

Abstract
Most DNA replication systems include a sliding clamp that encircles the genomic DNA and links the polymerase to the template to control polymerase processivity. A loading complex is required to open the clamp and place it onto the DNA. In phage T4 this complex consists of a trimeric clamp of gp45 subunits and a pentameric loader assembly of four gp44 and one gp62 subunit(s), with clamp loading driven by ATP binding. We measure this binding as a function of input ligand concentration and show that four ATPs bind to the gp44/62 complex with equal affinity. In contrast, the ATPase rate profile of the clamp-clamp loader complex exhibits a marked peak at an input ATP concentration close to the overall Kd (∼30 μm), with further increases in bound ATP decreasing the ATPase rate to a much lower level. Thus the progressive binding of the four ATPs triggers a conformational change in the complex that markedly inhibits ATPase activity. This inhibition is related to ring opening by using a clamp that is covalently cross-linked across its subunit interfaces and thus rendered incapable of opening. Binding of this clamp abolishes substrate inhibition of the ATPase but leaves ATP binding unchanged. We show that four ATP ligands must bind to the T4 clamp loader before the loader can be fully “activated” and the clamp opened, and that ATP hydrolysis is required only for release of the loader complex after clamp loading onto the replication fork has been completed.
In the DNA replication system of phage T4, as well as in those of most other organisms, the replication polymerase does not act alone in synthesizing the DNA product. Rather it requires a ring-shaped “sliding clamp” that confers processivity (and processivity control) onto the replication process by linking the replication polymerases to the genomic template DNA. This linkage is topological in nature, reflecting the assembly of the subunits of the clamp into a ring that encircles the DNA. When loaded onto the DNA of the replication fork at a primer-template (P/T)3 junction, this clamp (which in the T4 system is a trimer of gp45 subunits) is free to slide along the DNA but cannot fall off without opening at one of the subunit interfaces.
The assembly of the clamp around the DNA requires the action of a protein complex called the “clamp loader.” In T4 this complex is a pentamer of proteins comprising four identical gp44 subunits that each carry an ATP binding and hydrolysis site, together with one gp62 subunit that coordinates the clamp loading function of the complex. The ATP-dependent clamp loader binds to the ring-shaped trimer and “activates it” for loading onto the DNA of the replication fork at the P/T junction. The clamp loader complex then dissociates from the clamp and the DNA, leaving the newly closed clamp on the DNA where it is available to bind to DNA polymerase and regulate the processivity of the template-dependent synthesis of the nascent DNA strands.
The DNA replication complex is similarly structured in organisms ranging from eukaryotes to Archaea, as well as in prokaryotes and their bacteriophages, such as T4 and RB69. The sliding clamps (1) are all ring-shaped oligomers of two (Escherichia coli) or three (eukaryotes, Archaea,, and RB69) identical subunits with a central hole that can accommodate double-stranded (ds-) DNA. These rings do not have significant direct binding affinity for dsDNA; rather their topology (and positive charge on the inner surface) allows them to slide freely along the DNA while completely encircling it (2, 3). When a sliding clamp is attached to its cognate DNA polymerase, a replication holoenzyme is formed that can move freely along the DNA but is also prevented from falling off. As a consequence, the processivity required for the template-directed polymerization of thousands of nucleotide residues is achieved (1).
The DNA genomes of all organisms are both long and often locally circularly closed, meaning that the ring-shaped sliding clamps of the replication complex cannot be “threaded onto” the DNA, but must be transiently opened to permit the clamp loader complex to place them onto (and in some cases also to remove them from) the DNA replication fork. Furthermore, this process must be performed in a regulated fashion because, although leading strand DNA synthesis is generally continuous, lagging strand synthesis is not. Rather, the lagging strands of the nascent DNA are synthesized as discrete (∼1 kb) Okazaki fragments, meaning that the processivity of the leading and lagging strand polymerases must be tightly (and differentially) controlled. To exercise this control, the DNA replication complexes of all organisms that utilize sliding clamps also include a multisubunit (generally pentameric) clamp loader complex that (driven by cycles of ATP binding and hydrolysis) is used to open the sliding clamps and place them onto (and remove them from) the genomic DNA as necessary. These clamp loaders act catalytically, dissociating from both the clamp and the DNA once the clamp has been loaded (4–6). The clamp loader assemblies belong to the AAA+ family of ATPases, which are also often arranged as partially ring-shaped oligomers and are involved in NTP-dependent assembly and disassembly of protein and protein-DNA complexes (7, 8).
The functional mechanisms and control systems used by the clamp loader complex are best understood for E. coli. The crystal structure of the free E. coli clamp loader complex (the γ-complex) has been solved (9), as has the structure of the β-clamp bound to the δ subunit of the loader complex (10). These structures, combined with numerous biochemical studies, have led to detailed mechanistic proposals for how this clamp loader is likely to work (reviewed in Ref. 11). The E. coli γ-complex contains three γ, one δ, and one δ′ subunits. The γ subunits bind and hydrolyze ATP and thus form the “motor” of the clamp loader complex. The δ subunit is termed the “wrench” of the clamp loader for its ability to “crack open” the dimeric β-clamp at one interface when bound to it. Ring opening seems to be induced by a simple protein-protein interaction between one of the subunits of the β ring and the δ subunit. This component is thought to bind to a protomer of the β-dimer and either traps or induces a conformational change that distorts its structure, thus presumably rendering it incapable of reforming a closed ring. The δ′ subunit is termed the “stator,” based on its apparent rigidity compared with the other subunits.
It has been proposed that the β subunits of E. coli, which appear to be bent and under strain in the dimer, may “spring” into a more relaxed conformation when a subunit interface of the β-dimer opens to produce a gap large enough to permit dsDNA (or at least the single-/double-stranded P/T junction) to pass through. In the absence of ATP, the γ-complex appears to be in a conformation in which the binding site for the β-clamp on the δ subunit is “hidden inside” the complex. ATP binding to the γ subunits is postulated to drive a subtle conformational change that results in the exposure of this interaction surface of the δ subunit, permitting the clamp to bind to the complex and be opened. The ATP-bound clamp loader complex bearing the open clamp then binds to a P/T-DNA junction, triggering ATP hydrolysis and permitting the γ-complex to return to its original conformation, accompanied by release of the clamp onto (and closure of the clamp around) the DNA.
No structural information is available for the T4 gp44/62 clamp loader (where gp44/62 is the T4 clamp loader complex, containing four gp44 subunits and one gp62 subunit), although sequence similarities coupled with biochemical studies suggest that it may share the major features of the mechanism proposed for the E. coli system (12, 13). The T4 clamp loader consists of two types of subunits, which form a pentamer comprising one gp62 and four gp44 subunits (14). Only the gp44 subunits carry ATP binding and hydrolysis sites (14, 15). Thus, by analogy, the gp44 subunits should include the motor of the T4 clamp loader (11). We note that the ATPase activity of a tetramer of gp44 subunits is not responsive to the addition of the gp45 clamp in the absence of the gp62 subunit (15), whereas the clamp does stimulate the ATPase activity of the complete gp44/62 loader complex. This suggests that the gp62 subunit may be analogous to the δ subunit of E. coli and thus serve as the wrench of the T4 clamp loader complex. The role of the stator (δ′ in the E. coli complex) has been assigned to one of the four gp44 subunits, probably the one positioned next to the gp62 subunit, which would lack the “arginine finger” domain that spans the other subunit-subunit interfaces of the clamp loader and reaches from one gp44 subunit to the next to activate ATP hydrolysis in the adjacent subunit (11).
In a previous study (16), we examined the effects of the sliding clamp and of P/T-DNA on the steady-state and pre-steady-state ATPase activity of the T4 gp44/62 clamp loader at saturating ATP concentrations. Our results were consistent with a mechanism in which a slow conformational change in the clamp-clamp loader complex precedes the binding of the complex to the P/T-DNA, with ATP hydrolysis following to complete the loading of the clamp onto the DNA and the release (recycling) of the clamp loader. This mechanistic scheme is very comparable with that suggested for the clamp loading process in the E. coli DNA replication system on the basis of different kinds of experiments, and it supports the notion that the replication complex of T4 bacteriophage represents the simplest version (in terms of numbers and types of protein components and their interactions) of the more highly regulated replication systems of higher organisms.
These earlier pre-steady-state and steady-state studies of the ATP binding and hydrolysis cycle at physiological concentrations of ATP and in the presence and absence of the various modifying cofactors (i.e. the clamp and the P/T DNA loading target) led us to conclude that activation of the clamp loading process required only ATP binding, and that the hydrolysis of a single ATP molecule (or possibly two) was required only to release the activated clamp from the clamp loader and complete its loading onto the P/T DNA (see Ref. 16 and references therein). This model is consistent with the conclusions of O'Donnell and co-workers (17, 18) with respect to the clamp loading cycle in E. coli on the basis of other types of experiments, as well as with the more limited results available with the clamp-clamp loader complex of yeast (6, 19, 20).
However, these earlier findings with all the various clamp loading complexes have left unanswered several other questions of general interest as follows. (i) What role (if any) is played in the clamp loading process by the “other” ATP-binding sites of the clamp loader complex (a total of four in the T4 system)? (ii) How is the ATP binding and hydrolysis cycle of the clamp loader modified by the activation state (open or closed) of the clamp? (iii) How is the ATP binding and hydrolysis cycle further modified by the presence of the P/T DNA “loading target”? The experiments that follow were designed to address these questions.
EXPERIMENTAL PROCEDURES
Materials—Unlabeled ATP was purchased from Amersham Biosciences, and radiolabeled [α-32P]ATP was from PerkinElmer Life Sciences. The BMH and DPDPD cross-linkers were obtained from Pierce. The SYPRO-Orange protein stain was from Molecular Probes. Phosphoenolpyruvate, NADH, rabbit muscle pyruvate kinase, and rabbit muscle l-lactic dehydrogenase were obtained from Sigma.
Oligonucleotides—DNA oligonucleotides were purchased from Integrated DNA Technologies (Coralville, IA). The P/T (30–50-mer) DNA sequence used was as follows: 5′-ccgcagcctcgcagccgtccaaccaactca-3′ and 3′-ggcgtcggagcgtcggcaggttggttgagtaggtcttgttttgccggtca-5′. This P/T DNA construct was constructed by annealing equimolar concentrations of the single-stranded constituent DNA oligonucleotides; the completeness of the hybridization process was confirmed by native gel electrophoresis.
