

Abstract
CprK from Desulfitobacterium dehalogenans is the first characterized transcriptional regulator of anaerobic dehalorespiration and is controlled at two levels: redox and effector binding. In the reduced state and in the presence of chlorinated aromatic compounds, CprK positively regulates expression of the cpr gene cluster. One of the products of the cpr gene cluster is CprA, which catalyzes the reductive dehalogenation of chlorinated aromatic compounds. Redox regulation of CprK occurs through a thiol/disulfide redox switch, which includes two classes of cysteine residues. Under oxidizing conditions, Cys11 and Cys200 form an intermolecular disulfide bond, whereas Cys105 and Cys111 form an intramolecular disulfide. Here, we report that Cys11 is involved in redox inactivation in vivo. Upon replacement of Cys11 with serine, alanine, or aspartate, CprK loses its DNA binding activity. C11A is unstable; however, circular dichroism studies demonstrate that the stability and overall secondary structures of CprK and the C11S and C11D variants are similar. Furthermore, effector binding remains intact in the C11S and C11D variants. However, fluorescence spectroscopic results reveal that the tertiary structures of the C11S and C11D variants differ from that of the wild type protein. Thus, Cys11 plays a dual role as a redox switch and in maintaining the correct tertiary structure that promotes DNA binding.
Desulfitobacterium dehalogenans, one of the most extensively studied reductive dehalogenating bacteria, was identified 13 years ago (1). It is an anaerobic, Gram-positive bacterium with low G+C content (2). D. dehalogenans can use 3-chloro-4-hydroxyphenylacetate (CHPA)2 and several other chlorinated aromatic compounds, including polychlorinated biphenyls, as electron acceptors (3, 4) in an energy yielding process called dehalorespiration (1, 5). In the absence of chlorinated aromatics, this organism can also utilize sulfite, thiosulfate, fumarate, and nitrite as electron acceptors (1).
Genes that encode proteins involved in dehalorespiration are present within the cpr (chlorophenol reduction) gene cluster, which contains eight genes, cprT, cprK, cprZ, cprE, cprB, cprA, cprC, and cprD, which are located in five transcriptional units (cprK, cprT, cprZE, and cprBA or cprBACD) controlled by three promoters (6). CprK is constitutively expressed at low levels and acts as a transcriptional regulator for the cpr gene cluster (7). CprA catalyzes the reductive dehalogenation of chlorinated aromatic compounds and CprB is an integral membrane protein that binds CprA, whereas CprT, CprB, CprC, and CprD seem to be involved in maturation of CprA (2). The function of CprZ is unknown.
CprK belongs to the superfamily of CRP-FNR (cAMP-binding protein-fumarate nitrate reduction regulatory protein) transcription regulators (8). Like other family members, CprK has a C-terminal winged helix-turn-helix DNA-binding motif (9) and an N-terminal effector binding motif. CHPA binds to the effector domain in CprK, which triggers a conformational change (10) that promotes the interaction of CprK with a nearly palindromic DNA sequence called a “dehalobox” (11) that is located in the promoter regions of the cprT, cprZ, and cprB genes in the cpr gene cluster (7). Based on studies of a related transcriptional activator (CprK1) from Desulfitobacterium hafniense, the optimal and consensus dehalobox sequence appears to be a perfect inverted repeat, TTAATX4ATTAA (11), that resembles the FNR consensus sequence (TTGATX4ATCAA) (12). The effector (a chlorinated aromatic) can bind to CprK in both the reduced and oxidized states, but the protein must be in the reduced state to bind DNA (13, 14). Thus, dehalorespiration is regulated at the transcriptional level by effector binding and by redox state. As shown in Fig. 1, transcription only occurs under reducing conditions and when effector (the chlorinated aromatic substrate for the dehalogenase, CprA) is present.
FIGURE 1.

Regulation of CprK. CprK can exist in a thiol-oxidized state, containing an intermolecular disulfide linkage between Cys11 and Cys200 and an intramolecular disulfide linkage between Cys105 and Cys111, or a thiol-reduced state, in which all these Cys residues are in the thiol(ate) form. Oxidized and reduced CprK bind effector (CHPA) with similar affinity. The oxidized state of CprK has low affinity for DNA, whereas the reduced form with effector binds DNA with high affinity and positively regulates expression of the cpr gene cluster. The products of this gene cluster are involved in catalysis of dehalorespiration (10).
CprK from D. dehalogenans has five cysteine residues: Cys11, Cys105, Cys111, Cys161, and Cys200. All cysteines except Cys161 are involved in redox-regulation of CprK. Upon oxidation, CprK loses DNA binding activity (7) because of the formation of an intermolecular disulfide bond between Cys11 and Cys200 and/or an intramolecular disulfide bond between Cys105 and Cys111 (13). Based on the crystal structures of oxidized D. hafniense CprK1 and reduced D. dehalogenans CprK, an inactive state is stabilized by linkage of the effector domain to the DNA-binding domain by the intermolecular disulfide bond. In addition, mass spectrometric studies reveal that several regions of the structure exhibit enhanced dynamics upon reduction (14).
The physiological relevance of the disulfide bond between Cys11 and Cys200 in redox regulation has been questioned (11). Here, we show that the intermolecular disulfide bond between Cys11 and Cys200 is observed in vivo upon exposure of Escherichia coli cells overexpressing CprK to oxidative stress conditions. We also show that replacement of Cys11 with Ser or Asp results in a stable but inactive protein. The results of fluorescence spectroscopic studies and electrophoreic mobility shift assays (EMSA) reveal that the C11D and C11S variants can still bind effector with high affinity but are unable to bind DNA. Changes in the fluorescence spectrum of the variants reveal that the loss of DNA binding activity in the Cys11 variants is related to a change in the tertiary structure of the protein. Thus, Cys11 plays a dual role in maintaining the correct structure for DNA binding and in redox regulation of CprK.
