

Abstract
The fosfomycin resistance protein FomA inactivates fosfomycin by phosphorylation of the phosphonate group of the antibiotic in the presence of ATP and Mg(II). We report the crystal structure of FomA from the fosfomycin biosynthetic gene cluster of Streptomyces wedmorensis in complex with diphosphate and in ternary complex with the nonhydrolyzable ATP analog adenosine 5′-(β,γ-imido)-triphosphate (AMPPNP), Mg(II), and fosfomycin, at 1.53 and 2.2Å resolution, respectively. The polypeptide exhibits an open αβα sandwich fold characteristic for the amino acid kinase family of enzymes. The diphosphate complex shows significant disorder in loops surrounding the active site. As a result, the nucleotide-binding site is wide open. Binding of the substrates is followed by the partial closure of the active site and ordering of the α2-helix. Structural comparison with N-acetyl-l-glutamate kinase shows several similarities in the site of phosphoryl transfer: 1) preservation of architecture of the catalytical amino acids of N-acetyl-l-glutamate kinase (Lys9, Lys216, and Asp150 in FomA); 2) good superposition of the phosphate acceptor groups of the substrates, and 3) good superposition of the diphosphate molecule with the β- and γ-phosphates of AMPPNP, suggesting that the reaction could proceed by an associative in-line mechanism. However, differences in conformations of the triphosphate moiety of AMPPNP molecules, the long distance (5.1Å) between the phosphate acceptor and donor groups in FomA, and involvement of Lys18 instead of Lys9 in binding with the γ-phosphate may indicate a different reaction mechanism. The present work identifies the active site residues of FomA responsible for substrate binding and specificity and proposes their roles in catalysis.
Phosphorus-carbon bond-containing natural products (phosphonates and phosphinates) are of great medical importance because they exhibit a wide spectrum of antibacterial, antiviral, and antiparasitic activities in addition to high chemical stability. Fosfomycin ((1R,2S)-1,2-epoxy-propylphosphonic acid) (Fig. 1), a natural product of several species of Pseudomonas (1) and Streptomyces (2), is one of the best known representatives of the class. Since its introduction by Merck in 1969 (2, 3), the antibiotic has successfully been used for treatment of lower urinary tract infections (4). It is also very effective against methicillin- and vancomycin-resistant strains of Staphylococcus aureus (5, 6). In addition fosfomycin has the capacity to favor phagocystosis, act as an immunomodulator, and protect human cells from cisplatin, cyclosporine, aminoglycoside, vancomycin, amphotericin B, and polyximin toxicity (7). Although new, and in some instances better, urinary tract infection drugs have become available, fosfomycin remains a very useful drug. Studies of antibiotic prescribing patterns for cystitis during the period 1999–2002 show that fosfomycin trometamol (Monurol®) use increased dramatically, becoming the first choice for any type of cystitis (8). Fosfomycin trometamol is still the only drug approved by the Food and Drug Administration for treatment of acute cystitis during pregnancy (9).
FIGURE 1.
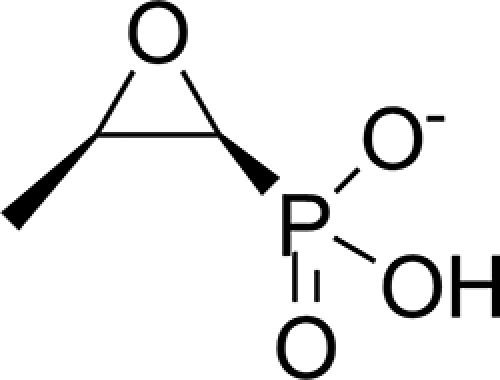
Chemical structure of the antibiotic fosfomycin.
