

Abstract
Proline residues play a fundamental and subtle role in the dynamics, structure, and function in many membrane proteins. Temperature derivative spectroscopy and differential scanning calorimetry have been used to determine the effect of proline substitution in the structural stability of the active site and transmembrane arrangement of bacteriorhodopsin. We have analyzed the Pro-to-Ala mutation for the helix-embedded prolines Pro50, Pro91, and Pro186 in the native membrane environment. This information has been complemented with the analysis of the respective crystallographic structures by the FoldX force field. Differential scanning calorimetry allowed us to determine distorted membrane arrangement for P50A and P186A. The protein stability was severely affected for P186A and P91A. In the case of Pro91, a single point mutation is capable of strongly slowing down the conformational diffusion along the denaturation coordinate, becoming a barrier-free downhill process above 371 K. Temperature derivative spectroscopy, applied for first time to study thermal stability of proteins, has been used to monitor the stability of the active site of bacteriorhodopsin. The mutation of Pro91 and Pro186 showed the most striking effects on the retinal binding pocket. These residues are the Pro in closer contact to the active site (activation energies for retinal release of 60.1 and 76.8 kcal/mol, respectively, compared to 115.8 kcal/mol for WT). FoldX analysis of the protein crystal structures indicates that the Pro-to-Ala mutations have both local and long-range effects on the structural stability of residues involved in the architecture of the protein and the active site and in the proton pumping function. Thus, this study provides a complete overview of the substitution effect of helix-embedded prolines in the thermodynamic and dynamic stability of a membrane protein, also related to its structure and function.
INTRODUCTION
Protein function is mostly characterized by the existence of an active site. The wide variety and complexity of active sites in proteins is mainly responsible for one thing: assuring a selective and effective function. In some specific cases, the active site is in charge of “listening” to a specific signal, and generating a response. How the signal is transmitted from the active site to the rest of the protein, is what we have called intraprotein signal transduction (1). Eventually, the signal is translated into functionality via conformational changes of the protein (the network), which may directly end up in function in the case of a single protein, or in interprotein signal transduction in the case of a complex of proteins. The most representative cases of signal-transduction are the GPCR, where rhodopsin is one of the most studied (2–5). The example of the complex of rhodopsin and transducin, which generates a second messenger cascade, is represented in the microbial rhodopsin world (including prokaryotes and eukaryotes) by different photochemically reactive membrane proteins.
One of the most interesting membrane proteins is embedded in the membrane of the archaeobacteria Halobacterium salinarum and is known as bR (Fig. 1). Bacteriorhodopsin is a seven transmembrane helix chromoprotein (in the same structural superfamily as rhodopsin; helices named from A to G), which has a retinal molecule covalently linked via a SB to the Lys216 (6). The retinal molecule provides the capacity of absorbing photons (as in rhodopsin), a “stimulus” that generates a proton pumping function (7). The pumped protons may be regarded as a second messenger for other proteins in this archaeobacteria, “regulating” functions such as energy production and motility. To transmit the signal to the protein, the retinal uses as a network the amino acids in its close contact, this is the so-called RBP (Fig. 1, b and c). Thus, the retinal molecule and the RBP should be considered the active site of bR, as it should be considered for rhodopsin and other microbial rhodopsins (7–11). A proof of this comes from directed mutagenesis studies of bR, which have been used to switch the proton pumping function to chloride pumping function (i.e., halo rhodopsin-like protein) (10) or to a sensory receptor protein (i.e., sensory rhodopsin-like protein) (12), mimicking other retinal proteins.
FIGURE 1.

Halobacterium salinarum PM. (a) Two-dimensional paracrystalline arrangement of the PM disposed in hexamers. (b) Trimer of bR molecules (PDB code 1brr). The chromophore molecule of retinal is shown in black. Helix-embedded prolines are indicated by Van der Waals spheres in green (Pro50), orange (Pro91), and red (Pro186). To identify the RBP, a molecular surface around the retinal with a 5 Å radius has been calculated. The color code for the molecular surface corresponds to the atom type (carbon, gray; nitrogen, blue; oxygen, red; sulfur, yellow). (c) Representation of the 3D structure of bR indicating the presence of the seven transmembrane α-helices and the β-strands in a cartoon visualization. The figure has been generated from the 1c3w PDB code following the same color code as for (b). (d) Overlay of the chain A of 1py6 and 1pxr crystal structures obtained at pH 3.5. (e) Overlay of the chain A of 1py6 and 1q5j crystal structures obtained at pH 3.5 and 3.7, respectively. (f) Overlay of the chain A of 1py6 and 1q5i crystal structures obtained at pH 3.5 and 4.0, respectively. For distinction, the carbon atoms in 1py6 (WT) are plotted in gray and carbon atoms in 1pxr (P50A), 1q5j (P91A), and 1q5i (P186A) are plotted in orange. Retinal and residues interacting with the chromophore are indicated in addition by a molecular surface. Molecular representations in (a), (b), and (c) have been plotted with the academic version of MAESTRO 8.0 software. Pictures in (d), (e), and (f) have been generated with BallView.
The architecture of the RBP is defined by the overall structure of the protein and its location in the membrane. Bacteriorhodopsin is a protein organized in a natural 2D crystal arrangement in the membrane. The so-called PM patches are ordered systems with a hexagonal disposition of trimers of bR embedded in the membrane (Fig. 1, a and b). Thus, the overall structure of this highly ordered system is based on lipid-protein, protein-protein, and protein-chromophore interactions, and of course, interactions with the solvent.
In the vicinity of the RBP, where the initial steps related to the signal transduction occur, there are three prolines (Fig. 1, b and c): Pro50, Pro91, and Pro186. Proline residues have been shown as α-helix breaker amino acids (13–15), with a less known but important effect in the structure dynamics (16). The cis-trans isomerization of the pyrrolidine ring and the steric clash introduced in the α-helix by proline, favor some conformations that allow the protein to optimally carry out its function. Moreover, the reduced conformational degrees of freedom allow for a faster structural diffusion, important for functions relaying on fast protein dynamics, as the bR photocycle (16). In a previous study (16), we determined the functionality and dynamics of bR when Pro50, Pro91, or Pro186 were mutated to alanine. These three prolines interact with the RBP in the following way: Pro50 (helix B) contact is through the interaction of Val49 with Lys216 (Fig. 1 d); Pro91 (helix C) interacts via Thr90 and Trp86 in the same helix C (Fig. 1 e); and Pro186 (helix F) contacts directly with the retinal (Fig. 1 f). Hence, these three helix-embedded prolines are probes to analyze the environment of the active site of bR.
Most of the studies on structural stability of membrane proteins have been done by spectroscopic techniques and DSC (17–19). Both spectroscopy and DSC are widespread techniques that yield information on the thermodynamics and dynamics of the structural stability of the protein (20–22). The interest on understanding the key elements governing the folding/unfolding of proteins to achieve a determined tertiary structure has been translated into the in silico world. Structural bioinformatics and computational biophysical methods have been developed to understand and predict folding, and to compute thermodynamic values of the folded structures (23–28). This last possibility is especially relevant for membrane proteins, because their common irreversible denaturation on heating, generally precludes their full thermodynamic characterization.