MATERIALS AND METHODS
Cloning, Overexpression, and Purification of CprK—DNA isolation and manipulation were performed by standard techniques (15). Plasmid DNA was purified with a QIAprep spin miniprep kit (Qiagen). Construction of the overexpression plasmid and purification of CprK were described previously (7).
Site-directed Mutagenesis of CprK—Cys11 was substituted with alanine, serine, and aspartate residues by performing site-directed mutagenesis according to the QuikChange protocol from Stratagene (La Jolla, CA). The pQE60:cprK plasmid was the template for PCR, and the primers were purchased from Integrated DNA Technologies. The DNA sequences of all PCR-generated DNA fragments were confirmed using dye terminator chemistry and automated sequencing of both DNA strands with a Beckman/Coulter CEQ2000XL 8-capillary DNA sequencer at the Genomics Core Research Facility (University of Nebraska, Lincoln, NE).
Construction of Strains for lacZ Reporter Assays—To measure in vivo activity of CprK, a two-plasmid reporter system was constructed in which an arabinose-inducible cprK gene on one plasmid and a cpr promoter-lacZ fusion on another plasmid are cotransformed into an ara- E. coli strain. Wild type CprK was cloned into the pBAD-Myc-HisA expression vector (Invitrogen), generating pBAD/Myc/HisA::cprK. To express the native protein lacking the His tag, the native stop codon was included before the sequence that encodes the His tag. Cys11 variants were generated by site-directed mutagenesis in the same construct. A cpr promoter-lacZ fusion was generated by cloning the promoter region of cprB into the EcoRI cloning site of pRS551 (16) to yield pRS551::PcprB, which was used to transform the ara- E. coli strain LMG194 (Invitrogen). Then the pBAD/Myc/HisA::cprK construct (and the corresponding C11A, C11S, and C11D variants) was transformed into the LMG194 cells containing pRS551::PcprB to generate the two-plasmid reporter system. The pBAD system provides tight control of CprK expression (17), and the E. coli strain provides a background that lacks dehalorespiration genes.
Assay of in Vivo Activity of CprK—Promoter activity was measured in vivo by the β-galactosidase assay. Overnight cultures were grown aerobically at 37 °C in RM medium plus 50 μm CHPA, 0.2% arabinose, 100 μg/ml ampicillin, and 34 μg/ml chloramphenicol. When required, diamide (1 mm final concentration) was added to the culture at an A600 of ∼0.2. Growth was stopped during exponential phase by placing the cultures on ice when the A600 reached 0.4-0.6. After 20 min, 500 μl of Z buffer (60 mm Na2HPO4·7H2O, 40 mm NaH2PO4·H2O, 10 mm KCl, 1 mm MgSO4, and 50 mm β-mercaptoethanol, pH 7.0), 50 ml of 0.1% SDS, and two drops of chloroform were added to 500 μl of culture, and the solution was incubated at 28 °C for at least 5 min when 200 μl of 4 mg/ml O-nitrophenyl-β-d-galactoside (final concentration, 0.8 mg/ml) was added. The reaction was stopped by adding 500 μl of 1 m Na2CO3 when the reaction mixture began to turn yellow, and the time of reaction was recorded. The absorbances at 420 and 550 nm were then measured to calculate the promoter activity in Miller units as described (18). Each reaction was performed in quadruplicate.
In Vivo Intermolecular Disulfide Bond Detection—E. coli strains containing the cprK gene were grown in LB medium at 37 °C. At an A600 of ∼0.6, a 0.5-ml sample was centrifuged, and the cell pellet was immediately frozen in liquid nitrogen. Diamide (1 mm, final concentration) was added to the rest of the culture, and 0.5 ml samples were collected from the culture after 5, 10, 30, and 60 min, centrifuged, and frozen as just described. Nonreducing loading buffer (60 mm Tris-Cl, pH 6.8, 1% SDS, 10% glycerol, 0.01% bromphenol blue) was then added to the cell pellets at varying volumes to provide samples with final equal cell density (based on the absorbance at 600 nm). Finally, 10 μl of each sample was loaded on a 15% SDS-PAGE gel and immunoblotted with an anti-CprK antibody. After electrophoresis, the proteins were transferred to a polyvinylidene difluoride membrane, which was first incubated with primary rabbit anti-CprK antibody (Cocalico Biologicals Inc., Reamstown, PA) that had been purified by chromatography on a CprK-actigel ALD Superflow affinity column (Sterogene Bioseparations Inc., Carlsbad, CA). The membrane was then treated with secondary antibody (goat anti-rabbit IgG conjugated to horseradish peroxidase; Sigma-Aldrich), and the presence of CprK was detected by chemiluminescence, using the protocol recommended by the manufacturer (Sigma-Aldrich).
Electrophoretic Mobility Shift Assay—EMSAs were performed as described (7) with increasing amounts of IRDye 700-labeled oligonucleotides (LI-COR, Inc., Lincoln). The data were analyzed by infrared imaging with an ODYSSEY infrared imager (LI-COR).