Bactericidal activity of fosfomycin is based on inhibition of UDP-N-acetyl-glucosamine-3-O-enolpyruvyl-transferase, MurA, the enzyme responsible for the first stage of peptidoglycan biosynthesis (10). Fosfomycin irreversibly inactivates MurA by alkylating an active site cysteine (10, 11). Upon introduction into the clinic, resistance to fosfomycin emerged rapidly and was mainly attributed to mutations affecting antibiotic transport (12, 13) or to mutations in the MurA target itself (14). Later, a plasmid-related mechanism of resistance was identified in clinical isolates (15, 16).
Three different fosfomycin resistance genes, fosA, fosB, and fosX, have been extensively described in the literature (16–19). All of the corresponding proteins open the epoxide ring of the antibiotic during the catalysis, but each in a distinct way. FosA utilizes the tripeptide glutathione as a thiol donor, Mn2+ and K+; FosB subgroup members perform a similar reaction using l-cysteine and Mg2+, whereas FosX produces a diol product, 1,2-dihydroxypropylphosphonic acid, with the addition of water, which is assisted by Mn2+. Despite the fact that FosA, FosB, and FosX proteins share modest (23–40%) sequence identity between the subgroups and perform different chemical reactions, they all belong to the vicinal oxygen chelate superfamily of metalloenzymes and share a common molecular fold. Two additional fosfomycin resistance genes, named fomA and fomB, that confer high level fosfomycin resistance on Escherichia coli have recently been discovered in the fosfomycin biosynthetic clusters of Streptomyces wedmorensis (20) and Streptomyces fradiae (21). The fomA and fomB genes encode 29- and 37-kDa proteins, respectively. In contrast to previously known types of fosfomycin resistance proteins, these novel proteins have partial homology to the Mg-ATP-binding domains of some eukaryotic kinases, and therefore they represent a novel mechanism of resistance to the antibiotic. FomA catalyzes phosphorylation of fosfomycin to fosfomycin monophosphate, and FomB, phosphorylation of fosfomycin monophosphate to fosfomycin diphosphate in the presence of ATP and magnesium ions (20) (Scheme 1). Fosfomycin monophosphate and fosfomycin diphosphate have not been shown to alkylate an active site cysteine of MurA.
SCHEME 1.

Enzymatic reactions catalyzed by FomA/FomB proteins.
Fosfomycin resistance kinases FomA and FomB are encoded in the fosfomycin biosynthetic gene clusters of different species of Streptomyces and Pseudomonas. The fomA and fomB genes are required for the production of fosfomycin (21), although their particular role in the biosynthesis still remains unclear. As is true for other antibiotic-producing organisms, fosfomycin producing bacteria must protect themselves from the lethal effect of the antibiotic. It is suggested that FomA and FomB proteins are responsible for the self-resistance of bacteria (20). Both proteins were also proposed to participate in the transport of the antibiotic (20).
The growing threat of antibiotic-resistant microorganisms accentuates the importance of understanding the mechanism of resistance to design effective inhibitors to combat the antibiotic resistance and reduce the further spread of resistant bacteria. ATP-dependent fosfomycin resistance has already been detected in Pseudomonas aeruginosa clinical isolates (22). However, it is not clear whether it is connected to the fomA and fomB genes, because an enzyme characterization was not performed. The understanding of the precise mechanism of the fosfomycin phosphorylation by FomA and FomB proteins will lead to the development of inhibitors specifically targeting FomA-dependent antibiotic resistance. In this paper we report high resolution crystal structure of the fosfomycin resistance kinase FomA from S. wedmorensis in complex with diphosphate and in ternary complex with the nonhydrolyzable ATP analog AMPPNP,3 Mg2+, and fosfomycin. The structure of the ternary complex, which approximates the initial step of the enzymatic reaction, provides a detailed picture of the interactions between both the substrates and the enzyme and reveals amino acids that could play important roles in catalysis.
