, the proportion of unblocked channels increases.
FIGURE 3.

The steady-state level of Ba2+ block is dependent on the stimulation frequency and the [Ba2+]. Test pulses to +50 mV for 10 ms were applied at either 30-s intervals (0.033 Hz (open circles)) or 1.43-s intervals (0.7 Hz (solid circles)) with 0 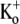 and a pHo of 7.4. Ba2+ was applied at various concentrations using a computer-controlled fast perfusion system. The steady-state residual current (1 − proportion of blocked (PB,SS)) at the end of the test pulse was normalized to the control test current before Ba2+ application and plotted against the concentration of Ba2+ applied (2 μM – 20 mM). The black solid lines represent the best fits of the two data sets to the Hill equation. Estimates of the apparent KBa,d and the Hill coefficient are 20.3 ± 2.6 μM and 1.12, respectively, for the data collected at 0.033 Hz, whereas the estimates from the 0.7 Hz data are 361 ± 151 μM and 1.16, respectively. Gray dashed lines represent the outcome of a simultaneous fit of the two data sets to Eq. 4. From the fit, k2, KBa,s, k−2, and τd were estimated to be 0.15 s−1, 0.8 mM, 0.0006 s−1, and 59 ms, respectively (see text). The dotted gray line represents the solution for the residual normalized current (1 − Pr), where Pr is the proportion of channels blocked after a prolonged rest at the holding potential, i.e., at 0 Hz. A fit of 1 – Pr to the Hill equation gave a Hill coefficient of 1 and a KBa,d of 4 μM, indicating that the apparent KBa,d is profoundly affected by the pulse frequency. Data points are from a total of 29 cells (2–8 cells/point) for the 0.7 Hz data; and 45 cells (3–9 cells/point) for the 0.033 Hz data.
and a pHo of 7.4. Ba2+ was applied at various concentrations using a computer-controlled fast perfusion system. The steady-state residual current (1 − proportion of blocked (PB,SS)) at the end of the test pulse was normalized to the control test current before Ba2+ application and plotted against the concentration of Ba2+ applied (2 μM – 20 mM). The black solid lines represent the best fits of the two data sets to the Hill equation. Estimates of the apparent KBa,d and the Hill coefficient are 20.3 ± 2.6 μM and 1.12, respectively, for the data collected at 0.033 Hz, whereas the estimates from the 0.7 Hz data are 361 ± 151 μM and 1.16, respectively. Gray dashed lines represent the outcome of a simultaneous fit of the two data sets to Eq. 4. From the fit, k2, KBa,s, k−2, and τd were estimated to be 0.15 s−1, 0.8 mM, 0.0006 s−1, and 59 ms, respectively (see text). The dotted gray line represents the solution for the residual normalized current (1 − Pr), where Pr is the proportion of channels blocked after a prolonged rest at the holding potential, i.e., at 0 Hz. A fit of 1 – Pr to the Hill equation gave a Hill coefficient of 1 and a KBa,d of 4 μM, indicating that the apparent KBa,d is profoundly affected by the pulse frequency. Data points are from a total of 29 cells (2–8 cells/point) for the 0.7 Hz data; and 45 cells (3–9 cells/point) for the 0.033 Hz data.
To model the frequency dependence of the concentration-response relationship, the data obtained at 0.033 Hz and 0.7 Hz were simultaneously fit to Eq. 4; the outcome of the fit is represented by the gray dashed lines in Fig. 3. There are six parameters in the fitting equation: td, tr, k2, k−2, KBa,s, and τd. The value for τr in Eq. 4 arises from
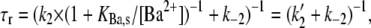 |
and the value for Pr arises from
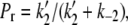 |
where τr is the time constant for the Ba2+ block of the deep pore site at the holding potential, and Pr is the proportion of channels with Ba2+ occupying the deep site at the steady state. To estimate td, defined as the average open time during a 10-ms test pulse to +50 mV, the proportion of maximal charge conducted during a pulse was first calculated by dividing the integral for a control pulse current by the integral for the current if activation had been instantaneous (i.e., Imax × 10 ms). The td was then obtained by multiplying the proportion of maximal charge by the maximal open probability (Po) of 0.8 measured from recordings of unitary Kv1.5 currents (5). The mean td calculated with this approach was 6.43 ± 0.25 ms (n = 7 cells). Ba2+ binding at the deep pore site is assumed to have no effect on td (but see Armstrong et al. (17)). For tr, the time spent at the holding potential during one cycle length, a fixed value of 1/stimulus frequency in Hz − 0.01 s was used. Consequently, there were four free parameters in the global fitting routine, and of these, the values for k2, k−2, and KBa,s were constrained to be in the range of the mean value ± 1 SD obtained from the fit of the data in Fig. 1 D. With these conditions, the best-fit values for the constrained parameters k2, KBa,s, and k−2 were 0.15 ± 0.08 s−1, 0.8 ± 0.3 mM, and 0.0006 ± 0.0009 s−1, respectively. For τd, an unconstrained free parameter, the best estimate obtained from the simultaneous fit to the 0.033 and 0.7 Hz data was 59 ± 19 ms.