Liquid Chromatography Mass Spectrometry Analysis—The intact protein was analyzed at the Mass Spectrometry Core Facility (University of Nebraska, Lincoln) by an liquid chromatography tandem mass spectrometry system comprised of a Shimadzu (SCL-10A) high pressure liquid chromatography system, a 4000 Qtrap (ABS) mass spectrometry system, and a turbo ion-spray source probe. Protein samples (20 ml) were loaded onto a Micro-Tech Scientific C18 column (1.0 (inner diameter) × 50 mm (length), 5 μm (particle size)) by a PE 200 Autosampler and eluted at a flow rate of 100 μl/min at room temperature with a gradient of 0.3% formic acid in H2O (Solvent A) and 0.3% formic acid in acetonitrile (Solvent B). The percentage of Solvent B was increased linearly from 10 to 70% over a 5-min elution time. The data were acquired and processed with Analyst 1.4.1 software. The data were acquired in the Q1 (quadrupole one) positive ion mode as the mass range (m/z) of 850-1150 atomic mass units was scanned in 4 s. The total run time for each sample was 20 min. The molecular mass of the protein was calculated by analyzing several multiply charged peaks with the Bayesian Protein Reconstruct option in Bioanalyst 1.4.1 software.
Intrinsic Fluorescence Quenching and CD Analysis—For the CHPA binding assay shown in Fig. 5, 180-500 nm CprK was analyzed by fluorescence spectroscopy with a Shimadzu RF-530 1 PC Spectrofluorophotometer (Columbia, MD) at room temperature as described (3, 13). All of the fluorescence data were corrected for the inner filter effect (19). The intrinsic tryptophan fluorescence spectra of wild type CprK, C11S, and C11D variants shown in Fig. 8 were recorded on an OLIS RSM1000F (OLIS, Inc., Bogart, GA).
FIGURE 5.

Effector binding affinity of wild type and Cys11 variants. Fluorescence intensities were measured, as described (10) for the oxidized (right) and reduced (left) wild type (WT, ⋄), C11D (○), and C11S (□) variants. The dashed and solid lines represent the fits to one-site binding models according to Equations 1 and 2, respectively, as described under “Materials and Methods.” Effector concentrations are given in micromolar units. The Kd values are given in Table 1.
FIGURE 8.

Intrinsic fluorescence spectra of CprK. Spectra were recorded for wild type (WT) CprK, C11S and C11D in 50 mm Tris-HCl, 300 mm NaCl, pH 7.5 (A), or the same buffer containing 50 mm DTT (B). Emission spectra were scanned from 300 to 400 nm. The data presented here are representative of three different experiments.
Binding of CHPA by CprK causes quenching of intrinsic tryptophan
fluorescence. Dissociation constants were calculated by fitting the
fluorescence quenching data to Equation 1 or 2, where Kd
represents the dissociation constant for the ligand (L, CHPA), [L] is
the total ligand concentration, [P] equals the total protein concentration,
ΔF is the observed fluorescence quenching,
F0 equals the initial fluorescence, and
ΔFmax is the maximum quenching that would be
observed at infinite ligand concentrations. These equations assume a single
binding site on CprK for CHPA. The [L] in Equation 1, which describes a
standard one-site binding isotherm, should be free ligand, and because the
free ligand concentration is unknown, we assumed that the free and total
ligand concentrations are equal. However, when the [L] is in the range of the
Kd, the fraction of bound ligand becomes significant;
therefore, the data were also fit to quadratic Equation 2, which accounts for
ligand depletion at low CHPA concentrations. We also estimated the free ligand
concentration by subtracting bound ligand ([P] *
ΔF/Fmax) from [L] and fit this data to
Equation 1.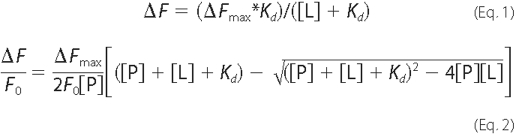
The different one-site treatments gave similar dissociation constants, whereas a two-site model gave a Kd1 in the low nanomolar range with high errors and a Kd2 that was similar to the Kd value obtained from the one-site model. The R2 values were also slightly better for the one-site model. Thus, we conclude that binding of CHPA to CprK follows a one-site binding model.
CD measurements were performed at 4 °C with a JASCO J-715 instrument (Jasco Inc.). The spectra were scanned from 260 to 190 nm at a speed of 50 nm/min with a data pitch of 1 nm. Forty scans at a 2-s response time and a 5-nm bandwidth were averaged for each sample. For secondary structure analysis, experiments were performed in a 0.1-mm-path length cell with 0.7 mg/ml CprK in 10 mm potassium phosphate buffer, pH 7.5. The melting temperature of CprK was determined by measuring the CD spectrum of 0.2 mg/ml protein (in 50 mm Tris, 300 mm NaCl, pH 7.5) over a temperature range of 20-80 °C in a 1-mm-path length cell. The data were obtained in millidegrees and converted into molar ellipticity by JASCO software. The Continll program (20) within the CDpro software (21) was used for secondary structure analysis.
RESULTS
Evidence for in Vivo Disulfide Bond Formation between Cys11 and Cys200—E. coli strains overexpressing CprK were grown in the presence or absence of oxidant (diamide or hydrogen peroxide), and samples were collected at 1, 5, 10, 30, and 60 min. When standard reducing and denaturing PAGE was performed, the wild type or variant (C11A, C11S, C200S, and C200A) proteins were observed at the position of the monomer (25 kDa); the samples were prepared for nonreducing SDS-PAGE analysis to identify the oligomeric state of CprK, omitting DTT, as described under “Materials and Methods.” When cells were treated in the absence of oxidant, CprK was predominantly in the monomeric state (Fig. 2A, first lane). However, when the cells were treated with 1 mm diamide, CprK was rapidly converted from the monomeric to the dimeric form (second through sixth lanes). Dimer formation was also observed when cells were treated with hydrogen peroxide (Fig. 2B).