EXPERIMENTAL PROCEDURES
Gene Amplification and Cloning—The open reading frames for FomA were amplified by PCR using pFBG1204 as a template. Each 5′ primer contained an NdeI site that overlapped the start codon, and each 3′ primer contained a HindIII site immediately downstream of the stop codon. The PCR products were A-tailed using Taq polymerase and dATP, ligated into pGEM-T Easy, and transformed into E. coli α-Select. The cells were plated on LB containing ampicillin and 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-gal). White colonies were picked for plasmid purification. The NdeI/HindIII-generated inserts were cloned into pET28b, digested with the same enzymes, and transformed into E. coli α-Select. Plasmid DNA was purified and used to transform E. coli BL21(DE3). The presence of inserts was demonstrated by digestion of plasmids with the cloning enzymes.
Protein Expression and Purification—FomA was expressed in E. coli BL21(DE3) cells by the autoinduction method (23). An overnight culture grown in MDG medium containing 100 μg/ml of kanamycin was diluted 1:1000 (v/v) into ZYM-5052 medium and incubated at 37 °C for 5 h, and then the temperature was reduced to 22 °C. The cells were pelleted after reaching saturation, frozen at -80 °C, subsequently resuspended in the binding buffer (50 mm Tris-HCl, pH 8.0, 20 mm imidazole, 10% glycerol), and sonicated for 5 min. After removal of debris by centrifugation (at 46,000 × g for 20 min at 4 °C), the supernatant was applied to a TALON metal affinity resin (Co2+ affinity, Clontech Inc.) preequilibrated with the binding buffer. Protein was extensively washed with the binding buffer and eluted with the elution buffer (50 mm Tris-HCl, pH 8.0, 200 mm imidazole, 10% glycerol). The final purification step was performed by size exclusion chromatography (Superdex 200 column; GE Healthcare). The column was preequilibrated with 10 mm Tris-HCl, pH 8.0, supplemented with 150 mm NaCl. A typical flow rate of 0.5 ml/min was maintained during elution. Absorbance at 280 nm was measured to monitor elution of the protein from the column. Purified protein was concentrated to 10 μg/μl.
Seleno-l-methionine (SeMet)-labeled FomA was expressed using E. coli BL21(DE3) cells. An overnight culture grown in MDG medium containing 100 μg/ml of kanamycin was diluted 1:100 (v/v) into M9 medium supplemented with 2 mm MgSO4, 100 nm vitamin B12, 0.4% glucose, and 0.2× trace metals mix (23). The cells were grown at 37 °C until A600 reached 0.5, at which point 100 mg/liter of amino acids threonine, lysine, phenylalanine, and 50 mg/liter of leucine, valine, and isoleucine were added to the medium to inhibit the methionine biosynthetic pathway. The cells were grown for additional 45 min to deplete residual cellular levels of l-methionine, and then 0.2 mm isopropyl β-d-thiogalactopyranoside and 50 mg/liter of SeMet were added, and the growth temperature was reduced to 26 °C. Production of SeMet FomA was allowed to proceed for 24 h before the cells were harvested. Purification of SeMet FomA was carried out as described for the native FomA, with the exception that all buffers were supplemented with 10 mm β-mercaptoethanol.
Crystallization—Crystals of FomA protein have been obtained by the hanging drop method at room temperature. The protein (6–9 mg/ml concentration in 10 mm Tris-HCl, pH 8.0, 150 mm NaCl) was incubated with 0.4 mm solution of ATP for at least 1 h prior to crystallization. The protein-ATP complex was mixed with an equal volume of the well solution (11–13% polyethylene glycol 3350, 0.1–0.15 m triammonium citrate, pH 7.0, 0.1 m MES, pH 5.2–6.2). The crystals appeared in a week and grew to the maximum dimensions in 3 weeks. They belong to the trigonal P3221 space group with a = b = 88.36 Å, c = 79.05 Å. Crystals of SeMet-FomA were grown from the same conditions, and they are isomorphous with the native crystals. Crystals of the FomA·MgAMPPNP·fosfomycin complex were obtained by soaking of FomA·ATP crystals in modified mother liquor consisted of 30% polyethylene glycol 3350, 0.1 m MES, pH 5.5, 10 mm AMPPNP, 50 mm MgCl2, and 10 mm fosfomycin for 5 h.
