

Abstract
Adenosine is a key endogenous molecule that regulates tissue function by activating four G-protein-coupled adenosine receptors: A1, A2A, A2B and A3. Cells of the immune system express these receptors and are responsive to the modulatory effects of adenosine in an inflammatory environment. Animal models of asthma, ischaemia, arthritis, sepsis, inflammatory bowel disease and wound healing have helped to elucidate the regulatory roles of the various adenosine receptors in dictating the development and progression of disease. This recent heightened awareness of the role of adenosine in the control of immune and inflammatory systems has generated excitement regarding the potential use of adenosine-receptor-based therapies in the treatment of infection, autoimmunity, ischaemia and degenerative diseases.
In 1929, Drury and Szent-Györgyi first reported the concept of adenosine acting as an extracellular signalling molecule1. The authors found that when simple extracts of heart muscle and other tissues were injected intravenously into the whole animal they produced decreased heart rate and increased coronary blood flow. The active constituent of the extracts turned out to be adenosine, and since the 1980s adenosine has been used to slow down the heart of patients suffering from excessively increased heart rate caused by supraventricular tachycardia2. In addition, adenosine has been used as a diagnostic agent, that is, a coronary vasodilator, to assess coronary artery function in conjunction with radionuclide myocardial perfusion imaging3. Following the original discovery of the effects of adenosine on cardiac function, adenosine has been found to have regulatory roles in virtually every organ system studied.
Adenosine accumulates in the extracellular space in response to metabolic stress and cell damage (BOX 1), and elevations in extracellular adenosine are found in conditions of ischaemia, hypoxia, inflammation and trauma4,5. The rapid release of adenosine in response to tissue-disturbing stimuli has a dual role in modulating homeostasis. First, extracellular adenosine represents a pre-eminent alarm molecule that reports tissue injury in an autocrine and paracrine manner to surrounding tissue. Second, extracellular adenosine generates a range of tissue responses that can be generally viewed as organ protective thereby mediating homeostasis. Adenosine elicits its physiological responses by binding to and activating one or more of the four transmembrane adenosine receptors, denoted A1, A2A, A2B and A3 (BOX 1). A prominent body of evidence supports the notion that the ability of adenosine, acting at its receptors, to control the immune and inflammatory systems plays a key role in the modulatory effects of adenosine in both health and disease. There are many promising emerging therapeutic approaches that are centred on the modulation of adenosine in the immune system. These encompass pharmacological compounds that interfere with the breakdown and generation of adenosine, as well as selective agonists and antagonists of various adenosine receptor subtypes (FIG. 1). Some of these compounds are in preclinical investigations, whereas others have already entered clinical trials for various indications. In this Review, we provide a general overview of adenosine receptors, covering aspects of cell biology, molecular biology and pharmacology. Separate sections focus on the multitude of roles that adenosine has in dictating the function of cell types that are considered basic constituents of the innate and adaptive immune systems. Finally, we discuss the therapeutic basis of harnessing the adenosine receptor system in managing patients suffering from inflammatory, autoimmune and ischaemic diseases.
Box 1. Adenosine-receptor metabolism and signalling.
Extracellular adenosine levels increase following the release of adenosine from cells or as a result of extracellular catabolism of released adenine nucleotides. Intracellular adenosine, which can be derived from increased intracellular metabolism of ATP during cellular stress or S-adenosyl homocysteine, is liberated mainly via equilibrative nucleoside transporters. Extracellular ATP and ADP are catabolized by a cascade of ectoenzymes comprising CD39 (ENTPD1; ectonucleoside triphosphate diphosphohydrolase 1) and CD73 (ecto-5′-nucleotidase). CD39 is an enzyme that hydrolyses ATP and ADP to AMP, and CD73, in turn, rapidly dephosphorylates AMP to adenosine126. Owing to the ubiquitous presence of equilibrative nucleoside transporters, adenosine generated from extracellular ATP is rapidly taken up by cells from the extracellular space. Adenosine in the cytosol is then metabolized either by adenosine kinase to form AMP or adenosine deaminase, which deaminates adenosine to inosine6,127. As a result of the rapid uptake and metabolism of adenosine, levels of this mediator are maintained low in unstressed, healthy tissues. However, under pathophysiological conditions adenosine removal can not keep pace with its generation, resulting in markedly increased extracellular adenosine concentrations.
All of the adenosine receptors contain seven transmembrane domains and couple to intracellular GTP-binding proteins (G proteins). Adenosine can activate A1, A2A and A3 receptors with EC50 values that are between 0.01 μM and 1 μM, whereas A2B receptor activation generally requires adenosine levels that exceed 10 μM (EC50 of 24 μM)128. Because physiological adenosine concentrations are lower than 1 μM, physiological levels of adenosine can activate A1, A2A and A3 receptors, whereas A2B receptor activation requires pathophysiological conditions129. Although the cellular responses to adenosine are highly dependent on the adenosine concentrations at the cell surface, several other factors, such as receptor density and the functionality of the intracellular signalling pathways coupled to adenosine receptors, are also key determinants in dictating the nature and magnitude of the effect of adenosine on the cell. For example, A2A receptor activation potently suppresses the production of the T helper 1-inducing cytokine interleukin 12 (IL12) by monocytes that are preincubated with the pro-inflammatory cytokine IL1 or tumour-necrosis factor-α, agents that also upregulate A2A receptors in these cells119. This example highlights the importance of receptor expression in defining the magnitude of the response to receptor stimulation. Furthermore, the effect of adenosine can also depend on the polarized localization of adenosine receptors. For example, A3 adenosine receptors accumulate at the leading edge of migrating neutrophils and have an important role in facilitating the directional movement of cells in response to chemotactic stimuli35. Finally, caution should be exercised in interpreting data assessing adenosine receptor function in different species, because sequence variations in cloned adenosine receptors have been shown to be associated with varying pharmacological responses to selective agonists and antagonists. This is best illustrated by the fact that A3 receptors remained undiscovered until they were cloned130, because the rodent A3 receptor is insensitive to xanthines such as theophylline and caffeine, antagonists that had been extensively used in identifying adenosine receptor-mediated effects.
Figure 1. Representative adenosine receptor ligands.

These ligands are the most widely used adenosine receptor agonists and antagonists in in vitro and in vivo studies assessing the function of adenosine receptors. IB-MECA (CF101) is currently undergoing testing in clinical trials for the treatment of rheumatoid arthritis.
Adenosine receptor signalling
A large body of evidence supports the view that adenosine receptors govern cell function by coupling to G proteins, although some G-protein-independent effects have also been reported6. Traditionally, adenosine receptor signalling is thought to occur through the inhibition or stimulation of adenylyl cyclase with a concomitant decrease or increase in intracellular cyclic AMP concentrations. Based on their ability to decrease or increase cAMP accumulation, adenosine receptors were initially classified as A1 or A2 receptors, respectively7. Subsequent studies have refined the classification of adenosine receptors and cAMP-increasing A2 receptors have been divided into two groups: high-affinity A2A receptors and low-affinity A2B receptors8. The more recent discovery and characterization of A3 receptors have made it clear that in addition to A1 receptors, A3 receptors dictate certain cellular responses such as rodent mast-cell degranulation, in part by decreasing intracellular cAMP concentrations9. This early picture of adenosine receptor signalling through the adenylyl cyclase–cAMP system has been substantially expanded, and it is now established that adenosine receptors can be linked to various other pathways.
A1 receptor activation had traditionally been linked to Gi-mediated inhibition of adenylyl cyclase. However, it is now known to be also linked to various kinase pathways including protein kinase C (PKC), phosphoinositide 3 (PI3) kinase and mitogen-activated protein (MAP) kinases10. In addition, A1 receptor activation can directly activate K+ channels and inhibit Q-, p- and N-type Ca2+ channels. It is noteworthy that most of these signalling pathways were uncovered in non-immune cell types, and A1 receptor signalling mechanisms in cells of the immune system are not known.
A2A receptors, similar to other Gs-protein-coupled receptors, signal mainly by the adenylate cyclase–cAMP–protein kinase A (PKA) canonical pathway, but they can also signal through the activation of an exchange factor that is directly activated by cAMP (Epac)6. Signalling downstream from PKA proceeds through phosphorylation of the transcription factor CREB on serine residue 133, resulting in its activation11. Activated CREB can mediate gene expression directly by interacting with gene promoters or indirectly by competing with nuclear factor-κB (NF-κB) or other transcription factors for an important cofactor, CBP6. In other cell types A2A receptors have been shown to signal for increased collagen production by activating MAP kinases12 and inhibiting neutrophil superoxide production by activating protein phosphatase13. Furthermore, recent results indicate that CEBPβ is responsible for the stimulatory effect of A2A receptor agonists on interleukin 10 (IL10) production by macrophages14 (FIG. 2). which of the outlined mechanisms mediate A2A receptor signalling in the various immune cell types remains to be defined in more detail.
Figure 2. Pattern-recognition receptor-mediated and A2A receptor-triggered pathways converge on CEBPβ to induce IL10 production by macrophages.
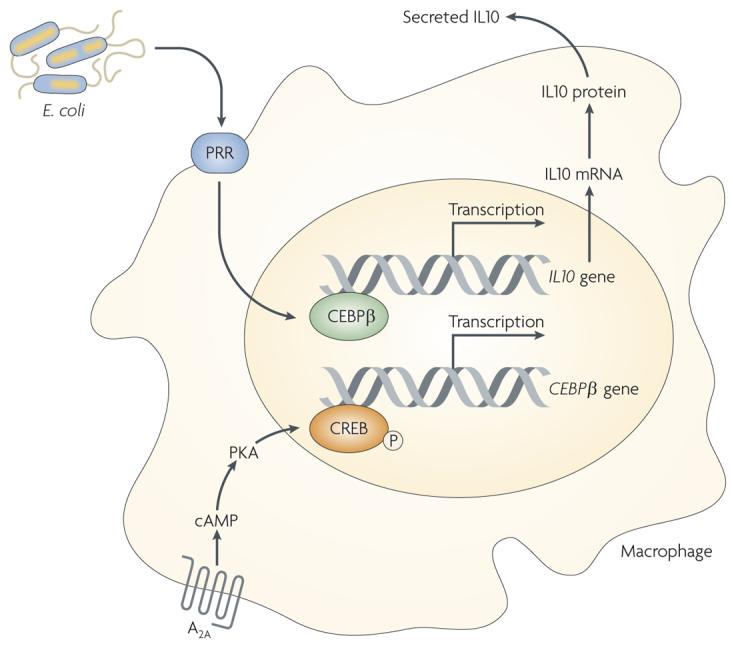
A2A receptor activation increases intracellular cyclic AMP (cAMP) levels resulting in increased protein kinase A (PKA) activation. PKA phosphorylates cAMP responsive element binding protein (CREB), which causes an increase in its transactivating potential leading to the transcription of the CEBPβ gene. CEBPβ protein binds to the IL10 gene promoter, which triggers IL10 transcription and subsequently leads to the release of IL10. Components of Escherichia coli trigger activation of pattern-recognition receptors (PRRs) and bring about increased activation of CEBPβ.
A2B receptor stimulation can trigger adenylyl cyclase activation via Gs and phospholipase C (PLC) activation via Gq (REF. 15). Cross-talk between these two pathways appears to be essential for the upregulation of IL4 production by human mast cells following A2B receptor activation16. In particular, Gq-mediated activation of PLCβ leads to calcium mobilization and an increase in NFATc1-dependent IL4 transcription, and this response is amplified by the Gs-mediated accumulation of NFATc1 protein.