In this study, we have investigated the structural behavior of the P50A, P91A, and P186A PM as a function of temperature, to understand the effect of replacing an α-helix breaker residue (Pro) by an α-helix inducer residue such as Ala. We have complemented this experimental data with the analysis of the crystal structures of WT and mutant bR (P50A, P91A, and P186A) by FoldX (29) to get detailed atomic information. In summary, we have analyzed the structural stability of native and proline transmembrane mutants of bR under H. salinarum membrane environment and in silico conditions.
METHODS
FoldX computing structural stability
For the stability analysis the crystal structures were preprocessed: water molecules were removed, and the crystal structures containing two chains (chains A and B for 1py6, 1pxr, 1q5i, and 1q5j) were split and analyzed as two different structures. The obtained values were later averaged. The analysis carried out were “Stability” providing the overall structural stability, and “SequenceDetail” for the structural stability energy contribution per residue. The analysis was carried out at 298 K, 0.05 M of ionic strength, small Van der Waals clashes were ignored, and the force field predicted the position for water molecules.
Expression of protein samples
The construction of BR mutants P50A, P91A, and P186A and the expression and purification of the PM in H. salinarum were carried out as described previously (30).
Differential scanning calorimetry
Differential scanning calorimetry experiments were carried out using a MicroCal MC2 instrument (MicroCal, Northampton, MA). Samples were dialyzed previously against water and adjusted to pH 7.0 and pH 4.0 with a final protein concentration of 2 mg/ml. Experiments were done under a nitrogen pressure of 1.7 atm to avoid sample evaporation at high temperatures. Scanning speed was set at 1.5 K/min (25 mK/s). Three consecutive thermograms were registered for each sample. The first one informs about the heat released or taken by the protein on temperature increase. After cooling down to room temperature, second and a third thermograms were run to check the reversibility of the transitions. Two corrections were applied to the first thermogram: a), subtraction of the second thermogram that acts as a blank; and b), subtraction of the chemical baseline using the method of Takahashi and Sturtevant (31). Tmax was defined as the temperature where the Cp value is maximal. The temperature corresponding to a 50% of the molecules in each the denatured and native state defines the Tm value (see Table 2 and Fig. 4). DSC thermograms were analyzed by the Sánchez-Ruiz (22) method represented by the following equation:
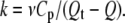 |
(1) |
k is a first-order kinetic constant, ν is the heating rate, and Qt and Q are the total heat of the process and the heat at a certain temperature respectively. The effective activation energy as a function of temperature was obtained from the relation (32):
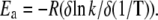 |
(2) |
TABLE 2.
Experimental thermodynamic parameters of the irreversible denaturing transition analyzed by DSC and UV-vis spectroscopy
| WT | P50A | P91A | P186A | ||
|---|---|---|---|---|---|
| DSC | |||||
| pH 7.0 | Tm (K)* | 370.6 | 369.3 | 367.9 | 372.4 |
| Ea (kcal/mol)† | 95.15 | 105.8 | 76.4‡ | 62.6 | |
| UV-Vis | |||||
| pH 7.0 | Tm (K)§ | 367.7 | 367.6 | 369.7 | 363.4 |
| Ea (kcal/mol)¶ | 115.8 | 121.2 | 60.1 | 76.8 |
Tm value obtained from the mid-point of the sigmoidal associated to the DSC data shown in Fig. 3.
Activation energy corresponding to the Tm of the DSC thermal transition, plotted in Fig. 3 c.
Activation energy temperature dependent behavior.
Tm value obtained from the midpoint of the sigmoidal associated to the UV-vis data shown in Fig. 4.
FIGURE 4.

Denaturation of the bR active site monitored by UV-vis spectroscopy. The upper panel of each indicated protein (WT or mutant) corresponds to the original spectra showing the absorbance changes as a function of wavelength and temperature. The lower panel for each indicated protein (WT or mutant) depicts the plot of the minus second derivative of the absorbance versus the wavelength at the measured temperatures. In the second derivative, irrelevant and undesirable baseline features are strongly removed, helping the visualization of the relevant spectral information. Moreover, the narrowing of the bands, allowed us to distinguish between spectral shift and band broadening, facilitating the identification of the initial state (thick black spectra), the intermediate state (thick light gray spectra), and the final state (gray spectra). The arrows in the plot indicate the direction of the main band in the thermal reaction.
Ultraviolet-visible thermal stability
Thermal stability of dark-adapted PM samples by ultraviolet-visible (UV-vis) spectroscopy was studied in H2O (0.75 × 10−5 M) at pH 7.0 and 4.0. Spectra from 800 nm to 250 nm were recorded from 293 to 393 K (20 to 100°C) in 5 K steps, in a Cary Bio3 spectrophotometer. Samples were allowed to equilibrate for 8 min at each temperature, with a 1 K/min heating rate between equilibrations, corresponding to an apparent linear heating rate of ∼0.38 K/min (6.3 mK/s). The spectra presented a baseline contribution, especially intense for some mutants, as well as a baseline drift to respect the temperature. Both effects had a negative effect in posterior analysis. As a consequence, to minimize/remove undesirable baseline contributions, the original data were preprocessed by carrying out the second derivative as a function of the wavelength. Besides removing baseline contributions, the second derivative has also the benefit of reducing the width of the UV-vis bands, although at the cost of introducing some negative side-lobules. The derivative was carried out using the generalized Fourier derivative method (33), with a derivative power of 2, a 0.05 nm−1 cut point (equivalent to an instrumental resolution of ∼20 nm), and a Sinc filter, in a homemade program implemented in MATLAB (Natick, MA) (34). The second derivative spectra were used instead of the original data in all the posterior analysis (unless otherwise stated).
UV-vis provides a thermal trace at each wavelength. Independently of its nature (reversible or irreversible) a thermal transition shows a sigmoidal-like shape. The first derivative of a spectral thermal trace to respect the temperature, also known as TDS, transforms the sigmoidal-shape into a band-shape. After this transformation, the band positions correspond to the Tm of the transitions, and the bandwidths are related to their cooperativity. Thermal denaturation of bacteriorhodopsin manifests in the UV-vis as the disappearance of the initial absorbance band at around 565 nm (actual position depends on the mutant and pH) and the appearance of a band at ∼385 nm, characteristic of a retinal in solution with a deprotonated SB. Because other pretransitions were observed at the 450–650 nm region (see 2D-TDS, Fig. 5), the analysis of the thermal trace at 565 nm does not report uniquely the retinal release from the protein. Instead, only the thermal trace at around 385 nm was analyzed to obtain the kinetic parameters associated to the release and deprotonation of the retinal. The thermal traces at 385 nm were fitted to a two-state irreversible model accounting for a linear dependence for the initial and final states:
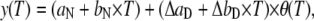 |
(3) |
where aN, bN, aD, and bD give the contribution of the native and denatured states with temperature, and θ (T) is the fraction of the denatured state with time. Assuming that the thermal denaturation follows an Arrhenius behavior, the fraction of the denatured state is given by (22):
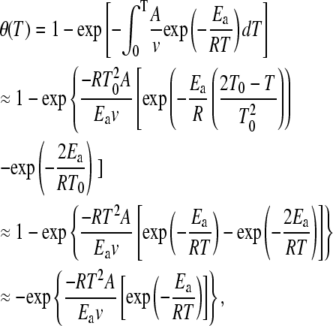 |
(4) |
where A is the temperature independent preexponential factor, Ea is the temperature independent activation energy, and v is the linear heating rate. The first approximation in Eq. 2 solves the integral by approximating 1/T by (2T0 − T)/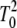 (22); the second approximation substitutes T0 by T; the final approximation neglects the lower component in the exponential (22). We confirmed the suitability of the approximations by comparing the denatured fractions generated by the last approximated equation with those obtained directly solving the integral equation numerically. Curve-fitting was carried out in Origin v6, using the Levenberg-Marquardt algorithm with a Tolerance of 0 and a Derivative Delta of 5 × 10−6. Once A and Ea were estimated, the fraction of the denatured state, and the corresponding Tm, was computed for a heating rate of 1.5 K/min, allowing for a mindful comparison of the DSC and UV-vis results.