Data Collection, Structure Solution, and Refinement—For the structure solution by multiple wavelength anomalous dispersion, three data sets were collected at 100 K from a single SeMet derivative crystal at the protein crystallography beamline at the Center for Advanced Microstructures and Devices at Louisiana State University. Prior to data collection, a suitable crystal was dipped for 30 s in a modified mother solution with the addition of 25% glycerol as a cryo-protectant. The images were processed and scaled using DENZO and SCALEPACK (24). Data collection and data processing statistics are summarized in Table 1. Five selenium sites were located by direct methods using the SnB program (25). Reflection phases to 1.6 Å resolution were calculated and improved with CNS (26), which was also used for density modification. The protein model was built into the electron density with program O (27). The structure was refined against 1.53 Å native data using the maximum likelihood refinement in REFMAC (28) with the TLS parameters generated by the TLSMD server (29). TLS tensors were analyzed, and anisotropic B-factors were derived with TLSANL (30). The program O (27) was used to build the models throughout the refinement. No significant electron density was observed for amino acid residues 57–68, 179–182, and 207–210 as well as the last three residues at the C terminus, indicating that these regions are highly mobile or disordered. A difference Fourier map revealed electron density for only a part of the ATP molecule in the active site (Fig. 2A). The diphosphate moiety (DPO) was modeled into the density. Nine residues (residues -8 to 0) of the N-terminal poly-His tail were visible and were included in the final model. Alternate conformations were built for residues His0, Met1, Leu6, Ile8, Arg19, Tyr43, Glu242, Ser249, and Ser250. A total of 231 water molecules were included into the final model. The protein molecule displays excellent stereochemistry with 93.3% of nonglycine residues in the most favored regions of a Ramachandran plot.
TABLE 1.
Data collection, phasing, and refinement statistics
|
FomA·DPO
|
FomA·MgAMPPNP· fosfomycin
|
SeMet FomA·DPO
|
|||
|---|---|---|---|---|---|
| λ1 (peak) | λ2 (inflection) | λ3 (remote) | |||
| Wavelength (Å) | 1.38079 | 1.38074 | 0.9793 | 0.9795 | 0.9537 |
| Resolution (Å) | 1.53 | 2.20 | 1.45 | 1.45 | 1.45 |
| Temperature (K) | 113 | 113 | 113 | 113 | 113 |
| Space group | P3221 | P3221 | P3221 | ||
| Cell dimensions | |||||
| a (Å) | 88.36 | 85.66 | 88.30 | ||
| c (Å) | 79.05 | 78.98 | 78.87 | ||
| Number of molecules/asymmetric unit | 1 | 1 | 1 | ||
| No. of unique reflections | 52 986 | 17 293 | 55 525 | 61 104 | 55 632 |
| Rsym (%)a,b | 4.0 (51.0) | 7.1 (55.7) | 6.2 (64.2) | 5.7 (53.6) | 6.8 (70.0) |
| Completeness (%) | 97.9 (85.3) | 99.6 (97.9) | 97.1 (100.0) | 96.6 (100.0) | 87.4 (90.8) |
| Redundancies | 5.2 (4.6) | 4.1 (3.4) | 7.3 (7.5) | 9.2 (9.4) | 12.8 (11.8) |
| I/σ(I) | 34.8 (2.9) | 19.2 (1.8) | 22.6 (3.0) | 32.0 (4.0) | 28.0 (3.4) |
| Phasing statistics | |||||
| Resolution range (Å) | 30–1.6 | ||||
| Number of sites | 5 | ||||
| Phasing power acentric/centricc | 2.68/2.91 | ||||
| Rc centricd | 0.39 | ||||
| Mean FOM after multiple wavelength anomalous dispersion phasing | 0.666 | ||||
| Refinement statistics | |||||