Proteins—The overexpression vector containing the gp44/62 gene under an isopropyl 1-thio-β-d-galactopyranoside-inducible promoter was kindly provided by Dr. James D. Karam (Tulane University Health Sciences Center, New Orleans, LA), and the gp45-K82C/K166C double mutant was provided by Dr. Feng Dong. Construction of the gp45-K82C/K166C double mutant has been described elsewhere (21). The gp45 clamps (WT and dm) and the gp44/62 clamp loader complexes were purified as described previously (22). Protein concentrations were determined by UV absorbance at 280 nm, using the following calculated molar extinction coefficients (23): for gp44/62 ε280 was 1.23 × 105 m-1 cm-1 and for gp45 ε280 was 5.72 × 104 m-1 cm-1.
Cross-linking of gp45-K82C/K166C with BMH and DPDPB—The interfaces of the gp45-K82C/K166C double mutant trimer were cross-linked as described previously (21). The cross-linker, either BMH or DPDPB, was dissolved in DMSO, and the equivalent of a 3–5-fold excess of cross-linker over interfaces of the gp45 double mutant (three interfaces in each gp45 mutant trimer) was added to a solution of gp45-K82C/K166C protein in 25 mm HEPES (pH 7.5), 0.16 m KOAc, and 6 mm Mg(OAc)2. The reaction was incubated for ∼2 h at room temperature or overnight at 4 °C. Unreacted cross-linker was removed by a BioSpin P6 column (Bio-Rad) that had been equilibrated with the reaction buffer. Cross-linking efficiency was checked by SDS-PAGE. The gel was stained with SYPRO orange (Molecular Probes), and the gel bands were quantified (relative to the closed and open trimer forms) using a Storm model 860 PhosphorImager (GE Healthcare). Under these conditions, all the dm-gp45 (where dm-gp45 or gp45-K82C/S166C is a double mutant of the gp45 trimer, in which lysine at position 82 and serine at position 166 have been site-specifically mutated to cysteine) was cross-linked to the trimer level, with ∼90% of the trimers being completely covalently closed and ∼10% cross-linked at only two of the three interfaces (see Ref. 21). The two different cross-linkers used (BMH and DPDPB) gave similar results. The disulfide bond in the DPDPB cross-linker was cleaved by overnight incubation of DPDPB-dm-gp45 in reaction buffer plus 10 mm DTT at 4 °C.
Steady-state Kinetic ATPase Assays—The steady-state ATPase rates of the various forms of the clamp loader complex were measured spectrophotometrically in a Cary 3E spectrophotometer, using a coupled enzyme system as described previously (16). The assay monitors ATPase activity by following the decrease in absorbance at 340 nm as NADH is converted to NAD during the regeneration of the ADP product of the ATP hydrolysis reaction. In a standard reaction, the regeneration system contained 3–6 mm phosphoenolpyruvate, 0.5–3 mm NADH, 0.025–0.27 units per μl of pyruvate kinase, and 0.025–0.27 units per μl of lactate dehydrogenase. Regeneration of ADP to ATP was never rate-limiting. In addition to the regeneration system, the reactions contained 1 μm gp44/62 and, when present, 1.5–10 μm gp45 or dm-gp45 (either cross-linked or not, as indicated), 0.02–24 μm P/T DNA, when present, as indicated, and the listed concentration of total ATP, ranging from 2.5 μm to 8 mm. ATP was always added in a 1:1 ratio with Mg(OAc)2. The reaction buffer contained 25 mm HEPES (pH 7.5), 0.16–3 m KOAc, as indicated, and 6 mm Mg(OAc)2. In addition the buffer contained 10 mm DTT when the ATPase activity of the gp44/62 complex was measured in the presence of DPDPB-dm-gp45 that had been incubated in 10 mm DTT overnight at 4 °C to cleave the disulfide bond present in this cross-linker. DTT had no appreciable effect on the ATPase activity of a control sample that contained the uncross-linked dm-gp45 (data not shown). The volume of the reaction sample was 30 μl and was contained in a 0.3-cm path length spectrophotometer cuvette. The assay temperature was maintained at 23 °C.
Detection of Steady-state Bound ATP by Ultrafiltration—The ultrafiltration reactions contained 30 μm gp44/62 and 35 μm gp45 or dm-gp45 (when present), either cross-linked or not cross-linked. The ATP regeneration system consisted of 6 mm phosphoenolpyruvate, 0.27 units μl-1 of pyruvate kinase, and the indicated amount of ATP, ranging from 2.5 μm to 1 mm (with a trace amount of [α-32P]ATP) in a total volume of 100 μl. The amount of pyruvate kinase used was set by trial to achieve a low percentage of free ADP, which ranged from about 5% at the lowest ATP concentration used to ∼0% at the higher ATP concentrations.
The reaction buffer was the same used in the ATPase assay and contained 25 mm HEPES (pH 7.5), 0.16–3 m KOAc, and 6 mm Mg(OAc)2. A 100-μl reaction sample was placed into an Amicon Microcon-30 ultrafiltration cell containing a 30,000 Mr cutoff filter and centrifuged at maximum speed (14,000 × g) for 3 min at room temperature. About one-half of the reaction volume passed through the filter.4 3 μl of the original solution and 3 μl of the filtrate were analyzed by liquid scintillation counting. The free nucleotide concentration (calculated from the sample counts in the filtrate) was corrected for the percentage of ATP retained by the filter in the absence of proteins (∼7.5% of the added counts). The bound nucleotide concentration was then obtained by subtracting the corrected free nucleotide concentration from the total ATP concentration, calculated from the original sample counts. Controls were done to show that neither the regeneration system nor the gp45 clamp proteins bound measurable amounts of ATP (data not shown).
To ascertain the proper working of the regeneration system (i.e. that the majority of free nucleotide was present as ATP and not ADP), 1 μl of filtrate was analyzed on a PEI-F cellulose TLC plate after each experiment. The TLC plates were developed in 0.35 m potassium phosphate buffer (pH 3). The radioactive ATP and ADP spots obtained on the TLC plates were quantified using a Storm model 860 PhosphorImager (GE Healthcare).
To determine the identities and concentrations of the nucleotide forms bound to the gp44/62 complex, 1 μl of the original solution was quenched into 15 μl of 1 n HCl, treated with phenol/chloroform, and then neutralized to pH 7.5 by the addition, within 2 min, of 2 μl of a solution containing 1 m Tris base and 3 m NaOH. 1 μl of the filtrate was treated similarly, and then 1 μl of the quenched and neutralized solutions containing either the original sample or the filtrate was analyzed on a TLC plate as described above. The identity of the bound nucleotide was determined by comparing the original sample (containing both bound and free nucleotide) with the filtrate (containing free nucleotide only).
Data Analysis and Simulation—The ATP binding curves obtained from the ultrafiltration experiments were fitted to a binding isotherm as shown in Equation 1,
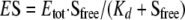 |
(Eq. 1) |
where ES is the concentration of ATP sites with bound ATP; Etot is the concentration of all the ATP-binding sites in solution (with and without bound ATP); Sfree is the concentration of the free ATP, and Kd is the apparent dissociation constant for bound ATP.
The ATPase rate data of the clamp-clamp loader complex were fitted to Equation 2,
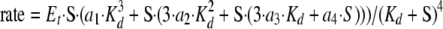 |
(Eq. 2) |
where S = (-4·Et - Kd + St + ((4·Et + Kd - St)2 + 4·Kd·St)0.5)/2 is the concentration of free ATP; Et is the total concentration of enzyme complex; Kd is the apparent dissociation constant for ATP binding to any of the four sites; St is the total concentration of ATP (bound and free); and a1, a2, a3, and a4 correspond to the rates at which ATP is hydrolyzed by the enzyme complex with one, two, three, and four ATP molecules bound, respectively.
Equation 2 was derived as follows. We assumed that only one ATP site was functionally catalytic (i.e. active in hydrolyzing ATP) and that the other three sites were inhibitory. We note, however, that the assumption of a different number of active catalytic sites would only change the absolute best fit values but not the relative best fit values for the hydrolysis rates per catalytic site of the different complexes, which are inversely proportional to the number of catalytic sites present (not shown). For example, if one assumes that all four sites are catalytically active, the best fit values obtained would be one-fourth of those listed here on the assumption that only one site is active.
With the assumption that only one site is catalytically active, the fractions of active enzyme species with one, two, three, and four ATP ligands bound are as follows: ES1 = p3q, ES2 = 3p2q2, ES3 = 3pq3, and ES4 = q4; and the overall rate is shown in Equation 3,
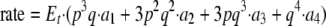 |
(Eq. 3) |
where p and q are the fractions of empty and filled ATP sites, respectively, p = Kd/(Kd + S); q = S/(Kd+S); and a1, a2, a3, and a4 are the rates at which ATP is hydrolyzed for complexes with one, two, three, and four ATPs bound, as above. Finally, S (the concentration of free ATP) can then be calculated from St (total concentration of ATP) as shown in Equation 4,
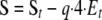 |
(Eq. 4) |
Given that the apparent dissociation constant (Kd) for ATP binding to all four ATP sites is known from the binding experiments, the fitting of the data to Equation 2 provides the best fit values for the ATPase rates for each of the four ATP-bound complexes.
RESULTS
Increasing ATP Concentration Inhibits the Steady-state ATPase Activity of the Clamp-Clamp Loader Complex—In Fig. 1A we plot the ATPase activity of a 1 μm solution of clamp-clamp loader complex (gp45-gp44/62) as a function of input ATP concentration. It is apparent that the rate profile obtained does not follow classical Michaelis-Menten enzyme kinetics. Rather a maximal ATPase rate of 7.1 μm ATP min-1 is reached with this concentration of complex at an input ATP concentration of ∼30 μm, and then the rate decreases as the concentration of ATP is increased, flattening out at a plateau hydrolysis rate of ∼3 μm min-1 (∼40% of the peak activity) at an ATP concentration of ∼300 μm (see Fig. 1A and Fig. 5, closed squares).
FIGURE 1.

Substrate inhibition is observed when the gp44/62 steady-state ATPase rates are measured in the presence of the gp45 clamp. The steady-state ATPase rates of gp44/62 in the presence (A) and in the absence (B) of gp45 were measured using an ATP regeneration system assay method as described under “Experimental Procedures.” Component concentrations were 1 μm gp44/62 and 1.5 μm gp45 (A) and 15 μm gp44/62 and no gp45 (B). Total input ATP concentrations are plotted on the x axis.
FIGURE 5.

The substrate inhibition effect on the ATPase rate profile for gp44/62-WT-gp45 is abolished as the monovalent salt concentration is increased. Component concentrations were 1 μm gp44/62, 1.5 μm gp45, and the indicated concentrations of ATP. The concentration of KOAc in the reaction buffer was 0.16 m (closed squares), 0.5 m (open squares), 1 m (closed circles), 1.5 m (open circles), 2 m (closed diamonds), or 3 m (open diamonds). Steady-state ATPase rates were measured in the presence of an ATP regeneration system (“Experimental Procedures”).