Apart from providing a good estimate of the shift of the concentration-response relationship at different frequencies, the best fit values derived in Fig. 3 can be used to approximate the proportion of unblocked channels at the deep site in the absence of test pulses. This is shown in Fig. 3 (gray dotted line) and represents the solution for 1 − Pr, where Pr is as defined above. A fit of 1 − Pr to the Hill equation gave a Hill coefficient of 1 and a KBa,d of 4 μM. In other words, with 4 μM Ba2+ the on-rate constant (k2′) is equal to the off-rate constant (k−2) and half of the channels have Ba2+ bound at the deep site. This suggests that even with 10-ms test pulses applied only once every 30 s the KBa,d is overestimated roughly fivefold, whereas at 0.7 Hz the KBa,d is overestimated roughly 90-fold, and emphasizes the potential importance of taking the pulse frequency and duration into account when comparing the relative sensitivities of channels to block by Ba2+. Thus, reported millimolar values of KBa,d for ShIR (20,21) describe the apparent affinity of the deep site at a specific voltage and pulse frequency.
Equation 4 has been shown to fairly faithfully describe the frequency dependence of the concentration-steady-state block relationship; however, the analysis of Fig. 3 provides no indication of how well the model replicates the time dependence of either the onset of, or the recovery from, block. Experimental data addressing that issue are shown in Fig. 4 A. For this representative experiment, at the end of several 10-ms control test pulses to +50 mV, a computer-controlled, rapid changeover from zero to 0.2 mM Ba2+ was made and that concentration was then maintained while test pulses were applied at 0.1 Hz (solid circles). Once a steady level of block was attained, the stimulus frequency was decreased to 0.033 Hz (open squares) to monitor the progression of the block to a new steady state. Complete reversal of the enhanced steady-state block observed with 0.033 Hz pulsing was then obtained after reverting to the 0.1-Hz stimulus frequency. The same approach was subsequently repeated with trains of 0.2- (open circles), 0.5- (open triangles), and 0.7-Hz (open diamonds) pulses. Fig. 4 B shows the output of a simulation of the proportion of unblocked current (1−PB,x) (see Eq. 5) at the end of the xth test pulse and was derived using the values for k2, KBa,s, k−2, and τd obtained from the fit to the data of Fig. 3. The values for td, tr, and τr were calculated as described in connection with Fig. 3; Pinitial of Eq. 5 is 1 − the proportion of current at the end of the preceding epoch. Fig. 4 B illustrates that the analytical solution reproduces the experimental data quite well not only in terms of the steady-state level of residual normalized current, as expected, but also from the standpoint of the time dependence of the relaxations to a new level of residual current after a change of the test-pulse frequency. These results also suggest that the values of k2, KBa,s, and k−2 are good estimates of the rate and equilibrium constants for Ba2+ binding to Kv1.5 channels, and they provide further support for the two-site binding model.
FIGURE 4.

A model of the frequency-dependent Ba2+ binding replicates the time dependence of block onset and reversal. (A) A representative experiment in which a cell was given four 10-ms test pulses to +50 mV in 0 mM Ba2+ before a rapid changeover to 0.2 mM Ba2+ while test pulses continued to be applied at 0.1 Hz (open circles). After steady-state block was achieved, the pulse frequency was decreased to 0.033 Hz (open squares) and the block was allowed to reach a new steady state. The enhanced steady-state block observed with 0.033-Hz stimulus frequency was reversed upon reverting to 0.1-Hz pulses. The effects of changing the pulse frequency to 0.2 (○), 0.5 (Δ) and 0.7 (◊) Hz were subsequently examined using the same approach. Data points show current amplitudes normalized to the peak current at the end of the initial control test pulses. (B) Plot of simulated proportion of residual normalized current (1 – PB,x) at the end of each test pulse shown in A, calculated using Eq. 5 and the values for k2, KBa,s,, k−2, and τd derived from fits to the data of Fig. 3. The values for td, tr, and τr were those derived from Fig. 3 (see text), whereas Pinitial of Eq. 5 is 1 − the proportion of current at the end of the preceding pulse train. At each stimulus frequency, the numerical simulation provides a good estimate both of the steady-state level of block and the time course of the relaxation to that steady state.
Ba2+ causes a weak fast block of open Kv1.5 channels
As noted above, the time course of Ba2+ block onset in Kv1.5 lacks the fast phase seen in ShIR channels (20,21) and instead exhibits a slow, monoexponential component (Fig. 1 C). However, our analysis of the [Ba2+] dependence of the blocking rate (1/τblock; Fig. 1 D) suggested that, as with ShIR, Ba2+ binds first to a low-affinity (KBa,s ≅ 1 mM) superficial site and then to a high-affinity (KBa,d = 4 μM) deep site. Given the low value of KBa,s compared to the concentration of Ba2+ used in experiments such as those of Fig. 1 C, it was somewhat surprising that a fast phase of block onset related to Ba2+ binding to a superficial site was not seen. This motivated experiments where a brief pulse of 20 mM Ba2+was applied during a 10-s pulse to +50 mV (Fig. 5). At the onset of the Ba2+ application, the current amplitude rapidly decreased by ∼10%, with a time constant that we assume was governed primarily by the kinetics of the solution change. Upon the switch back to Ba2+-free solution, the current recovered to the same amplitude as that of the control current. These results indicate a fast but comparatively weak block of Kv1.5 by 20 mM Ba2+, likely by binding to the superficial pore binding site. If so, the affinity of this site for Ba2+ in the open state is apparently much lower than that estimated for the superficial site in the closed state (Fig. 1 D) and appears to account for the lack of a fast phase of block in experiments such as those shown in Fig. 1 C.
FIGURE 5.