| Resolution range (Å) | 27.49–1.53 | 29.04–2.20 | |||
| No. of reflections used in refinement | 50 201 | 15 587 | |||
| σ cut-off used in refinement | none | none | |||
| R/Rfree (%)e | 16.86/19.80 | 19.26/25.20 | |||
| Number of refined atoms | |||||
| Protein | 1953 | 2034 | |||
| Heterogen atoms | 9 | 40 | |||
| Water | 231 | 60 | |||
| Average B-factors (Å2) | |||||
| Protein | 25.8 | 43.2 | |||
| Water | 35.8 | 42.7 | |||
| DPO | 41.9 | ||||
| AMPPNP | 44.1 | ||||
| Magnesium | 44.7 | ||||
| Root mean square deviations | |||||
| Bonds (Å) | 0.016 | 0.014 | |||
| Angles (°) | 1.738 | 1.727 | |||
| Ramachandran plot (%) | |||||
| Favored | 93.3 | 90.7 | |||
| Allowed | 6.7 | 8.8 | |||
| Generous | 0 | 0.4 | |||
| Disallowed | 0 | 0 | |||
The values in parentheses are for the highest resolution shell
Rsym = Σ|Ii – <Ii>|/ΣIi, where Ii is the intensity of the ith observation and <Ii> is the mean intensity of the reflection
Phasing power = Σ<Fh>/E, where <Fh> is the root mean square structure factor, and E is the residual lack of closure error
Rc = Σ||FpH ± Fp| – FHcalc|/Σ FpH ± Fp, where FpH and Fp are the structure factor amplitudes for the data collection on the selenium absorption edge
R = Σ||Fo| – |Fc||/Σ|Fo|, where Fo and Fc are the observed and calculated structure factors amplitudes. Rfree is calculated using 1.6 and 1.4% of reflections omitted from the refinement for the FomA·DPO and FomA·MgAMPPNP·fosfomycin structures, respectively
FIGURE 2.

Fo - Fc electron density omit map contoured at 3 σ for DPO in the FomA·DPO complex (A) and MgAMPPNP and fosfomycin in the FomA·MgAMPPNP·fosfomycin complex (B). Mg2+ cation is shown as a sphere.
FomA·MgAMPPNP·fosfomycin Complex—The structure of the complex was determined by the molecular replacement procedure as implemented in AMoRe (28) with the monomer of FomA from the FomA·DPO structure as a search model. The positioned molecular replacement model was refined essentially the same way as described above. A Fo - Fc electron density map unambiguously revealed the presence of AMPPNP, Mg2+, and fosfomycin in the active site (Fig. 2B). The final protein model consists of protein residues -8–178 and 183–263. Alternate conformations have been built for Cys106, Ser249, and Ser250. A total of 60 water molecules were included into the final model. The protein molecule has good stereochemistry with 90.7% of nonglycine residues in the most favored regions of a Ramachandran plot. Refinement statistics for both structures are listed in Table 1. All of the figures except Figs. 1 and 3B were prepared using PyMOL (31).
FIGURE 3.

The structure of the FomA·MgAMPPNP·fosfomycin complex. A, ribbon diagram of a monomer of FomA withα-helices andβ-strands labeled. The AMPPNP molecule is shown as a space filled model. B, structure based sequence alignment between FomA and NAGK. The secondary structure elements of FomA defined by DSSP (47) are given on top. Triangles indicate amino acids that line the AMPPNP-binding site, whereas stars indicate fosfomycin-binding site residues. The image was generated using the program ESPript (48). C, cartoon representation of the dimer with the dyadic axis perpendicular to the paper. The N- (cyan) and the C-lobe (green) domains are shown. AMPPNP and fosfomycin substrate molecules are shown in stick representation.