This result shows that the ATPase activity of the complex is progressively inhibited at increased ATP concentrations and that this unusual inhibitory behavior is dependent on the presence of the gp45 clamp. As shown in Fig. 1B, in the absence of the clamp the ATPase activity of the free clamp loader complex does follow a classical Michaelis-Menten rate profile. The ATPase rate measured for the clamp loader complex alone is significantly smaller than that of clamp loader complex bound to the clamp (note that the gp44/62 concentration used to collect the data shown in Fig. 1B is 15-fold higher than that of the gp44/62-gp45 complex used in Fig. 1A). Thus we find that binding of the clamp greatly increases the ATPase rate of the clamp loader complex and switches the activity into the “substrate inhibition” mode at higher ATP concentrations.
Four ATP Molecules Bind to the Clamp Loader Complex with Equal Affinity—The substrate-dependent inhibition observed in the ATPase rate profiles for the gp44/62 complex bound to the gp45 clamp (Fig. 1A) is consistent with the binding of more than one ATP molecule to the functional clamp loading complex. To measure the ATP binding directly, we used an ultrafiltration method (see “Experimental Procedures”) to monitor the number of ATP molecules bound to the gp44/62 complex as a function of input ATP concentration under the steady-state hydrolysis conditions used in Fig. 1, permitting us to establish the relation between ATP hydrolysis rates and ATP-binding site occupancy.
In a typical experiment, a solution containing the relevant protein complex was mixed with the indicated amount of ATP and a trace amount of [α-32P]ATP (to serve as label). The system also contained an ATP regeneration system to hold constant the concentration of free ATP. Samples were then partially filtered through a 30,000 Mr cutoff ultrafiltration membrane that retains the protein complexes and also any ATP molecules bound to them. The total amount of bound ATP was calculated as the difference between the total ATP in the original samples and the free ATP in the flow-through samples (see “Experimental Procedures” for details). Because this difference could be small, high concentrations of protein were used to limit the experimental error. For this reason we used a 30 μm solution of gp44/62 loader complex, together (when present) with a 35 μm concentration of clamp (gp45 trimers). Neither the ATP regeneration system nor the gp45 clamp bind ATP at the component concentrations used (data not shown).
Fig. 2 shows the results of such binding experiments conducted with the gp44/62 complex in the presence (closed circles) and in the absence (open circles) of WT-gp45 clamp. We found that a steady-state binding plateau of ∼4 ATP molecules per clamp loader complex was approached in both experiments, with 3.7 (±0.2) ATP molecules bound per free gp44/62 complex and 3.6 (±0.1) ATPs bound per gp44/gp62-gp45 complex. In addition, the binding curves displayed no sigmoid character, thus ruling out binding cooperativity between the four ATP-binding sites of the gp44/62 complex. Rather, both curves can be represented by simple binding isotherms (Equation 1, under “Experimental Procedures”), showing that four ATP molecules bind independently, and with equal affinity, to the four gp44 subunits of the complex in both the presence and the absence of the clamp. The measured Kd values for these binding experiments were 94 (±15) μm for gp44/62 alone and 27 (±3) μm for the gp44/62-gp45 complex. The simple shape of the binding isotherms also ruled out a sequential ATP-binding mechanism (i.e. binding of the first ATP being required to permit the binding of the second, and so on) to the four ATP-binding sites. This follows because a sequential mechanism, in contrast to random binding, would appear cooperative, with an apparent binding affinity that increases for each sequentially bound ATP.5
FIGURE 2.

The gp44/62 clamp loader complex binds four ATP molecules with similar affinities, both in the absence and in the presence of the gp45 clamp. The ATP bound to gp44/62 under steady-state conditions was measured (data points) using an ultrafiltration method as described under “Experimental Procedures.” Component concentrations were 30 μm gp44/62 alone (open symbols) or in the presence of 35 μm WT-gp45 (closed symbols). An ATP regeneration system was present in the reaction mixture. The curves were obtained by fitting the data points to a binding model (Equation 1) with four equal and independent ATP-binding sites.
The monotonic shape of the binding profile for the clamp-clamp loader complex (Fig. 2, closed symbols) clearly contrasts with the “substrate-inhibited” form of the ATPase rate plot measured with the loader complex carrying a bound clamp (Fig. 1A). In contrast, the inhibited rate profile is not seen for the binding of ATP to the free gp44/62 clamp loader complex, where ATP binding increases in parallel with ATPase rate in “standard” fashion (compare Fig. 2, open symbols, with Fig. 1B). Thus the inhibited (and peaked) form of the ATPase rate profile can be considered to be diagnostic of a conformational state of the clamp loader complex that has been “activated” by clamp binding. In the balance of this paper we investigate further various aspects of the activated state(s) of the clamp-clamp loader complex as a function of ATP concentration to further understand the loading process.
Because the ATPase kinetics (Fig. 1) and the steady-state ATP-binding isotherms (Fig. 2) were typically measured at very different protein concentrations, we performed control kinetic experiments at the elevated protein concentrations used in the binding studies (30 μm gp44/62 and 35 μm gp45). The ATPase rate profiles obtained at high complex concentrations (data not shown) closely resembled those of Fig. 1A, with the position of the peak in the rate profile at ∼25 μm free ATP for the concentrated protein solution, compared with a peak at ∼30 μm for the 1 μm gp44/62-gp45 complex concentrations used in the kinetic studies. The plateau under both concentration conditions was at ∼40% of the rate at the peak.
Only ATP Is Bound to the gp44/62-gp45 Complex under Steady-state Hydrolysis Conditions—Our earlier pre-steady-state studies (16) had suggested that only ATP binding (not hydrolysis) is required to permit the gp44/62 complex to bind to gp45 clamp and to “activate” the system for clamp loading onto the P/T DNA junction. These results suggested that in the complete system (clamp, clamp loader, and P/T DNA), the ATP hydrolysis reaction that follows serves only to release the loaded clamp and the DNA from the loader complex, although the present results show that in the absence of P/T DNA the clamp still binds to the clamp loader and forms an activated (ring-opened) complex. However, in the absence of DNA, the open clamp is simply released back into solution (and recloses) in an abortive ATP binding and hydrolysis cycle (Fig. 1A).
The ATP binding experiments described above were performed with [α-32P]ATP as the radioactive tracer. Because the α-phosphate of ATP was labeled, both the ATP and the ADP forms of the nucleotide that might bind to the gp44 subunits under steady-state hydrolysis conditions would be detected. To discriminate between the two forms, samples from the original reaction mixture and from the filtrate were acid-quenched (to release all of the bound nucleotide species), and the resulting solutions were run on a TLC plate to separate ATP and ADP. The reaction samples contain the nucleotide species bound to the enzyme, together with the unbound nucleotide (the latter should be entirely ATP because of the presence of the ATP regeneration system; see “Experimental Procedures”). The filtrate samples should contain only unbound ATP.
Table 1 shows the concentrations of ADP and ATP bound to the gp44/62-gp45 complex (30 μm) at the different input ATP concentrations. We note that the amount of ADP bound to the enzyme complex is always less than 4% of the total bound nucleotide, far less than the 25% expected if even one of the four ATP sites of the clamp loader were occupied by ADP. Table 1 also shows that no measurable ADP is bound to the gp44/62-gp45 complexes at the higher ATP concentrations used. This result is consistent with our earlier demonstration that ATP hydrolysis (or a step before ATP hydrolysis) is rate-limiting in the T4 clamp loading process under steady-state conditions and that the subsequent dissociation of the ADP and Pi reaction products is fast (16).
TABLE 1.
The nucleotide species bound to the clamp-clamp loader complex under steady-state hydrolysis conditions is almost entirely ATP The concentrations of ADP and ATP bound to the clamp-clamp loader complex were determined using the ultrafiltration method described under “Experimental Procedures.” The component concentrations were 30 μm gp44/62, 35 μm gp45, and the indicated amounts of input ATP (column 1). An ATP regeneration system was also present. The concentration of bound ADP and bound ATP are shown in columns 2 and 3, respectively. Since each gp44/62 complex contains four gp44 subunits that bind ATP, and the solution contains 30 μm of complex, binding should saturate at a bound ATP concentration of 120 μm.
| Input [ATP] | [ADP] bound | [ATP] bound |
|---|---|---|
| μm | μm | μm |
| 10 | 0.3 | 8 |
| 25 | 0.5 | 18.6 |
| 50 | 0.7 | 35 |
| 100 | 0.5 | 61 |
| 150 | 2 | 75.8 |
| 200 | 3.3 | 86.9 |
| 300 | 0 | 91.8 |
| 400 | 0 | 96.3 |
| 500 | 0 | 104.1 |
Binding of ATP to All Four gp44 Sites Decreases the ATPase Activity of the gp44/62-gp45 Complex ∼10-fold—To understand and model the mechanistic basis of the observed substrate inhibition at higher input ATP concentrations in binding terms, we fit ATPase rate data measured in the presence of a great excess of gp45 (10 μm; to ensure saturation of the loading complex with clamp and thus “full activation” of the loader system)6 to a simple rate equation for multiple independent ATP binding and hydrolysis sites (Equation 2; see under “Experimental Procedures”). The results are shown in Fig. 3A, where the points represent the total (measured) ATP hydrolysis rate at each input ATP concentration, and the curve was calculated using the individual (fitted) ATPase rates for each component (see below). The measured Kd value for ATP binding to the gp44/62-gp45 complex and the fact that all four ATPs bind with equal affinity (Fig. 2) permit us to determine the fractions of the total clamp-clamp loader complex that bind one, two, three, or four ATP molecules, respectively, as a function of input ATP concentration. Fig. 3B presents a plot of these fractions of total complex present in each of the four ATP-bound forms as a function of input ATP concentration.
FIGURE 3.

Fit of the overall ATPase rates of the gp44/62-gp45 complex to the sum of the activities of species carrying one, two, three, and four bound ATPs. A, measured ATPase rates obtained with 1 μm gp44/62 in the presence of excess (10 μm) WT-gp45 (data points) are plotted together with the fit (solid curve) of the data to Equation 2 (see “Experimental Procedures”). A Kd value of 27 μm was used for the binding of the ATP ligands, and the best fit values (see “Experimental Procedures”) for the rates at which ATP is hydrolyzed by complexes carrying one, two, three, and four bound ATP molecules are 30.5 (±0.6) μm min-1, 24.1 (±0.5) μm min-1, 7.4 (±0.2) μm min-1, and 2.87 (±0.3) μm min-1, respectively. B, calculated fractions of the different ATP-bound enzyme species in solution are plotted as a function of ATP concentration. The enzyme binds four ATP molecules with a Kd value of 27 μm. ES1, ES2, ES3, and ES4 represent the concentrations of enzyme species with one, two, three, and four ATPs bound, respectively, as a function of input ATP.
