

Abstract
Highly efficient solid-phase synthesis of 2-substituted-3-hydroxy-4(1H)-quinolinone-7-carboxamides was developed using anthranilates and bromoketones as the key synthons. Primary amines immobilized to acid-cleavable BAL linker were acylated with 1-methyl-2-aminoterephtalate. Following cleavage of the methyl ester, bromoketones were used to form resin-bound phenacyl esters. Acid-mediated cleavage and subsequent cyclization in solution afforded 3-hydroxy-4(1H)-quinolinones in high purity and yield. Highly efficient solid-phase synthesis (purity >90%, yield >80%, synthetic time 2 days using commercially available synthons) is amenable to high throughput/combinatorial synthesis to match the high throughput screening capability.
Keywords: hydroxyquinolinones, quinolonones, solid-phase synthesis, highly efficient solid-phase organic synthesis, anticancer activity
Hydroxycoumarine skeleton of flavonols, a group of natural compounds with wide range of different biological activities, has become a focus of intense studies aimed at preparation of novel compounds with pharmacologically relevant properties.1 We became interested in the less commonly studied 3-hydroxy-4(1H)-quinolinones2, the aza-analogues of hydroxycoumarines. Hradil and Jirman reported the first synthetic route to 2-phenyl-3-hydroxy-4(1H)-quinolinones by heating phenacylanthranilates in polyphosphoric acid.3 Phenacylanthranilates are accessible from commercially available anthranilic acids and bromoketones, making this synthetic route amenable to preparation of a number of different derivatives. Later, they extended the synthesis to 3-amino-4(1H)-quinolinones4 and in 2006 reported the synthesis of 2-phenylsubstituted-3-hydroxy-4(1H)-quinolinone-7-carboxylic acids and their phenacyl esters in solution.5
Biological screening of hydroxyquinolinones revealed anticancer activity of chloro derivatives of 2-phenyl-3-hydroxy-4(1H)-quinolinones6 as well as anticancer activity of some 2-phenylsubstituted-3-hydroxy-4(1H)-quinolinone-7-carboxylic acids phenacylesters.5 In addition, inhibition of DNA gyrase and topoisomearse II by hydroxyquinolinones has been reported by Sui et al.7 At present, the mode of action of hydroxyquinolinones in cellular systems is not understood and thus makes it a subject of study.
Promising pharmacological properties of 3-hydroxy-4-quinolinones (cytotoxicity to cell lines A549, K562, K562-Tax, CEM, CEM-DNRB) prompted us to carry out a detailed study of the structure-activity relationship. In order to effectively address the SAR we developed a highly efficient solid-phase synthesis amenable to high throughput/combinatorial synthesis of hydroxyquinolinone analogs. An inherent simplicity of isolation of intermediates and target compounds synthesized by solid-phase was combined with a highly efficient sequence of transformations that yield target products in high purity (> 90%) and high yield (> 80%).
Results and discussion
Solid-phase synthesis of quinolinones followed the synthetic route developed for solution-phase synthesis with anthranilic acids and bromoketones being the key synthones. Combination of those two building blocks allowed preparation of quinolinones with two diversity positions: one on the carbocyclic benzene ring originating from anthranilates and the second one on position 2 of the quinolinones from bromoketones. For the solid-phase synthesis we decided to immobilize the anthranilate component.
Polyphosphoric acid, used for the cyclization of phenacyl esters to hydroxyquinolinones in solution-phase syntheses,3;5;6;8;9 is not well-suited for high throughput synthesis due to difficulty in isolation of products. We evaluated different cyclization conditions and found that cyclization of acyclic precursors of target compounds can be accomplished by heating in trifluoroacetic acid (TFA), which would enable a simplified and straight forward isolation of products.
Since the cyclization of linear intermediate phenacylanthranilates to quinolinones could be advantageously carried out by heating in TFA, a reagent commonly used to release target compounds from acid-labile linkers, we synthesized the phenacylanthranilates on an acid-cleavable linker and carried out the cyclization in a TFA solution after releasing the acyclic intermediates from the resin by the same reagent.