Fast application of Ba2+ during a long depolarizing pulse reveals very weak open-channel block by 20 mM Ba2+. Current traces in response to depolarizing pulses to +50 mV for 10 s in 0 mM 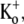 Ba2+-free solution (black trace) and with a rapid application and withdrawal of a 0 mM
Ba2+-free solution (black trace) and with a rapid application and withdrawal of a 0 mM 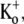 20 mM Ba2+ solution during a subsequent pulse (gray trace). Down and up arrows indicate the start and end of the Ba2+ application, respectively. Exposure to Ba2+ caused a rapid fall in current amplitude of ∼10%. A similar effect was observed in six other cells.
20 mM Ba2+ solution during a subsequent pulse (gray trace). Down and up arrows indicate the start and end of the Ba2+ application, respectively. Exposure to Ba2+ caused a rapid fall in current amplitude of ∼10%. A similar effect was observed in six other cells.
Ba2+ block of Kv1.5 at rest and open-channel dissociation
As mentioned in connection to Fig. 1 B, when Ba2+ is applied for a prolonged period to resting channels, the first 10-ms pulse shows a slow activation time course mainly reflecting a slow unblock of channels. To characterize this unbinding of Ba2+ ions from open channels and determine whether it was dependent on factors such as the test voltage or [K+]o, experiments were performed where Ba2+ was applied for 2 min to closed Kv1.5 channels at −80 mV in 0.5 mM 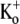 (see experimental protocol in Fig. 6 A). After a 4-min wash to remove all Ba2+, a 1.5-s step to +50 mV was used to assess the time course of Ba2+ unbinding from the open channel. In agreement with findings in other K+ channels (17,19,21) and our estimate of k−2, Ba2+ remained bound to the channels even after the prolonged wash-out period, such that the first pulse elicited current with a much slower rise time than that seen before Ba2+ application (Fig. 6 B). The lack of either an initial delay or fast phase, which would be indicative of current moving through channels that were not blocked at the start of the pulse, together with the single exponential time course of the “post-Ba2+” current rising phase (τact,app = 22.56 ± 1.25 ms; n = 11) suggest that with the loading protocol used, all of the channels were Ba2+-bound before the pulse. (With 0
(see experimental protocol in Fig. 6 A). After a 4-min wash to remove all Ba2+, a 1.5-s step to +50 mV was used to assess the time course of Ba2+ unbinding from the open channel. In agreement with findings in other K+ channels (17,19,21) and our estimate of k−2, Ba2+ remained bound to the channels even after the prolonged wash-out period, such that the first pulse elicited current with a much slower rise time than that seen before Ba2+ application (Fig. 6 B). The lack of either an initial delay or fast phase, which would be indicative of current moving through channels that were not blocked at the start of the pulse, together with the single exponential time course of the “post-Ba2+” current rising phase (τact,app = 22.56 ± 1.25 ms; n = 11) suggest that with the loading protocol used, all of the channels were Ba2+-bound before the pulse. (With 0 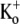 there was, in addition to a slow component with τact,app = 15.30 ± 0.99 ms, which we take to reflect Ba2+ unbinding, a very slow component with τ = 1.08 ± 0.14 s (n = 10; data not shown). Investigation of the slower component of current was beyond the scope of this study, but we do note that confounding results with 0
there was, in addition to a slow component with τact,app = 15.30 ± 0.99 ms, which we take to reflect Ba2+ unbinding, a very slow component with τ = 1.08 ± 0.14 s (n = 10; data not shown). Investigation of the slower component of current was beyond the scope of this study, but we do note that confounding results with 0 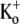 solutions and Ba2+ have also been reported by others (21).) The first sweep after Ba2+ loading showed little or no decay over the course of the pulse, perhaps because of the overlap of inactivation and Ba2+ unblocking. From the rising phase of the current, the Ba2+ unbinding rate (1/τact,app) was calculated to be 45.79 ± 3.08 s−1, although this may be an underestimate if substantial inactivation, and hence Ba2+ trapping, occurs during the rising phase. Additional depolarizations (not shown) evoked currents with activation kinetics similar to that of control, with a small (<10%) slow component, perhaps associated with residual Ba2+ that did not unbind during the initial 1.5-s pulse. With subsequent pulses, the contribution of the slow component diminished and current amplitude recovered toward control values.
solutions and Ba2+ have also been reported by others (21).) The first sweep after Ba2+ loading showed little or no decay over the course of the pulse, perhaps because of the overlap of inactivation and Ba2+ unblocking. From the rising phase of the current, the Ba2+ unbinding rate (1/τact,app) was calculated to be 45.79 ± 3.08 s−1, although this may be an underestimate if substantial inactivation, and hence Ba2+ trapping, occurs during the rising phase. Additional depolarizations (not shown) evoked currents with activation kinetics similar to that of control, with a small (<10%) slow component, perhaps associated with residual Ba2+ that did not unbind during the initial 1.5-s pulse. With subsequent pulses, the contribution of the slow component diminished and current amplitude recovered toward control values.
As reported by others (19,21), we found that in Ba2+-free medium the dissociation rate of Ba2+ depended on both the step potential and [K+]o. Ba2+ unbinding was faster at more positive potentials (Fig. 6, C and D), suggestive of outward, voltage-dependent dissociation. Using the Woodhull model (28), the electrical distance (δBa,off) between the deep Ba2+ binding site and the rate-limiting barrier for exit was estimated to be 0.51 ± 0.04. This value is similar to those calculated for ShIR (δBa,off = 0.37) and BKCa channels (δBa,off = 0.45) (18,19,21). When [K+]o was changed the properties of control currents were unaffected (not shown), which is consistent with previous reports that at pHo 7.4, Kv1.5 open probability is insensitive to changes in [K+]o (9). After a 2-min loading period with 5 mM Ba2+ (in 0.5 mM 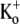 ) to block all channels (see Fig. 6 B), the cells were washed with solutions of varying [K+]o and test pulses to +50 mV were repeated. As shown in Fig. 6, E and F, Ba2+ unbinding was slowed by increasing [K+]o. A fit of the unbinding rates and [K+]o to the Hill equation (see Methods) gave an apparent KD of 1.00 ± 0.08 mM [K+]o for this effect, with a Hill coefficient of 1.02 ± 0.06. We take this to be the dissociation constant at +50 mV for a K+ binding site external to the deep Ba2+ ion binding site that needs to be vacant for Ba2+ unbinding, and that would be analogous, if not homologous, to the “lock-in” site described in BK and ShIR channels (19–21).