FIGURE 2.

In vivo Cys11-Cys200 disulfide bond formation upon treatment of cells with oxidants. Exposure of cells containing wild type CprK to 1 mm DA (A) or 1 mm hydrogen peroxide (B) or after treatment of the C11S variant (C) and the C200S variant (D) with 1 mm DA followed by separation on non reducing SDS-PAGE and Western blot analysis. The membranes were immunoblotted with anti-CprK antibody and stained as described under “Materials and Methods.” WT, wild type.
Treatment of the C11S variant with diamide did not result in dimer formation (Fig. 2C). Similarly, the C200S variant remained predominantly in the monomeric state after treatment with diamide (Fig. 2D), with a small proportion of the dimeric form, as was observed in earlier in vitro experiments (13). Another band in the 15-20-kDa region appeared in the C200S variant, which might reflect a degradation product. In summary, these results indicate that in vivo Cys11 and Cys200 exist as the free thiol(ates) under normal growth conditions and that they form an intermolecular disulfide bond when exposed to oxidants.
Mass spectrometric analyses of the purified Cys11 variants revealed that the C11S and C11D variants are full-length proteins just like the wild type CprK (supplemental Fig. S1), with the only observed differences being the site-directed amino acid replacements.
Substitutions of Cys11 Inactivate CprK—To analyze the role of Cys11, in vivo β-galactosidase and in vitro DNA binding assays were performed with C11S and C11D variants. Attempts to purify the C11A variant failed, whereas the C11D and C11S variants could be purified at levels of 10-20 mg/liter of culture. EMSA assays were performed on the wild type and variant forms of CprK in the presence of effector. Neither the C11S nor C11D variants showed any mobility shift of pCprB (Fig. 3).
FIGURE 3.

InvitroDNAbindingactivityofCys11 variants.A, EMSA experiments after incubation of wild type (WT) CprK (lanes 1-4) or C11S (lanes 5-7) with DNA in the presence of DA (lanes 2 and 5), DTT (lanes 3 and 6), or DTT+DA (lanes 4 and 7). B, EMSA experiments comparing wild type CprK with C11D. Lane 2 has wild type CprK with 50 mm DTT. Lanes 3 and 4 have the C11D variant with 50 mm DTT and 5 mm DA, respectively. Lane 1 in A or B is a control that lacks protein.
Similarly, in vivo analysis by the cprB promoter-lacZ fusion assay demonstrated that the C11S, C11A, and C11D variants were severely compromised in DNA binding, with 2-3% β-galactosidase activity of the wild type protein (Fig. 4). Diamide-treated wild type CprK showed 10% activity, indicating that oxidative stress inactivates CprK in vivo and that the reducing conditions within the cell compete with diamide oxidation to restore the active protein. Diamide treatment of C11A, C11S, and C11D variants further decreased the DNA binding activity to background levels. Because formation of the Cys11-Cys200 disulfide bond is prevented in these variants, the additional decrease in activity could result from oxidation of Cys105 and Cys111 to form an intramolecular disulfide bond.
FIGURE 4.

In vivo DNA binding activity of Cys11 variants. E. coli strains expressing wild type (WT) CprK or Cys11 variants were incubated with no (filled bar) or 1 mm DA (open bar) and promoter activity was measured by the β-galactosidase assay as described under “Materials and Methods.” Activity is expressed as a percentage of Miller units exhibited by the variant compared with that of wild type CprK. The data are shown as the means ± S.D. and are representative of three separate experiments, each performed in quadruplicate.
Loss of DNA Binding Activity but Unimpaired Effector Binding of Cys11 Variants—To determine whether inactivity of the C11S and C11D variants is due to loss of effector or of DNA binding, effector binding affinity was measured by intrinsic tryptophan fluorescence (Fig. 5). As shown in Table 1 and Fig. 5, the oxidized and reduced states of CprK, as well as the Cys11 variants bind CHPA with high affinity, and the data for the wild type and Cys11 variant proteins are virtually superimposable. The binding data for the wild type protein and the Cys11 muteins best fit a single binding site model. The ranges of Kd values are 0.29-0.49 and 0.34-0.68 μm for the oxidized and reduced proteins, respectively. In fact, composite fits that include all the data for the oxidized proteins or the reduced proteins give standard deviations that are similar to those for the individual fits. In addition, similar Kd values are obtained by fitting the data to the standard binding isotherm given in Equation 1 (Fig. 5, dashed line) as with the more rigorous quadratic Equation 2 (solid line) that takes ligand depletion at low CHPA concentrations into account. Therefore, based on the composite fits, the Kd values for CHPA of the oxidized and reduced states of CprK are between 0.3 and 0.5 μm.
TABLE 1.
Dissociation constants for the complex between CprK (and variants) and CHPA
| Protein | Redox state | Kda |
|---|---|---|
| μm | ||
| Wild type | Reduced | 0.52 ± 0.18 |
| Oxidized | 0.49 ± 0.16 | |
| C11D | Reduced | 0.68 ± 0.11 |
| Oxidized | 0.31 ± 0.14 | |
| C11S | Reduced | 0.34 ± 0.11 |
| Oxidized | 0.29 ± 0.11 | |
| Combinedb | Reduced | 0.50 ± 0.09 |
| Oxidized | 0.30 ± 0.09 |
These values were calculated by fits to Equation 2, which accounts for ligand depletion at low concentrations of CHPA.
These values were obtained by fitting the combined data sets for wild type, C11S, and C11D.