(22); the second approximation substitutes T0 by T; the final approximation neglects the lower component in the exponential (22). We confirmed the suitability of the approximations by comparing the denatured fractions generated by the last approximated equation with those obtained directly solving the integral equation numerically. Curve-fitting was carried out in Origin v6, using the Levenberg-Marquardt algorithm with a Tolerance of 0 and a Derivative Delta of 5 × 10−6. Once A and Ea were estimated, the fraction of the denatured state, and the corresponding Tm, was computed for a heating rate of 1.5 K/min, allowing for a mindful comparison of the DSC and UV-vis results.
FIGURE 5.

2D-TDS contour plot. The first derivative of the absorbance as a function of temperature for the different bR variants is shown as a 2D-TDS contour plot. The process was carried out in the second derivative data (Fig. 4, lower panel) for a better accuracy/resolution. The transitions are represented by positive (onset) and negative values (disappearance). At the bottom of each contour plot there is an absolute value representation summarizing the transitions occurring in the denaturation process. The dotted lines indicate the position for the different transitions.
RESULTS
Sequence detailed structural stability: FoldX analysis
Protein folding has become an indispensable discipline to understand both protein structure and function. Experimental data has been used to setup reliable algorithms and force fields. One of the most interesting ones is the FoldX force field (29). Based mainly in a wide database of point mutation crystal structures, this method determines the structural stability of a protein from their crystallographic coordinates. In addition, it computes the individual amino acid contribution to the overall stability of the protein. In our case, we have applied FoldX to the corresponding crystal structures of bR, to examine the effect of the point mutation of the helix-embedded prolines (Table 1). In WT (PDB code 1py6), the three helix-embedded prolines have different but stabilizing contributions to the overall structural stability of the protein (ΔG = −95.10 kcal/mol). As indicated in Table 1, Pro186 is the most stabilizing with a ΔG = −2.50 kcal/mol, followed by Pro91 (−2.14 kcal/mol) and Pro50 (−1.86 kcal/mol). The crystal structures of the Pro-to-Ala mutants show an overall free energy (ΔG) slightly lower (ranging from a ΔΔG of ∼11 kcal/mol for P186A and P50A to 6 kcal/mol for P91A) than WT (−95.10 kcal/mol) as shown in Table 1.
TABLE 1.
Structural stability values obtained from FoldX analysis at 298 K
| PDB code* | Protein | Resolution (Å) | ΔG (kcal/mol) | ΔΔGmut-WT (kcal/mol) | ΔGPro-50 (kcal/mol) | ΔGPro-91 (kcal/mol) | ΔGPro-186 (kcal/mol) |
|---|---|---|---|---|---|---|---|
| 1py6 | bR WT | 1.80 | −95.10 | – | −1.86 | −2.14 | −2.50 |
| 1pxr | bR P50A | 1.70 | −84.15 | 10.95 | −1.10† | −1.34 | −2.50 |
| 1q5j | bR P91A | 2.10 | −89.10 | 6.00 | −1.82 | −0.02† | −2.50 |
| 1q5i | bR P186A | 2.30 | −83.80 | 11.30 | −1.80 | −2.10 | −0.82† |
ΔG values shown correspond to the average of the analysis of FoldX applied separately to chain A and B of the respective structure.
The residue indicated in the respective structure corresponds to Ala.
The local/distant effects of the mutation on the overall structure of the protein are shown in Fig. 2. To make a direct comparison between the mutation and the native protein the residual stability of the mutant has been subtracted to the WT. We have considered the absolute value of ±0.46 kcal/mol (23) as a threshold to determine a significant stabilization (− value) or destabilization (+ value) in the energy assigned to each amino acid.
FIGURE 2.

FoldX residual stability of helix-embedded prolines. Calculated residual stabilities from the available crystal structures (average of the chains A and B) for WT (PDB code 1py6), P50A (PDB code 1pxr), P91A (PDB code 1q5j), and P186A (PDB code 1q5i). To determine the changes in the WT bR stability, each residual stability value of the mutation was subtracted to the corresponding value in the structure of the native protein. Values beyond ±0.46 kcal/mol23 indicate significant stabilization (negative values) or destabilization (positive values) induced by the mutation. A linearization of the 3D structure is shown on the top of the figure for a clearer visualization. For the overall structure stabilities calculated with FoldX for the different mutations, refer to Table 1.
The contribution of each amino acid to the overall difference in structural stability for P50A is shown in Fig. 2, upper panel. As a summary, the evident difference to WT comes from Ala50 as a destabilizing amino acid. In addition, some amino acids related to the stabilization of the RBP are affected, such as Tyr83. The stability of the cytoplasmic loops between helices is also affected (Asp104 in C-D loop and Arg164 in E-F loop are stabilized residues and Asp102 in C-D loop and Pro165 in E-F loop are destabilized). Concerning the proton transport function, a striking destabilizing effect induced by P50A mutation is observed on relevant amino acids such as Asp96 (see Fig. 1 d for further detail), Glu194, and Glu204 (35). Pro91 is also affected by the Ala50 mutation.
For P91A (Fig. 2, middle panel), the main change in the structural stability of the protein comes directly from the Pro-to-Ala substitution. Taking into account the relevance of this residue in bR intraprotein signal transduction (1), the ∼2 kcal/mol destabilizing effect originated from this residue alone should not be considered trivial. Other structurally destabilized residues are Asp96 and Glu194, related closely to the proton transport function.
The P186A mutation stabilizes Tyr185, although Ala186 is a destabilizing residue for the overall structural stability, being both residues (Tyr185-Pro186 in WT bR) constituents of the RBP (see Figs. 1 e and 2, lower panel, for further details). The loops between helices E-F (cytoplasmic, destabilized Phe156) and F-G (extracellular, stabilized Ser193 and Gly195; destabilized Glu194) are affected by this mutation. Concerning the proton transport function, the mutation exerts a negative effect on Thr46 and Asp96 stability. These two residues are normally interacting by a hydrogen bond relevant for the bR function (36). Glu194 is also affected by this mutation (see Fig. 1 e for a WT vs. P186A structural comparison). The effect exerted over the residues involved in the proton transport function is translated in some way in a less efficient proton pumping of these mutants (16).