The classical signalling pathways associated with A3 receptor activation comprise Gi-mediated inhibition of adenylyl cyclase and Gq-mediated stimulation of PLC17. In addition, A3 receptors can utilize the PLD, RhoA, WNT, MAP kinase and PI3 kinase pathways in dictating cell function. In fact, A3 receptor-dependent enhancement of histamine release in sensitized murine mast cells was prevented by inhibition of Gi proteins using pertussis toxin and by pharmacological inhibition of PI3 kinase18.
- Gi
An α-subunit of a heterotrimeric GTP-binding and GTP-hydrolysing protein (G protein) that inhibits the activity of a downstream enzyme such as adenylyl cyclase.
- Gs
An α-subunit of a heterotrimeric GTP-binding and GTP-hydrolysing protein (G protein) that stimulates the activity of a downstream enzyme such as adenylyl cyclase.
Adenosine receptors on immune cells
Macrophages
The effect of adenosine on cytokine production by macrophages has attracted considerable attention, because macrophage-derived cytokines are crucial initiators and orchestrators of immune responses. As tumour necrosis factor-α (TNF-α) was one of the first cytokines to be discovered, a substantial body of information has accumulated regarding the ability of adenosine receptor activation to limit TNF-α production following macrophage activation. Recent studies using adenosine-receptor knockout mice have painted a detailed but still not complete picture of the receptors involved. Several studies agree that the A2A receptor is the primary and dominant adenosine receptor subtype that mediates inhibition of TNF-α19-21. A role for other receptors was postulated based on the observation that adenosine, and the agonists NECA and IB-MECA (CF-101) can each inhibit, albeit to a lesser extent, TNF-α production even in A2A-receptor knockout mice19,20. A study using a combined approach of using A2A-receptor knockout mice and the A2B receptor antagonist MRS 1754 supports a role for A2B receptors as the other inhibitory receptor20. However, it appears that A2B receptors become operational only when their effect is not masked by A2A receptors, because both MRS 1754 and genetic deletion of A2B receptors in the presence of functional A2A receptors fails to affect the suppression of TNF-α production20,21.
- Gq
An α-subunit of a heterotrimeric GTP-binding and GTP-hydrolysing protein (G protein) that stimulates the activity of a downstream enzyme such as phospholipase C.
A similar picture is emerging regarding how adenosine receptor activation augments IL10 production. A key role for A2A receptors was recently documented when adenosine failed to upregulate Escherchia coli-induced IL10 release by macrophages lacking A2A receptors but not wild-type macrophages14. In the RAW264.7 macrophage cell line, which expresses negligible levels of A2A receptors, adenosine upregulates IL10 production via an A2B-receptor-mediated mechanism22. while A2B receptor activation has a minor impact on TNF-α and IL10 production, it is key to the stimulatory effect of adenosine on IL6 production, as NECA is unable to induce the release of IL6 in A2B-receptor knockout but not wild-type macrophages21.
Finally, it is important to emphasize that the issue of which adenosine receptors regulate cytokine production by human monocytes/macrophages is even more contentious with evidence pointing to the involvement of all four receptors23. These results, however, must be interpreted with caution, because they are based entirely on pharmacological approaches, and several of the ligands used, especially in early studies, are not particularly selective. Nevertheless, the recent observations that human newborn plasma contains elevated concentrations of adenosine when compared with adult plasma, and degrading adenosine with adenosine deaminase or interrupting adenosine signalling by adenosine receptor blockade augments TNF-α production by neonatal but not adult blood, highlight the importance of the inhibitory effect of adenosine on TNF-α production in human immunity24. Based on these results it was speculated that adenosine might protect the fetus from excessive inflammatory responses that drive alloimmune reactions and premature delivery. However, this protective effect would come at a price of increased susceptibility to infections following birth.
Dendritic cells
Although dendritic cell responses from adenosine-receptor knockout mice have yet to be characterized, available data from human studies support a consistent role for adenosine receptors in orchestrating dendritic cell function. Gi-coupled (A1, A3 or both depending on the experimental system) adenosine receptors are expressed on both immature myeloid25 and plasmocytoid dendritic cells26, and their activation results in the mobilization of intracellular calcium from intracellular stores and reorganization of the actin cytoskeleton. Consistent with these adenosine-induced intracellular responses, immature dendritic cells migrate along different concentration gradients of adenosine, indicating that adenosine receptor activation induces chemotaxis in immature dendritic cells25,26. These stimulatory responses induced by Gi-coupled adenosine receptors can be observed only in immature dendritic cells, as these receptors undergo downregulation during dendritic cell maturation25,26.
Although A2A receptors are present on immature dendritic cells, they are expressed at low levels and appear to be silent, as their activation is unable to elicit downstream signalling events such as accumulation of intracellular cAMP25. However, dendritic cell maturation is accompanied by the emergence of A2A-receptor-mediated signalling responses, owing to both increased expression and coupling of A2A receptors25,26. A2A receptor activation on mature dendritic cells shifts their cytokine profile from a pro-inflammatory to an anti-inflammatory one, with reduced IL12, IL6 and interferon-α (IFN-α) production and augmented IL10 production25-27. It is likely that dendritic cells in the presence of adenosine have a reduced capacity to induce T helper 1 (TH1) cell versus TH2 cell polarization of naive CD4+ cells27. This is due to the adenosine-induced switch in dendritic cell cytokine production away from the TH1-inducing IL12 towards the TH2-inducing IL10.
In summary, the available data support a dual role for adenosine in dictating dendritic cell function. Adenosine promotes the recruitment of immature dendritic cells to sites of inflammation and injury via A1 or A3 receptors. At these sites adenosine produces, via A2A receptors, an anti-inflammatory dendritic cell phenotype driving T-cell responses towards a TH2 profile.
Neutrophils
Adenosine is a potent modulator of neutrophil function and it has long been appreciated that adenosine, by activating its receptors, regulates stimulated production of reactive oxygen species by these cells and phagocytosis28-30.
Individual neutrophils do not produce large quantities of cytokines; however, because of the large numbers of accumulated neutrophils the cumulative contribution to pro-inflammatory cytokine levels at a given site is large. Adenosine, acting at A2A receptors, regulates the production of a range of cytokines including TNF-α, macrophage inflammatory protein (MIP)-1α (also known as CCL3), MIP-1β (CCL4), MIP-2α (CXCL2) and MIP-3α (CCL20)31. Neutrophils are recruited to inflammatory sites by the post-capillary venular endothelium, which alters the expression of adhesive molecules on its surface to capture neutrophils from the circulation. Adenosine, via A2A receptors, inhibits the adhesion of neutrophils to the endothelium by decreasing the expressionand stickiness of the adhesion molecules expressed on neutrophils32-34.
- NFATc1
NFATc1 is a member of the nuclear factor of activated T cells (NFAT) protein family, which are a family of transcription factors whose activation is controlled by calcineurin, a Ca2+-dependent phosphatase. They were originally identified in T cells as inducers of cytokine gene expression.
- Pertussis toxin
A compound that inhibits the guanine nucleotide binding proteins Gi and Go via ADP ribosylation.
- Macrophage
One of the main types of professional phagocytes. Macrophages are long-lived and detrimental for many microbial pathogens. Intracellular bacteria can survive within the macrophages. They can mediate antibody-dependent cellular cytotoxicity through phagocytosis.
By contrast, A1 receptors promote neutrophil adhesion to different adhesive molecules on the endothelium and on other surfaces32. Once in the tissue neutrophils migrate along gradients of chemoattractants. There are many chemoattractants including activated complement components (C5a), bacterial products (formylated peptides) and chemokines, and studies have shown that adenosine promotes directed migration of neutrophils via A1 and A3 receptors35-37. In addition, neutrophils cluster their A3 receptors at the leading edge of the cell and release ATP, which is converted at the cell surface to adenosine, which then acts in an autocrine manner to stimulate migration35. At inflamed sites neutrophils undergo apoptosis and adenosine, acting at A2A receptors, prevents neutrophils from undergoing apoptosis38-40. Thus, virtually every function carried out by neutrophils is regulated by adenosine and its receptors.
Mast cells
The observation that inhaled adenosine provokes bronchoconstriction in individuals suffering from asthma but not in normal volunteers has propelled adenosine and adenosine receptors into the forefront of asthma research41. Although the mechanisms of how adenosine mediates bronchoconstriction are still elusive, there is overwhelming evidence that mast-cell mediators such as cytokines and histamine may have a key role in evoking airway constriction in response to adenosine. Although A2A, A2B and A3 receptors are clearly present on the cell membrane of mast cells42, there is uncertainty regarding which receptor(s) account for the increased mast-cell activation following adenosine receptor activation.
Adenosine was recently documented to stimulate release of the major asthma-promoting cytokine IL13 by mouse mast cells that were obtained from wild-type but not A2B-receptor-knockout mice. This result implies that A2B receptors are the receptors involved in the pro-inflammatory effects of adenosine43. In addition, it is clear from genetic studies with knockout mice that A3 but not A2B receptor agonism can produce histamine release of lung mast cells in the absence of specific antigenic stimulation18. Thus, A2B and A3 receptors subserve different pro-inflammatory roles in naive mouse mast cells. The role of A2B receptors in regulating antigen-stimulated histamine release in mouse mast cells is controversial. Mast cells isolated from wild-type animals display decreased histamine release following stimulation with antigen when compared with mast cells obtained from A2B-receptor knockout mice44. This observation was interpreted as indicating that constitutive activation of A2B receptors on cultured mast cells suppresses the release of histamine. A subsequent study confirmed the increased degranulation of A2B-deficient mast cells, but based on additional approaches the authors questioned this interpretation of A2B-receptor-mediated decrease of degranulation of mast cells43. The reason for the different interpretation in the latter study was that exogenous adenosine maintained its stimulatory effect on histamine release in A2B-receptor knockout mast cells43. In addition, no evidence of constitutive activity of A2B receptors was revealed by studies using inverse agonists and measuring intracellular cAMP levels. Thus, it appears likely that the decreased release of histamine by mast cells isolated from A2B receptor wild-type versus knockout mice reflects an altered responsiveness of A2B receptor wild-type versus knockout cells to antigen. This is possibly from altered development of mast cells in A2B-receptor-knockout mice. Unlike A2B receptors, A3 receptors directly control histamine release by antigen-stimulated mouse mast cells, because the stimulatory effect of exogenous adenosine noted in wild-type mast cells is not observed in A3-receptor-knockout mast cells45.
In canine and human mast cells, degranulation or cytokine release appears to be mediated primarily by A2B receptor activation46,47. A2B receptor stimulation in human mastocytoma HMC-1 cells induces secretion of the TH2 cytokines IL4 and IL13, as well as a number of other pro-inflammatory cytokines such as IL1β and IL8. Although it is not clear whether adenosine receptor activation is able to stimulate the degranulation of human mast cells44, the observation of increased IL4 and IL13 production following adenosine receptor activation strongly implicates human mast-cell adenosine receptors as important players in the pathophysiology of human asthma.