) to block all channels (see Fig. 6 B), the cells were washed with solutions of varying [K+]o and test pulses to +50 mV were repeated. As shown in Fig. 6, E and F, Ba2+ unbinding was slowed by increasing [K+]o. A fit of the unbinding rates and [K+]o to the Hill equation (see Methods) gave an apparent KD of 1.00 ± 0.08 mM [K+]o for this effect, with a Hill coefficient of 1.02 ± 0.06. We take this to be the dissociation constant at +50 mV for a K+ binding site external to the deep Ba2+ ion binding site that needs to be vacant for Ba2+ unbinding, and that would be analogous, if not homologous, to the “lock-in” site described in BK and ShIR channels (19–21).
Ba2+ does not block Kv1.5 at low pHo
The results presented thus far are consistent with Ba2+ binding sequentially to two sites in Kv1.5, with the stable block of the channel resulting from Ba2+ occupancy of the deeper, higher-affinity site. Having established that Ba2+ can access and bind within the pore of Kv1.5 at rest, we proceeded to examine the hypothesis that extracellular acidification results in outer pore constriction and an altered ability of Ba2+ to bind to the pore. Fig. 7 A illustrates the results of experiments monitoring the current amplitude with 10-ms pulses to +50 mV applied at 1 Hz during the fast application of Ba2+ at low pHo. When the pHo was decreased from pH 7.4 to pH 5.5, current amplitude fell quickly to ∼6% of the control level. The small residual current is consistent with our previous findings that at low pHo, an equilibrium exists between the available and unavailable states of the channel (5); it also likely includes a small contribution from endogenous K+ currents in the HEK293 cell line (3). Subsequent application of 20 mM Ba2+ at pHo 5.5 caused a small additional decrease of the current amplitude. Upon the rapid washout of Ba2+ and return to pHo 7.4, the recovery time course was fast (τrecov = 4.01 ± 0.85 s; n = 4) and was not significantly different (p > 0.05) from that after pHo 5.5 treatment alone (τ = 2.60 ± 0.18 s; n = 5; not shown). If lowering pH had no effect on Ba2+ accessibility, we would have expected to see ∼70% block of current under these conditions and, thus, a significant slowing of the recovery time course upon the return to pHo 7.4. These results suggest that lowering pHo before and during Ba2+ application largely prevents Ba2+ from accessing its blocking site in most of the channels, allowing them to quickly return to a conducting state after changing back to pHo 7.4.
FIGURE 7.

Low pHo inhibits both Ba2+ binding and unbinding from Kv1.5. (A) Diary plot of normalized current amplitudes at the end of 10-ms pulses to +50 mV applied at 1 Hz. Experiments were started in 0 mM  pH 7.4 bath solution. Rapid switching to pH 5.5 solution caused a rapid fall of the current amplitude. Subsequent application of 20 mM Ba2+ (down arrow), still at pH 5.5, caused a further small decline. The rapid return (up arrow) to Ba2+-free, pHo 7.4 solution resulted in a quick recovery of current amplitude (τrecov = 3.1 s), which would not be expected had Ba2+ accessed the deep pore site. (B) In the converse of the experiment shown in A, 20 mM Ba2+ was applied (down arrow) at pHo 7.4, resulting in 71% block of current, and was followed by a rapid wash-off of Ba2+ (up arrow) with pH 5.5 solution that caused an almost complete loss of current. Fast change of the bath solution back to pH 7.4 caused a rapid component of current recovery, followed by a large slow component (64% of total recovery) with a time constant of 23.6 s. These results are consistent with the interpretation that the proportion of Ba2+-loaded channels is not affected by high-frequency pulsing when the pHo is low.
pH 7.4 bath solution. Rapid switching to pH 5.5 solution caused a rapid fall of the current amplitude. Subsequent application of 20 mM Ba2+ (down arrow), still at pH 5.5, caused a further small decline. The rapid return (up arrow) to Ba2+-free, pHo 7.4 solution resulted in a quick recovery of current amplitude (τrecov = 3.1 s), which would not be expected had Ba2+ accessed the deep pore site. (B) In the converse of the experiment shown in A, 20 mM Ba2+ was applied (down arrow) at pHo 7.4, resulting in 71% block of current, and was followed by a rapid wash-off of Ba2+ (up arrow) with pH 5.5 solution that caused an almost complete loss of current. Fast change of the bath solution back to pH 7.4 caused a rapid component of current recovery, followed by a large slow component (64% of total recovery) with a time constant of 23.6 s. These results are consistent with the interpretation that the proportion of Ba2+-loaded channels is not affected by high-frequency pulsing when the pHo is low.