Because the CprK variants with substitutions at Cys11 retain high affinity for CHPA, the inactivity of these variants was presumed to result from loss of DNA binding ability. We compared the affinity of the Cys11 variants for DNA with that of the wild type protein by performing EMSA at increasing DNA concentrations. The C11S variant requires greater than 50-fold more DNA to obtain the same amount of CprK-DNA complex as with the wild type protein (Fig. 6). Thus, only marginal activity is observed when the thiol of Cys11 is replaced by a hydroxide (in C11S). With the C11D variant (data not shown), no gel shift was observed even at 500 nm DNA, indicating either that the negative charge or the increased steric bulk of Asp at residue 11 strongly inhibits DNA binding. Thus, CprK promoter binding activity appears to require the Cys11 thiol(ate) functionality.
FIGURE 6.

Effect of increasing DNA concentration on DNA binding activity of C11S. Wild type (WT) CprK (first three lanes) and the C11S variant (last three lanes) were incubated with 10 nm to 500 nm DNA containing the cpr promoter. The middle lane was a control that lacked protein.
Loss of DNA Binding Activity May Reflect a Change in Tertiary Structure of Cprk—Although the first 18 amino acid residues were disordered and not seen in the crystal structure of the reduced protein, the structure of oxidized CprK reveals Cys11 to be positioned near the DNA-binding domain (Fig. 7) (10). To determine whether the loss of DNA binding activity in the Cys11 variants is associated with a change in the structure of CprK, intrinsic tryptophan fluorescence spectra were recorded of oxidized and DTT-reduced CprK and of the Cys11 variants (Fig. 8).
FIGURE 7.

Cys11 in the crystal structure of oxidized CprK. The crystal structure of oxidized D. hafniense CprK (left), focusing on the helix-turn-helix DNA-binding domain (boxed and expanded to the right). Cys11 and other residues predicted to be in the recognition helix, based on the structure of the CRP-DNA complex (10), are shown as space-filling diagrams and colored according to element. This figure was generated from Protein Data Bank code 2h6b in CHIMERA.
As observed earlier, reduction of CprK leads to quenching of intrinsic tryptophan fluorescence (13), indicating that the single Trp at position 106 becomes more solvent-exposed upon reduction (Fig. 8). The fluorescence spectra of the DTT-reduced forms of C11S and C11D coincide, with a significantly lower intensity than that of the reduced wild type protein (Fig. 8B). These results indicate that the tertiary structures of the reduced Cys11 variants (at least in the region of Trp106) are different from that of wild type CprK.
The fluorescence spectrum of the oxidized C11S variant is similar to that of the reduced state of wild type CprK (Fig. 8A), indicating that Trp106 in C11S is already rather solvent-exposed in the oxidized protein. The fluorescence spectrum of oxidized C11D is similar to that of oxidized wild type CprK (Fig. 8A), whereas reduction of C11D leads to a marked (∼40%) quenching of fluorescence (Fig. 8B).
Based on CD analysis, substitutions at Cys11 cause only marginal changes in secondary structure (supplemental Fig. S2 and Table 2), which are within or near the error limits of the experiment. Table 2 shows the results obtained from the secondary structure analysis for wild type CprK and the Cys11 variants. The normalized root mean squared deviation values for all of the fits are below 0.1, which indicates that the experimental data are well fit by parameters calculated by the Continll program.
TABLE 2.
Secondary structure analysis of wild type CprK and variants
Secondary structure fractions are shown here as analyzed by the Continll program (CDPro software) in comparison with the secondary structure fractions based on the x-ray structure (Protein Data Bank entry 2h6b) of wild type CprK. Variation from the secondary structure content of wild type is given in parentheses after the measured secondary structure contents of variants.
| Protein | α-Helix | β-Sheet | Turn | Unordered | NRMSDa |
|---|---|---|---|---|---|
| Wild type | 0.421 | 0.167 | 0.154 | 0.258 | 0.018 |
| C11S | 0.409 (2.8%) | 0.159 (4.8%) | 0.150 (2.6%) | 0.281 | 0.026 |
| C11D | 0.415 (1.4%) | 0.139 (16.7%) | 0.159 (3.2%) | 0.296 | 0.028 |
| X-ray | 0.42 | 0.26 |
NRMSD, normalized root mean square difference.
CD Measurements of Stabilities of Cys11 Variants—Stabilities of the Cys11 variants in the oxidized state were measured by recording the CD spectrum at 222 nm in the temperature range between 20 °C and 80 °C (supplemental Fig. S3). The actual melting temperature (Tm) could not be accurately determined because the thermal unfolding curves were irreversible. The wild type protein gave an approximate Tm of 54 °C, whereas the variants exhibited transitions at slightly lower temperatures, indicating that they have similar or only slightly decreased stability.
DISCUSSION
Upon binding its effector (CHPA), CprK induces the expression of genes involved in the use of chlorinated aromatic compounds as electron acceptors. Besides providing energy for the microbe, dehalorespiration helps to rid the environment of xenobiotics (like polychlorinated biphenyls) that constitute a significant health risk. Previous in vitro studies indicate that CprK contains two thiol/disulfide redox switches that regulate transcriptional activity of CprK: one involving Cys11 and Cys200 that, in the oxidized state, forms an intermolecular disulfide linkage, and another involving Cys105 and Cys111 that forms an intramolecular disulfide bond (13).