Lymphocytes
In addition to regulating lymphocyte function indirectly by stimulating adenosine receptors on innate immune cells such as dendritic cells, adenosine can also directly affect lymphocyte responses by binding and activating adenosine receptors on lymphocytes. A number of recent studies using adenosine-receptor-knockout mice have evaluated the effect of adenosine receptor activation on various lymphocyte functions. The consensus emerging from these studies, as well as pharmacological studies, is that A2A receptors are the dominant adenosine receptors in dictating lymphocyte responses.
Studies using A2A-knockout models have shown that A2A receptor activation inhibits IL2 secretion48 by naive CD4+ T cells thereby reducing their proliferation49 following T-cell receptor stimulation. A2A receptor activation also suppresses the production of both IFN-γ and IL4 by both naive CD4+ T cells48,50 and polarized TH1 and TH2 cells51, thus challenging the hypothesis that A2A receptors on lymphocytes might shift TH cell responses towards a TH2 profile. Further immunosuppressive effects of A2A receptor stimulation include the upregulation of the expression of negative co-stimulatory molecules such as cytotoxic T-lymphocyte antigen 4 (CTLA4) and programmed cell death 1 (PD1), and downregulation of the positive co-stimulatory molecule CD-40L49.
Similar to CD4+ cells, adenosine inhibits IL2 production by both polarized type 1 cytotoxic T (TC1) and TC2 CD8+ cells, the effect of which was proposed to proceed through A2A receptors based on pharmacological evidence52. However, the production of neither TC1 (IFN-γ) nor TC2 (IL4 and IL5) cytokines was influenced by A2A receptor activation. In addition, pharmacological A2A receptor activation failed to reduce TC1 or TC2 cell cytolytic function52, which suggests that another subtype, possibly the A3 receptor, may mediate the anti-cytotoxic effect of adenosine noted in prior studies53,54. In contrast to these results with CD8+ cytotoxic cells, recent data with A1-, A2A- and A3-receptor-knockout mice support a primary role for A2A receptors in preventing the cytolytic activity of IL2-activated natural killer (NK) cells55.
- Adenosine deaminase
Adenosine deaminase irreversibly deaminates adenosine, converting it to the related nucleoside inosine by the removal of an amino group.
- Alloimmune reaction
Alloimmunity is an immune reaction against non-self material from the same species.
- Dendritic cell
These professional antigen-presenting cells are increasingly recognized as having crucial immunoregulatory functions. They are found in various tissues where they take up antigens, process them, migrate to the lymph nodes and present the antigens to T cells.
- CD4+ cells
Cells expressing the CD4+ glycoprotein that recognises major histocompatibility class II molecules.
- TH1 and TH2 cells
The TH1/TH2 hypothesis came to prominence in the late 1980s, indicating that mouse T-helper (TH) cells broadly express differing cytokine profiles. T helper 1 (TH1) cells secrete interferon-γ and tumour necrosis factor-α. TH2 cells secrete interleukin 4 (IL4), IL5 and IL13. In addition, TH3 and regulatory CD4+ CD25+ T cells exist that produce transforming growth factor-α and IL10, respectively.
- Mast cell
A bone marrow-derived cell that is present in various tissues; they are important contributors to allergic disease and possibly arthritis. They are granular cells that bear Fc receptors for immunoglobulin E (IgE), which, when crosslinked by IgE and antigen, causes degranulation and release of mediators such as histamine, leukotrienes and PGD2.
- Antigenic stimulation
When the body's immune system responds to a foreign substance.
Recent studies have revealed an important role for adenosine in mediating the immune suppressive properties of regulatory T (TReg) cells, a cell type that is thought to have a key role in keeping the immune system at bay, thereby preventing excessive tissue injury (FIG. 3). It was found that TReg cells — as defined by their expression of CD4+/CD25+/FoXp3+ and an ability to suppress the proliferation of CD4+/CD25− cells — express high levels of both CD39 (REF. 56) and CD73 (REFs 56,57), cell surface ectoenzymes that convert extracellular ATP and ADP to adenosine (BOX 1, FIG. 3). The finding that TReg cells selectively co-express CD39 and CD73 prompted the authors to propose that CD39 and CD73 could represent new specific markers of TReg cells. The mechanistic link between the transcription factor FOXP3+ and CD39 is suggested by recent studies showing that FOXP3+ actually drives the expression of CD39 (REF. 58).
Figure 3. Mechanisms of TReg cell-mediated suppression of T-effector cells.

Regulatory T (TReg) cells produce adenosine following sequential degradation of ATP/ADP via CD39 (ENTPD1; ectonucleoside triphosphate diphosphohydrolase 1) and CD73 (ecto-5′-nucleotidase) (a). Adenosine activates A2A receptors on T-effector cells to inhibit T-cell receptor (TCR)-mediated signalling by preventing ZAP70 phosphorylation and activation of the transcription factor activator protein 1 (AP1) (b). This decreased TCR signalling leads to decreased interleukin 2 (IL2) production and CD25 expression resulting in decreased T effector cell proliferation. In addition, the development of both T helper 1 (TH1) and TH2 cells, as well as the generation of TH17 lymphocytes is inhibited following A2A receptor stimulation. A2A receptor stimulation on T-effector cells increases expression of negative co-stimulatory molecules such as cytotoxic T-lymphocyte-associated antigen 4 (CTLA4) and programmed cell death 1 (PD1). A2A receptor stimulation on TReg cells augments FOXP3 expression in these cells.
The mechanisms of the immune suppressive effects of TReg cells have been elusive. In this regard, it was recently discovered that TReg cells can efficiently hydrolyse exogenous ADP to generate immune suppressive adenosine via CD39 and CD73 (REF. 56). Furthermore, the regulatory function of TReg cells depended largely on their ability to produce adenosine, because TReg cells isolated from CD39-knockout mice lost their potential to suppress proliferation of CD4+/CD25− cells, the effect of which was reconstituted by addition of soluble exogenous forms of CD39 (REF. 56). Further support to highlight an important role for adenosine was provided by the observation that A2A-receptor-knockout target cells (CD4+/CD25−) proliferated more than wild-type cells when cultured with wild-type TReg cells. It is noteworthy that CD4+/CD25− cells increased their expression of A2A receptor by day 4 in culture, at a time when TReg-cell-mediated suppression of target cell proliferation is highest, again confirming that adenosine generation is an important component of the TReg-cell armamentum. In addition to target cells, TReg cells also express A2A receptors and the activation of these receptors upregulates FOXP3+ expression in TReg cells59. The functional significance of A2A receptors on TReg cells was demonstrated in a mouse model of colitis48. In this model, adoptively transferred TReg cells lacking A2A receptors were found to be defective in their ability to suppress colitis. Hence, in an adenine-nucleotide-rich environment, TReg cells generate adenosine via ectoenzymes and are responsive to adenosine via A2A receptors.
Conventional T cells are activated via major histocompatability complex (MHC) molecules on antigen-presenting cells that present antigens to T-cell receptors. The subsequent expansion of activated T cells is responsible for adaptive immunity. A subset of T cells known as invariant NKT (iNKT) cells plays an important role in the innate immune response that triggers rapid host responses to infection. These cells express an invariant α,β T-cell receptor (vα14Jα18 in mice) in addition to molecular markers found on NK cells, such as NK1.1 (REF. 60). Unlike conventional T cells that recognize peptides, iNKT cells recognize lipid antigens that are derived from pathogens or injured host tissue. These lipid antigens are presented by antigen presenting cells to iNKT cells by the MHC class I-related molecule CD1d61. A2A receptors are found on iNKT cells and strongly suppress the release of pro-inflammatory cytokines such as INF-γ62. The presence of A2A receptors on iNKT cells suggests that there could be novel therapeutic applications of A2A agonists or antagonists as activation of iNKT cells has been implicated in a number of disease processes including atherosclerosis, type 1 diabetes, arthritis and various allergic diseases63.
In summary, lymphocyte function is potently regulated by A2A receptors, suggesting that the anti-inflammatory effects of A2A receptor agonists in animal models of autoimmunity and ischaemia are mediated, in part, by targeting lymphocytes.
Overall effect of adenosine in inflammation
Asthma and COPD
Several lines of clinical and preclinical evidence support the notion that adenosine and its receptors are intimately involved in defining the pathophysiology of asthma and chronic obstructive pulmonary disorder (COPD). Adenosine receptors on immune cells contained in the lung appear to have particularly important roles. Most compelling are the observations that inhaled adenosine can induce bronchoconstriction in patients suffering from asthma or COPD, but not in healthy individuals, and adenosine receptor blockade can prevent this bronchoconstrictive response42. These observations, combined with the demonstration that both adenosine levels and adenosine receptor expression on immune cells in the lung are elevated in patients with asthma and COPD, implicate endogenous adenosine as a prominent signalling molecule in lung diseases. As the bronchoconstrictive response to adenosine can be blocked by mast-cell membrane-stabilizing agents that can prevent the release of injurious mast-cell mediators, and because histamine receptor antagonists are also effective against adenosine, one major hypothesis explaining the mechanism of adenosine-mediated bronchoconstriction centres around a key role played by mast cells42. This general scheme of the injurious role of adenosine receptor activation on mast cells has been reinforced by studies using rodent models of asthma. These studies have also identified A2B (REF. 43) and A3 (REFS 18,45) receptors as the main contributors that mediate the stimulatory effect of adenosine on mast-cell activation. Although there is a paucity of human in vivo studies detailing the role of adenosine receptor subtypes in asthma and COPD, in vitro studies using human mast cells have narrowed down the candidate adenosine receptors to the A2B receptor (FIG. 4). In addition to mast cells, pro-inflammatory effects of A2B stimulation have also been observed with human bronchial smooth-muscle cells64, human bronchial epithelial cells65 and human lung fibroblasts66, which respond to adenosine by increased release of Il6 (REFS 64,66) and IL19 (REF. 65). A2B receptor activation on human lung fibroblasts promotes their differentiation into myofibroblasts that are capable of overproducing extracellular matrix66, which suggests that adenosine may participate in the fibrosis and remodelling of the lung during asthma and COPD.
Figure 4. A2B receptor activation has broad pro-inflammatory actions by stimulating the pro-inflammatory functions of a variety of cell types that mediate asthma.

A2B receptor activation increases interleukin 6 (IL6) production by pulmonary fibroblasts, which in turn leads to increased generation of myofibroblasts, which are capable of depositing extracellular matrix. A2B receptor activation promotes the production of pro-inflammatory factors by mast cells and stimulates mast-cell degranulation. Increased IL4 production following mast cell A2B receptor activation leads to increased immunoglobulin E (IgE) production by B cells. bFGF, basic fibroblast growth factor; MCP1, monocyte chemoattractant protein 1; VEGF, vascular endothelial growth factor.
- Inverse agonists
Inverse agonists reverse constitutive receptor activity, and are proposed to show selectively higher affinity for the inactive versus the active conformation of the receptor. In the absence of constitutive activity, inverse agonists function as competitive antagonists.
- Lymphocyte
White blood cells of lymphoid origin that function as part of the immune system.
- TC1 and TC2 CD8+ cells
CD8+ T cells have been subdivided into CD8+ T cells secreting a TH1-like cytokine pattern, which are defined as TC1 (T cytotoxic type 1) cells, versus CD8+ T cells secreting a TH2-like pattern (TC2 cells).