Membrane arrangement and protein denaturation: DSC analysis
Differential scanning calorimetry is a suitable technique to determine the thermodynamic and/or dynamic stability of proteins (21,22,37). Protein denaturation is achieved by gradual heating of the sample at a constant pressure, while recording the heat capacity of the sample with respect to a reference. This results in a thermogram (Fig. 3 a) with peaks reflecting any thermal transition/denaturation process taking place in the protein with a measurable associated enthalpy change. As shown in Fig. 3, the denaturation of WT bR at pH 7.0 showed a two-peak curve; the one occurring at lower temperature (Tmax = 351 K) corresponded to the loss of arrangement of the paracrystalline structure of the PM, i.e., the disappearance of the typical 2D arrangement of hexamers and trimers disposed in the PM (Fig. 1, a and b) (18,38,39). This process was reversible (40,41). The second transition (Tmax = 371 K) corresponded to the denaturation of the protein (irreversible process), with an Ea of 95.15 kcal/mol (see Fig. 3 b for Arrhenius plot and Table 2 for values). The thermal denaturation of bR (and any membrane protein in general) is not a mere unfolding process as in many soluble proteins; the denaturation leads to a re-folding process in which the protein adopts a functionally inactive new fold. This denatured folding is characterized by the loss of interhelix interactions, and the aggregation of cytoplasmic loops, but a high degree of secondary structure is conserved (18,42).
FIGURE 3.

(a) DSC thermograms of bR. DSC experimental values are shown as open circles. The gray line shows the trend of the thermogram. The pretransition corresponding to the loss of the paracrystalline arrangement of the PMs is indicated by an asterisk. The arrows indicate the selected temperature range for the Arrhenius plot. (b) Arrhenius plot for WT and the Pro-to-Ala mutants. Notice the convex Arrhenius plot for P91A. (c) Plot of the Ea versus temperature, showing a temperature dependent behavior for P91A.
How the introduction of a single mutation affected the thermodynamics and dynamics of the PM and the protein is shown in Fig. 3 a. The proline substitution by alanine, in the middle of an α-helix of a transmembrane protein such as bR, had different effects.
At pH 7.0, in the case of the P50A mutation, the reversible transition corresponding to the paracrystalline arrangement was not present, which can be explained by the absent cooperativity of this transition induced by this mutation (Fig. 3 a). The main irreversible transition Tmax of this mutant was barely affected compared to WT (370 K), but it was different in the shape, narrower, and more intense, indicating higher cooperativity.
P91A presented a decreased Tmax of the main irreversible transition (367.6 K) and the transition was narrower but less intense than WT. The paracrystalline arrangement of this mutant was not affected (Tmax = 352 K and similar area than WT). The main transition associated with the protein denaturation depicted a very unusual convex Arrhenius plot (Fig. 3 b) (32), which can be phenomenologically interpreted as a decrease of the effective activation energy barrier for the thermal denaturation as the temperature increases (Fig. 3 c). The activation energy nearly reached zero at 371 K, and so above this temperature the rate of denaturation is no longer limited by a thermal activated barrier. The rate constant of denaturation reaches a maximum rate constant slightly lower than 1 min−1, i.e., the protein can not diffuse from the native to the denatured state faster than 1 min at any temperature. Note for comparison that the maximal rate constant of diffusion along the reactive coordinate for the folding of small proteins is considered to be in the order of 106 s−1 (43,44). We suggest that this unusual low diffusion time constant along the denaturation coordinate may allow for a better characterization of downhill relaxation processes in a system as complex as a membrane protein, for instance, by recording a time-resolved denaturation experiment at a fixed temperature above 371 K.
P186A (Fig. 3 a) showed a Tmax similar to WT (372 K), but it also showed the least cooperative (broader) transition reflected in an Ea of 66.5 kcal/mol, almost 30 kcal/mol less than WT bR (Table 2). The temperature of the transition for the paracrystalline arrangement transition was slightly lower (Tmax = 347 K) and the cooperativity of this reaction was significantly lower.
Active site denaturation: UV-vis and TDS spectroscopy
Spectral changes in the visible absorption band of the chromoprotein on temperature increase were monitored by UV-vis spectroscopy (for original spectra and minus second derivative spectra of WT and proline mutants see Fig. 4). For WT, the absorbance of the retinal under native conditions was characterized by an unusual high wavelength, caused both from the protonation of the retinal SB and the interactions of the retinal with the RBP (Fig. 4, black lines). On thermal denaturation, the SB was hydrolyzed and the retinal was released from the protein to the solution, with a characteristic retinal absorbance at 384 nm. The proline mutants, especially P91A, showed under native conditions a small peak at 415 nm (in the minus second derivative plot) suggesting that in these mutants a small but significant fraction of the retinal SB could be deprotonated. For the P91A mutant the absorbance of the retinal after thermal denaturation was 392 nm and not 384 nm like in WT and the other mutants. This suggested that for P91A, the retinal was not fully released to the solution on thermal denaturation, and it still interacted with the protein.
For the analysis of the temperature dependence of the experimental data we applied the TDS method to all the thermal traces of the second derivative spectra. This transformation generates a 2D plot (2D-TDS) with bands distributed in the wavelength-temperature dimensions (45,46), providing a qualitative visual but powerful way to observe transitions present in a thermal experiment (Fig. 5). To our knowledge, 2D-TDS has only been used in the study of ligand rebinding in proteins (45–47), and this represents to the best of our knowledge its first use to study thermal stability of proteins. For bR, when membrane suspensions are gradually heated, the structural dynamics of the protein varies and a set of events occur before the denaturation of the RBP.
WT showed a high temperature resistance with only one completely resolved transition at around 348 K, and another partially resolved transition above 365 K. The first transition was close to 351 K, the Tmax for the transition involving the paracrystalline arrangement of the membrane observed by DSC. For UV-vis, the onset of this initial transition was around 320 K, preceding that observed in DSC, and it overlapped with the second and main transition, also in contrast to the transition observed by DSC. The corresponding spectral changes associated to the initial transition were the decrease of absorbance at ∼570 nm, with a concomitant increase at ∼625 nm. These spectral changes were caused by a spectral broadening, restricted to the red part of the spectrum (compare the corresponding back and thick light gray lines in Fig. 4 for WT to confirm this interpretation). The lost of the trimer arrangement with temperature is likely to affect the vibrational state of the retinal, which is known to be coupled to the trimeric arrangement as observed by circular dichroism in PM (48). The transition at ∼630 nm may also be contributed by the bR blue form, coming from the release of cations and the subsequent partial protonation of Asp85 due to a change in its pKa (49). The complete release of retinal was not achieved even at 365 K showing an incomplete transition by wavelengths 680 (+), 565 (−), and 383 (+) nm. The bands at 680 nm and 565 nm originated from the lost retinal with protonated SB, and the band at 383 nm from the newly appearing retinal with deprotonated SB generated as the retinal was released from the protein during the denaturation of the RBP.