With the assumption (discussed below) that only one ATP-binding site is catalytic and that the other three ATP molecules are stably bound and inhibitory, the total ATPase rate produced by this distribution of complexes at each ATP concentration is obtained by using Equation 2 to sum the ATPase rates of the different active species present. A fit of our ATPase rate data to this equation permits us to calculate the contribution of each active enzyme species (i.e. each species with one or more ATP bound) to the overall ATPase rate at each input ATP concentration. We designate the ATPase activities of each component as a1, a2, a3, and a4, corresponding to the hydrolysis rates of the loader species carrying one, two, three or four bound ATP molecules, respectively.
Based in part on our previous pre-steady-state kinetic analysis of this system (16), we have assumed in these calculations that only one ATP site in each complex is catalytically active and (based on the present results) that the three remaining ATP-bound sites are inhibitory. Of course, if more than one site hydrolyzes ATP, the calculation can be modified by dividing the best fit values for the ATPase rates obtained using Equation 2 by the number of catalytic sites present to obtain the rate per site (see “Experimental Procedures” for details).
The fit (solid curve) obtained for the ATPase rates is superimposed on the experimental ATPase rates plotted as a function of total input ATP concentration (data points) in Fig. 3A. For these calculations, the Kd value for ATP binding by the system was fixed at 27 μm, as measured in the binding experiment shown in Fig. 2 (closed symbols). The best fit values for the ATPase rates of the complexes containing different numbers of bound ATP ligands were as follows: a1 = 30.5 (±0.6) μm min-1; a2 = 24.1 (±0.5) μm min-1; a3 = 7.4 (±0.2) μm min-1; and a4 = 2.87 (±0.03) μm min-1. We note, as expected, that the ATPase rate per gp44/62-gp45 complex decreases progressively as the ATP-binding sites are filled, with an ∼10-fold decrease in apparent ATPase rate in going from the species that binds only one ATP (a1) to the species with all four ATP sites filled (a4). The latter species is expected to be the dominant form of the clamp-clamp loader complex at physiological concentrations of ATP (∼1 mm or higher), and thus the ATPase rate at lower ATP concentrations (and its relationship to binding) serves here primarily as a tool to understand the molecular mechanisms of the clamp loading complex.
Inhibition of the ATPase Activity of the Clamp-Clamp Loader Complex Is Linked to Clamp Opening—In the preceding sections we have shown that the ATPase rate of the clamp loader complex is substrate-inhibited when bound to the gp45 clamp at physiological concentrations of ATP, and that this inhibition is relieved in the absence of the clamp. If indeed this ATP concentration-dependent inhibition reflects some aspect of the function of the clamp loader, it is reasonable to suggest that it might be related to activation of the clamp for loading and is likely dependent on whether the clamp is open or closed. We here test this hypothesis directly.
Crystallographic studies have suggested that in its free form the gp45 clamp is normally closed (24) or, if slightly open, the opening is too small to admit P/T DNA (25). In earlier work we (21) developed a site-specifically modified double mutant form of the trimeric T4 clamp that can be covalently cross-linked across its interfaces and thus rendered “permanently closed.” We therefore refer to this doubly mutated subunit, gp45-K82C/S166C as the double mutant (or dm) form of gp45. It carries two cysteine residues per subunit that are located at opposite ends of the protomer. On assembly into a trimeric ring, these site-specifically introduced cysteine residues are positioned as contiguous pairs at each of the clamp interfaces. Therefore, the use of this dm form of gp45 permits the trimer ring to be covalently closed by treating the clamp with a homo-bifunctional cross-linking agent that reacts with the cysteine pairs aligned across each subunit-subunit interface. We have shown previously that these covalently cross-linked rings cannot be loaded onto P/T DNA (21).
In that study, which was performed at saturating (1 mm) concentrations of ATP, we also observed that the covalently closed clamps appeared, when bound, to “hyper-stimulate” the ATPase activity of the clamp loader complex, relative to the rate measured under the same conditions with the uncross-linked double mutant (or wild-type) clamp as cofactor, although the mechanistic basis of this hyper-stimulation was unclear. Given the substrate inhibition of the ATPase of the clamp loader complex seen at higher ATP concentrations in the present study with the WT-gp45 clamp as cofactor, it seems likely that the hyper-stimulation of the ATPase activity with the covalently closed clamp seen previously at 1 mm ATP reflects a decrease in (or even the disappearance of) the substrate inhibition at high ATP concentration as a consequence of the binding of WT-gp45 to the clamp loader that we have observed in this study.
To examine this possibility, we compared (in Fig. 4) the ATPase rate profiles obtained with uncross-linked (open symbols) and cross-linked (with BMH, closed circles, or DPDPB, closed squares) dm-gp45 clamps. Fig. 4 shows, as expected, that the ATPase activity of the gp44/62-gp45 complex, plotted as a function of ATP concentration, displays a peaked rate profile when bound to the uncross-linked double mutant clamp (open symbols) that is very similar to the profile observed for the complex bound to the WT-gp45 clamp (Fig. 1A). Furthermore, the final plateau value falls to ∼40% of the activity seen at the peak, as also observed previously for the clamp loader bound to WT-gp45 (Fig. 1A). Thus we conclude that the essential features of the inhibited ATPase rate profile of the loader complex seen with the WT-gp45 clamp are also observed with the uncross-linked double mutant clamp.7
FIGURE 4.

Substrate inhibition is almost absent when the gp44/62 steady-state ATPase rates are measured in the presence of a covalently closed clamp. The steady-state ATPase rates of gp44/62 in the presence of uncross-linked double mutant gp45 (open symbols) and in the presence of cross-linked double mutant gp45 (closed symbols) cross-linked with either BMH (circles) or DPDPB (squares) were measured in the presence of an ATP regeneration system as described under “Experimental Procedures.” Component concentrations were 1 μm gp44/62 and 1.5 μm dm-gp45 uncross-linked (open symbols) or cross-linked (closed symbols), and the indicated concentrations of ATP. Also shown is the ATPase rate measured in the presence of 1 μm gp44/62 and 1.5 μm DPDPB-dm-gp45 after the covalent cross-links had been cleaved by incubation overnight with 10 mm DTT (× symbol).
The ATPase rate profile obtained when the gp45 double mutant is covalently closed by cross-linking with either BMH (Fig. 4, closed circles) or DPDPB (closed squares) is quite different. With the cross-linked clamp as cofactor, a peak is no longer evident in the kinetic profile, and the shape of the curve more closely resembles that expected for an enzyme system displaying simple Michaelis-Menten kinetics.8
These major changes in ATPase rate must reflect one or more conformational differences within the two protein complexes. The only structural difference between the two complexes is the presence of a processivity clamp that cannot be opened in one and a clamp that can be opened in the other. Because the cross-linked gp45 mutant is permanently closed, we suggest that the uncross-linked gp45 mutant ring must be open at the higher (inhibitory) ATP concentrations and that both rings are closed at low ATP concentrations. As additional support for this hypothesis, we show that cleaving the cross-link (by reducing the internal disulfide bond of the DPDPB cross-linker with DTT so that the formerly cross-linked ring can now be opened) results in a drop in ATPase activity to the level observed with the uncross-linked dm clamp as cofactor (Fig. 4, × symbol).
The ATP binding studies described in Fig. 2 (above) were also performed in the presence of the uncross-linked and cross-linked dm clamp. Binding isotherms similar to those shown for the clamp loader alone and in the presence of WT-gp45 were obtained. These binding plots showed plateau levels of 3.7 (±0.1) and 3.4 (±0.2) bound ATP molecules per clamp loader complex, and Kd values for ATP binding of 34 (±4) μm and 42 (±6) μm, for complexes containing uncross-linked and cross-linked dm-gp45, respectively (data not shown). This control shows that the differences in the ATPase rate profiles, observed under the different conditions tested (in the absence and presence of WT-gp45, dm-gp45, or cross-linked dm-gp45), do not reflect differences in the levels of ATP bound to the clamp loader complex under steady-state hydrolysis conditions. Rather the gp44/62 complex appears to be capable of binding four ATPs under all conditions, regardless of the presence or absence of the clamp (either wild type or mutant) or of whether the dm-gp45 clamp is cross-linked or not. Furthermore, the ATP-binding sites of the four gp44 subunits show similar affinities for this ligand (i.e. no binding cooperativity) under all conditions tested.
The ATPase rates measured for the clamp loader complex in the presence of excess uncross-linked dm-gp45 clamp (10 μm, to avoid making the concentration of clamp rate-limiting)5 were fit to Equation 2 as described above for WT-gp45, using here a fixed Kd value of 34 μm (measured in the ATP binding experiments with the dm-gp45 clamp). The best fit values of the activities of the various ATP-bound clamp loader species complexed to the dm-gp45 clamp cofactor were as follows: a1 = 53 ± 1.6 μm min-1; a2 = 25 ± 1.5 μm min-1; a3 = 15 ± 1 μm min-1; and a4 = 4.7 ± 0.1 μm min-1. Here again the ATPase rate decreases as the four ATP sites are filled, and there is an ∼11-fold decrease in activity when four ATPs are bound, compared with the ATPase activity displayed by complexes with only one bound ATP. These results are very similar to those described above for the clamp loader bound to the wild-type clamp, and lead us to conclude that the clamp loader activation process reflected in the substrate (ATP) inhibition profile shown in Fig. 1A tracks the opening of the clamp, and that this inhibition is abolished when the clamp cannot be opened.
High Concentrations of Salt Also Banish the ATP Concentration-dependent ATPase Inhibition of the Clamp-Clamp Loader Complex—We next tested whether other changes might also alter the distribution of conformational forms of the clamp-clamp loader complex between activated (clamp open) and unactivated (clamp closed) forms. Fig. 5 shows, again as a function of input ATP concentration, the effect of increasing the concentration of monovalent salt, here potassium acetate (KOAc), on the ATPase activity of the complex. We show that the ATPase rate at the activity plateau increases greatly as the salt concentration is increased from 0.16 to 3 m KOAc. At 3 m KOAc, the ATPase activity at the plateau has increased ∼14-fold relative to that seen in 0.16 m KOAc. At the highest concentration of ATP tested (8 mm), the ATPase rate in 3 m KOAc is about 40 μm min-1 (compared with ∼2.8 μm min-1 for the clamp-clamp loader complex in 0.16 m KOAc), and the rate increase is nearly as large at physiological concentrations of ATP (∼1 mm; see Fig. 5).