This substantial evidence documenting the pro-inflammatory effects of selective A2B receptor activation in human and rodent cellular systems combined with the utility of A2B receptor antagonism in preventing disease progression in rodent animal models67,68 suggest that A2B antagonists may be a viable treatment option for the management of asthma and COPD in human patients. Indeed, it is now well established that enprofylline, an anti-asthmatic agent, is a relatively selective (albeit not potent) A2B receptor antagonist69. The selective A2B receptor antagonist CVT-6883 (REFS 67,68), which has shown efficacy in preventing disease in rodent models of asthma and COPD, appeared to be safe and well tolerated in a recent Phase I study, raising hopes that this agent might have utility in the treatment of individuals suffering from asthma and COPD (TABLE 1).
Table 1.
Adenosine receptor ligands in clinical studies for treating inflammatory diseases
| Drug | Target receptor |
Agonist or antagonist |
Disease | Status | Company | References |
|---|---|---|---|---|---|---|
| CVT-6883 | A2B | Antagonist | COPD | Phase I | CV Therapeutics | Company web site‡ |
| GW328267X | A2A | Agonist | COPD | Phase II* | GlaxoSmithKline | 72 |
| UK-432097 | A2A | Agonist | COPD | Phase II | Pfizer | ClinicalTrials.gov Identifier NCT00430300‡ |
| IB-MECA (CF-101) |
A3 | Agonist | Rheumatoid arthritis |
Phase II | CanFite BioPharmaceuticals |
99 |
| Sonedenoson (MRE-0094) |
A2A | Agonist | Diabetic foot ulcer |
Phase II | King Pharmaceuticals |
Company web site‡ |
Discontinued.
See Further information for more details. COPD, chronic obstructive pulmonary disorder.
Lung inflammation results in the accumulation of neutrophils and other leukocytes in the interstitial space and in the airway. This is accompanied by an increased microvascular permeability and the release of chemotactic cytokines. A2A agonists have been found to reverse these effects. To determine which pulmonary cells respond to A2A receptor activation, pulmonary inflammation has been assessed in wild-type and A2A-receptor-knockout mice70. To differentiate the role of A2A receptors on haematopoietic and parenchymal cells, chimeric mice were created by transfer of bone marrow between wild-type and A2A receptor knockout mice. In wild-type mice, A2A receptor activation reduces lipopolysaccharide-induced neutrophil recruitment and release of cytokines. Pretreatment, but not post-treatment, also reduces the increase in vascular permeability. A2A receptor activation only reduced lung inflammation when the A2A receptor was present on bone-marrow-derived cells. In addition, using A2A-receptor-knockout mice, the adenosine–A2A receptor–cAMP axis was identified as a potent endogenous anti-inflammatory signalling pathway that reduces airway reactivity and inflammatory-cell infiltration following sensitization with ragweed71. Thus, A2A agonists appear to be effective at curbing inflammatory lung tissue damage. However, in clinical trials, the utility of the A2A agonist GW328267X was limited by cardiovascular side effects72 (TABLE 1).
Ischaemia
During reperfusion following ischaemic injury, many tissues have been shown to be protected from reperfusion injury owing to the activation of A2A receptors on bone-marrow-derived cells (FIG. 5). These include liver73, kidney74, heart75, skin76, spinal cord77,78 and lung79. In liver and kidney tissues iNKT cells have been found to be sensitive to A2A receptor activation and to have a crucial role in initiating an inflammatory cascade during reperfusion injury. These cells are rapidly stimulated to produce IFN-γ within 2 hours after the initiation of reperfusion, and the use of antibodies to deplete NK1.1-positive cells (NK and iNKT) or to block CD1d-mediated lipid-antigen presentation to iNKT cells replicates, but is not additive to the protection from reperfusion injury afforded by A2A adenosine receptor activation62,80. Liver reperfusion injury is also reduced in Rag1-knockout mice that lack mature lymphocytes, and can be restored to the level seen in wild-type mice by adoptive transfer of iNKT cells purified from wild-type or A2A receptor knockout mice, but not IFN-γ-knockout mice62. Additionally, animals with transferred A2A-knockout iNKT cells are not protected from hepatic reperfusion injury by A2A receptor activation. In vitro, A2A receptor activation potently inhibits activation of iNKT cells. These findings suggest that reperfusion injury is initiated by CD1d-dependent activation of iNKT cells, and the activation of these cells is inhibited by A2A receptor activation. The activation of iNKT cells results in the recruitment and transactivation of other immune cells such as macrophages and neutrophils that propagate tissue inflammation and injury.
Figure 5. A2A receptor activation protects organs from ischaemia–reperfusion injury by widely inactivating the ischaemia–reperfusion-induced inflammatory response.
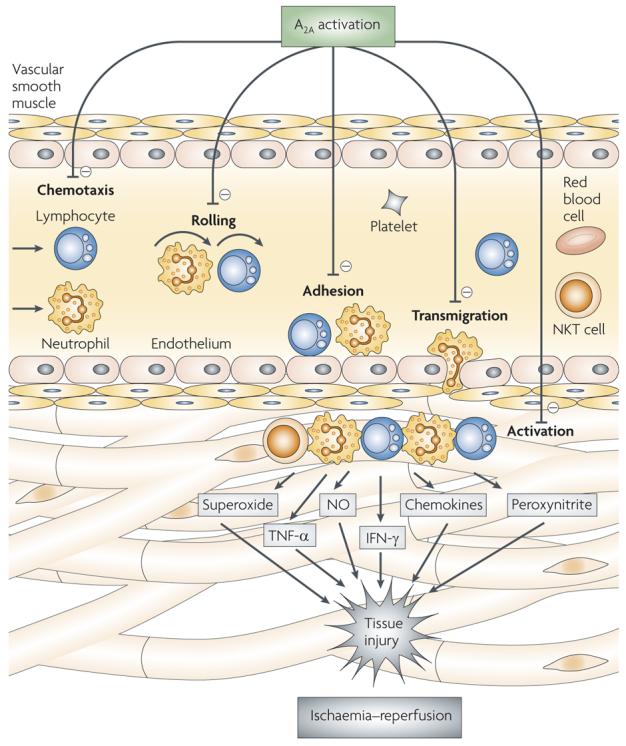
A2A receptor activation reduces ischaemia–reperfusion-induced rolling, adhesion and transmigration of various inflammatory cells, including natural killer T (NKT) cells, lymphocytes and neutrophils. A2A receptor stimulation also limits inflammatory cytokine and chemokine production, superoxide release and interferon-γ (IFN-γ) secretion by activated immune cells. NO, nitric oxide; TNF-α, tumour-necrosis factor-α.
- Natural killer (NK) cells
A lymphocyte subset that is part of the innate immune response and is able to recognize virus-infected or transformed cells that lack major histocompatibility class I expression. In contrast to T cells, NK cells do not require activation but are able to immediately kill these cells.
- Regulatory T (TReg) cells
T cells are a CD4+ T-cell subset that are characterized by the expression of CD25 (interleukin 2 receptor-(IL2R) subunit) and FOXP3. TReg cells are powerful suppressors of adaptive immune responses.
- MHC molecules
Originally named because they function as transplantation antigens. MHC molecules have a crucial role in antigen presentation, and serve as accessory binding proteins for T-helper and T-killer cells.
Arthritis
Despite the introduction of a number of effective biological agents for the treatment of rheumatoid arthritis over the past decade methotrexate remains one of the most effective and most commonly used therapies for inflammatory arthritis. Although originally introduced for the treatment of cancer as a folic acid analogue, the mechanism by which methotrexate, at very low dosages (average dosage in the United States, 17.5 mg per week for rheumatoid arthritis versus as much as 5 g per week for cancer), diminishes inflammation differs from the anti-proliferative mechanism of the drug. At low doses methotrexate is taken up by cells and polyglutamated to a long-lasting metabolite81. Methotrexate Polyglutamates have a different spectrum of enzyme inhibition and AICAR transformylase, an enzyme that is part of the de novo purine synthetic pathway, appears to be most sensitive to inhibition by methotrexate polyglutamates82. At low doses of methotrexate, this leads to the intracellular accumulation of AICAR83,84. AICAR is a competitive inhibitor of AMP deaminase leading, ultimately, to enhanced adenosine release from cells83. Although it is difficult to measure adenosine levels in biological fluids owing to the short half-life (2–8 seconds) of adenosine in blood and other fluids85 in patients with rheumatoid arthritis treated with methotrexate there is strong evidence that methotrexate therapy promotes adenosine release86,87. Studies in mice and rats demonstrate that the anti-inflammatory effects of methotrexate are mediated by adenosine and that the anti-inflammatory effect of methotrexate is lost if animals are treated with adenosine receptor antagonists or if their adenosine A2A or A3 receptors have been deleted88-94. Similar loss of methotrexate efficacy has been noted in patients with rheumatoid arthritis who ingest significant quantities of caffeine, an adenosine receptor antagonist95. Thus, by promoting adenosine release at inflamed sites methotrexate diminishes inflammation in patients with rheumatoid arthritis. Building on the observation that methotrexate exerts some of its beneficial effects via A3 receptors, coupled with the finding that the selective A3 receptor agonist IB-MECA ameliorates the course of arthritis in mouse models96-98, IB-MECA was recently tested in a phase II trial in patients with rheumatoid arthritis. IB-MECA was safe and well tolerated, and patients treated with this drug achieved a moderate reduction of symptoms99 (TABLE 1). Methotrexate and IB-MECA have additive anti-inflammatory effects in rat adjuvant arthritis, which are mediated by an upregulation of the expression of A3 receptors on inflammatory cells100.
Sepsis
Preclinical studies using both knockout and pharmacological approaches have provided insights into the role of the various adenosine receptors in the physiological response of the organism to sepsis101. Inactivation of A1 receptors increased mortality in intra-abdominal sepsis provoked by cecal ligation and puncture in mice, an effect that was correlated with heightened inflammation-induced hepatic and renal injury102. These results suggest that A1 receptor activation may have a beneficial effect and additional studies testing the efficacy of A1 receptor agonists are warranted. Similar to results with A1 receptor blockade in cecal ligation and puncture, both knockout and pharmacological antagonism of A3 receptors increased mortality, whereas stimulation of A3 receptors with IB-MECA was protective103. In contrast to observations with blockade of A1 and A3 receptors, A2A receptor inactivation by either gene deletion or administration of ZM241385 prevented cecal ligation and puncture-induced mortality by a mechanism that involved decreased bacterial dissemination that appeared to be secondary to sustained immune system function104. Paradoxically, A2A receptor activation, when combined with antibiotics, has been noted to reduce mortality from sepsis induced by injecting with live E. coli, possibly by suppressing an exaggerated inflammatory response that can result from the rapid drug-induced killing of large numbers of bacteria105. Recent preliminary studies evaluating the role of A2B receptors have demonstrated an anti-inflammatory and protective role of A2B receptor stimulation in cecal ligation and puncture (C. Csoka, G. Hasko, Z. H. Nemeth and p. Pacher, unpublished observations).
- Invariant NKT (iNKT) cells
A rare subset of lymphocytes that expresses an invariant T-cell receptor that recognizes certain glycolipids when bound to the major histocompatibility complex class I-like molecule, CD1d. Through secretion of cytokines they are powerful modulators of adaptive immune responses.
- Cecal ligation and puncture
An experimental model of polymicrobial sepsis that is generally considered more relevant to the human disease than rodents injected with bacterial lipopolysaccharide (endotoxin).