Ba2+ unbinding is inhibited at low pHo
To test whether the lack of Ba2+ block at pHo 5.5 is due to H+-induced unbinding of Ba2+, which would be indistinguishable from an inability of Ba2+ to bind in the experiments shown in Fig. 7 A, experiments were also performed where the pH of the bath solution was decreased after Ba2+ loading. Fig. 7 B shows the results from a representative experiment where the current level was allowed to reach a steady state (72 ± 2% block; n = 6) in 20 mM Ba2+ with pulses applied at 1 Hz, after which the cell was returned to Ba2+-free medium first at pHo 5.5 and then at pHo 7.4. Changing to the pHo 5.5 solution caused a further, and almost complete, reduction in current magnitude. In contrast to the situation where Ba2+ was applied after switching to pHo 5.5, the return to pHo 7.4 saw current recovery proceed with a fast phase and a slow phase. The amplitude of the fast phase was similar to that of the residual current recorded at the end of the Ba2+ loading protocol at pHo 7.4 (39 ± 4% vs. 28 ± 2% of control amplitude, respectively) and, thus, reflects the relatively rapid recovery of Ba2+-free channels from the action of pH 5.5 solution. Had significant Ba2+ unbinding occurred at the same rate as at pH 7.4 (Fig. 1 C), then 63% recovery from the Ba2+ block would have occurred. In addition, the time constant of the slow recovery phase (τslow = 25.59 ± 5.32 s) was not significantly different (p > 0.05) from that observed for current recovery at pHo 7.4 after Ba2+ loading (Fig. 1 C). These results are consistent with an inability of Ba2+ to unbind from Kv1.5 at low pHo. Together with the findings from the previous section, this suggests that the conduction pathway between the deep pore binding site and the external solution is changed sufficiently under acidic conditions to restrict free ingress and egress of Ba2+ to and from its binding sites and is supportive of a low-pHo-mediated constriction of the outer pore of resting channels.
DISCUSSION
Ba2+ block at pH 7.4
The first series of experiments presented in this article show that in Kv1.5, at pHo 7.4, external Ba2+ ions cause a high-affinity, slow block of closed channels (Figs. 1, 3, and 4) and a low-affinity, fast block of open channels (Fig. 5). Although the time course of the onset of closed-channel block (τblock) followed a single exponential, rather than showing distinct fast and slow phases as in ShIR, the dependence of τblock on [Ba2+]o with 0 mM  was still well described by a sequential two-site binding model (Fig. 1 D) in which the loading of a deep site with an apparent equilibrium dissociation constant (KBa,d) in the low micromolar range depends on the occupancy of a superficial site with an apparent dissociation constant (KBa,s) of ∼1 mM. Near-saturation of the superficial site with 5–20 mM Ba2+ explains why τblock was not linearly related to [Ba2+]o (Fig. 1 D), as would occur for a simple bimolecular reaction. It is unlikely that the slow Ba2+ block and/or lack of an apparent fast block are due to nonspecific effects of charge screening since 1), the block induced by either 1 mM or 20 mM Ba2+ is qualitatively similar; and 2), charge screening would be expected to cause a fast change in current amplitude both when Ba2+ is applied and when it is removed (see Hurst et al. (20)).
was still well described by a sequential two-site binding model (Fig. 1 D) in which the loading of a deep site with an apparent equilibrium dissociation constant (KBa,d) in the low micromolar range depends on the occupancy of a superficial site with an apparent dissociation constant (KBa,s) of ∼1 mM. Near-saturation of the superficial site with 5–20 mM Ba2+ explains why τblock was not linearly related to [Ba2+]o (Fig. 1 D), as would occur for a simple bimolecular reaction. It is unlikely that the slow Ba2+ block and/or lack of an apparent fast block are due to nonspecific effects of charge screening since 1), the block induced by either 1 mM or 20 mM Ba2+ is qualitatively similar; and 2), charge screening would be expected to cause a fast change in current amplitude both when Ba2+ is applied and when it is removed (see Hurst et al. (20)).
Using three different approaches, a very rough estimate of ∼0.001 s−1 (with a large standard deviation) was obtained for k−2, the rate constant for unblocking of the deep site from closed channels at −80 mV with nominally zero  (Figs. 1, D and E, and 3). The apparent off rate from the deep site was substantially increased either by raising the frequency of 10-ms test steps to +50 mV (Fig. 1 E), by increasing the test-pulse duration (Fig. 6 B), or by increasing the intensity of depolarizing pulses (Fig. 6 D). Indeed, extrapolation of the open-channel dissociation rate (Fig. 6 D) for Ba2+ to −80 mV, assuming δBa,off = 0.51, gives an estimate that is >200 times faster than the measured closed-state off rate at −80 mV (Fig. 1 E). This facilitation of Ba2+ unbinding by depolarization, which is consistent with results from studies on K+ channels of the squid giant axon (17), BK channels (19) and ShIR (21), is likely due to a combination of several factors: 1), the deep site is in the pore and therefore “senses” part of the electric field so that depolarization decreases the on rate and/or increases the off rate; and 2), channel opening, which for Kv1.5 is maximal at voltages ≥+20 mV (3), may cause a conformational change of the binding site and may also permit a knock-off effect by intracellular K+ ions entering the pore.