In the case of the mutants at pH 7.0, all of them achieved a complete denaturation of the active site before 373 K. For P50A (Figs. 4 and 5), there was a previous transition starting at 308 K and with a maximum intensity occurring at 322 K. This transition may originate from a distorted RBP and Asp85 environment compared to WT (26 K difference) and may be related to the absence of pretransition observed by DSC for P50A. This transition was very similar to the pretransition observed for the WT, corresponding to a spectral broadening in the red (compare back and thick light gray lines in Fig. 4). The spectral similarity of the pretransition observed by UV-vis for WT and P50A (also P186A as described below) argues for a common origin and thermal path for this pretransition, which is not related to the pretransition observed by DSC, but may still be related with the trimeric arrangement. The areas at 668, 569, and 382 nm showed a complete transition, starting at 355 K and finishing at 370 K, reflecting the deprotonation of the SB as the RBP was denatured.
For P91A (Figs. 4 and 5), both the pre- and the main transitions were overlapped. The final denaturation indicated by the retinal release, occurred at 361 K, but represented by the areas at 614 (+), 548 (−), 419 (−), and 388 (+) nm. The new band at 419 nm, absent for WT and visible for P50A (and slightly visible for P186A), represented a fraction of the retinal in the RBP with a deprotonated SB (see also the second derivative spectra in Fig. 4, which clearly resolved this band). Here, the transition occurring at 630 nm consisted in a blue spectral shift more than in a broadening of the red (Fig. 4, thick light gray line spectrum).
For P186A, the initial transition started around 305 K reaching its maximum at 325 K, corresponding to a very intense red broadening at 635 nm. The active site total denaturation is now evidenced by the intense bands at 703 (+), 630 (−), 578 (−), and 381 (+) nm bands at ∼360 K.
Protein versus active site denaturation
DSC reports both the contacts that allow the paracrystalline ordination (the pretransition) and the contacts (involving retinal or not) that maintain the native protein architecture (main transition). UV-vis experiments points mainly on the contacts affecting the retinal environment and the retinal SB protonation state. Parameters obtained by both techniques allowed us to establish some relationship between the protein and active site denaturation for the Pro-to-Ala mutants (Table 2). This relationship can be extended trying to relate the denatured fraction of both protein and active site (retinal plus RBP).
Integrating and normalizing the DSC curves for the main transition directly provided the protein denatured fraction (Fig. 6), without fitting the data to any model (i.e., the two-state model assumed in sodium dodecyl sulfate (SDS) unfolding experiments (50)). For RBP denaturation we monitored the absorbance at ∼385 nm (corresponding to the deprotonation of the retinal SB and its release to the solution), and fit it to a two-state irreversible model to obtain the fraction of the denatured state as a function of temperature. A comparison between the protein and the active site melting temperature (Tm) is possible (Table 2). The Tm given in Table 2 corresponds to the midpoint of the transition between the native and denatured state. In general, the active site denatured at lower temperatures compared to the full protein denaturation for all the proteins in this study, but for P91A (369.7 and 367.9 K for active site and protein denaturation, respectively; Fig. 6 and Table 2). Concerning the Ea barrier for P91A, the comparison was not possible because the Ea by DSC showed a temperature dependent behavior. In particular, P50A showed a barely affected Tm for both denaturations compared to WT (≈370 K for protein and ≈368 K for active site), nevertheless it presented a higher Ea barrier for both denaturations (105.8 and 121.2 kcal/mol for protein and active site denaturation respectively, compared to 95.15 and 115.8 kcal/mol for WT; Table 2). P186A showed both an affected Tm and Ea when compared to WT. Despite having a higher Tm for the protein denaturation, the Ea was decreased severely (372 K and ≈63 kcal/mol; Table 2). The parameters for the active site denaturation were also affected compared to WT, showing a Tm of 363 K and an Ea of ≈77 kcal/mol (Table 2).
FIGURE 6.

Temperature dependence on the fractions of denatured bR (DSC, black line) and RBP (UV-vis spectroscopy, gray line). Curves showing the denatured fraction for the different Pro to Ala substitutions compared to WT at pH 7.0 are shown.
DISCUSSION
Despite the absence of sequence homology between microbial rhodopsins and rhodopsin, the structural relationship among them is evident as seven helices transmembrane proteins. Furthermore, a hypothetical evolutive relationship between bR, rhodopsin and microbial rhodopsins from the folding energies perspective points toward a rhodopsin-like folding model (51). The typical architecture determines a series of intermolecular (lipid-protein, protein-protein) and intramolecular interactions (protein-ligand (retinal), helix-helix) making bR a unique protein to model transmembrane proteins. From the thermodynamics point of view, the challenge appears in correlating the structure-function relationship of bR to the energetic patterns of its interactions. Calorimetry used as a denaturation tool has given us the chance to analyze and correlate the energy landscapes (experimental data shown in this study) with the computational analysis of the structure (crystal structures) (50,52) and finally with the function (experimental data shown in Perálvarez-Marín et al. (16)).
Considering Pro50, located in the middle of helix B, we have observed that the introduction of the mutation severely affects the lipid-protein and protein-protein interactions responsible of maintaining the proper paracrystalline arrangement (shown by the lack of pretransition observed by DSC, Fig. 3 a). It is worthy to note the disposition of helix B (Fig. 1 b) to understand that helix B participates directly with helices D and E in the protein-protein interactions stabilizing the trimeric arrangement. The overall protein and the active site structural stability are slightly increased in P50A with respect to WT (Table 2), but this does not favor a better function of the protein (30% decreased pumping function (16)) despite a more rigid structure (see Fig. 1 d for local structural details).
The mutation of Pro91 promotes more localized effects. There is no effect on the paracrystalline arrangement (i.e., intermolecular interactions), but the active site and the protein are severely affected. P91A mutation slows down the protein dynamics making the protein conformational diffusion along the denaturation coordinate slow enough to be no longer barrier-limited; i.e., the transition between the native and the denatured conformation becomes an “exotic” downhill process, which may be related to diffusion dependent phenomena affecting the folding kinetics, kinetic routes, and the thermodynamic free-energy barrier (53). Thus, the observed altered structural dynamics relates with the altered function (65% decreased pumping function (16)) and gives a clear idea why Pro91 and its vicinity are so important in the signal transduction from the retinal to the rest of the protein (1,16,54,55). The P91A mutation disrupts the polar cluster constituted by Pro91, Thr90, and Asp115 embedded in the transmembrane helices of bR and forming part of the active site of the protein (see Fig. 1 e for further details). This is reflected in the analysis of the structural stability of the active site by the TDS spectroscopy approach, showing the most distorted behavior for the P91A mutation.