As the plateau rate increases, the peak shifts to higher concentrations of ATP and also becomes less and less pronounced, until it almost vanishes in 3 m KOAc. The shift of the position of the peak toward higher ATP concentrations likely reflects the weakened affinity of the gp44-binding sites for the highly charged ATP ligand as the salt concentration is increased. A higher Kd value means a higher concentration of ATP is required to fill the sites, and therefore the inhibition profile (and the peak) should move toward higher ATP concentrations as the salt concentration is increased. Control experiments show that the clamp remains bound to the clamp loader at the high salt concentrations used because the ATPase rate at the plateau remains ∼15-fold (or more) higher than that seen at the same salt concentrations with the clamp loader complex in the absence of clamp.
The increase of the plateau ATPase activity with increasing salt concentration, and the simultaneous disappearance of the substrate inhibition profile (i.e. the peak in the ATP concentration-dependent rate profile), could have a variety of origins. One possibility is that at high salt concentrations the ATP-binding sites are not equivalent, with the inhibitory sites being more sensitive to salt concentration and therefore remaining empty as the salt concentration is increased. This hypothesis was ruled out by carrying out ATP binding experiments at higher salt concentrations (Fig. 6), which revealed that the same plateau (four ATP molecules bound with equal affinity) was approached at all salt concentrations and that binding continued to be independent and noncooperative, but that the overall binding affinity decreased significantly as the salt concentration was increased.
FIGURE 6.

The gp44/62-gp45 clamp loader-clamp complex binds four ATP molecules with similar affinities in salt concentrations up to 3 m KOAc. The ATP bound to gp44/62 under steady-state conditions in the presence of gp45 was measured using an ultrafiltration method as described under “Experimental Procedures.” Component concentrations were 30 μm gp44/62 and 35 μm WT-gp45. An ATP regeneration system was present in the reaction mixture. The concentration of KOAc in reaction buffer was 0.16 m (open circles), 1 m (closed circles), 2 m (open squares), and 3 m (closed squares). The data for 0.16 m KOAc (open circles) are the same as in Fig. 2. The curves were obtained by fitting the data points to a binding model (Equation 1) with four equal and independent ATP-binding sites, with Kd and the number of ATP sites per complex as floating parameters (continuous lines), or with Kd as the only floating parameter and the number of ATP sites per complex fixed at four (dashed line). The best fit values for Kd were 27 (±4) μm (0.16 m KOAc, open circles), 200 (±15) μm (1 m KOAc, closed circles), 500 (±240) μm (2 m KOAc, open squares), and 600 (±200) μm when both Kd and the number of ATP sites per complex in Equation 1 were allowed to float (3 m KOAc, closed squares, continuous line) or 1,300 (±60) μm when the number of ATP sites per complex in Equation 1 was fixed at four (3 m KOAc, closed squares, dotted line). The best fit values for the number of ATP sites per complex were 3.6 (±0.1) (0.16 m KOAc, open circles), 4.4 (±0.2) (1 m KOAc, closed circles), 3.7 (±1) (2 m KOAc, open squares), and 2.3 (±0.5) when both Kd and the number of ATP sites per complex in Equation 1 were allowed to float (3 m KOAc, closed squares, continuous line).
The most likely explanation is that high salt concentrations “lock” the clamp-clamp loader complex into the more active (clamp closed) conformation seen at low ATP concentrations, so that the shift to the functionally activated (clamp open) conformation as the ATP sites are filled cannot occur. We also found that the substrate inhibition peak disappears when the ATPase rates were measured at lower temperature (5 °C, rather than room temperature) in 0.16 m KOAc, arguing that low temperature also inhibits the shift of the clamp-clamp loader conformation to the functionally activated form.
Clamp Bound to the Clamp Loader Complex Is in the Closed (Unactivated) Form at High Salt Concentrations—Increasing the salt concentration had the same effect on the ATPase rate profile of the gp44/62-dm-gp45 complex as that seen when the wild-type clamp served as cofactor. At 3 m KOAc an ∼6.6-fold increase of the ATPase rate at the plateau was observed relative to the rate in 0.16 m KOAc (data not shown). High salt concentration appears, at least qualitatively, to have the same effect on the ATPase activity of the clamp-clamp loader complex as does covalently closing the clamp by cross-linking (Fig. 4). We reasoned that if high salt prevents the ring from being opened by the clamp loader, then the ATPase rate profile seen at high salt concentrations with the clamp loader bound to either a cross-linked or a noncross-linked clamp should be the same. Fig. 7 shows that the ATPase rates, measured in the presence of the uncross-linked and cross-linked forms of dm-gp45 in 3 m KOAc, are indeed indistinguishable. This is very different from the situation observed in 0.16 m KOAc, where at higher ATP concentrations the ATPase rates were much greater when the cross-linked clamp serves as cofactor instead of the uncross-linked clamp. We conclude that at high salt concentrations the clamp loader complex is unable to open the clamp, and thus the ATP concentration-rate profile characteristic of the closed clamp dominates.
FIGURE 7.

The clamp in the clamp-clamp loader complex is closed in the presence of 3 m KOAc at all ATP concentrations. The steady-state ATPase rates were measured in the presence of an ATP regeneration system (“Experimental Procedures”). Component concentrations were 1 μm gp44/62, 1.5 μm uncross-linked dm-gp45 (closed symbols), or BMH-dm-gp45 (open symbols), and the indicated amount of ATP. The KOAc concentration in the reaction buffer was 3 m.
Primer-Template DNA Binding to the gp44/62-gp45 Complex Is Rapid and Catalytic When the Clamp Is Activated (Opened) at Higher ATP Concentrations—Finally, we return to the issue of whether the abortive clamp-loading cycle seen in the absence of DNA is the same as the productive cycle observed in the presence of P/T DNA (the clamp loading target). Previously, in our pre-steady-state experiments with this system (16), we had shown that the P/T DNA cofactor works catalytically to increase the ATPase activity of the complex by speeding the turnover of the open clamp (and thus the rate of the ATPase cycle) as a consequence of the transient loading of the open ring onto the P/T DNA.9 Fig. 8A shows the ATPase rates (as a function of input ATP concentration) for a 1 μm concentration of gp44/62 loader complex bound to both the gp45 (1.5 μm) and the P/T DNA (1.2 μm) cofactors. Here the response of the ATPase rate to added ATP is opposite that observed in the presence of the clamp (without P/T DNA) (Fig. 1A). With only the clamp present, we observed an inhibition of the ATPase activity as the ATP concentration was increased, whereas in the presence of P/T DNA a progressive activation of the complex was seen as the ATP concentration was increased, with the kinetic profile appearing slightly sigmoidal. This is consistent with our demonstration in this study that an open clamp complex is present only at physiological ATP and salt concentrations, because the open clamp-containing complex is presumably the activated conformation in the loading process and therefore manifests P/T DNA-dependent stimulation of the ATP binding and hydrolysis cycle.
FIGURE 8.

The DNA-stimulated gp44/62 ATPase rates suggest that the ring opens up as more ATP is added. Steady-state ATPase rates were measured in the presence of an ATP regeneration system, as described under “Experimental Procedures.” A, gp44/62 ATPase activity measured as a function of ATP in the presence of gp45 and P/T DNA cofactors displays a sigmoidal rate profile. Component concentrations were 1 μm gp44/62, 1.5 μm gp45, 1.2 μm P/T DNA, and the indicated amounts of ATP. B and C, binding of P/T DNA to the gp44/62-gp45 complex is slow at low ATP concentrations and fast at high ATP concentrations. Component concentrations were 1 μm gp44/62, 1.5 μm gp45, and 2.5 μm (B) or 1 mm (C) ATP, plus the indicated amount of P/T DNA. D, DNA-stimulated gp44/62 ATPase rates measured in the presence of an open and closed dm clamp. Component concentrations were 1 μm gp44/62, 1.2 μm P/T DNA, 1.5 μm uncross-linked dm-gp45 (open symbols), or BMH-cross-linked dm-gp45 (closed symbols), and the indicated amount of ATP.
P/T DNA Is Catalytic for Clamp Loading at High ATP Concentrations but Not at Low ATP Concentrations—We show in Fig. 8, B and C, that DNA binding is at least partially rate-limiting at low ATP concentrations, whereas it is not rate-limiting at high ATP concentrations. Thus Fig. 8B shows, at an input ATP concentration of 2.5 μm, that the ATPase activity of the gp44/62-gp45 complex increases with increasing P/T DNA concentration and that a plateau is not reached even in the presence of a 24-fold excess of P/T DNA over enzyme complex. In contrast (Fig. 8C), at 1 mm ATP the ATPase activity approaches a maximal value at P/T DNA concentrations at levels well below 1:1 with the clamp loader complex (note difference in the scale of the x axes of Fig. 8, B and C). This outcome is consistent with our earlier observations that had also suggested, on the basis of pre-steady-state kinetic measurements (16), that P/T DNA acts catalytically on the ATPase rate of the clamp-containing complex at 1 mm ATP concentrations and that the rate of binding of the P/T DNA cofactor to the loading complex is fast relative to the rates of the other processes driven by ATP binding and hydrolysis.
Increasing ATP Concentrations Do Not Increase the Rate of Loading of the Covalently Closed Clamp—Finally, Fig. 8D shows the ATPase rate of the clamp loader complex as a function of ATP concentration in the presence of P/T DNA and the double mutant clamp (either uncross-linked or cross-linked). As observed previously, for ATPase rates measured in the presence of dm-gp45 but in the absence of P/T DNA (Fig. 4), the complexes formed with either uncross-linked or cross-linked clamps at low ATP concentrations show the same ATPase rate, suggesting that under these conditions both the cross-linked and the uncross-linked clamps are closed. Fig. 8D shows that the two kinetic profiles begin to diverge at ATP concentrations greater than 10 μm, although they diverge differently than in the absence of P/T DNA (compare with Fig. 4).
In Fig. 8D we see that, in the presence of P/T DNA, the ATPase rate with the uncross-linked ring continues to increase with increasing ATP concentrations, although the rate with the cross-linked clamp soon reaches a plateau (Fig. 8D). This indicates again that the uncross-linked ring complex undergoes a conformational change with increasing ATP concentrations, which is manifested here by an increase in the ATPase rate of the complex. The ATPase profile appears slightly sigmoidal, as observed previously in the presence of the WT-gp45 clamp (Fig. 8A). As shown for WT-gp45 (Fig. 8, B and C), this increase in ATPase activity is most likely because of an increased rate of binding of the P/T DNA cofactor to the clamp-clamp loader complex, which at higher ATP concentrations carries an open clamp.