(Figs. 1, D and E, and 3). The apparent off rate from the deep site was substantially increased either by raising the frequency of 10-ms test steps to +50 mV (Fig. 1 E), by increasing the test-pulse duration (Fig. 6 B), or by increasing the intensity of depolarizing pulses (Fig. 6 D). Indeed, extrapolation of the open-channel dissociation rate (Fig. 6 D) for Ba2+ to −80 mV, assuming δBa,off = 0.51, gives an estimate that is >200 times faster than the measured closed-state off rate at −80 mV (Fig. 1 E). This facilitation of Ba2+ unbinding by depolarization, which is consistent with results from studies on K+ channels of the squid giant axon (17), BK channels (19) and ShIR (21), is likely due to a combination of several factors: 1), the deep site is in the pore and therefore “senses” part of the electric field so that depolarization decreases the on rate and/or increases the off rate; and 2), channel opening, which for Kv1.5 is maximal at voltages ≥+20 mV (3), may cause a conformational change of the binding site and may also permit a knock-off effect by intracellular K+ ions entering the pore.
The Ba2+ unbinding rate from open channels at +50 mV was slowed by increasing [K+]o (see Fig. 6, E and F). It is interesting to note that the apparent KD (1 mM) for the inhibition of Ba2+ unbinding by  is similar to the KD (1 mM) for the antagonism by
is similar to the KD (1 mM) for the antagonism by 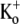 of the low pHo-induced loss of channel availability (3), suggesting that the same K+-binding site may be involved in both situations. This site is perhaps homologous to the “lock-in” binding site for the K+-dependent inhibition of Ba2+ unbinding in ShIR and BK channels (19–21). Indeed, the affinity of the lock-in site for K+ in ShIR (KD = 0.75 mM (21)) is quantitatively similar to what we observed in Kv1.5. It should be noted that we measured the KD of the lock-in site in Kv1.5 with a Ba2+ ion already bound to the deep site, as was done in ShIR. The actual affinity of the lock-in site for K+ may therefore be underestimated due to the possible coulombic interaction with a bound Ba2+ ion.
of the low pHo-induced loss of channel availability (3), suggesting that the same K+-binding site may be involved in both situations. This site is perhaps homologous to the “lock-in” binding site for the K+-dependent inhibition of Ba2+ unbinding in ShIR and BK channels (19–21). Indeed, the affinity of the lock-in site for K+ in ShIR (KD = 0.75 mM (21)) is quantitatively similar to what we observed in Kv1.5. It should be noted that we measured the KD of the lock-in site in Kv1.5 with a Ba2+ ion already bound to the deep site, as was done in ShIR. The actual affinity of the lock-in site for K+ may therefore be underestimated due to the possible coulombic interaction with a bound Ba2+ ion.
Ba2+ block of Kv1.5 versus ShIR
The divergence of the KBa,d estimates for Kv1.5 and ShIR can probably be accounted for, at least in part, by the test-pulse frequency and duration, but there are other significant differences in the Ba2+ block of Kv1.5 and ShIR channels. The fast phase of Ba2+ block, which is not seen in Kv1.5, arises because Ba2+ (KBa,s (0 mV) = 19 mM (20)) causes a rapidly equilibrating (flickery) block of open channels during test pulses used to monitor the development of block. We attribute the absence of a fast phase of Ba2+ block in Kv1.5 to the comparatively much weaker open-channel block.
Given the estimate of 1 mM for the KBa,s of the superficial site, this weak open channel block is perhaps unexpected. For example, if this binding site is assumed to be outside the electric field, then 20 mM Ba2+ would be expected to cause 95% inhibition when applied, as in Fig. 5, to open channels. If it is also assumed, for simplicity, that the superficial site that contributes indirectly to the slow block also accounts for the fast open-channel block, then possible explanations for the weak open-channel block are that the transition to the open conformation changes the site's affinity for Ba2+ or that the efflux of K+ through the open pore competitively antagonizes the fast Ba2+ block. In support of the K+ inhibition of Ba2+ block, with 3.5 mM 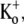 only ∼40% of channels were blocked at rest by 1 mM Ba2+, compared to ∼90% with 0
only ∼40% of channels were blocked at rest by 1 mM Ba2+, compared to ∼90% with 0 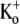 (measured using 10-ms pulses to +50 mV at 0.033 Hz; data not shown). The effect of K+ is likely to be even greater when the channels are open, since, it has been suggested, even when the bulk [K+]o is zero, outward K+ current through ShIR channels leads to an accumulation of K+ in the outer pore mouth, resulting in a [K+] of ∼15 mM (29).
(measured using 10-ms pulses to +50 mV at 0.033 Hz; data not shown). The effect of K+ is likely to be even greater when the channels are open, since, it has been suggested, even when the bulk [K+]o is zero, outward K+ current through ShIR channels leads to an accumulation of K+ in the outer pore mouth, resulting in a [K+] of ∼15 mM (29).
Although Kv1.5 and Shaker have identical primary sequences from the N-terminus of the pore helix through to the GYGDM sequence of the selectivity filter (Fig. 2), the much weaker open-channel Ba2+ block of Kv1.5 points to significant structural differences between Kv1.5 and Shaker. The residue at position 449 in the outer pore mouth, which is threonine in wild-type Shaker, can be an important molecular determinant of Ba2+ block in ShIR (30) and it seems reasonable to propose that the presence in Kv1.5 of arginine (R487) at the position homologous to Shaker T449 contributes to their divergent responses to Ba2+. It is interesting to note that in Kv1.3, the mutation H399K, which is the positional equivalent of Kv1.5 R487, causes a marked decrease of τblock (13,14), implying, albeit counterintuitively, that a basic side chain enhances Ba2+ loading. In light of the latter observation, the fact that onset of the slow block in Kv1.5 (τblock ≈ 6 s at −80 mV with 0 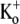 and 20 mM Ba2+; Fig. 1 D) is more than an order of magnitude faster than in ShIR (τblock ≈ 100 s at −90 mV with 2 mM
and 20 mM Ba2+; Fig. 1 D) is more than an order of magnitude faster than in ShIR (τblock ≈ 100 s at −90 mV with 2 mM 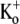 and 20 mM Ba2+ (Fig. 9 of Hurst et al. (20)) might also be due to the arginine at position 487.
and 20 mM Ba2+ (Fig. 9 of Hurst et al. (20)) might also be due to the arginine at position 487.