Inflammatory bowel disease
Crohn's disease and ulcerative colitis are chronic, relapsing inflammatory bowel diseases (IBDs) that are characterized by a dysfunction of mucosal T cells, imbalanced cytokine production and cellular inflammation leading to damage of the intestinal mucosa. Although the aetiology of IBD remains to be determined, recent studies suggest that disease results from an inappropriately regulated immune response to luminal antigens in a genetically susceptible host106. CD4+CD25+ TReg cells have emerged as master regulators of the inflammatory response of the mucosa in IBD and have also been documented to offer a therapeutic option in severe inflammatory colitis107. Studies have highlighted the protective effects of A2A receptor activation in various animal models of colitis, and these protective effects can be ascribed to two major mechanisms: decrease of inflammatory-cell infiltration and function in the mucosa, and increased activity of TReg cells. The first mechanism is supported by evidence that A2A receptor activation by ATL146e (an A2A receptor agonist) decreases both leukocyte infiltration and the production of inflammatory cytokines by disease-inducing T-effector cells108. The role of A2A receptors on TReg cells was inferred by results showing that co-transfer of CD4+CD25+ TReg cells obtained from A2A receptor wild-type mice was able to prevent disease induction in severe combined immunodeficiency mice that were transferred adoptively with disease-inducing CD45RBhigh CD4+ T cells, a rodent model of IBD, whereas co-administration of CD4+CD25+ TReg cells isolated from A2A-receptor-knockout mice in conjunction with disease-inducing CD45RBhigh CD4+ T cells was inefficient in blocking the development of disease48.
The observation that activation of A2B receptors on intestinal epithelial cells can augment Il6 production by these cells resulting in increased neutrophil activation109 combined with increased detection of A2B receptors in epithelial cells isolated from mouse or human colitis110 prompted recent studies assessing the effect of A2B receptor blockade in IBD. Administering ATL-801, a selective A2B antagonist, to mice suffering from colitis markedly decreased IL6 production and neutrophil infiltration, and reduced the extent of mucosal damage thereby ameliorating the course of disease111. This pro-inflammatory role of A2B receptor activation by endogenous adenosine was recently confirmed using A2B-receptor-knockout mice112, confirming that blockade of A2B receptor represents a potentially advantageous treatment option that selectively targets the gut for patients afflicted with IBD. This proposition is predicated on the findings that A2B receptor expression is highest in the gut when compared with other organs and that levels of endogenous adenosine that appear to perpetuate the disease process through A2B receptors are elevated mostly locally in the gut111.
Wound healing
In the skin, inflammation is part of a continuum in which injured tissue is repaired and replaced resulting in wound healing. Montesinos et al. first reported that topical application of adenosine A2A receptor agonists increases the rate of wound healing in normal mice and in diabetic rats113. Increases in matrix and vessel formation were induced by adenosine receptor agonists and the improvement in the rate of wound healing was significantly greater than that induced by recombinant platelet-derived growth factor113-116. A2A and A2B receptor stimulation increases angiogenesis both directly and indirectly: adenosine receptors directly stimulate microvascular endothelial cell-proliferation as well as the autocrine production of vascular endothelial growth factor (VEGF), a central stimulus for endothelial proliferation and angiogenesis117-119, and inhibit the production of thrombospondin 1, an anti-angiogenic matrix protein120. In addition, adenosine stimulates VEGF production by macrophages116,121,122. Moreover, A2A and A2B receptor stimulation promotes matrix production by skin fibroblasts, an essential step in tissue repair66,123-125. In this context, it is noteworthy that A2A-receptor-deficient mice do not form granulation tissue indicating that endogenous adenosine plays a central role in wound healing114. Sonedenoson (MRE0094), an A2A agonist that regulates the inflammatory response and enhances tissue regeneration, is currently undergoing trials for the treatment of diabetic foot ulcers(TABLE 1).
- Angiogenesis
The growth of new blood vessels from pre-existing vessels. Angiogenesis is a normal process in growth and development but it also a fundamental process required for the growth of tumours.
Conclusions
The adenosine receptor system has evolved as both a rapid sensor of tissue injury and the major ‘first-aid’ machinery of tissues and organs. Adenosine receptor activation thus preserves tissue function and prevents further tissue injury following an acute injurious insult, such as reperfusion injury, actions in which the immune system has a paramount role. This primordial protective function of the adenosine receptor system following acute insults can, however, be overshadowed by its reduced ability to protect against chronic insults. In addition, in certain chronic disease states, such as asthma, the adenosine receptor system can even exacerbate tissue dysfunction.
Recent advances in understanding the role of the distinct adenosine receptors and the complex network of cellular players that determine the adenosine response to tissue injury have helped identify novel pharmacological targets to restore tissue function in various diseases(BOX 2). Despite growing enthusiasm regarding these targets, it is important to emphasize that because most of the data were obtained using animal models, caution should be exercised in translating results to the clinic. potential difficulties in translating results may stem from, in part, species differences and the fact that animal studies generally are done in young, relatively healthy animals over a short time-span. Of particular relevance is species differences in adenosine signalling in asthma studies. In rodents, primed mast-cell degranulation appears to be mediated primarily by A3 receptors, whereas in humans A2B receptors are more important.
Box 2. Targeting adenosine receptors for treating inflammatory diseases.
Asthma and chronic obstructive pulmonary disease (COPD)
A2A receptor agonists prevent inflammatory-cell infiltration into the lung and thus may be of therapeutic use. Potential problems may result from cardiovascular side effects with these agents.
A2B receptor antagonists prevent mast-cell degranulation and the overproduction of pro-inflammatory cytokines and extracellular matrix by smooth-muscle cells, bronchial epithelial cells and lung fibroblasts.
Ischaemia
A2A receptor agonists potently downregulate inflammatory-cell infiltration into tissues, production of deleterious free radicals and pro-inflammatory cytokines. Potential issues relate to their cardiovascular side effects.
Arthritis
A2A receptor agonists have a wide range of anti-inflammatory effects in the inflamed joint, but can exhibit cardiovascular side effects.
A3 receptor agonists decrease tumour necrosis factor-α production by monocytes and synoviocytes. Development of more selective and potent A3 receptor agonists is needed.
Sepsis
A1 receptor agonists can prevent inflammation-mediated organ injury in animal models; however, their utility is limited by cardiovascular depressive effects.
A2A receptor antagonists are beneficial in sepsis by boosting eradication of bacteria. One potential problem is that sepsis is a heterogeneous group of diseases and inflammatory tissue injury rather then infection dominates the clinical picture in a smaller group of patients. A2A receptor antagonists may exacerbate organ injury in these patients.
Inflammatory bowel disease
A2A receptor agonists attenuate inflammatory-cell sequestration in the gut and increase regulatory T-cell activity thereby ameliorating the course of disease. They may have cardiovascular side effects.
A2B receptor antagonists prevent intestinal epithelial-cell-mediated inflammatory events and thereby prevent mucosal inflammation.
Wound healing
Topical administration of A2A receptor agonists increases the rate of wound healing, in part, by stimulating angiogenesis in the skin. Thus, A2A receptor agonists are good candidates for the treatment of diabetic foot ulcer.
Although adenosine receptor agonists have powerful immunomodulatory actions, the wide tissue distribution of adenosine receptors may limit their usefulness in the treatment of inflammatory diseases. Adenosine receptor antagonists, however, represent an ideal target for the therapy of certain immune-related disorders because their action is selectively targeted to the site of injury, where endogenous adenosine is released. Similarly, interfering with the function of enzymes and transporters that are responsible for the accumulation of extracellular adenosine allows local targeting of adenosine receptors, providing an opportunity for interventions with limited side effects. It is clear that a better understanding of adenosine receptor function will be required before the enormous potential of adenosine-based therapies can be realized to ease human suffering.
Acknowledgements
This work was supported by the National Institutes of Health (NIH) Grant R01GM66189 and the Intramural Research Program of NIH, National Institute on Alcohol Abuse and Alcoholism.
Footnotes
Competing interests statement
The authors declare competing financial interests: see web version for details.