This increase of the ATPase activity of the complex as more ATP is added is not observed when the clamp is cross-linked (Fig. 8D, closed symbols). The cross-linked clamp cannot form an open ring, and therefore binding of the clamp-clamp loader complex to P/T DNA cannot result in normal clamp loading. As a consequence, the kinetic profile obtained with the cross-linked ring follows simple Michaelis-Menten kinetics. It appears that binding to P/T DNA is at least partially rate-limiting when the ring is cross-linked, because the ATPase activity increases as more DNA is added (data not shown). We speculate that the closed ring, either alone or in complex with the clamp loader, has some probability of reaching the P/T DNA junction (and thus stimulating the ATPase of the complex), perhaps by sliding onto the linear P/T DNA molecule from the ends. This process is clearly slower than the active gp44/62-directed loading of the open clamp onto the P/T DNA and, in any case, could not occur with the real genome where “open ends” of the DNA are not available.
DISCUSSION
Roles of ATP Binding and Hydrolysis in the Function of Macromolecular Machines—Most macromolecular machines of gene expression are driven by ATP binding and hydrolysis. Enzymologists have traditionally focused on the catalytic efficiency of enzymes, with increased rates of product formation corresponding to “better” function. This is clearly a reasonable point-of-view for enzymes in metabolic pathways, where turnover rates directly control the rate of appearance of the reaction product needed in the next step. However, it represents a less appropriate orientation in considerations of molecular motors, where the physiological objective is to produce directed motion (muscle), cargo transfer (ion channels, cytoplasmic motors, clamp loaders), or nucleic acid unwinding (helicases). ATP is the “fuel” that drives these reactions, and thus the ATP binding and hydrolysis cycle must be coupled to the ultimate function of these systems, just as fuel consumption in a motor car must be coupled to the drive wheels. However, maximizing the output of the products of ATP hydrolysis is clearly not the central issue in systems of this kind; rather, as seen here, effective functional coupling may actually decrease the overall rate of ATP hydrolysis.
In this study we have investigated the behavior of the steady-state ATP cycle in the clamp loading complex of the DNA replication system of bacteriophage T4 at three different levels of function as follows: (i) ATP binding and hydrolysis by the isolated clamp loader complex; (ii) ATP binding and hydrolysis by the clamp loader bound to the clamp co-factor and using either a functional (“openable”) or a non-functional (permanently closed) clamp; and (iii) ATP binding and hydrolysis by the clamp loader in the presence of the clamp co-factor and a DNA loading “target” cofactor (here a short oligomeric P/T DNA construct). All of these represent partial model systems that undergo “futile reaction cycles,” futile because the processivity clamp (when present) is released back into solution after being opened (activated), rather than fulfilling its “final function” of loading onto a replication fork located on a long (usually closed circular) ds-DNA genome from which it cannot readily dissociate, and then moving along the DNA to reach and bind a DNA polymerase molecule. After binding to the polymerase in the “real” cycle, the clamp subsequently interacts with other regulatory components to control the processivity of DNA synthesis during the various phases of the replication cycle.
What Have We Learned from the Three “Futile Cycle” Models of the T4 Clamp Loader System?—Our measurements with the pentameric T4 clamp loader complex alone (gp44/62 only; no clamp) show that four ATPs bind to the gp44 subunits of the clamp loader with reasonably strong affinity (Kd = 94 μm) and with no cooperativity (Fig. 2). Furthermore, the basal steady-state rate of hydrolysis of the ATP substrate by gp44/62 is low and follows standard Michaelis-Menten kinetics, as expected for independent ATPase active sites (Fig. 1B). Addition of a wild-type gp45 clamp leaves the ATP-binding isotherm of the clamp loader virtually unchanged (although with a slightly increased ATP binding affinity; Kd = 27 μm), but still displaying independent and equal binding of the four ATPs (Fig. 2) to their gp44 subunit sites. This means that the ATP ligands bind to the gp44 sites of the loader complex independently and at random (that is not sequentially) in both of these model systems.
In contrast, the ATPase kinetics of the clamp loader complex change appreciably on binding the gp45 clamp, with binding to the second through the fourth ATP sites of the loader progressively and significantly inhibiting the ATPase activity of the complex (Figs. 1A and 3). Thus binding of the wild-type clamp to the clamp loader activates (changes the conformation of) the gp44/62-gp45 complex, and this change manifests itself as a major decrease in the turnover rate of the ATP substrate, although substrate binding is unchanged. We show in this study that this conformational change involves the opening of the bound processivity clamp, because the inhibition rate profile of the overall complex is effectively abolished if a covalently cross-linked (i.e. non-openable) gp45 trimer is substituted for the wild-type clamp. Furthermore, the conformational change driven by ATP binding to the loader subunits appears to reflect interactions of all the ATP ligands with the other components of the complex, but these interactions affect only the rate of ATP hydrolysis and are only manifested in the presence of the bound and open processivity clamp (Fig. 3).
We note that this “activation” of the clamp-clamp loader complex (activation of the clamp loading function, not the ATPase activity) is likely driven by electrostatic interactions because the inhibition rate profile (Fig. 5), but not the binding of the clamp to the loader complex, is suppressed at very high salt concentrations that also diminish the binding affinities of the ATPs for their gp44-binding sites (Fig. 6). Furthermore, at very high salt concentrations, which limit the range of electrostatic interactions, the inhibitory rate profile is abolished, and the rate profiles of complexes containing either a cross-linked or an uncross-linked clamp are the same and revert to a simple Michaelis-Menten shape with identical Km parameters (Fig. 7), showing that, in the absence of the open clamp, the individual gp44 subunits again bind and hydrolyze ATP independently.
Finally, using our most complex futile reaction cycle system containing clamp, clamp loader, and an oligomeric P/T junction DNA construct (Fig. 8), we have also shown that the bound and open (Fig. 8D) clamp is very rapidly loaded onto the P/T junction. We have additionally shown that clamp loading, and the subsequent release back into solution of the loader complex and the clamp (as a consequence of slippage of the closed clamp over the ends of the P/T DNA construct), is catalyzed by the DNA construct at high ATP concentrations (Fig. 8, B and C), as shown by further DNA-dependent increases in the ATP hydrolysis rate.
Binding the Open Clamp Drives the Clamp Loader into a Special Conformation—The simplest interpretation of these results and those of our earlier pre-steady-state kinetics study (16), taken together with the structural models derived from studies with the E. coli and yeast systems (20, 26), is that, on binding, the open T4 sliding clamp “traps” its homologous clamp loader platform into a particular (activated) conformation. We note that even though all four ATP-binding subunits of the T4 system are identical, the individual subunits are not structurally equivalent in this conformation because in the tightly associated gp44/62 complex (14) both the open clamp and the single gp62 subunit (consistent with the δ subunit of E. coli and the RFC5 subunit of yeast) must have a specific (and different) structural relationship to each of the otherwise identical gp44 subunits.
Thus, as in the model proposed by Bowman et al. (26) and Johnson et al. (20) for the clamp loading complexes of higher organisms, we suggest that the T4 clamp binds in a specific structural relationship to the individual subunits of the overall clamp loader and that this binding distorts the clamp loader and simultaneously distorts and opens the clamp. The positively charged interior of the clamp can then bind to (and around) the negatively charged P/T junction (27, 28), facilitating the release of the clamp onto, and its closure around, the P/T DNA. This clamp release process is also facilitated by the proximity of the open clamp to a particular subunit-subunit interface of the distorted clamp loader, which proximity we assume triggers the hydrolysis of the ATP ligand bound at that interface by facilitating interaction of the γ-phosphate of the bound ATP with the arginine finger of the adjacent subunit, as proposed (on structural grounds) for the E. coli and yeast complexes (11, 26).
This hydrolysis, and the subsequent rapid release into solution of the ADP and Pi products at the affected ATP-binding site (Table 1), decreases the electrostatic potential of the overall complex that is crucial for holding the clamp open and for maintaining the distorted conformation of the clamp loader. This change, in turn, reduces the binding affinity of the distorted clamp for the distorted clamp loader sufficiently to permit both to relax to their ground-state conformations. The result is release of the closed clamp from the clamp loader, either directly back into solution or via loading onto and sliding off of the P/T DNA construct in the ternary model reaction.
The present study has shown that the binding of all four ATP ligands to the T4 clamp loader is required to distort the bound gp45 trimer into the open clamp form, and suggests also that only when the clamp is bound in the open form do the four ATP sites become structurally (and hydrolytically) distinct. As a consequence, ATP binding occurs at random to the four active sites, as observed, but the effect of the binding of the open clamp to the clamp loader is to structurally differentiate the ATP-binding sites and trigger ATP hydrolysis at a discrete ATP site within the loading complex.
This model is presented schematically in Fig. 9 and is completely compatible with the assumptions of the ATP binding and hydrolysis analysis presented in Fig. 3. Furthermore, because the T4 open clamp-clamp loader complex is particularly unstable, the hydrolysis of only one ATP appears here to be sufficient to release the clamp from the clamp loader (either into solution or onto the P/T DNA construct) and to permit the clamp to close, as well as permitting the loader to relax back into its undistorted form in which all four ATP sites again become thermodynamically and kinetically equivalent. We note that the E. coli and yeast clamp-clamp loader complexes are likely to be more stable than the T4 complex, and that the hydrolysis of several ATPs, perhaps as proposed (20, 29–31) in a particular sequence, may be necessary to destabilize these binary or ternary complexes, although parallel binding and ATPase hydrolysis studies of the sort described here have not been done with these more complicated and more heterogeneous (in terms of subunits) clamp-clamp loader complexes.
FIGURE 9.

Schematic model of the clamp loading cycle of the T4 system. The individual mechanistic reaction steps are numbered and illustrated as described in the text.
Clamp Loader Complexes Contain Multiple ATP Binding and Hydrolysis Sites—Such studies of the clamp loader system of phage T4 and its direct analogues in higher organisms are rendered complicated because the clamp loader complexes contain multiple ATP-binding sites, and these ATP binding and hydrolysis sites are heterogeneous in function, in that only some (and probably only one in T4) of these sites actually complete the ATP binding and hydrolysis cycle as a part of the clamp loading process. What then are the roles, as well as the physiological functions, of the other (enzymatically “silent”) ATP-binding sites in these systems? These questions are of interest beyond clamp loaders alone, because multiple ATP sites of possibly heterogeneous function occur also in a variety of molecular motors (e.g. ATP synthase, hexameric helicases, etc.).