RESULTS AND DISCUSSION
Overall Fold and Quaternary Structure—According to the classification based on sequence similarity, FomA belongs to the amino acid kinase family (AAKF) of enzymes (PFAM group PF00696) that includes N-acetyl-l-glutamate kinase (NAGK), glutamate-5 kinase (GK), carbamate kinase, the N-terminal domain of aspartokinase, and uridylate kinase (UMPK). The best amino acid identity, however, is only about 24% for the NAGKs. The representatives from the first four classes of the AAKF transfer the γ-phosphate of ATP to a carboxylate or carbamate group of the substrates making acylphosphate bonds, whereas the UMPK enzymes phosphorylate a phosphate group of uridilate. The crystal structure of FomA protein confirmed the assignment of FomA to the AAKF because the enzyme exhibits the polypeptide fold found in all members of the AAKF for which crystal structures have been determined: N-acetyl-l-glutamate kinases (32–34), glutamate-5-kinase (35), carbamate kinase (36), aspartokinases (37–39), and uridylate kinases (40–43). The search was made by the secondary structure matching data base (44) using FomA coordinates from the FomA·MgAMPPNP·fosfomycin complex, because that structure is more ordered and complete in comparison to the FomA·DPO one. Root mean square deviations between the equivalent CA atoms of FomA and all these structures lie in the range of 2.21–2.92 Å (over the range of 185–214 CA atoms).
The molecule of FomA consists of an open αβα sandwich fold with seven α-helices and 14 β-strands (Fig. 3, A and B). The central core is comprised by eight β-strands (β1, β2, β3, β6, β9, β12, β13, and β14) that are sandwiched between two layers of α-helices (α1 and α3 flank one side of the central β-sheet core, and the helices α4, α5, α6, and α7 lie on the opposite side). Similar to the members of the AAKF, the monomer of FomA could be divided into two lobes: the N-terminal lobe (residues 1–160) and the C-terminal lobe (residues 161–263) with a large crevice between them where the active site of the enzyme is located (Fig. 3C). The N-terminal lobe residues participate in binding fosfomycin. The C-terminal lobe binds the nucleotide molecule.
As evidenced by size exclusion chromatography, FomA is a homodimer in solution. The asymmetric unit of FomA in the crystal, however, is represented by a monomer. The homodimer is generated by the crystallographic 2-fold axis. The dimer interface is solely mediated by the interactions between the N-lobes (Fig. 3C). It buries a total of 2875 Å2 of solvent-accessible area, as calculated using the PISA interface server (45) or 1437.5 Å2/monomer (11.8% of the surface) and renders a complexation significance score of 0.407, which accounts for a medium-tight complex. The interdimer interface, which is predominantly hydrophobic, mainly involves: 1) antiparallel interactions between the symmetry related α3-helixes (13 participating residues; most of the dimer interface), 2) interactions of the α4 helix with the symmetry related β7-β8 hairpin, and 3) antiparallel interactions between the 310 helix η3 and the connection loop β5-α4 with the same elements of the other dimer subunit. In addition to numerous hydrophobic interactions, primarily provided by Phe, Thr, Ala, and Val, the interface is stabilized by 18 hydrogen bonds and 18 salt bridges. It should be noted that although roughly the same surface of the N-lobe is used for the dimer formation in FomA, NAGK, GK, and UMPK, the mode of dimerization is significantly different in every case (see Ref. 35 for the detailed description of the NAGK, GK, and UMPK dimers). The list of dimer interface interactions is provided in the supplemental Table S1.
Initially we attempted to obtain a crystal structure of the FomA·ATP complex. However, cocrystallization of FomA with ATP was unsuccessful because the resulting structure (FomA·DPO complex) contains only the diphosphate moiety of the ATP molecule, presumably because of hydrolysis of ATP during crystallization. Soaking of the FomA·DPO crystals in modified mother liquor with the addition of AMPPNP, MgCl2, and fosfomycin (see “Experimental Procedures” for details) prior to data collection proved to be sufficient to obtain the ternary complex (FomA·MgAMPPNP·fosfomycin). Thus, the current paper reports structures of two different reaction states: the FomA·DPO complex, where the active site of the enzyme does not contain any of the required substrates, and the FomA·MgAMPPNP·fosfomycin complex, the approximation of the early stage of the phosphorylation reaction, when all the substrates are bound, and the enzyme is ready to proceed with the catalytical reaction.