DATABASES
IUPHAR Receptor Database: http://www.iuphar-db.org A1 | A2A | A2B | A3
FURTHER INFORMATION
ClinicalTrials.gov: http://clinicaltrials.gov/ct2/show/NCT00430300
CV Therapeutics: http://www.cvt.com/a_pipeline.html
King Pharmaceuticals: http://www.kingpharm.com/kingpharm/AboutUs/fastFacts.asp
References
- 1.Drury AN, Szent-Gyorgyi A. The physiological activity of adenine compounds with especial reference to their action upon the mammalian heart. J. Physiol. 1929;68:213–237. doi: 10.1113/jphysiol.1929.sp002608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.diMarco JP, et al. Diagnostic and therapeutic use of adenosine in patients with supraventricular tachyarrhythmias. J. Am. Coll. Cardiol. 1985;6:417–425. doi: 10.1016/s0735-1097(85)80181-9. [DOI] [PubMed] [Google Scholar]
- 3.Travain MI, Wexler JP. Pharmacological stress testing. Semin. Nucl. Med. 1999;29:298–318. doi: 10.1016/s0001-2998(99)80018-x. [DOI] [PubMed] [Google Scholar]
- 4.Linden J. Molecular approach to adenosine receptors: receptor-mediated mechanisms of tissue protection. Annu. Rev. Pharmacol. Toxicol. 2001;41:775–787. doi: 10.1146/annurev.pharmtox.41.1.775. [DOI] [PubMed] [Google Scholar]
- 5.Fredholm BB, AP IJ, Jacobson KA, Klotz KN, Linden J. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol. Rev. 2001;53:527–552. [PMC free article] [PubMed] [Google Scholar]
- 6.Fredholm BB, Chern Y, Franco R, Sitkovsky M. Aspects of the general biology of adenosine A2A signaling. Prog. Neurobiol. 2007;83:263–276. doi: 10.1016/j.pneurobio.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 7.van Calker D, Muller M, Hamprecht B. Adenosine regulates via two different types of receptors, the accumulation of cyclic AMP in cultured brain cells. J. Neurochem. 1979;33:999–1005. doi: 10.1111/j.1471-4159.1979.tb05236.x. [DOI] [PubMed] [Google Scholar]
- 8.Bruns RF, Lu GH, Pugsley TA. Characterization of the A2 adenosine receptor labeled by [3H]NECA in rat striatal membranes. Mol. Pharmacol. 1986;29:331–346. [PubMed] [Google Scholar]
- 9.Jin X, Shepherd RK, Duling BR, Linden J. Inosine binds to A3 adenosine receptors and stimulates mast cell degranulation. J. Clin. Invest. 1997;100:2849–2857. doi: 10.1172/JCI119833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jacobson KA, Gao ZG. Adenosine receptors as therapeutic targets. Nature Rev. Drug Discov. 2006;5:247–264. doi: 10.1038/nrd1983. Comprehensive review of the role of adenosine receptors in regulating the disease processes of various organs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nemeth ZH, et al. Adenosine stimulates CREB activation in macrophages via a p38 MAPK-mediated mechanism. Biochem. Biophys. Res. Commun. 2003;312:883–888. doi: 10.1016/j.bbrc.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 12.Che J, Chan ES, Cronstein BN. Adenosine A2A receptor occupancy stimulates collagen expression by hepatic stellate cells via pathways involving protein kinase A, Src, and extracellular signal-regulated kinases 1/2 signaling cascade or p38 mitogen-activated protein kinase signaling pathway. Mol. Pharmacol. 2007;72:1626–1636. doi: 10.1124/mol.107.038760. [DOI] [PubMed] [Google Scholar]
- 13.Revan S, Montesinos MC, Naime D, Landau S, Cronstein BN. Adenosine A2A receptor occupancy regulates stimulated neutrophil function via activation of a serine/threonine protein phosphatase. J. Biol. Chem. 1996;271:17114–17118. doi: 10.1074/jbc.271.29.17114. [DOI] [PubMed] [Google Scholar]
- 14.Csoka B, et al. A2A adenosine receptors and C/EBPβ are crucially required for IL-10 production by macrophages exposed to Escherichia coli. Blood. 2007;110:2685–2695. doi: 10.1182/blood-2007-01-065870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feoktistov I, Biaggioni I. Adenosine A2B receptors. Pharmacol. Rev. 1997;49:381–402. [PubMed] [Google Scholar]
- 16.Ryzhov S, Goldstein AE, Biaggioni I, Feoktistov I. Cross-talk between Gs- and Gq-coupled pathways in regulation of interleukin-4 by A2B adenosine receptors in human mast cells. Mol. Pharmacol. 2006;70:727–735. doi: 10.1124/mol.106.022780. [DOI] [PubMed] [Google Scholar]
- 17.Gessi S, et al. The A3 adenosine receptor: an enigmatic player in cell biology. Pharmacol. Ther. 2008;117:123–140. doi: 10.1016/j.pharmthera.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 18.Zhong H, et al. Activation of murine lung mast cells by the adenosine A3 receptor. J. Immunol. 2003;171:338–345. doi: 10.4049/jimmunol.171.1.338. [DOI] [PubMed] [Google Scholar]
- 19.Hasko G, et al. Adenosine inhibits IL-12 and TNF-α production via adenosine A2a receptor-dependent and independent mechanisms. FASEB J. 2000;14:2065–2074. doi: 10.1096/fj.99-0508com. First use of adenosine receptor (A2A) knockout mice in investigating the effect of adenosine on the immune system. [DOI] [PubMed] [Google Scholar]
- 20.Kreckler LM, Wan TC, Ge ZD, Auchampach JA. Adenosine inhibits tumor necrosis factor-α release from mouse peritoneal macrophages via A2A and A2B but not the A3 adenosine receptor. J. Pharmacol. Exp. Ther. 2006;317:172–180. doi: 10.1124/jpet.105.096016. [DOI] [PubMed] [Google Scholar]
- 21.Ryzhov S, et al. Effect of A2B adenosine receptor gene ablation on adenosine-dependent regulation of proinflammatory cytokines. J. Pharmacol. Exp. Ther. 2008;324:694–700. doi: 10.1124/jpet.107.131540. This paper demonstrates using A2B receptor knockout mice that A2B receptors mediate the stimulatory effect of adenosine on IL13 and VEGF secretion by mast cells. [DOI] [PubMed] [Google Scholar]
- 22.Nemeth ZH, et al. Adenosine augments IL-10 production by macrophages through an A2B receptor-mediated posttranscriptional mechanism. J. Immunol. 2005;175:8260–8270. doi: 10.4049/jimmunol.175.12.8260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hasko G, Pacher P, Deitch EA, Vizi ES. Shaping of monocyte and macrophage function by adenosine receptors. Pharmacol. Ther. 2007;113:264–275. doi: 10.1016/j.pharmthera.2006.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Levy O, et al. The adenosine system selectively inhibits TLR-mediated TNF-α production in the human newborn. J. Immunol. 2006;177:1956–1966. doi: 10.4049/jimmunol.177.3.1956. Highlights the importance of the physiological role of adenosine in human immunity by showing that human newborn plasma contains elevated concentrations of adenosine when compared with adult plasma. Also degradation of adenosine with adenosine deaminase or interrupting adenosine signaling by adenosine receptor blockade augments TNF-α production by neonatal but not adult blood. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Panther E, et al. Expression and function of adenosine receptors in human dendritic cells. FASEB J. 2001;15:1963–1970. doi: 10.1096/fj.01-0169com. First demonstration for a role of adenosine receptors in shaping dendritic cell function. [DOI] [PubMed] [Google Scholar]
- 26.Schnurr M, et al. Role of adenosine receptors in regulating chemotaxis and cytokine production of plasmacytoid dendritic cells. Blood. 2004;103:1391–1397. doi: 10.1182/blood-2003-06-1959. [DOI] [PubMed] [Google Scholar]
- 27.Panther E, et al. Adenosine affects expression of membrane molecules, cytokine and chemokine release, and the T-cell stimulatory capacity of human dendritic cells. Blood. 2003;101:3985–3990. doi: 10.1182/blood-2002-07-2113. [DOI] [PubMed] [Google Scholar]
- 28.Cronstein BN, Kramer SB, Weissmann G, Hirschhorn R. Adenosine: a physiological modulator of superoxide anion generation by human neutrophils. J. Exp. Med. 1983;158:1160–1177. doi: 10.1084/jem.158.4.1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cronstein BN, Rosenstein ED, Kramer SB, Weissmann G, Hirschhorn R. Adenosine; a physiologic modulator of superoxide anion generation by human neutrophils. Adenosine acts via an A2 receptor on human neutrophils. J. Immunol. 1985;135:1366–1371. [PubMed] [Google Scholar]
- 30.Varani K, Gessi S, Dionisotti S, Ongini E, Borea PA. [3H]-SCH 58261 labelling of functional A2A adenosine receptors in human neutrophil membranes. Br. J. Pharmacol. 1998;123:1723–1731. doi: 10.1038/sj.bjp.0701758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McColl SR, et al. Immunomodulatory impact of the A2A adenosine receptor on the profile of chemokines produced by neutrophils. FASEB J. 2006;20:187–189. doi: 10.1096/fj.05-4804fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cronstein BN, et al. Neutrophil adherence to endothelium is enhanced via adenosine A1 receptors and inhibited via adenosine A2 receptors. J. Immunol. 1992;148:2201–2206. [PubMed] [Google Scholar]
- 33.Sullivan GW, et al. Activation of A2A adenosine receptors inhibits expression of α4/β1 integrin (very late antigen-4) on stimulated human neutrophils. J. Leukoc. Biol. 2004;75:127–134. doi: 10.1189/jlb.0603300. [DOI] [PubMed] [Google Scholar]
- 34.Zhao ZQ, Sato H, Williams MW, Fernandez AZ, Vinten-Johansen J. Adenosine A2-receptor activation inhibits neutrophil-mediated injury to coronary endothelium. Am. J. Physiol. 1996;271:H1456–H1464. doi: 10.1152/ajpheart.1996.271.4.H1456. [DOI] [PubMed] [Google Scholar]
- 35.Chen Y, et al. ATP release guides neutrophil chemotaxis via P2Y2 and A3 receptors. Science. 2006;314:1792–1795. doi: 10.1126/science.1132559. Describes that A3 receptors clustered at the leading edge of neutrophils are instrumental in promoting directed migration of these cells. [DOI] [PubMed] [Google Scholar]
- 36.Cronstein BN, Daguma L, Nichols D, Hutchison AJ, Williams M. The adenosine/neutrophil paradox resolved: human neutrophils possess both A1 and A2 receptors that promote chemotaxis and inhibit O2 generation, respectively. J. Clin. Invest. 1990;85:1150–1157. doi: 10.1172/JCI114547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rose FR, Hirschhorn R, Weissmann G, Cronstein BN. Adenosine promotes neutrophil chemotaxis. J. Exp. Med. 1988;167:1186–1194. doi: 10.1084/jem.167.3.1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mayne M, et al. Adenosine A2A receptor activation reduces proinflammatory events and decreases cell death following intracerebral hemorrhage. Ann. Neurol. 2001;49:727–735. doi: 10.1002/ana.1010. [DOI] [PubMed] [Google Scholar]
- 39.Walker BA, Rocchini C, Boone RH, Ip S, Jacobson MA. Adenosine A2a receptor activation delays apoptosis in human neutrophils. J. Immunol. 1997;158:2926–2931. [PubMed] [Google Scholar]
- 40.Yasui K, et al. Theophylline induces neutrophil apoptosis through adenosine A2A receptor antagonism. J. Leukoc. Biol. 2000;67:529–535. doi: 10.1002/jlb.67.4.529. [DOI] [PubMed] [Google Scholar]
- 41.Cushley MJ, Tattersfield AE, Holgate ST. Inhaled adenosine and guanosine on airway resistance in normal and asthmatic subjects. Br. J. Clin. Pharmacol. 1983;15:161–165. doi: 10.1111/j.1365-2125.1983.tb01481.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Polosa R, Holgate ST. Adenosine receptors as promising therapeutic targets for drug development in chronic airway inflammation. Curr. Drug Targets. 2006;7:699–706. doi: 10.2174/138945006777435236. [DOI] [PubMed] [Google Scholar]