To evaluate the scope and limitations of the solution-phase synthesis on solid phase, we prepared a model compound, the nonsubstituted amide (5), using Rink amide resin10 (Scheme 1). Briefly, Rink amide resin was acylated by commercially available 1-methyl-2-aminoterephtalate through activation by 1,3-diisopropylcarbodiimide (DIC) in the presence of 1-hydroxybenzotriaziole (HOBt). 1-Methyl-2-aminoterephtalate is a suitable building block, because it allowed immobilization via its unprotected carboxylate while the carboxylic group ortho to the amino group was esterified and did not interfere during acylation reaction. The resin-bound methylester (1) was saponified using potassium trimethylsilanolate (KOTMS) in THF11 and the resin-bound carboxylic acid (2) was treated with a solution of 2-bromo-4′-methyl-acetophenone and triethylamine (TEA) in N,N-dimethylformamide (DMF). Because the nucleophilicity of the aniline is decreased by an electron withdrawing effect of both the carboxylic group and carboxamide, an excess of bromoketone could be used without risk of any side reactions. Esterification was completed at room temperature in 2 h. The ester (4) was then cleaved from the polymer support by TFA in dichloromethane (DCM) followed by evaporation of the cleavage cocktail. Cyclization of hydroxyquinolinone (5) was then carried out by heating the cleaved ester (4) in neat TFA at 80 °C for 2 h. The cyclization was not accompanied by any noticeable side-reactions and the crude product was isolated in greater than 90% purity as judged from analytical HPLC traces. The product was characterized by MS and NMR spectra.
Scheme 1.

The preparation of nonsubstituted carboxamide (5)a
a Reagents: (i) 1-methyl-2-aminoterephtalate, DIC, HOBt, DCM, DMF, rt ,overnight; (ii) TMSOK, THF, rt, 7 h.; (iii) 2-bromo-4′-methyl-acetophenone, TEA, DMF, rt, 2 h.; (iv) 50% TFA, DCM, 30 min.; (v) TFA, 80 °C, 2 h.
In order to introduce a diversity position on the phenyl ring, we prepared N-derivatized carboxamides (11) using the resin-bound BAL linker.12 Polymer-supported secondary amines (6) were prepared using already described procedure.13 Subsequently, four chemical transformations were completed using the same procedures developed for unsubstituted amide (5) (Scheme 2).
Scheme 2.

Preparation of N-derivatized carboxamidesa
aReagents: (i) amine, 10% AcOH/DMF, overnight, NaBH(OAc)3, 5 h; (ii) 1-methyl-2-aminoterephtalate, DIC, HOBt, DCM, DMF, rt,overnight; (iii) TMSOK, THF, rt, 7 h; (iv) 2-bromo-4′-methyl-acetophenone, TEA, DMF, rt, 2 h; (v) 50% TFA, DCM, 30 min; (vi) TFA, 80 °C, 2 h.
The outcome of the reaction sequence was evaluated for a set of the six amines (Figure 1). n-Propyl (6a) and benzyl (6c) amines were included as representative examples of amines without any side-chain functional groups and the corresponding target compounds were obtained in high purity. In the case of the polymer supported ethanolamine (6b), we observed a side reaction caused by acylation of the hydroxy group by 1-methyl-2-aminoterephtalate (about 30 % of double acylated side product, HPLC). The ester of the side product was cleaved in the next step, thus it was not necessary to protect the hydroxy group before the acylation. Lastly, three amines (6d-f) were selected to introduce a heterocyclic moiety into the target molecule. All building blocks provided the expected quinolinones in high purity.
Figure 1.

Six amines for the first diversity position.
In the case of derivatives prepared from polymer-supported N-ethylaminopiperidin (6f), different conditions had to be used for cleavage from the resin. We observed only marginal cleavage under standard conditions (50% TFA in DCM, rt, 30 min). We speculate that the strongly basic tertiary amine of the piperidine moiety was protonated by TFA and the resulting quaternary ammonium ion in close proximity to the cleavage site decreased the electron density and decelerated the cleavage rate. Thus, heating in neat TFA was required for quantitative cleavage. The target hydroxyquinolinone (11f) was prepared by heating its polymer-supported intermediate in neat TFA; the purity of the product was not influenced by presence of the resin (HPLC, NMR).