The PM arrangement becomes affected by the Pro186 substitution. The positioning of this residue in the helix F is relevant for the relationship between trimers and the lipidic component of the 2D paracrystalline arrangement (Fig. 1 a). The DSC data argue for a less compact arrangement compared to WT and the overall protein stability is strikingly decreased (activation barrier ∼30 kcal/mol below that of WT). The direct contact of Pro186 with the retinal molecule is evidenced by the effect over the function of the substitution of this residue for Ala. This also affects the structural stability of important residues related to the function (such as Glu194 and Glu204; Fig. 1 f) and the most striking effect is exerted over the retinal environment (translated into a 60% decreased proton pumping function (16)).
The crystal structures of helix embedded prolines mutated to alanine, have shown no dramatic changes in the structure (50,52). The FoldX analysis of these monomeric structures points to a destabilizing effect of the Pro-to-Ala mutation. The crystal structures were obtained under extreme conditions (pH ≤4.0), where the protein is not functional. Our results at pH 4.0 did not show significant differences between the WT and the mutant proteins, either for the paracrystalline arrangement, the overall protein stability, or the active site (supporting information). However, the information contained in crystal structures is extremely valuable because the best energy-structure-function correlations are obtained when high resolution x-ray studies are available (56). The experimental conditions for these crystal structures are similar enough to allow direct comparison between the native and mutated proteins. SDS unfolding experiments of monomeric bR for these proline mutants showed contribution of Pro91 and Pro186 to the structural stability of the protein, but not for Pro50 (50,52). Previous work on SDS protein folding kinetics of these same mutants on Escherichia coli membranes, indicated that formation of a seven-transmembrane helical intermediate state in the absence of retinal was slower for P50A and P91A compared to WT, however, at final stages of folding, when retinal was incorporated, all three Pro residues contributed to retinal binding and final folding of bR (57). A recent study by single-molecule force spectroscopy on the same Pro-to-Ala mutations has focused on the point mutation effects on the unfolding and structural stability of bR in response to mechanical pulling (58).
The achievement of this study consists in the dissection of the structural stability of bR from different perspectives. The computational analysis of crystal structures with FoldX cross-linked with experimental data has allowed us to relate detailed atomic information of the crystal structures to the active site, protein and PM structural stability.
Facing the study of protein stability from the overall protein and active site point of view makes it easier to understand the relevance of certain residues in determined positions. In addition, we have observed how a single mutation of a Pro residue is capable of reducing the protein conformational diffusion enough to transform the thermal denaturation into a barrier-free transition above 371 K. This reduced dynamics is likely to affect also the folding/unfolding of bR. It is also possible that similar “frozen” dynamics can be induced by Pro mutations in other membrane proteins.
Our study suggests that the overrepresentation of prolines included in transmembrane helices, where proline is a well known α-helix breaking amino acid (actually imino acid), comes from the optimal energy/structure/dynamics/function relationship of this amino acid. Its presence in transmembrane helices is important not only for signal transduction as happens in bR or GPCR (1,16,59–61), but also for other transmembrane proteins such as transporters (62), carriers (63), bacterial membrane proteins (64), and viral glycoproteins (65,66) among others.
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Fig. S1 in Supplementary Material, Data S1 shows the fitting of the UV-Vis experimental data to Eqs. 3 and 4 (Methods) to get the values shown in Table 2. Fig. S2 in Data S1 shows denatured fractions obtained by DSC and UV-Vis spectroscopy for the samples at pH 4.0. Table S1 in Data S1 lists Tm and Ea values obtained for the WT and mutants at pH 4.0 by DSC and UV-Vis spectroscopy.
Acknowledgments
The authors thank Dr. Michael Bennett for valuable suggestions in the DSC analysis and Dr. Sebastian Wärmländer and Prof. Gunnar von Heijne for critical reading of the manuscript. The authors also thank Itxaso Apraiz for valuable help in the preparation of Figure 1 and Elodia Serrano and Alex Fernández for skillful technical assistance.
This work was supported by the Catalan Government “Beatriu de Pinos” Postdoctoral Fellowship (2005 BP-A 10085 to A.P.-M.). E.P. acknowledges financial support from the Dirección General de Investigación (MEC) grant BFU2006-04656BMC.
Abbreviations used: Cp, apparent heat capacity; bR, bacteriorhodopsin; DSC, differential scanning calorimetry; GPCR, G-protein coupled receptors; PM, purple membrane; RBP, retinal binding pocket; SB, Schiff base; TDS, temperature derivative spectroscopy.
Editor: Kathleen B. Hall.
References
- 1.Perálvarez-Marín, A., V. A. Lórenz-Fonfría, J. L. Bourdelande, E. Querol, H. Kandori, and E. Padrós. 2007. Inter-helical hydrogen bonds are essential elements for intra-protein signal transduction: the role of Asp115 in bacteriorhodopsin transport function. J. Mol. Biol. 368:666–676. [DOI] [PubMed] [Google Scholar]
- 2.Ballesteros, J., and K. Palczewski. 2001. G protein-coupled receptor drug discovery: implications from the crystal structure of rhodopsin. Curr. Opin. Drug Discov. Devel. 4:561–574. [PMC free article] [PubMed] [Google Scholar]
- 3.Maeda, T., Y. Imanishi, and K. Palczewski. 2003. Rhodopsin phosphorylation: 30 years later. Prog. Retin. Eye Res. 22:417–434. [DOI] [PubMed] [Google Scholar]
- 4.Menon, S. T., M. Han, and T. P. Sakmar. 2001. Rhodopsin: structural basis of molecular physiology. Phys. Rev. 81:1659–1688. [DOI] [PubMed] [Google Scholar]
- 5.Park, P. S., S. Filipek, J. W. Wells, and K. Palczewski. 2004. Oligomerization of G protein-coupled receptors: past, present, and future. Biochemistry. 43:15643–15656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haupts, U., J. Tittor, and D. Oesterhelt. 1999. Closing in on bacteriorhodopsin: progress in understanding the molecule. Annu. Rev. Biophys. Biomol. Struct. 28:367–399. [DOI] [PubMed] [Google Scholar]