- 43.Ryzhov S, et al. Effect of A2B adenosine receptor gene ablation on proinflammatory adenosine signaling in mast cells. J. Immunol. 2008;180:7212–7220. doi: 10.4049/jimmunol.180.11.7212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hua X, et al. Enhanced mast cell activation in mice deficient in the A2b adenosine receptor. J. Exp. Med. 2007;204:117–128. doi: 10.1084/jem.20061372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Salvatore CA, et al. Disruption of the A3 adenosine receptor gene in mice and its effect on stimulated inflammatory cells. J. Biol. Chem. 2000;275:4429–4434. doi: 10.1074/jbc.275.6.4429. [DOI] [PubMed] [Google Scholar]
- 46.Feoktistov I, Biaggioni I. Adenosine A2b receptors evoke interleukin-8 secretion in human mast cells. An enprofylline-sensitive mechanism with implications for asthma. J. Clin. Invest. 1995;96:1979–1986. doi: 10.1172/JCI118245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Auchampach JA, Jin X, Wan TC, Caughey GH, Linden J. Canine mast cell adenosine receptors: cloning and expression of the A3 receptor and evidence that degranulation is mediated by the A2B receptor. Mol. Pharmacol. 1997;52:846–860. doi: 10.1124/mol.52.5.846. [DOI] [PubMed] [Google Scholar]
- 48.Naganuma M, et al. Cutting edge: Critical role for A2A adenosine receptors in the T cell-mediated regulation of colitis. J. Immunol. 2006;177:2765–2769. doi: 10.4049/jimmunol.177.5.2765. [DOI] [PubMed] [Google Scholar]
- 49.Sevigny CP, et al. Activation of adenosine 2A receptors attenuates allograft rejection and alloantigen recognition. J. Immunol. 2007;178:4240–4249. doi: 10.4049/jimmunol.178.7.4240. [DOI] [PubMed] [Google Scholar]
- 50.Lappas CM, Rieger JM, Linden J. A2A adenosine receptor induction inhibits IFN-γ production in murine CD4+ T cells. J. Immunol. 2005;174:1073–1080. doi: 10.4049/jimmunol.174.2.1073. [DOI] [PubMed] [Google Scholar]
- 51.Csoka BH, et al. Adenosine A2A receptor activation inhibits T helper 1 and T helper 2 cell development and effector function. FASEB J. 2008 Jul 14; doi: 10.1096/fj.08-107458. (doi:10.1096/fj.08-107458) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Erdmann AA, et al. Activation of Th1 and Tc1 cell adenosine A2A receptors directly inhibits IL-2 secretion in vitro and IL-2-driven expansion in vivo. Blood. 2005;105:4707–4714. doi: 10.1182/blood-2004-04-1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Koshiba M, Kojima H, Huang S, Apasov S, Sitkovsky MV. Memory of extracellular adenosine A2A purinergic receptor-mediated signaling in murine T cells. J. Biol. Chem. 1997;272:25881–25889. doi: 10.1074/jbc.272.41.25881. One of the early studies describing the inhibitory effects of A2A receptor stimulation on T-cell activation. [DOI] [PubMed] [Google Scholar]
- 54.Hoskin DW, Butler JJ, Drapeau D, Haeryfar SM, Blay J. Adenosine acts through an A3 receptor to prevent the induction of murine anti-CD3-activated killer T cells. Int. J. Cancer. 2002;99:386–395. doi: 10.1002/ijc.10325. [DOI] [PubMed] [Google Scholar]
- 55.Raskovalova T, et al. Gs protein-coupled adenosine receptor signaling and lytic function of activated NK cells. J. Immunol. 2005;175:4383–4391. doi: 10.4049/jimmunol.175.7.4383. [DOI] [PubMed] [Google Scholar]
- 56.Deaglio S, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J. Exp. Med. 2007;204:1257–1265. doi: 10.1084/jem.20062512. Demonstrates that TReg cells release adenosine, which then suppresses T-effector cell activation via A2A receptors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kobie JJ, et al. T regulatory and primed uncommitted CD4 T cells express CD73, which suppresses effector CD4 T cells by converting 5′-adenosine monophosphate to adenosine. J. Immunol. 2006;177:6780–6786. doi: 10.4049/jimmunol.177.10.6780. [DOI] [PubMed] [Google Scholar]
- 58.Borsellino G, et al. Expression of ectonucleotidase CD39 by Foxp3+ Treg cells: hydrolysis of extracellular ATP and immune suppression. Blood. 2007;110:1225–1232. doi: 10.1182/blood-2006-12-064527. [DOI] [PubMed] [Google Scholar]
- 59.Zarek PE, et al. A2A receptor signaling promotes peripheral tolerance by inducing T-cell anergy and the generation of adaptive regulatory T cells. Blood. 2008;111:251–259. doi: 10.1182/blood-2007-03-081646. This paper shows that A2A receptor signalling induces T-cell anergy in vivo. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zajonc DM, et al. Structural basis for CD1d presentation of a sulfatide derived from myelin and its implications for autoimmunity. J. Exp. Med. 2005;202:1517–1526. doi: 10.1084/jem.20051625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kaneko S, et al. Melatonin scavenges hydroxyl radical and protects isolated rat hearts from ischemic reperfusion injury. Life Sci. 2000;67:101–112. doi: 10.1016/s0024-3205(00)00607-x. [DOI] [PubMed] [Google Scholar]
- 62.Lappas CM, Day YJ, Marshall MA, Engelhard VH, Linden J. Adenosine A2A receptor activation reduces hepatic ischemia reperfusion injury by inhibiting CD1d-dependent NKT cell activation. J. Exp. Med. 2006;203:2639–2648. doi: 10.1084/jem.20061097. Describes a crucial role for NKT cells in instigating tissue injury following ischaemia–reperfusion and as central targets of the anti-inflammatory effects of adenosine. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yamamura T, Sakuishi K, Illes Z, Miyake S. Understanding the behavior of invariant NKT cells in autoimmune diseases. J. Neuroimmunol. 2007;191:8–15. doi: 10.1016/j.jneuroim.2007.09.014. [DOI] [PubMed] [Google Scholar]
- 64.Zhong H, et al. A2B adenosine receptors increase cytokine release by bronchial smooth muscle cells. Am. J. Respir. Cell. Mol. Biol. 2004;30:118–125. doi: 10.1165/rcmb.2003-0118OC. [DOI] [PubMed] [Google Scholar]
- 65.Zhong H, Wu Y, Belardinelli L, Zeng D. A2B adenosine receptors induce IL-19 from bronchial epithelial cells, resulting in TNF-α increase. Am. J. Respir. Cell. Mol. Biol. 2006;35:587–592. doi: 10.1165/rcmb.2005-0476OC. [DOI] [PubMed] [Google Scholar]
- 66.Zhong H, Belardinelli L, Maa T, Zeng D. Synergy between A2B adenosine receptors and hypoxia in activating human lung fibroblasts. Am. J. Respir. Cell. Mol. Biol. 2005;32:2–8. doi: 10.1165/rcmb.2004-0103OC. [DOI] [PubMed] [Google Scholar]
- 67.Sun CX, et al. Role of A2B adenosine receptor signaling in adenosine-dependent pulmonary inflammation and injury. J. Clin. Invest. 2006;116:2173–2182. doi: 10.1172/JCI27303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mustafa SJ, et al. Effect of a specific and selective A2B adenosine receptor antagonist on adenosine agonist AMP and allergen-induced airway responsiveness and cellular influx in a mouse model of asthma. J. Pharmacol. Exp. Ther. 2007;320:1246–1251. doi: 10.1124/jpet.106.112250. [DOI] [PubMed] [Google Scholar]
- 69.Holgate ST. The Quintiles Prize Lecture 2004. The identification of the adenosine A2B receptor as a novel therapeutic target in asthma. Br. J. Pharmacol. 2005;145:1009–1015. doi: 10.1038/sj.bjp.0706272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Reutershan J, Cagnina RE, Chang D, Linden J, Ley K. Therapeutic anti-inflammatory effects of myeloid cell adenosine receptor A2a stimulation in lipopolysaccharide-induced lung injury. J. Immunol. 2007;179:1254–1263. doi: 10.4049/jimmunol.179.2.1254. [DOI] [PubMed] [Google Scholar]
- 71.Nadeem A, Fan M, Ansari HR, Ledent C, Jamal Mustafa S. Enhanced airway reactivity and inflammation in A2A adenosine receptor-deficient allergic mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007;292:L1335–L1344. doi: 10.1152/ajplung.00416.2006. [DOI] [PubMed] [Google Scholar]
- 72.Luijk B, et al. Effect of an inhaled adenosine A2A agonist on the allergen-induced late asthmatic response. Allergy. 2008;63:75–80. doi: 10.1111/j.1398-9995.2007.01557.x. [DOI] [PubMed] [Google Scholar]
- 73.Day YJ, et al. A2A adenosine receptors on bone marrow-derived cells protect liver from ischemia-reperfusion injury. J. Immunol. 2005;174:5040–5046. doi: 10.4049/jimmunol.174.8.5040. [DOI] [PubMed] [Google Scholar]
- 74.Day YJ, et al. Renal protection from ischemia mediated by A2A adenosine receptors on bone marrow-derived cells. J. Clin. Invest. 2003;112:883–891. doi: 10.1172/JCI15483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yang Z, et al. Myocardial infarct-sparing effect of adenosine A2A receptor activation is due to its action on CD4+ T lymphocytes. Circulation. 2006;114:2056–2064. doi: 10.1161/CIRCULATIONAHA.106.649244. [DOI] [PubMed] [Google Scholar]
- 76.Peirce SM, Skalak TC, Rieger JM, Macdonald TL, Linden J. Selective A2A adenosine receptor activation reduces skin pressure ulcer formation and inflammation. Am. J. Physiol. Heart Circ. Physiol. 2001;281:H67–H74. doi: 10.1152/ajpheart.2001.281.1.H67. [DOI] [PubMed] [Google Scholar]
- 77.Li Y, et al. Mouse spinal cord compression injury is reduced by either activation of the adenosine A2A receptor on bone marrow-derived cells or deletion of the A2A receptor on non-bone marrow-derived cells. Neuroscience. 2006;141:2029–2039. doi: 10.1016/j.neuroscience.2006.05.014. [DOI] [PubMed] [Google Scholar]
- 78.Reece TB, et al. Functional and cytoarchitectural spinal cord protection by ATL-146e after ischemia/reperfusion is mediated by adenosine receptor agonism. J. Vasc. Surg. 2006;44:392–397. doi: 10.1016/j.jvs.2006.04.032. [DOI] [PubMed] [Google Scholar]
- 79.Reece TB, et al. Adenosine A2A receptor activation reduces inflammation and preserves pulmonary function in an in vivo model of lung transplantation. J. Thorac. Cardiovasc. Surg. 2005;129:1137–1143. doi: 10.1016/j.jtcvs.2004.11.042. [DOI] [PubMed] [Google Scholar]
- 80.Li L, et al. NKT cell activation mediates neutrophil IFN-γ production and renal ischemia-reperfusion injury. J. Immunol. 2007;178:5899–5911. doi: 10.4049/jimmunol.178.9.5899. [DOI] [PubMed] [Google Scholar]
- 81.Chabner BA, et al. Polyglutamation of methotrexate. Is methotrexate a prodrug? J. Clin. Invest. 1985;76:907–912. doi: 10.1172/JCI112088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Allegra CJ, Drake JC, Jolivet J, Chabner BA. Inhibition of phosphoribosylaminoimidazole-carboxamide transformylase by methotrexate and dihydrofolic acid polyglutamates. Proc. Natl Acad. Sci. USA. 1985;82:4881–4885. doi: 10.1073/pnas.82.15.4881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Baggott JE, Vaughn WH, Hudson BB. Inhibition of 5-aminoimidazole-4-carboxamide ribotide transformylase, adenosine deaminase and 5′-adenylate deaminase by polyglutamates of methotrexate and oxidized folates and by 5-aminoimidazole-4-carboxamide riboside and ribotide. Biochem. J. 1986;236:193–200. doi: 10.1042/bj2360193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cronstein BN, Naime D, Ostad E. The anti-inflammatory mechanism of methotrexate. Increased adenosine release at inflamed sites diminishes leukocyte accumulation in an in vivo model of inflammation. J. Clin. Invest. 1993;92:2675–2682. doi: 10.1172/JCI116884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Moser GH, Schrader J, Deussen A. Turnover of adenosine in plasma of human and dog blood. Am. J. Physiol. 1989;256:C799–C806. doi: 10.1152/ajpcell.1989.256.4.C799. [DOI] [PubMed] [Google Scholar]
- 86.Baggott JE, Morgan SL, Sams WM, Linden J. Urinary adenosine and aminoimidazolecarboxamide excretion in methotrexate-treated patients with psoriasis. Arch. Dermatol. 1999;135:813–817. doi: 10.1001/archderm.135.7.813. [DOI] [PubMed] [Google Scholar]