Redox regulation of CprK activity has been proposed to prevent expression of CprA under oxidative conditions (7). CprA catalyzes reductive dehalogenation through a mechanism involving oxygen-sensitive iron-sulfur clusters and the low valent Co(I) state of cobalamin (22). Here we describe further studies indicating that the intermolecular thiol/disulfide switch is formed in the E. coli cytoplasm under oxidative stress conditions. Exposure of D. dehalogenans to such oxidative conditions would likely occur commonly in this microaerophile, as it does in the facultative anaerobe E. coli. Because it would be wasteful to produce CprA under oxidative conditions, it would be advantageous for the cells to exert redox control of transcription of the cpr gene cluster.
Thus, as shown in Fig. 1, CprK is proposed to exert a two-tier mode of regulation of dehalorespiration involving a redox switch at one level and an effector-binding switch at another level. The studies described here also provide evidence that Cys11 plays a dual role in transcriptional activation: as part of the intermolecular redox switch and in maintaining an optimal structure of the DNA-binding domain of CprK.
Formation of either the intramolecular or the intermolecular disulfide bond appears to be sufficient to inhibit transcriptional activation, because variants containing substitutions at Cys105, Cys111, or Cys200 (or Cys161, which does not appear play a redox role) retain redox sensitivity and bind DNA and effector with affinities similar to those of the wild type protein (data not shown). Thus, formation of either the intramolecular or the intermolecular disulfide bond appears to reversibly inactivate CprK. Surprisingly, as shown in this paper, substitution of Cys11 with Ala, Ser, or Asp results in a protein that is unable to activate transcription in vivo or to bind the cpr promoter DNA in vitro, even under reducing conditions and at saturating concentrations of effector. Thus, beyond its involvement in the intermolecular redox switch, Cys11 plays an additional role in transcriptional activation. These results differ from studies in which C11S of the related CprK1 from D. hafniense was shown to retain DNA binding activity (11). Based on DNA sequence and mass spectrometric analysis, we are certain that the only mutation in these variants is at Cys11, and the redox inactivity is observed in forms of CprK containing (in vivo) and lacking (in vitro) the His tag sequence. Thus, it is difficult to harmonize our results with those described earlier (11), except to suggest that CprK1 in D. hafniense may exhibit different redox properties than CprK from D. dehalogenans. For example, Cys105 is absent in D. hafniense.
As shown in Fig. 7, Cys11 (in the oxidized protein) is very close to the recognition helix of the winged helix-turn-helix DNA-binding domain in CprK that binds to the dehalobox promoter sequence. Furthermore, binding of DNA protects the recognition helix (a 14-amino acid peptide including residues 182-196) from limited proteolysis (14). Although Cys11 is not observed in the structure of the reduced protein, reduction of CprK increases the overall structural dynamics of CprK in the region of Cys11 (14). Other regions that were shown to undergo local changes upon effector binding include the long C-helix that connects the DNA and effector binding domains and Arg152, which is in a proposed hinge region that may transmit the effector-binding signal to the DNA-binding domain.
By comparing the activities of CprK variants with Ala, Ser, or Asp substitution at Cys11, we expected to be able to determine whether Cys11 is involved in hydrophobic, H-bonding, or ionic interactions that are important for transcriptional activation. However, all three variants share similarly low abilities to activate transcription (Fig. 4). Although the C11A variant could not be purified for in vitro studies, C11S, C11D, and wild type CprK are all stable proteins that can be purified in large quantities and studied by various biochemical methods. As with the wild type protein, both oxidized and reduced states of the C11S and C11D variants have a single high affinity (0.3-0.7 μm) binding site for the effector CHPA (Fig. 5) Similarly, the crystal structure revealed a single CHPA molecule bound at the effector binding site of CprK (10). A single site binding model was used to fit the effector binding data for the related CprK from D. hafniense (10). Previous fluorescence quenching and isothermal calorimetry data on the wild type protein also demonstrated high affinity binding of CHPA to CprK; however, the previous data fit two- and three-binding site models better than a single-site model (13). On the other hand, we observe that, at the higher protein concentrations used in earlier experiments (1-3 μm), the data best fit a two-site (and, in some cases, three-site) model. In addition, corrections for inner filter effects in the present paper removed an apparent quenching component from the data. In summary, our results indicate that Cys11 does not play a major role in effector binding.
Upon replacing Cys11 with serine, the DNA binding activity of CprK decreases more than 50-fold (Fig. 6). Because the thiol and hydroxyl groups in the side chains of Cys and Ser are similar in size and H-bonding ability, a H-bonding interaction(s) involving the neutral thiol of Cys11 does not explain the importance of this residue in transcriptional activation. Some redoxactive thiols have unusually low pKa values; thus, to test the hypothesis that Cys11 may function as an ionized thiolate, the DNA binding activity of the C11D variant was tested. Although C11D retains high effector binding capacity, it exhibits no detectable DNA binding (Fig. 5). These results suggest that the thiol(ate) function of Cys11 plays a crucial and very specific role in DNA binding.