Role of Bound ATPs in Clamp Loader Activation—What can we conclude from these studies about the role(s) of the multiple bound, but unhydrolyzed, ATP molecules in facilitating the binding and activation (opening) of the previously closed clamp? Because all four of its ATP-binding sites are on identical subunits and because all four sites bind ATP with the same affinity under all circumstances tested, the T4 clamp loader system is particularly well suited to addressing such questions. We have done so here by examining ATP binding and hydrolysis as a function of input ATP concentration in the three model “ATP turnover” systems defined above.
The simplest interpretation of our results is that the presence of four bound ATP molecules is necessary to put the clamp loader into a conformation that permits it to both bind and open the clamp. Furthermore, the demonstration that this process can be suppressed by high salt suggests that the interactions driving the relevant conformation change are electrostatic in nature and reflect a shift in the charge balance within the clamp loader system as a consequence of the binding of the four highly negatively charged ATP ligands.
The binding of these ATPs is clearly central to the activation process, and the subsequent hydrolysis of some (probably one) of these ligands is involved only in the release of the clamp loader from the clamp and the DNA after the clamp has been activated (opened) and loaded. The fact that only binding of ATP is required for loading has been shown previously in our pre-steady-state kinetic studies (16) and has now also been directly confirmed by using Ca2+-ATP rather than Mg2+-ATP as the clamp loader substrate. In this latter work we have shown that both substrates bind to the ATP sites of the T4 clamp loader with equal affinity, but that hydrolysis is significantly inhibited when the Ca2+-ATP substrate is used.10 This approach has provided us with a method to cleanly separate in time the processes that are dependent on ATP binding from those that are dependent on ATP hydrolysis. Some general comments on the use of ATP binding as a conformational regulator have recently been published elsewhere (32).
Multiple ATP Binding in Clamp Loader Activation Is Seen in a Variety of Organisms—The clamp loader complexes of most organisms contain five subunits, including several that bind ATP. Thus three of the five clamp loader subunits of E. coli bind ATP (29), four or five subunits bind ATP in the yeast complex (19, 26), and four bind ATP in the T4 system (14, 22). It has been suggested for the E. coli complexes that ATP binding may expose the binding site for the clamp on the surface of the clamp loader complex, and that a particular ATP site may be centrally involved in modulating the interaction with the clamp and therefore presumably in driving clamp opening as well (31).
In contrast, in the T4 system we have shown that more than one ATP site (and probably all four) are involved in clamp opening. Our results show that the “open clamp” loader complex conformation is present only at high ATP concentrations and is responsible for the substrate inhibition profile of the ATPase rates (Fig. 1A and Fig. 4). We note that if only one particular ATP site were responsible for binding and opening the clamp in the T4 system, the effect (whether inhibition or stimulation) of ring opening on the ATPase activity would be proportional to ATP binding to that site only, and because ATP binding at one any site must follow a single-site isotherm (Fig. 2), simple Michaelis-Menten ATPase kinetics would have been observed.
From the present data we cannot determine exactly how many ATP-binding sites are required to activate clamp opening in the T4 system, although we have calculated the relative concentrations of the four different ATP-bound species present throughout the ATP titration. That calculation was based on the apparent Kd for ATP binding obtained from the binding experiments and the fact that the ATP sites are equal and independent, as suggested by the simple binding isotherms obtained (Fig. 2). Fitting our data to Equation 2 revealed the rates at which ATP is hydrolyzed by the different binding species of the clamp loader complex. We showed that the four ATP-bound species hydrolyze ATP progressively more slowly as the number of bound ATPs increases (Fig. 3). One explanation for this would be that clamp opening becomes progressively easier as two, three, and four ATPs are bound, and that the inhibition of the ATPase activity is proportional to the extent of ring opening. Alternatively, and more likely, there may be only two possible conformations of the complex, one that binds a closed clamp and the other an open clamp, and the equilibrium between these two conformations shifts progressively toward the open clamp conformation as the ATP sites are filled.
This hypothesized conformational shift appears to be impeded when the salt concentration is increased. We found, under very high (and nonphysiological) salt concentrations (3 m KOAc), that four ATP molecules could be bound to the complex, and yet the conformational shift to an open ring did not occur (Fig. 5). The ATPase profile obtained at this high salt concentration closely follows simple (Michaelis-Menten) kinetics, and the ATPase rate plateau increased ∼14-fold relative to the plateau obtained at physiological salt concentration (160 mm). In addition, no difference in the clamp loader ATPase activity was observed, regardless of whether a cross-linked or an uncross-linked mutant clamp was bound. Thus at high salt the bound clamp remains closed, regardless of the number of ATPs bound to the clamp loader complex (Fig. 7).
This latter finding may cast light on the puzzling observation that the crystal structure for the yeast clamp-clamp loader-DNA complex was obtained with effectively nonhydrolyzable ATPγS ligands bound to its ATP-binding sites, and yet the clamp appeared to be closed around the P/T DNA (26). Our results suggest that the high salt concentrations in which these crystals were grown might be responsible for the otherwise difficult-to-explain observation that the sliding clamp was present in its closed conformation within this structure.
Possible Regulatory Implications of the Effect of Clamp Conformation on the ATPase Activity of the Clamp Loader—Under physiological conditions it is clearly desirable that a clamp-clamp loader complex be formed that contains an open clamp, and at the same time hydrolyzes ATP at a low rate. The “open ring” conformation allows rapid binding of the clamp to the P/T DNA junction when it is present on the replication fork, and the low ATPase rate reduces ATP consumption when a P/T DNA junction is not available for clamp loading. On the other hand, it may be advantageous to trigger ATP hydrolysis (to drive clamp loader release) once this open clamp-clamp loader complex binds to the P/T DNA junction and the clamp has closed around the DNA of the replication fork. Our results are consistent with such an hypothesis.
Although the gp45 clamp, in the absence of DNA, stimulates the ATPase activity of gp44/62 at any substrate concentration, this stimulation is reduced to a minimum at the physiological ATP concentrations at which the bound gp45 clamp exists in the open conformation. Our data fitting results suggest that at lower ATP concentrations, where only one ATP is bound and the clamp is most likely closed, the complex hydrolyzes ATP at a rate that is ∼10-fold faster than when the clamp is open at higher levels of bound ATP (Fig. 3). When the double mutant gp45 clamp is forced to remain closed by cross-linking its interfaces, the ATPase rate at physiological ATP concentrations is also higher than the rate observed with the uncross-linked double mutant clamp (Fig. 4). In addition, we found that at high salt concentration (3 m KOAc), where the loader-bound clamp appears to be closed (Fig. 7), the ATPase rate is ∼14-fold higher than that at physiological salt concentration (160 mm KOAc) where the ring is open (Fig. 5). All these observations suggest that the ATP hydrolysis is favored under “closed ring” conditions. At physiological ATP and salt concentrations, DNA binding stimulates the ATPase activity of the clamp loader more than 100-fold (compare Fig. 8A and Fig. 1A), suggesting that the presence of a P/T DNA junction induces the clamp-clamp loader complex to assume the closed ring conformation (after the clamp has been loaded onto the DNA). This stimulates ATP hydrolysis, which then triggers the release of the clamp loader from the replication complex (Fig. 9).
Final Thoughts and Evolutionary Speculations—The results presented in this study, together with earlier findings on the T4 system (16), are consistent with results on other systems and suggest that the basic mechanism for clamp loading is evolutionarily conserved across much of the biological spectrum, including phage T4. In this study we have shown that, in the T4 system, the binding of multiple ATP molecules to the clamp loader complex is required to open the bound processivity clamp. This has not been tested directly for the clamp loading systems of higher organisms, but might well apply there also. Finally, it seems reasonable to speculate that the observation that the T4 clamp can be opened only when multiple ATP molecules are bound to the clamp loader, which requires relatively high ATP concentrations, could also serve as an effective feedback mechanism to prevent processive DNA replication from proceeding in the absence of sufficient NTPs to support the replication and transcription activities required for cell function.
Acknowledgments
We are grateful to our laboratory colleagues for many useful discussions of this work.
This work was supported, in whole or in part, by National Institutes of Health Grants GM-15792 and GM-29158. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: P/T DNA, the primer-template junction within the replication fork; BMH, bismaleimido hexane; DPDPB, 1,4-di-[3′-(2′-pyridyldithio)-propionamido]butane; DTT, dithiothreitol; ds, double-stranded; dm, double mutant; ATPγS, adenosine 5′-O-(thiotriphosphate).
It is not always understood that the volume of filtrate used is not a critical parameter in these measurements. Thus, the use of 10, 50, or nearly 100% of the original sample volume as filtrate does not alter the result obtained. This follows because, even if the protein concentration increases in the solution that is retained after centrifugation, the concentration of free ligand that is in equilibrium with the protein-bound complex remains the same. We can model the filtration process as two steps as follows: (i) a rapid centrifugation step, during which the equilibrium is frozen; followed by (ii) a re-equilibration step during which the components can reach a new equilibrium if necessary. We begin with the solution components at equilibrium, “freeze” the equilibrium, and then centrifuge until part of the solution has passed through the membrane. This step decreases the volume of the retained fraction, and therefore increases the concentration of both the free protein and the fraction of the protein that is complexed with the ligand. It does not change the concentration of free ligand because the ligand passes freely through the membrane. We now ask whether the components are still in equilibrium or whether a new equilibrium has to be established? Before centrifugation, the concentrations of the components were related as follows: [P][X]/[PX] = Kd. Here P represents free protein; X is free ligand; PX is protein-ligand complex, and Kd is the dissociation constant for the ligand. If centrifugation decreased the volume of the retained fraction by factor a, it increased the concentration of both P and PX by the same factor a, whereas the concentration of X was not changed. The a coefficients cancel, and we write the following: a[P][X]/a[PX] = [P][X]/[PX] = Kd. Thus the system is still in equilibrium, and no adjustment of the component concentrations needs be made to validate the equilibrium expression. This shows that during centrifugation, even if the concentration of the protein in the retained fraction is increased, the ratio of free to bound protein and consequently the concentration of free ligand remain constant.