Low pHo prevents movement of Ba2+ to and from its deep binding site
In support of an inactivation-mediated constriction of the outer pore, the movement of Ba2+ from its deep pore binding site to the bulk solution is significantly slowed in P/C-type inactivated ShIR channels, to the extent that Ba2+ is effectively trapped within the pore of P/C-type inactivated ShIR (21,27). We have shown here that with external acidification, external Ba2+ cannot access the deep pore binding site at rest (Fig. 7 A), nor can Ba2+ that is already bound at the deep site in the pore leave that binding site (Fig. 7 B). These results are therefore consistent with the hypothesis that an outer pore constriction, perhaps similar to that proposed to underlie P/C-type inactivation, also underlies the 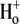 -mediated decrease in Ba2+ access to, or egress from, the pore.
-mediated decrease in Ba2+ access to, or egress from, the pore.
Is the H+-induced pore constriction indicative of closed-state P/C-type inactivation?
It remains an open question whether the proposed constriction of the outer pore of a closed Kv1.5 channel under resting, acidic conditions is the same conformation the channel enters during P/C-type inactivation, which is typically thought to be strongly coupled to channel activation. Unfortunately, we were unable to assess the Ba2+ block of P/C-type inactivated channels at pHo 7.4, because the inactivation is slow and incomplete, even with long depolarizing pulses (3). Although the molecular mechanism for the low-pHo-induced block of Kv1.5 is still unclear, we (3) and others (8,9) have proposed that the H463 residue in the outer turret acts as the putative pHo sensor, since both the decrease in channel availability and enhanced depolarization-induced inactivation are substantially diminished in Kv1.5 H463Q. The ability of the noninactivating Kv1.5 R487V mutant, which is analogous to Shaker T449V, to mitigate the effects of low pHo block are suggestive of an inactivation mechanism involving this residue, but the nature of the coupling mechanism between protonation of H463 and channel inactivation is unknown at this time.
Acidosis of cardiac muscle is a common outcome of a number of pathological conditions, such as myocardial ischemia, and can induce arrhythmias (31). Although the extent of extracellular acidification in native cardiac tissue is seldom likely to be as low as pHo 5.5, the pKa of the 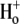 -block of Kv1.5 in 5 mM
-block of Kv1.5 in 5 mM 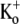 is ∼6.2 (3,5), such that even small decreases in pHo can result in a decrease in channel availability and faster macroscopic inactivation. Thus, pathological cardiac acidosis may be associated with reduced availability of Kv1.5 channels due to closed-state inactivation, which, coupled with faster inactivation of the remaining channels and a right shift of the activation curve (32), results in reduced IKur.
is ∼6.2 (3,5), such that even small decreases in pHo can result in a decrease in channel availability and faster macroscopic inactivation. Thus, pathological cardiac acidosis may be associated with reduced availability of Kv1.5 channels due to closed-state inactivation, which, coupled with faster inactivation of the remaining channels and a right shift of the activation curve (32), results in reduced IKur.
Editor: Richard W. Aldrich.
References
- 1.Fedida, D., B. Wible, Z. Wang, B. Fermini, F. Faust, S. Nattel, and A. M. Brown. 1993. Identity of a novel delayed rectifier current from human heart with a cloned K+ channel current. Circ. Res. 73:210–216. [DOI] [PubMed] [Google Scholar]
- 2.Feng, J., B. Wible, G. R. Li, Z. Wang, and S. Nattel. 1997. Antisense oligodeoxynucleotides directed against Kv1.5 mRNA specifically inhibit ultrarapid delayed rectifier K+ current in cultured adult human atrial myocytes. Circ. Res. 80:572–579. [DOI] [PubMed] [Google Scholar]
- 3.Kehl, S. J., C. Eduljee, D. C. Kwan, S. Zhang, and D. Fedida. 2002. Molecular determinants of the inhibition of human Kv1.5 potassium currents by external protons and Zn2+. J. Physiol. 541:9–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fedida, D., S. Zhang, D. C. Kwan, C. Eduljee, and S. J. Kehl. 2005. Synergistic inhibition of the maximum conductance of Kv1.5 channels by extracellular K+ reduction and acidification. Cell Biochem. Biophys. 43:231–242. [DOI] [PubMed] [Google Scholar]
- 5.Kwan, D. C., D. Fedida, and S. J. Kehl. 2006. Single channel analysis reveals different modes of Kv1.5 gating behavior regulated by changes of external pH. Biophys. J. 90:1212–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Claydon, T. W., M. Vaid, S. Rezazadeh, D. C. Kwan, S. J. Kehl, and D. Fedida. 2007. A direct demonstration of closed-state inactivation of K+ channels at low pH. J. Gen. Physiol. 129:437–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pérez-Cornejo, P. 1999. H+ ion modulation of C-type inactivation of Shaker K+ channels. Pflugers Arch. 437:865–870. [DOI] [PubMed] [Google Scholar]