- 87.Riksen NP, et al. Methotrexate modulates the kinetics of adenosine in humans in vivo. Ann. Rheum. Dis. 2006;65:465–470. doi: 10.1136/ard.2005.048637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cronstein BN, Eberle MA, Gruber HE, Levin RI. Methotrexate inhibits neutrophil function by stimulating adenosine release from connective tissue cells. Proc. Natl Acad. Sci. USA. 1991;88:2441–2445. doi: 10.1073/pnas.88.6.2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Morabito L, et al. Methotrexate and sulfasalazine promote adenosine release by a mechanism that requires ecto-5′-nucleotidase-mediated conversion of adenine nucleotides. J. Clin. Invest. 1998;101:295–300. doi: 10.1172/JCI1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Montesinos MC, et al. Reversal of the antiinflammatory effects of methotrexate by the nonselective adenosine receptor antagonists theophylline and caffeine: evidence that the antiinflammatory effects of methotrexate are mediated via multiple adenosine receptors in rat adjuvant arthritis. Arthritis Rheum. 2000;43:656–663. doi: 10.1002/1529-0131(200003)43:3<656::AID-ANR23>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 91.Montesinos MC, et al. Adenosine A2A or A3 receptors are required for inhibition of inflammation by methotrexate and its analog MX-68. Arthritis Rheum. 2003;48:240–247. doi: 10.1002/art.10712. [DOI] [PubMed] [Google Scholar]
- 92.Delano DL, et al. Genetically based resistance to the antiinflammatory effects of methotrexate in the air-pouch model of acute inflammation. Arthritis Rheum. 2005;52:2567–2575. doi: 10.1002/art.21208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Montesinos MC, Desai A, Cronstein BN. Suppression of inflammation by low-dose methotrexate is mediated by adenosine A2A receptor but not A3 receptor activation in thioglycollate-induced peritonitis. Arthritis Res. Ther. 2006;8:R53. doi: 10.1186/ar1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Montesinos MC, et al. The antiinflammatory mechanism of methotrexate depends on extracellular conversion of adenine nucleotides to adenosine by ecto-5′-nucleotidase: findings in a study of ecto-5′-nucleotidase gene-deficient mice. Arthritis Rheum. 2007;56:1440–1445. doi: 10.1002/art.22643. [DOI] [PubMed] [Google Scholar]
- 95.Nesher G, Mates M, Zevin S. Effect of caffeine consumption on efficacy of methotrexate in rheumatoid arthritis. Arthritis Rheum. 2003;48:571–572. doi: 10.1002/art.10766. [DOI] [PubMed] [Google Scholar]
- 96.Baharav E, et al. Antiinflammatory effect of A3 adenosine receptor agonists in murine autoimmune arthritis models. J. Rheumatol. 2005;32:469–476. [PubMed] [Google Scholar]
- 97.Bar-Yehuda S, et al. The anti-inflammatory effect of A3 adenosine receptor agonists: a novel targeted therapy for rheumatoid arthritis. Expert Opin. Investig. Drugs. 2007;16:1601–1613. doi: 10.1517/13543784.16.10.1601. [DOI] [PubMed] [Google Scholar]
- 98.Szabo C, et al. Suppression of macrophage inflammatory protein (MIP)-1α production and collagen-induced arthritis by adenosine receptor agonists. Br. J. Pharmacol. 1998;125:379–387. doi: 10.1038/sj.bjp.0702040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Silverman MH, et al. Clinical evidence for utilization of the A3 adenosine receptor as a target to treat rheumatoid arthritis: data from a phase II clinical trial. J. Rheumatol. 2008;35:41–48. [PubMed] [Google Scholar]
- 100.Ochaion A, et al. Methotrexate enhances the anti-inflammatory effect of CF101 via up-regulation of the A3 adenosine receptor expression. Arthritis Res. Ther. 2006;8:R169. doi: 10.1186/ar2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Law WR. Adenosine receptors in the response to sepsis: what do receptor-specific knockouts tell us? Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006;291:R957–R958. doi: 10.1152/ajpregu.00412.2006. [DOI] [PubMed] [Google Scholar]
- 102.Gallos G, Ruyle TD, Emala CW, Lee HT. A1 adenosine receptor knockout mice exhibit increased mortality, renal dysfunction, and hepatic injury in murine septic peritonitis. Am. J. Physiol. Renal Physiol. 2005;289:F369–F376. doi: 10.1152/ajprenal.00470.2004. [DOI] [PubMed] [Google Scholar]
- 103.Lee HT, et al. A3 adenosine receptor activation decreases mortality and renal and hepatic injury in murine septic peritonitis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006;291:R959–R969. doi: 10.1152/ajpregu.00034.2006. [DOI] [PubMed] [Google Scholar]
- 104.Nemeth ZH, et al. Adenosine A2A receptor inactivation increases survival in polymicrobial sepsis. J. Immunol. 2006;176:5616–5626. doi: 10.4049/jimmunol.176.9.5616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sullivan GW, Fang G, Linden J, Scheld WM. A2A adenosine receptor activation improves survival in mouse models of endotoxemia and sepsis. J. Infect. Dis. 2004;189:1897–1904. doi: 10.1086/386311. [DOI] [PubMed] [Google Scholar]
- 106.Bamias G, Cominelli F. Immunopathogenesis of inflammatory bowel disease: current concepts. Curr. Opin. Gastroenterol. 2007;23:365–369. doi: 10.1097/MOG.0b013e3281c55eb2. [DOI] [PubMed] [Google Scholar]
- 107.Izcue A, Coombes JL, Powrie F. Regulatory T cells suppress systemic and mucosal immune activation to control intestinal inflammation. Immunol. Rev. 2006;212:256–271. doi: 10.1111/j.0105-2896.2006.00423.x. [DOI] [PubMed] [Google Scholar]
- 108.Odashima M, et al. Activation of A2A adenosine receptor attenuates intestinal inflammation in animal models of inflammatory bowel disease. Gastroenterology. 2005;129:26–33. doi: 10.1053/j.gastro.2005.05.032. [DOI] [PubMed] [Google Scholar]
- 109.Sitaraman SV, et al. Neutrophil-epithelial crosstalk at the intestinal lumenal surface mediated by reciprocal secretion of adenosine and IL-6. J. Clin. Invest. 2001;107:861–869. doi: 10.1172/JCI11783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kolachala V, et al. TNF-α upregulates adenosine 2b (A2b) receptor expression and signaling in intestinal epithelial cells: a basis for A2b R overexpression in colitis. Cell. Mol. Life Sci. 2005;62:2647–2657. doi: 10.1007/s00018-005-5328-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kolachala VL, Bajaj R, Chalasani M, Sitaraman SV. Purinergic receptors in gastrointestinal inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 2008;294:G401–G410. doi: 10.1152/ajpgi.00454.2007. [DOI] [PubMed] [Google Scholar]
- 112.Kolachala VL, et al. A2B adenosine receptor gene deletion attenuates murine colitis. Gastroenterology. 2008 May 22; doi: 10.1053/j.gastro.2008.05.049. (doi:10.1053/j.gastro.2008.05.049) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Montesinos MC, et al. Wound healing is accelerated by agonists of adenosine A2 (Gαs-linked) receptors. J. Exp. Med. 1997;186:1615–1620. doi: 10.1084/jem.186.9.1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Victor-Vega C, Desai A, Montesinos MC, Cronstein BN. Adenosine A2A receptor agonists promote more rapid wound healing than recombinant human platelet-derived growth factor (Becaplermin gel) Inflammation. 2002;26:19–24. doi: 10.1023/a:1014417728325. [DOI] [PubMed] [Google Scholar]
- 115.Montesinos MC, et al. Adenosine promotes wound healing and mediates angiogenesis in response to tissue injury via occupancy of A2A receptors. Am. J. Pathol. 2002;160:2009–2018. doi: 10.1016/S0002-9440(10)61151-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Macedo L, et al. Wound healing is impaired in MyD88-deficient mice: a role for MyD88 in the regulation of wound healing by adenosine A2A receptors. Am. J. Pathol. 2007;171:1774–1788. doi: 10.2353/ajpath.2007.061048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Feoktistov I, et al. Differential expression of adenosine receptors in human endothelial cells: role of A2B receptors in angiogenic factor regulation. Circ. Res. 2002;90:531–538. doi: 10.1161/01.res.0000012203.21416.14. [DOI] [PubMed] [Google Scholar]
- 118.Merighi S, et al. Caffeine inhibits adenosine-induced accumulation of hypoxia-inducible factor-1α, vascular endothelial growth factor, and interleukin-8 expression in hypoxic human colon cancer cells. Mol. Pharmacol. 2007;72:395–406. doi: 10.1124/mol.106.032920. [DOI] [PubMed] [Google Scholar]
- 119.Khoa ND, et al. Inflammatory cytokines regulate function and expression of adenosine A2A receptors in human monocytic THP-1 cells. J. Immunol. 2001;167:4026–4032. doi: 10.4049/jimmunol.167.7.4026. [DOI] [PubMed] [Google Scholar]
- 120.Desai A, et al. Adenosine A2A receptor stimulation increases angiogenesis by down-regulating production of the antiangiogenic matrix protein thrombospondin 1. Mol. Pharmacol. 2005;67:1406–1413. doi: 10.1124/mol.104.007807. [DOI] [PubMed] [Google Scholar]
- 121.Leibovich SJ, et al. Synergistic up-regulation of vascular endothelial growth factor expression in murine macrophages by adenosine A2A receptor agonists and endotoxin. Am. J. Pathol. 2002;160:2231–2244. doi: 10.1016/S0002-9440(10)61170-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Pinhal-Enfield G, et al. An angiogenic switch in macrophages involving synergy between Toll-like receptors 2, 4, 7, and 9 and adenosine A2A receptors. Am. J. Pathol. 2003;163:711–721. doi: 10.1016/S0002-9440(10)63698-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Chan ES, et al. Adenosine A2A receptors in diffuse dermal fibrosis: pathogenic role in human dermal fibroblasts and in a murine model of scleroderma. Arthritis Rheum. 2006;54:2632–2642. doi: 10.1002/art.21974. [DOI] [PubMed] [Google Scholar]
- 124.Chunn JL, et al. Partially adenosine deaminase-deficient mice develop pulmonary fibrosis in association with adenosine elevations. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006;290:L579–L587. doi: 10.1152/ajplung.00258.2005. [DOI] [PubMed] [Google Scholar]
- 125.Chen Y, et al. Functional effects of enhancing or silencing adenosine A2b receptors in cardiac fibroblasts. Am. J. Physiol. Heart Circ. Physiol. 2004;287:H2478–H2486. doi: 10.1152/ajpheart.00217.2004. [DOI] [PubMed] [Google Scholar]
- 126.Yegutkin GG. Nucleotide- and nucleoside-converting ectoenzymes: important modulators of purinergic signalling cascade. Biochim. Biophys. Acta. 2008;1783:673–694. doi: 10.1016/j.bbamcr.2008.01.024. [DOI] [PubMed] [Google Scholar]
- 127.Hasko G, Cronstein BN. Adenosine: an endogenous regulator of innate immunity. Trends Immunol. 2004;25:33–39. doi: 10.1016/j.it.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 128.Fredholm BB, Irenius E, Kull B, Schulte G. Comparison of the potency of adenosine as an agonist at human adenosine receptors expressed in Chinese hamster ovary cells. Biochem. Pharmacol. 2001;61:443–448. doi: 10.1016/s0006-2952(00)00570-0. [DOI] [PubMed] [Google Scholar]
- 129.Fredholm BB. Adenosine, an endogenous distress signal, modulates tissue damage and repair. Cell Death Differ. 2007;14:1315–1323. doi: 10.1038/sj.cdd.4402132. [DOI] [PubMed] [Google Scholar]
- 130.Zhou QY, et al. Molecular cloning and characterization of an adenosine receptor: the A3 adenosine receptor. Proc. Natl Acad. Sci. USA. 1992;89:7432–7436. doi: 10.1073/pnas.89.16.7432. [DOI] [PMC free article] [PubMed] [Google Scholar]