There is a sequential ATP binding model that does not result in a sigmoid titration curve. In this model the intrinsic affinities of the ATP sites become progressively lower as each site (here the four gp44 subunits) is filled. As a consequence the resulting “negative cooperativity” exactly balances the apparent positive cooperativity because of sequential binding. More generally, the binding of a ligand to n independent sites with the same Kd value is indistinguishable from sequential binding for situations in which the dissociation constants are related as Kd,i = (Kd)(i/(n - i + 1)), where i refers to the i-th site that is filled with ligand (here ATP); n is the total number of binding sites, and Kd,i is the apparent Kd at each occupancy level. However, such a special progression of sequential binding affinities is rendered particularly unlikely for our system because we obtain the same simple binding isotherm for this highly charged ligand, with ∼4 ATPs bound at the plateau over salt concentrations ranging from 0.16 to 3 m (Fig. 6). We therefore conclude that the ATP sites of the gp44/62 complex (whether or not clamp is bound) fill independently and at random and that the remaining open sites are equally accessible to the ligand at each level of occupancy.
These data were obtained with excess gp45 because we have shown that the binding of the gp45 clamp (wild-type or double mutant, discussed below) to the gp44/62 clamp loader at lower input ATP concentrations is partially rate-limiting for ATPase rates measured in the presence of a 1 μm concentration of the gp44/62 clamp loader containing only a small excess of clamp (1.5 μm). Increasing the clamp concentration to 10 μm abolished this rate limitation, indicating that at such high clamp concentrations the loader complex is fully saturated with clamp at all input ATP concentrations, and clamp binding is no longer rate-limiting.
We note that the concentration of added ATP at the peak of the rate profile obtained with the uncross-linked dm-gp45 clamp falls at a slightly higher ATP concentration (∼40 μm) than does the peak for the complex bound to WT-gp45 (∼30 μm; Fig. 1A). The maximal activity at the peak is also slightly higher than that seen with WT-gp45 (10.6 μm min-1 versus 7.1 μm min-1 measured with the WT-gp45 clamp, see Fig. 1A), and the rate plateau is approached slightly more slowly with increasing concentrations of ATP. However, these minor quantitative differences do not obscure the essential similarity of the ATPase rate profiles of the gp44/62 complex with WT- and dm-gp45 as a function of ATP concentration.
A small inhibition is still detectable in this profile. The highest ATPase rate is attained at an added ATP concentration of ∼200 μm, and beyond this concentration, the activity decreases slightly before a final plateau is reached at ∼86% of the maximal activity (14.4 and 12.8 μm min-1 at 200 μm ATP and 12.4 and 10.9 μm min-1 at the plateau, for BMH-cross-linked and DPDPB-cross-linked dm-gp45, respectively). In addition, the ATPase activity at the plateau for the complex bound to the covalently closed ring is 2.4- to 2.7-fold higher than that of the complex with the uncross-linked clamp (12.4 and 10.9 μm min-1 for BMH-dm-gp45 and DPDPB-dm-gp45, respectively, versus 4.5 μm min-1 for the uncross-linked mutant clamp). However, at added ATP concentrations up to 20–30 μm, the ATPase activity measured in the presence of uncross-linked dm-gp45 and that measured in the presence of cross-linked dm-gp45 are essentially the same, suggesting that both clamps are effectively in the same conformation at low ATP levels. The ATPase rate profiles begin to diverge at input ATP concentrations greater than ∼30 μm, with the profile for the cross-linked ring continuing to increase and then decreasing only slightly (perhaps reflecting a residual contaminant of the solution of cross-linked clamps with uncross-linked gp45?, whereas the rate for the complex containing the uncross-linked clamp decreases much more rapidly. We do note that the increase in ATPase activity that results from closing the dm clamp is only ∼2.5-fold, which is significantly less than the 11-fold that the fitting results lead us to expect if the substrate inhibition effect is indeed removed totally by cross-linking the clamp. As noted above, cross-linking of the gp45 double mutant is not 100% efficient, and ∼10% of the dm-gp45 clamps remain partially open at one interface (see “Experimental Procedures”). If cross-linking the clamp eliminated completely the 11-fold substrate inhibition, then in the presence of 10% open rings one would expect an ∼10-fold increase in the ATPase activity (0.1 + 0.9 × 11-fold = 10-fold), instead of the smaller increase that is actually observed. Clearly, some substrate inhibition still occurs with cross-linked dm-gp45, although much less than seen with the wild-type or the dm-clamp. We speculate that this may reflect the fact that the cross-links introduced are longer than the direct residue-to-residue distance across the subunit interfaces, meaning that the trimeric clamp may retain some conformational flexibility, even though none of the interfaces can “open” to admit DNA. If true, this suggests that the clamp loader responds to the exact geometry of the clamp, rather than sensing directly whether one interface (or more) is completely open.
The P/T DNA construct we used in these studies was relatively short, and therefore clamp loading was transient (and could be shown to be catalytic), because as soon as the closed clamp was released onto the P/T DNA construct by the clamp loader, it slipped off the ends of the DNA and could thus be recycled (see Ref. 16 and “Discussion”).
P. Pietroni and P. H. von Hippel, manuscript in preparation.
References
- 1.Hingorani, M. M., and O'Donnell, M. (2000) Curr. Biol. 10 R25-R29 [DOI] [PubMed] [Google Scholar]
- 2.Huang, C. C., Hearst, J. E., and Alberts, B. M. (1981) J. Biol. Chem. 256 4087-4094 [PubMed] [Google Scholar]
- 3.Stukenberg, P. T., Studwell-Vaughan, P. S., and O'Donnell, M. (1991) J. Biol. Chem. 266 11328-11334 [PubMed] [Google Scholar]
- 4.Maki, S., and Kornberg, A. (1988) J. Biol. Chem. 263 6561-6569 [PubMed] [Google Scholar]
- 5.Kaboord, B. F., and Benkovic, S. J. (1995) Curr. Biol. 5 149-157 [DOI] [PubMed] [Google Scholar]
- 6.Gomes, X. V., and Burgers, P. M. (2001) J. Biol. Chem. 276 34768-34775 [DOI] [PubMed] [Google Scholar]
- 7.Neuwald, A. F., Aravind, L., Spouge, J. L., and Koonin, E. V. (1999) Genome Res. 9 27-43 [PubMed] [Google Scholar]
- 8.Ogura, T., and Wilkinson, A. J. (2001) Genes Cells 6 575-597 [DOI] [PubMed] [Google Scholar]
- 9.Jeruzalmi, D., O'Donnell, M., and Kuriyan, J. (2001) Cell 106 429-441 [DOI] [PubMed] [Google Scholar]
- 10.Jeruzalmi, D., Yurieva, O., Zhao, Y., Young, M., Stewart, J., Hingorani, M., O'Donnell, M., and Kuriyan, J. (2001) Cell 106 417-428 [PubMed] [Google Scholar]
- 11.Davey, M. J., Jeruzalmi, D., Kuriyan, J., and O'Donnell, M. (2002) Nat. Rev. Mol. Cell Biol. 3 826-835 [DOI] [PubMed] [Google Scholar]
- 12.O'Donnell, M., Onrust, R., Dean, F. B., Chen, M., and Hurwitz, J. (1993) Nucleic Acids Res. 21 1-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cullmann, G., Fien, K., Kobayashi, R., and Stillman, B. (1995) Mol. Cell. Biol. 15 4661-4671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jarvis, T. C., Paul, L. S., and von Hippel, P. H. (1989) J. Biol. Chem. 264 12709-12716 [PubMed] [Google Scholar]
- 15.Rush, J., Lin, T. C., Quinones, M., Spicer, E. K., Douglas, I., Williams, K. R., and Konigsberg, W. H. (1989) J. Biol. Chem. 264 10943-10953 [PubMed] [Google Scholar]
- 16.Pietroni, P., Young, M. C., Latham, G. J., and von Hippel, P. H. (2001) J. Mol. Biol. 309 869-891 [DOI] [PubMed] [Google Scholar]
- 17.Hingorani, M. M., and O'Donnell, M. (1998) J. Biol. Chem. 273 24550-24563 [DOI] [PubMed] [Google Scholar]
- 18.Turner, J., Hingorani, M. M., Kelman, Z., and O'Donnell, M. (1999) EMBO J. 18 771-783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gomes, X. V., Schmidt, S. L., and Burgers, P. M. (2001) J. Biol. Chem. 276 34776-34783 [DOI] [PubMed] [Google Scholar]
- 20.Johnson, A., Yao, N. Y., Bowman, G. D., Kuriyan, J., and O'Donnell, M. (2006) J. Biol. Chem. 281 35531-35543 [DOI] [PubMed] [Google Scholar]
- 21.Latham, G. J., Dong, F., Pietroni, P., Dozono, J. M., Bacheller, D. J., and von Hippel, P. H. (1999) Proc. Natl. Acad. Sci. U. S. A. 96 12448-12453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Young, M. C., Weitzel, S. E., and von Hippel, P. H. (1996) J. Mol. Biol. 264 440-452 [DOI] [PubMed] [Google Scholar]
- 23.Jarvis, T. C., Ring, D. M., Daube, S. S., and von Hippel, P. H. (1990) J. Biol. Chem. 265 15160-15167 [PubMed] [Google Scholar]
- 24.Moarefi, I., Jeruzalmi, D., Turner, J., O'Donnell, M., and Kuriyan, J. (2000) J. Mol. Biol. 296 1215-1223 [DOI] [PubMed] [Google Scholar]
- 25.Alley, S. C., Shier, V. K., Abel-Santos, E., Sexton, D. J., Soumillion, P., and Benkovic, S. J. (1999) Biochemistry 38 7696-7709 [DOI] [PubMed] [Google Scholar]
- 26.Bowman, G. D., O'Donnell, M., and Kuriyan, J. (2004) Nature 429 724-730 [DOI] [PubMed] [Google Scholar]
- 27.Georgescu, R. E., Kim, S. S., Yurieva, O., Kuriyan, J., Kong, X. P., and O'Donnell, M. (2008) Cell 132 43-54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jarvis, T. C., Paul, L. S., Hockensmith, J. W., and von Hippel, P. H. (1989) J. Biol. Chem. 264 12717-12729 [PubMed] [Google Scholar]
- 29.Johnson, A., and O'Donnell, M. (2003) J. Biol. Chem. 278 14406-14413 [DOI] [PubMed] [Google Scholar]
- 30.Williams, C. R., Snyder, A. K., Kuzmic, P., O'Donnell, M., and Bloom, L. B. (2004) J. Biol. Chem. 279 4376-4385 [DOI] [PubMed] [Google Scholar]
- 31.Snyder, A. K., Williams, C. R., Johnson, A., O'Donnell, M., and Bloom, L. B. (2004) J. Biol. Chem. 279 4386-4393 [DOI] [PubMed] [Google Scholar]
- 32.von Hippel, P. H. (2007) Annu. Rev. Biophys. Biomol. Struct. 36 79-105 [DOI] [PMC free article] [PubMed] [Google Scholar]