A set of bromoketones was evaluated using the n-propylamide derivative (6a) and 2-substituted-3-hydroxy-4(1H)-quinolinone-7-carboxylic acid propylamides were prepared. Six phenylbromoketones were selected that contain various combinations of electron withdrawing as well as electron donating substituents. One heterocyclic bromoketone and chloroacetone were also included. Bromoketones (B), (G), (H) were obtained from commercially available sources. Bromoketones (A), (C), (D), (E), (F) were prepared by bromination from corresponding acetylderivatives,14 although it should be noted that they are also commercially available.
After finishing the solid-supported synthesis, the linear precursors were cleaved from the resin and cyclized to target quinolinones in TFA. Since the reaction sequence was high yielding and provided crude products of high purity it enabled very simple isolation of pure target compounds. After evaporation of TFA, the residual oil was sonified for five minutes in diethylether. Precipitated products were collected by suction filtration, washed with fresh diethylether and dried. This simple procedure removed minor impurities detected in crude products as they remained dissolved in diethylether during sonification. This isolation was very fast, effective and amenable to high throughput synthesis. For the isolation of derivatives (11D) and (11F), suction filtration and drying under a nitrogen atmosphere was necessary. It was observed that derivatives (11D) and (11F) smoothly turned into an oily material when the procedure describe was not followed. Typical purity of crude hydroxyquinolinones was 90 - 95% as judged from the analytical HPLC traces (integration of diode array 200-450 nm traces) with the exception of (12G). During the cyclization of (12G), partial cleavage of thiophenylmethyl group was observed and the product was contaminated by the carboxamide (5) (Scheme 3).
Scheme 3.

The cyclization of 12G
It is worth mentioning that the ammonium acetate buffer, typically used as the HPLC aqueous buffer, did not provide satisfactory separation. Peaks corresponding to target compounds were tailing, occasionally spreading over several munutes in a 10 min gradient. Interestingly, acyclic substrates provided sharp separation. Thus, for evaluation of the purity of target compounds by HPLC we changed the mobile phase and found satisfactory separation using a 0.1% aqueous TFA. Purification by preparative HPLC was not necessary because of the high purity of the isolated products (except in the case of 12G). However, if needed, purification by preparative HPLC in the described TFA containing buffer is possible.
In order to create the third diversity place in target structures, we evaluated alkylation of the amino group of intermediate (7a). The cyclization of N-methyl and N-phenyl phenacylanthranilates in polyphosphoric acid to corresponding N-substituted-hydroxyquinolinones in solution has been already described15. The alkylation of the resin-bound aniline (7a) was difficult because of a double electron withdrawing effect that reduced the nucleophlicity of the aniline nitrogen and stronger reagents were required to complete the alkylations at elevated temperature. 2-tert-Butylimino-2-diethylamino-1,3-dimethyl-perhydro-1,3,2-diazaphosphorine was found to be a suitable base for these reactions (attempts with sodium hydride or potassium carbonate failed). We observed clean alkylation with reactive electrophiles such as benzylbromide and ethyl-bromoacetate. Attempts at alkylation using either phenethylbromide or isopropyliodide were not successful. After saponification and esterification with bromoketone, N-alkylated esters (16a,c) did not cyclize to hydroxyquinolinone under the TFA conditions. Finally, the amino group was acylated using benzoylchloride in pyridine, but the benzoyl derivative (16b) did not cyclize to the corresponding hydroxyquinolinone in TFA (Scheme 4).
Scheme 4.

Attempts to prepare N-substituted derivativesa
aReagents: (i) benzylbromide, BMP, NMP, 50 °C, overnight or benzoylchloride, pyridine, rt, 7 h, or ethyl-bromoaceate, BMP, NMP, 50 °C, 48 h; (ii) TMSOK, THF, rt, 7 h; (iii) 2-bromo-4′-methyl-acetophenone, TEA, DMF, rt, 2 h; (iv) 50% TFA, DCM, 30 min.; (v) TFA, 80 °C, 2 h.
In conclusion, we have developed a highly efficient solid-phase synthesis of 2-substituted-3-hydroxy-4(1H)-quinolinone-7-carboxamides with two diversity positions. All reaction steps were completed in high purity with no significant side-reactions observed. Almost quantitative conversion of all reaction steps resulted in high yields of crude target products. Isolated yields, after simple purification by precipitation with ether, were high and the yields were influenced only by solubility of target compounds in diethylether. After optimization of all steps, the entire synthetic sequence, including isolation of products, can be accomplished in 2 days using commercially available synthons.