Based on studies of a related transcriptional activator (CprK1) from D. hafniense and two variants (C11S and C200S), the disulfide bond between Cys11 and Cys200 in redox regulation was concluded to lack physiological relevance (11). Gabor et al. (11) hypothesized that, if the Cys11-Cys200 disulfide bond was important in vivo, replacement of either Cys11 or Cys200 with serine would result in a redox-insensitive active state of CprK. Because, in aerobically grown E. coli, the level of transcriptional activation mediated by the C11S variant was similar to that of wild type CprK1, it was concluded that the Cys11-Cys200 disulfide bond was not present in the cytoplasm of aerobically cultivated E. coli cells. However, the results described here clearly demonstrate the formation of the Cys11-Cys200 disulfide bond under oxidative stress conditions in the cytoplasm of E. coli, with an ambient redox potential of approximately -250 mV (23, 24), which is similar to the redox potential of the ox/red couple for glutathione (i.e. between -240 (25) and -263 mV (26), the major redox buffer in most cells. Although the redox potentials for the intramolecular and intermolecular thiol/disulfide switches in CprK have not yet been determined, the redox potential of disulfide bonds in proteins vary over a wide range (-122 mV for DsbA to -470 mV) (23). Thus, we suggest that transcriptional control by CprK is subject to thiol/disulfide regulation, like a number of processes in prokaryotes and eukaryotes, including transcriptional regulation by OxyR (27), glutathione biosynthesis (28), cell cycle progression (29), heme binding by heme oxygenase-2 (30), iron metabolism by FurS (31), DNA binding by NF-κB (32), and thioredoxin/thioredoxin reductase-dependent reactions (33, 34).
Crystallographic (35) and site-directed mutagenesis (36) studies of bovine papillomavirus-1 E2 demonstrated that Cys340 is required for transcriptional activation through interactions between its sulfhydryl group and two bases in its target DNA. Like CprK, the E2 protein is subject to redox regulation, with its transcriptional activity lost upon oxidation (37). Similar results were obtained with the DNA-binding domain of NF-κB, where it loses DNA binding activity upon alkylation of a Cys residue in the DNA-binding domain (38).
Fluorescence spectroscopic results demonstrate that the tertiary structures of the Cys11 variants are different from that of wild type CprK, in both the reduced and oxidized states. Furthermore, the thiol/disulfide transition in CprK results in a substantial change in conformation, especially affecting the DNA-binding domain, as observed in the crystal structures of the oxidized and reduced proteins (10), by limited proteolysis studies (14) and by fluorescence spectroscopy (Fig. 8). An important structural role for Cys11 is also supported by the observations that the C11A variant cannot be purified and that the C11S and C11D variants are slightly less stable than the wild type protein. Functional and structural roles for cysteine have been demonstrated in various proteins (39-43) where cysteine plays a role in activity, in maintaining an active dimer, and in thermal stability (44). In the case of CprK, perhaps the charged Asp, being larger than the cysteine residue, interferes with protein-DNA interactions by either direct stereoelectronic effects or by indirect effects on protein packing near the recognition helix. However, the position of Cys11 in active reduced CprK is not known (10). It is hoped that further structural studies will lead to a better understanding of the nature of interactions involving Cys11 that promote DNA binding.
Supplementary Material
Acknowledgments
We are enormously indebted to Dr. Ari Gafni for providing advice and for letting us use his CD instrument, to Dr. Joseph A. Schauerte and Kathleen Wisser for assistance with CD experiments, and Dr. Bruce Palfrey for helpful discussions.
This work was supported, in whole or in part, by National Institutes of Health Grant 1P20RR17675. This work was also supported by National Science Foundation Grant GM 39451 (to S. W. R.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. S1-S3.
Footnotes
The abbreviations used are: CHPA, 3-chloro-4-hydroxyphenylacetate; DA, diamide; DTT, dithiothreitol; EMSA, electrophoretic mobility shift assay.
References
- 1.Utkin, I., Woese, C., and Wiegel, J. (1994) Int. J. Syst. Bacteriol. 44 612-619 [DOI] [PubMed] [Google Scholar]
- 2.Villemur, R., Lanthier, M., Beaudet, R., and Lepine, F. (2006) FEMS Microbiol. Rev. 30 706-733 [DOI] [PubMed] [Google Scholar]
- 3.Wiegel, J., Zhang, X., and Wu, Q. (1999) Appl. Environ. Microbiol. 65 2217-2221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wiegel, J., and Wu, Q. Z. (2000) FEMS Microbiol. Ecol. 32 1-15 [DOI] [PubMed] [Google Scholar]
- 5.Smidt, H., and de Vos, W. M. (2004) Annu. Rev. Microbiol. 58 43-73 [DOI] [PubMed] [Google Scholar]
- 6.Smidt, H., van Leest, M., van der Oost, J., and deVos, W. M. (2000) J. Bacteriol. 182 5683-5691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pop, S. M., Kolarik, R. J., and Ragsdale, S. W. (2004) J. Biol. Chem. 279 49910-49918 [DOI] [PubMed] [Google Scholar]