The protein in both complexes exhibits the same polypeptide fold and dimeric architecture. However, in contrast to the FomA·DPO structure, which has several disordered parts, the FomA·MgAMPPNP·fosfomycin structure is mainly ordered. The main differences between the two complexes lie in conformations of several structural elements surrounding the active site: 1) a loop stretching from residues 18 to 23 has slightly different conformation; 2) the amino acid residues 57–68 are completely disordered in the FomA·DPO complex, whereas they are ordered in the FomA·MgAMPPNP·fosfomycin structure. Moreover, residues 57–63 are a part of the α2-helix, and 3) residues 207–211 are not visible in the FomA·DPO structure, the helix α6 (residues 195–199) is four residues shorter in the ternary complex (Fig. 4A). As a result, residues 201–205 are no longer part of an α-helix, and they move closer to the active site so that the side chain of Trp202 participates in stacking interactions with the adenine ring of AMPPNP. Such interactions limit access to the active site (Fig. 4A). The conformational change observed in the ternary complex is obviously connected to the movements of loops during the binding of both substrates
FIGURE 4.

A, a stereo view of the superposition of FomA·DPO (magenta) and the FomA·MgAMPPNP·fosfomycin (blue) structures. The polypeptide chains are shown as ribbon representations, AMPPNP, DPO, and fosfomycin molecules are shown in stick representation. B, a superposition of the bound substrates in the FomA·DPO (magenta), FomA·MgAMPPNP·fosfomycin (blue), and NAGK·MgAMPPNP·NAG (orange) complexes. Mg2+ cations are shown as crosses.
The Nucleotide-binding Site—The nucleotide-binding site is located in the groove between the N- and C-lobes. The triphosphate part of AMPPNP binds deeply in the catalytic cleft on the interface between the two lobes (Fig. 3C) and is surrounded by the α6-α7 and β9-β10 connection loops and the η1 helix. The other side of the nucleotide-binding site (the adenosine moiety) is formed by the β9-β10 and α6-α7 connection loops, the β10 sheet, and the N-terminal part of the α7 helix. Two polypeptide segments containing glycine-rich sequence motifs are deeply buried in the catalytic cleft: a segment of the β1-α1 junction (residues 9–15; participates in binding with the γ-phosphate of the nucleotide) and the β2-α2 connection loop (residues 51–55; binds the phosphonate group of fosfomycin). These motifs are well conserved throughout the AAKF and are considered to be the some of main factors for neutralization of the accumulated negative charge during the transfer of a phosphoryl group.
The superposition of the active sites in both structures shows that the DPO molecule is positioned closer to the fosfomycin-binding site than the γ-phosphate of AMPPNP (Fig. 4B). Moreover, the overall position of the DPO molecule more resembles the position of the β- and γ-phosphate groups of AMPPNP in the NAGK structure (33). A different orientation of the γ-phosphate is observed in the case of the ternary complex, where the γ-phosphate group is positioned farther away from the fosfomycin-binding site (Fig. 4B). As a result of the different orientations of the phosphate groups, the modes of interaction of the β- and γ-phosphates differ in both FomA complexes. The diphosphate in the FomA·DPO complex is bound to protein via H-bonds from the nonbridging oxygen atoms of the γ-phosphate to the main chain NH atoms of Gly12 and Gly53, to the side chain NZ of Lys9 (from nonbridging oxygen atoms of both phosphates), and indirectly via a water molecule from the γ-phosphate to OG of Ser148 (Fig. 5A) (Thus we label the phosphate groups of diphosphate as β and γ). In the FomA·MgAMPPNP·fosfomycin complex, a water molecule occupies the position approximately equivalent to the γ-phosphate of DPO or the γ-phosphate of AMPPNP in the NAGK.
FIGURE 5.