- 7.Lanyi, J. K. 1999. Bacteriorhodopsin. Int. Rev. Cytol. 187:161–202. [DOI] [PubMed] [Google Scholar]
- 8.Radding, W., T. Romo, and G. N. Phillips, Jr. 1999. Protein-assisted pericyclic reactions: an alternate hypothesis for the action of quantal receptors. Biophys. J. 77:2920–2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shibata, M., M. Yoshitsugu, N. Mizuide, K. Ihara, and H. Kandori. 2007. Halide binding by the D212N mutant of Bacteriorhodopsin affects hydrogen bonding of water in the active site. Biochemistry. 46:7525–7535. [DOI] [PubMed] [Google Scholar]
- 10.Tittor, J., U. Haupts, C. Haupts, D. Oesterhelt, A. Becker, and E. Bamberg. 1997. Chloride and proton transport in bacteriorhodopsin mutant D85T: different modes of ion translocation in a retinal protein. J. Mol. Biol. 271:405–416. [DOI] [PubMed] [Google Scholar]
- 11.Vogel, R., T. P. Sakmar, M. Sheves, and F. Siebert. 2007. Coupling of protonation switches during rhodopsin activation. Photochem. Photobiol. 83:286–292. [DOI] [PubMed] [Google Scholar]
- 12.Sudo, Y., and J. L. Spudich. 2006. Three strategically placed hydrogen-bonding residues convert a proton pump into a sensory receptor. Proc. Natl. Acad. Sci. USA. 103:16129–16134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chang, D. K., S. F. Cheng, V. D. Trivedi, and K. L. Lin. 1999. Proline affects oligomerization of a coiled coil by inducing a kink in a long helix. J. Struct. Biol. 128:270–279. [DOI] [PubMed] [Google Scholar]
- 14.Cordes, F. S., J. N. Bright, and M. S. Sansom. 2002. Proline-induced distortions of transmembrane helices. J. Mol. Biol. 323:951–960. [DOI] [PubMed] [Google Scholar]
- 15.Tieleman, D. P., I. H. Shrivastava, M. R. Ulmschneider, and M. S. Sansom. 2001. Proline-induced hinges in transmembrane helices: possible roles in ion channel gating. Proteins. 44:63–72. [DOI] [PubMed] [Google Scholar]
- 16.Perálvarez-Marín, A., J. L. Bourdelande, E. Querol, and E. Padrós. 2006. The role of proline residues in the dynamics of transmembrane helices: the case of bacteriorhodopsin. Mol. Membr. Biol. 23:127–135. [DOI] [PubMed] [Google Scholar]
- 17.Brouillette, C. G., D. D. Muccio, and T. K. Finney. 1987. pH dependence of bacteriorhodopsin thermal unfolding. Biochemistry. 26:7431–7438. [DOI] [PubMed] [Google Scholar]
- 18.Cladera, J., M. L. Galisteo, M. Sabés, P. L. Mateo, and E. Padrós. 1992. The role of retinal in the thermal stability of the purple membrane. Eur. J. Biochem. 207:581–585. [DOI] [PubMed] [Google Scholar]
- 19.Shnyrov, V. L., and A. L. Berman. 1988. Calorimetric study of thermal denaturation of vertebrate visual pigments. Biomed. Biochim. Acta. 47:355–362. [PubMed] [Google Scholar]
- 20.Freire, E. 1995. Thermal denaturation methods in the study of protein folding. Methods Enzymol. 259:144–168. [DOI] [PubMed] [Google Scholar]
- 21.Sánchez-Ruiz, J. M. 1995. Differential scanning calorimetry of proteins. Subcell. Biochem. 24:133–176. [DOI] [PubMed] [Google Scholar]
- 22.Sánchez-Ruiz, J. M., J. L. López-Lacomba, M. Cortijo, and P. L. Mateo. 1988. Differential scanning calorimetry of the irreversible thermal denaturation of thermolysin. Biochemistry. 27:1648–1652. [DOI] [PubMed] [Google Scholar]
- 23.Guerois, R., J. E. Nielsen, and L. Serrano. 2002. Predicting changes in the stability of proteins and protein complexes: a study of more than 1000 mutations. J. Mol. Biol. 320:369–387. [DOI] [PubMed] [Google Scholar]
- 24.Hilser, V. J., and E. Freire. 1996. Structure-based calculation of the equilibrium folding pathway of proteins. Correlation with hydrogen exchange protection factors. J. Mol. Biol. 262:756–772. [DOI] [PubMed] [Google Scholar]
- 25.Rader, A. J., B. M. Hespenheide, L. A. Kuhn, and M. F. Thorpe. 2002. Protein unfolding: rigidity lost. Proc. Natl. Acad. Sci. USA. 99:3540–3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fang, Q., and D. Shortle. 2006. Protein refolding in silico with atom-based statistical potentials and conformational search using a simple genetic algorithm. J. Mol. Biol. 359:1456–1467. [DOI] [PubMed] [Google Scholar]
- 27.Isogai, Y., M. Ota, A. Ishii, M. Ishida, and K. Nishikawa. 2002. Identification of amino acids involved in protein structural uniqueness: implication for de novo protein design. Protein Eng. 15:555–560. [DOI] [PubMed] [Google Scholar]
- 28.Tartaglia, G. G., A. Cavalli, and M. Vendruscolo. 2007. Prediction of local structural stabilities of proteins from their amino acid sequences. Structure. 15:139–143. [DOI] [PubMed] [Google Scholar]
- 29.Schymkowitz, J., J. Borg, F. Stricher, R. Nys, F. Rousseau, and L. Serrano. 2005. The FoldX web server: an online force field. Nucleic Acids Res. 33:W382–W388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sanz, C., T. Lazarova, F. Sepulcre, R. González-Moreno, J. L. Bourdelande, E. Querol, and E. Padrós. 1999. Opening the Schiff base moiety of bacteriorhodopsin by mutation of the four extracellular Glu side chains. FEBS Lett. 456:191–195. [DOI] [PubMed] [Google Scholar]
- 31.Takahashi, K., and J. M. Sturtevant. 1981. Thermal denaturation of streptomyces subtilisin inhibitor, subtilisin BPN′, and the inhibitor-subtilisin complex. Biochemistry. 20:6185–6190. [DOI] [PubMed] [Google Scholar]
- 32.Truhlar, D., and A. Kohen. 2001. Convex Arrhenius plots and their interpretation. Proc. Natl. Acad. Sci. USA. 98:848–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cameron, D. G., and D. J. Moffatt. 1987. A generalized approach to derivative spectroscopy. Appl. Spectrosc. 41:539–544. [Google Scholar]
- 34.Lórenz-Fonfría, V. A., and E. Padrós. 2004. Curve-fitting of Fourier manipulated spectra comprising apodization, smoothing, derivation and deconvolution. Spectrochim. Acta [A]. 60:2703–2710. [DOI] [PubMed] [Google Scholar]
- 35.Lazarova, T., C. Sanz, F. Sepulcre, E. Querol, and E. Padrós. 2002. Specific effects of chloride on the photocycle of E194Q and E204Q mutants of bacteriorhodopsin as measured by FTIR spectroscopy. Biochemistry. 41:8176–8183. [DOI] [PubMed] [Google Scholar]
- 36.Maeda, A., R. B. Gennis, S. P. Balashov, and T. G. Ebrey. 2005. Relocation of water molecules between the Schiff base and the Thr46-Asp96 region during light-driven unidirectional proton transport by bacteriorhodopsin: an FTIR study of the N intermediate. Biochemistry. 44:5960–5968. [DOI] [PubMed] [Google Scholar]