- 8.Korner, H., Sofia, H. J., and Zumft, W. G. (2003) FEMS Microbiol. Rev. 27 559-592 [DOI] [PubMed] [Google Scholar]
- 9.Aravind, L., Anantharaman, V., Balaji, S., Babu, M. M., and Iyer, L. M. (2005) FEMS Microbiol. Rev. 29 231-262 [DOI] [PubMed] [Google Scholar]
- 10.Joyce, M. G., Levy, C., Gabor, K., Pop, S. M., Biehl, B. D., Doukov, T. I., Ryter, J. M., Mazon, H., Smidt, H., van den Heuvel, R. H. H., Ragsdale, S. W., van der Oost, J., and Leys, D. (2006) J. Biol. Chem. 28318-28325 [DOI] [PubMed]
- 11.Gabor, K., Verissimo, C. S., Cyran, B. C., Ter Horst, P., Meijer, N. P., Smidt, H., de Vos, W. M., and van der Oost, J. (2006) J. Bacteriol. 188 2604-2613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Spiro, S., and Guest, J. R. (1990) FEMS Microbiol. Rev. 6 399-428 [DOI] [PubMed] [Google Scholar]
- 13.Pop, S. M., Gupta, N., Raza, A. S., and Ragsdale, S. W. (2006) J. Biol. Chem. 281 26382-26390 [DOI] [PubMed] [Google Scholar]
- 14.Mazon, H., Gabor, K., Leys, D., Heck, A. J., van der Oost, J., and van den Heuvel, R. H. (2007) J. Biol. Chem. 282 11281-11290 [DOI] [PubMed] [Google Scholar]
- 15.Sambrook, J., Fritsch, E. F., and Maniatis, T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Ed., Cold Spring Harbor Laboratory, Cold Spring Harbor, NY
- 16.Simons, R. W., Houman, F., and Kleckner, N. (1987) Gene (Amst.) 53 85-96 [DOI] [PubMed] [Google Scholar]
- 17.Guzman, L. M., Belin, D., Carson, M. J., and Beckwith, J. (1995) J. Bacteriol. 177 4121-4130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miller, J. H., ed (1972) Experiments in Molecular Genetics, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY
- 19.Kubista, M., Sjoback, R., Eriksson, S., and Albinsson, B. (1994) Analyst 119 417-419 [Google Scholar]
- 20.Provencher, S. W., and Glockner, J. (1981) Biochemistry 20 33-37 [DOI] [PubMed] [Google Scholar]
- 21.Sreerama, N., and Woody, R. W. (2000) Anal. Biochem. 287 252-260 [DOI] [PubMed] [Google Scholar]
- 22.Krasotkina, J., Walters, T., Maruya, K. A., and Ragsdale, S. W. (2001) J. Biol. Chem. 276 40991-40997 [DOI] [PubMed] [Google Scholar]
- 23.Ostergaard, H., Henriksen, A., Hansen, F. G., and Winther, J. R. (2001) EMBO J. 20 5853-5862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Taylor, M. F., Boylan, M. H., and Edmondson, D. E. (1990) Biochemistry 29 6911-6918 [DOI] [PubMed] [Google Scholar]
- 25.Schafer, F. Q., and Buettner, G. R. (2001) Free Radic. Biol. Med. 30 1191-1212 [DOI] [PubMed] [Google Scholar]
- 26.Millis, K. K., Weaver, K. H., and Rabenstein, D. L. (1993) J. Org. Chem. 58 4144-4146 [Google Scholar]
- 27.Georgiou, G. (2002) Cell 111 607-610 [DOI] [PubMed] [Google Scholar]
- 28.Vitvitsky, V., Mosharov, E., Tritt, M., Ataullakhanov, F., and Banerjee, R. (2003) Redox Rep. 8 57-63 [DOI] [PubMed] [Google Scholar]
- 29.Savitsky, P. A., and Finkel, T. (2002) J. Biol. Chem. 277 20535-20540 [DOI] [PubMed] [Google Scholar]
- 30.Yi, L., and Ragsdale, S. W. (2007) J. Biol. Chem. 282 20156-21067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ortiz de Orue Lucana, D., Troller, M., and Schrempf, H. (2003) Mol. Genet. Genomics 268 618-627 [DOI] [PubMed] [Google Scholar]
- 32.Nishi, T., Shimizu, N., Hiramoto, M., Sato, I., Yamaguchi, Y., Hasegawa, M., Aizawa, S., Tanaka, H., Kataoka, K., Watanabe, H., and Handa, H. (2002) J. Biol. Chem. 277 44548-44556 [DOI] [PubMed] [Google Scholar]
- 33.Staples, C. R., Gaymard, E., Stritt-Ette, A. L., Telser, J., Hoffman, B. M., Schurmann, P., Knaff, D. B., and Johnson, M. K. (1998) Biochemistry 37 4612-4620 [DOI] [PubMed] [Google Scholar]
- 34.Zhong, L., Arner, E. S., and Holmgren, A. (2000) Proc. Natl. Acad. Sci. U. S. A. 97 5854-5859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hegde, R. S., Grossman, S. R., Laimins, L. A., and Sigler, P. B. (1992) Nature 359 505-512 [DOI] [PubMed] [Google Scholar]
- 36.Grossel, M. J., Sverdrup, F., Breiding, D. E., and Androphy, E. J. (1996) J. Virol. 70 7264-7269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McBride, A. A., Klausner, R. D., and Howley, P. M. (1992) Proc. Natl. Acad. Sci. U. S. A. 89 7531-7535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lambert, C., Li, J., Jonscher, K., Yang, T. C., Reigan, P., Quintana, M., Harvey, J., and Freed, B. M. (2007) J. Biol. Chem. 282 19666-19675 [DOI] [PubMed] [Google Scholar]
- 39.Giese, N. A., Robbins, K. C., and Aaronson, S. A. (1987) Science 236 1315-1318 [DOI] [PubMed] [Google Scholar]
- 40.Pajares, M. A., Corrales, F. J., Ochoa, P., and Mato, J. M. (1991) Biochem. J. 274 225-229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rotrekl, V., Nejedla, E., Kucera, I., Abdallah, F., Palme, K., and Brzobohaty, B. (1999) Eur. J. Biochem. 266 1056-1065 [DOI] [PubMed] [Google Scholar]
- 42.Waterman, M. R. (1974) Biochim. Biophys. Acta 371 159-167 [DOI] [PubMed] [Google Scholar]
- 43.Yang, Y., Chen, M., Kesterson, R. A., Jr., and Harmon, C. M. (2007) Am. J. Physiol. 293 R1120-R1126 [DOI] [PubMed] [Google Scholar]
- 44.Nath, M. D., and Peterson, D. L. (2001) Arch. Biochem. Biophys. 392 287-294 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.