Supplementary Material
Figure 2.
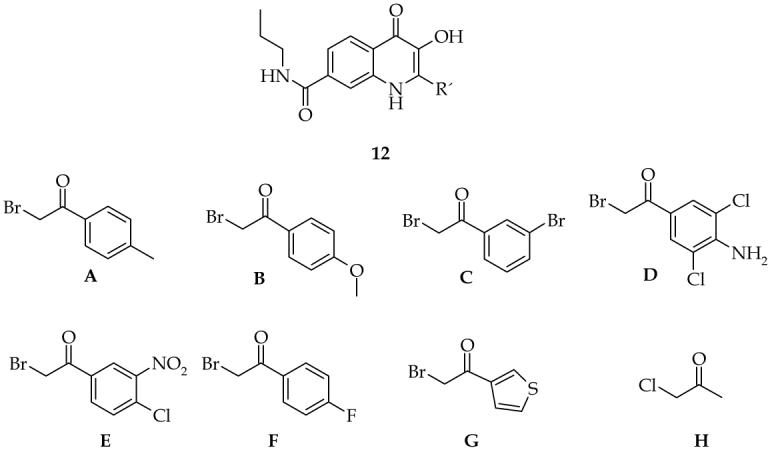
Bromoketones used for the synthesis of 2-substituted-3-hydroxy-4(1H)-quinolinone-7-carboxylic acid propylamides (12).
ACKNOWLEDGMENT
The work was supported by the Department of Chemistry and Biochemistry University of Notre Dame, the NIH (GM079576) and the Ministry of Education, Youth and Sport of the Czech Republic (MSM6198959216). The authors thank Katherine Waring for critically reading the manuscript.
REFERENCES
- 1.Brahmachari G, Gorai D. Curr. Org. Chem. 2006;10:873–898. [Google Scholar]
- 2.Kappe T. Farmaco. 1999;54:309–315. [Google Scholar]
- 3.Hradil P, Jirman J. Collect. Czech. Chem. Commun. 1995;60:1357–1366. [Google Scholar]
- 4.Hradil P, Grepl M, Hlaváč J, Soural M, Maloň M, Bertolasi V. J. Org. Chem. 2006;71:819–822. doi: 10.1021/jo051303k. [DOI] [PubMed] [Google Scholar]
- 5.Soural M, Hlaváč J, Hradil P, Frysova I, Hajduch M, Bertolasi V, Malon M. Eur. J. Med. Chem. 2006;41:467–474. doi: 10.1016/j.ejmech.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 6.Hradil P, Krejčí P, Hlaváč J, Wiedermannová I, Lyčka A, Bertolasi V. J. Heterocyclic Chem. 2004;41:375–379. [Google Scholar]
- 7.Sui ZH, Nguyen VN, Altom J, Fernandez J, Hilliard JJ, Bernstein JI, Barrett JF, Ohemeng KA. Eur. J. Immunol. 1999;34:381–387. [Google Scholar]
- 8.Yushchenko DA, Bilokin MD, Pyvovarenko OV, Duportail G, Mely Y, Pivovarenko VG. Tetrahedron Lett. 2006;47:905–908. [Google Scholar]
- 9.Yushchenko DA, Shvadchak VV, Klymchenko AS, Duportail G, Mely Y, Pivovarenko VG. New J. Chem. 2006;30:774–781. [Google Scholar]
- 10.Rink H. Tetrahedron Lett. 1987;28:3787–3790. [Google Scholar]
- 11.Minta E, Boutonnet C, Boutard N, Martinez J, Rolland V. Tetrahedron Lett. 2005;46:1795–1797. [Google Scholar]
- 12.Jensen KJ, Alsina J, Songster MF, Vagner J, Albericio F, Barany G. J. Am. Chem. Soc. 1998;120:5441–5452. [Google Scholar]
- 13.Krchňák V, Smith J, Vagner J. Collect. Czech. Chem. Commun. 2001;66:1078–1106. [Google Scholar]
- 14.Rather JB, Rfid E. Emmet. J. Am. Chem. Soc. 1919;41:75–83. [Google Scholar]
- 15.Hradil P, Hlaváč J, Lemr K. J. Heterocyclic Chem. 1999;36:141–144. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.