- 37.Hinz, H. J. 1986. Thermodynamic parameters for protein-protein and protein-ligand interaction by differential scanning microcalorimetry. Methods Enzymol. 130:59–79. [DOI] [PubMed] [Google Scholar]
- 38.Shnyrov, V. L., A. I. Azuaga, and P. L. Mateo. 1994. The nature of thermal transitions in purple membranes from Halobacterium halobium. Biochem. Soc. Trans. 22:367S. [DOI] [PubMed] [Google Scholar]
- 39.Shnyrov, V. L., and P. L. Mateo. 1993. Thermal transitions in the purple membrane from Halobacterium halobium. FEBS Lett. 324:237–240. [DOI] [PubMed] [Google Scholar]
- 40.Jackson, M. B., and J. M. Sturtevant. 1978. Phase transitions of the purple membranes of Halobacterium halobium. Biochemistry. 17:911–915. [DOI] [PubMed] [Google Scholar]
- 41.Taneva, S. G., R. Koynova, and B. Tenchov. 1994. Thermal stability of lipid-depleted purple membranes at neutral and low pH values. FEBS Lett. 345:154–158. [DOI] [PubMed] [Google Scholar]
- 42.Azuaga, A. I., F. Sepulcre, E. Padrós, and P. L. Mateo. 1996. Scanning calorimetry and Fourier-transform infrared studies into the thermal stability of cleaved bacteriorhodopsin systems. Biochemistry. 35:16328–16335. [DOI] [PubMed] [Google Scholar]
- 43.Naganathan, A. N., U. Doshi, A. Fung, M. Sadqi, and V. Muñoz. 2006. Dynamics, energetics, and structure in protein folding. Biochemistry. 45:8466–8475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang, W. Y., and M. Gruebele. 2003. Folding at the speed limit. Nature. 423:193–197. [DOI] [PubMed] [Google Scholar]
- 45.Mourant, J. R., D. P. Braunstein, K. Chu, H. Frauenfelder, G. U. Nienhaus, P. Ormos, and R. D. Young. 1993. Ligand binding to heme proteins: II. Transitions in the heme pocket of myoglobin. Biophys. J. 65:1496–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Berendzen, J., and D. Braunstein. 1990. Temperature-derivative spectroscopy: a tool for protein dynamics. Proc. Natl. Acad. Sci. USA. 87:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nienhaus, K., P. Deng, J. S. Olson, J. J. Warren, and G. U. Nienhaus. 2003. Structural dynamics of myoglobin: ligand migration and binding in valine 68 mutants. J. Biol. Chem. 278:42532–42544. [DOI] [PubMed] [Google Scholar]
- 48.Mukhopadhyay, A. K., S. Bose, and R. W. Hendler. 1994. Membrane-mediated control of the bacteriorhodopsin photocycle. Biochemistry. 33:10889–10895. [DOI] [PubMed] [Google Scholar]
- 49.Sanz, C., M. Márquez, A. Perálvarez, S. Elouatik, F. Sepulcre, E. Querol, T. Lazarova, and E. Padrós. 2001. Contribution of extracellular Glu residues to the structure and function of bacteriorhodopsin. Presence of specific cation-binding sites. J. Biol. Chem. 276:40788–40794. [DOI] [PubMed] [Google Scholar]
- 50.Yohannan, S., S. Faham, D. Yang, J. P. Whitelegge, and J. U. Bowie. 2004. The evolution of transmembrane helix kinks and the structural diversity of G protein-coupled receptors. Proc. Natl. Acad. Sci. USA. 101:959–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tastan, O., E. Yu, M. Ganapathiraju, A. Aref, A. J. Rader, and J. Klein-Seetharaman. 2007. Comparison of stability predictions and simulated unfolding of rhodopsin structures. Photochem. Photobiol. 83:351–362. [DOI] [PubMed] [Google Scholar]
- 52.Faham, S., D. Yang, E. Bare, S. Yohannan, J. P. Whitelegge, and J. U. Bowie. 2004. Side-chain contributions to membrane protein structure and stability. J. Mol. Biol. 335:297–305. [DOI] [PubMed] [Google Scholar]
- 53.Chahine, J., R. J. Oliveira, V. B. Leite, and J. Wang. 2007. Configuration-dependent diffusion can shift the kinetic transition state and barrier height of protein folding. Proc. Natl. Acad. Sci. USA. 104:14646–14651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Perálvarez-Marín, A., M. Márquez, J. L. Bourdelande, E. Querol, and E. Padrós. 2004. Thr-90 plays a vital role in the structure and function of bacteriorhodopsin. J. Biol. Chem. 279:16403–16409. [DOI] [PubMed] [Google Scholar]
- 55.Perálvarez, A., R. Barnadas, M. Sabés, E. Querol, and E. Padrós. 2001. Thr90 is a key residue of the bacteriorhodopsin proton pumping mechanism. FEBS Lett. 508:399–402. [DOI] [PubMed] [Google Scholar]
- 56.Hinz, H. J. 1983. Thermodynamics of protein-ligand interactions: calorimetric approaches. Annu. Rev. Biophys. Bioeng. 12:285–317. [DOI] [PubMed] [Google Scholar]
- 57.Lu, H., T. Marti, and P. J. Booth. 2001. Proline residues in transmembrane alpha helices affect the folding of bacteriorhodopsin. J. Mol. Biol. 308:437–446. [DOI] [PubMed] [Google Scholar]
- 58.Tanuj Sapra, K., G. Prakash Balasubramanian, D. Labbude, J. U. Bowie, and D. J. Muller. 2008. Point mutations in membrane proteins reshape energy landscape and populate different unfolding pathways. J. Mol. Biol. 376:1076–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gether, U. 2000. Uncovering molecular mechanisms involved in activation of G protein-coupled receptors. Endocr. Rev. 21:90–113. [DOI] [PubMed] [Google Scholar]
- 60.Sansom, M. S., and H. Weinstein. 2000. Hinges, swivels and switches: the role of prolines in signaling via transmembrane alpha-helices. Trends Pharmacol. Sci. 21:445–451. [DOI] [PubMed] [Google Scholar]
- 61.Stitham, J., K. A. Martin, and J. Hwa. 2002. The critical role of transmembrane prolines in human prostacyclin receptor activation. Mol. Pharmacol. 61:1202–1210. [DOI] [PubMed] [Google Scholar]
- 62.Joshi, A. D., and A. M. Pajor. 2006. Role of conserved prolines in the structure and function of the Na+/dicarboxylate cotransporter 1, NaDC1. Biochemistry. 45:4231–4239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Orzáez, M., J. Salgado, A. Giménez-Giner, E. Pérez-Payá, and I. Mingarro. 2004. Influence of proline residues in transmembrane helix packing. J. Mol. Biol. 335:631–640. [DOI] [PubMed] [Google Scholar]
- 64.Goemaere, E. L., E. Cascales, and R. Lloubes. 2007. Mutational analyses define helix organization and key residues of a bacterial membrane energy-transducing complex. J. Mol. Biol. 366:1424–1436. [DOI] [PubMed] [Google Scholar]
- 65.Gomara, M. J., P. Mora, I. Mingarro, and J. L. Nieva. 2004. Roles of a conserved proline in the internal fusion peptide of Ebola glycoprotein. FEBS Lett. 569:261–266. [DOI] [PubMed] [Google Scholar]
- 66.Will, C., E. Muhlberger, D. Linder, W. Slenczka, H. D. Klenk, and H. Feldmann. 1993. Marburg virus gene 4 encodes the virion membrane protein, a type I transmembrane glycoprotein. J. Virol. 67:1203–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]