- 8.Steidl, J. V., and A. J. Yool. 1999. Differential sensitivity of voltage-gated potassium channels Kv1.5 and Kv1.2 to acidic pH and molecular identification of pH sensor. Mol. Pharmacol. 55:812–820. [PubMed] [Google Scholar]
- 9.Jager, H., and S. Grissmer. 2001. Regulation of a mammalian Shaker-related potassium channel, hKv1.5, by extracellular potassium and pH. FEBS Lett. 488:45–50. [DOI] [PubMed] [Google Scholar]
- 10.Claydon, T. W., M. R. Boyett, A. Sivaprasadarao, and C. H. Orchard. 2002. Two pore residues mediate acidosis-induced enhancement of C-type inactivation of the Kv1.4 K+ channel. Am. J. Physiol. Cell Physiol. 283:C1114–C1121. [DOI] [PubMed] [Google Scholar]
- 11.Starkus, J. G., Z. Varga, R. Schonherr, and S. H. Heinemann. 2003. Mechanisms of the inhibition of Shaker potassium channels by protons. Pflugers Arch. 447:44–54. [DOI] [PubMed] [Google Scholar]
- 12.Trapani, J. G., and S. J. Korn. 2003. Effect of external pH on activation of the Kv1.5 potassium channel. Biophys. J. 84:195–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Somodi, S., Z. Varga, P. Hajdu, J. G. Starkus, D. I. Levy, R. Gáspár, and G. Panyi. 2004. pH-dependent modulation of Kv1.3 inactivation: role of His399. Am. J. Physiol. Cell Physiol. 287:C1067–C1076. [DOI] [PubMed] [Google Scholar]
- 14.Somodi, S., P. Hajdu, R. Gaspar, G. Panyi, and Z. Varga. 2008. Effects of changes in extracellular pH and potassium concentration on Kv1.3 inactivation. Eur. Biophys. J. 37:1145–1156. [DOI] [PubMed] [Google Scholar]
- 15.Zhang, S., H. T. Kurata, S. J. Kehl, and D. Fedida. 2003. Rapid induction of P/C-type inactivation is the mechanism for acid-induced K+ current inhibition. J. Gen. Physiol. 121:215–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kurata, H. T., and D. Fedida. 2006. A structural interpretation of voltage-gated potassium channel inactivation. Prog. Biophys. Mol. Biol. 92:185–208. [DOI] [PubMed] [Google Scholar]
- 17.Armstrong, C. M., R. P. Swenson, Jr., and S. R. Taylor. 1982. Block of squid axon K channels by internally and externally applied barium ions. J. Gen. Physiol. 80:663–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Neyton, J., and C. Miller. 1988. Discrete Ba2+ block as a probe of ion occupancy and pore structure in the high-conductance Ca2+-activated K+ channel. J. Gen. Physiol. 92:569–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Neyton, J., and C. Miller. 1988. Potassium blocks barium permeation through a calcium-activated potassium channel. J. Gen. Physiol. 92:549–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hurst, R. S., R. Latorre, L. Toro, and E. Stefani. 1995. External barium block of Shaker potassium channels: evidence for two binding sites. J. Gen. Physiol. 106:1069–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harris, R. E., H. P. Larsson, and E. Y. Isacoff. 1998. A permanent ion binding site located between two gates of the Shaker K+ channel. Biophys. J. 74:1808–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gibor, G., D. Yakubovich, A. Rosenhouse-Dantsker, A. Peretz, H. Schottelndreier, G. Seebohm, N. Dascal, D. E. Logothetis, Y. Paas, and B. Attali. 2007. An inactivation gate in the selectivity filter of KCNQ1 potassium channels. Biophys. J. 93:4159–4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiang, Y., and R. Mackinnon. 2000. The barium site in a potassium channel by x-ray crystallography. J. Gen. Physiol. 115:269–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jonas, P. 1995. Fast application of agonists to isolated membrane patches. In Single-Channel Recording. B. Sakmann and E. Neher, editors. Plenum Press, New York. 231–243.
- 25.Panyi, G., and C. Deutsch. 2006. Cross talk between activation and slow inactivation gates of Shaker potassium channels. J. Gen. Physiol. 128:547–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Starmer, C. F., J. Z. Yeh, and J. Tanguy. 1986. A quantitative description of QX222 blockade of sodium channels in squid axons. Biophys. J. 49:913–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Basso, C., P. Labarca, E. Stefani, O. Alvarez, and R. Latorre. 1998. Pore accessibility during C-type inactivation in Shaker K+ channels. FEBS Lett. 429:375–380. [DOI] [PubMed] [Google Scholar]
- 28.Woodhull, A. M. 1973. Ionic blockage of sodium channels in nerve. J. Gen. Physiol. 61:687–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baukrowitz, T., and G. Yellen. 1995. Modulation of K+ current by frequency and external [K+]: a tale of two inactivation mechanisms. Neuron. 15:951–960. [DOI] [PubMed] [Google Scholar]
- 30.Hurst, R. S., L. Toro, and E. Stefani. 1996. Molecular determinants of external barium block in Shaker potassium channels. FEBS Lett. 388:59–65. [DOI] [PubMed] [Google Scholar]
- 31.Orchard, C. H., and H. E. Cingolani. 1994. Acidosis and arrhythmias in cardiac muscle. Cardiovasc. Res. 28:1312–1319. [DOI] [PubMed] [Google Scholar]
- 32.Andalib, P., J. F. Consiglio, J. G. Trapani, and S. J. Korn. 2004. The external TEA binding site and C-type inactivation in voltage-gated potassium channels. Biophys. J. 87:3148–3161. [DOI] [PMC free article] [PubMed] [Google Scholar]