Stereo views of the active site. A, FomA·DPO complex. B, FomA·MgAMPPNP·fosfomycin complex. The ligand molecules are shown in ball-and-stick format. The Mg2+ (green) and coordinated water molecules are represented as spheres. The interacting protein residues are shown in stick format. C, a stereo view of the superposition of the AMPPNP binding sites in FomA·MgAMPPNP·fosfomycin (blue) and NAGK·MgAMPPNP·NAD (red) structures. Mg2+ cations are shown as spheres, and water molecules are shown as crosses.
In the ternary complex, the nonbridging oxygen atom O-2 of the α-phosphoryl group of AMPPNP is indirectly hydrogen-bonded via water molecules to the carbonyl oxygen of Ala222 and via the bridging oxygen atom to OG of Thr170. The nonbridging oxygens of the β-phosphoryl group make hydrogen bonds to NZ of Lys216 and OG of Thr170 (Fig. 5B). The bridging N is hydrogen-bonded to OG of Ser13. The γ-phosphoryl group participates in hydrogen bonding with NH of Ser13 and NZ of Lys18 (Fig. 5B). Lys9 and Lys216 correspond to fully conserved lysine residues in NAGKs and GKs. It was proposed for NAGK that these residues are centrally involved in catalysis of phosphoryl transfer by stabilizing the transition state (34). In the FomA·MgAMPPNP·fosfomycin complex, Lys9 does not participate in hydrogen bonding with theγ-phosphoryl group. Instead, NZ of Lys18 is hydrogen-bonded to the nonbridging O-3 of the γ-phosphoryl group. All three phosphoryl groups are complexed with the Mg+2 cation via nonbridging oxygen atoms. Three water molecules complete the hexacoordination sphere of magnesium. One of them is H-bonded to the phosphonate oxygen O-3 of fosfomycin.
The AMPPNP molecule is bound in the extended conformation. An eight-residue insertion loop (residues 203–210) with respect to that in NAGK makes the adenine-binding site less solvent-accessible. The adenine ring sits in the hydrophobic pocket formed by Met213, Val172, Val176, Leu20, Ile175, and Trp202. In the absence of nucleotide (FomA·DPO complex), the pocket is more open to the solvent because of flexibility of the loop 203–211 (Fig. 4A). The adenine ring is in the syn configuration and closely resembles the conformation of AMPPNP in the NAGK structure (33). Interactions with the protein involve hydrogen bonds between N-1 and the main chain NH of Val176, N-6 and the carbonyl oxygens of Val176 and Ala200, N-7 and the main chain NH of Trp202 (Fig. 5B), and stacking interactions in quite a spectacular way. First, the adenine ring makes classical π-π stacking interactions with the side chain of Trp202. Trp202 in turn participates in the additional stacking interaction with the side chain of His254 of a symmetry related subunit. Met213, which is highly conserved throughout the entire AAKF, completes the hydrophobic interactions from the other side of the adenine ring (Fig. 5B). The only hydrophilic interaction between the ribose ring and the protein includes hydrogen bonding between the 3′-hydroxyl group of the ribose ring and the carboxyl oxygen of Asp171.
The Fosfomycin-binding Site—A fosfomycin molecule binds in the N-terminal lobe. The binding pocket is comprised of the N-terminal parts of the helices α2 and α3, the β7-β8 hairpin, the β2-α2 glycine-rich connection loop, the β8-α5 connection, and the α6-α7 junction, which makes a “lid” over the fosfomycin-binding site just as the β3-β4 hairpin does in the NAGK over the NAG-binding site. The α2 helix and the α6-α7 junction (extra residues in comparison to other members of the AAKF) are not visible in the electron density map in the FomA·DPO complex, suggesting that the conformational change and ordering of those structural elements should occur during the binding of fosfomycin.
The phosphonate group of fosfomycin superimposes well on the γ-carboxylate group of NAG in the NAGK and points toward the triphosphate tail of AMPPNP (Fig. 5C). The interactions of fosfomycin with protein are provided by the phosphonate group of the antibiotic via hydrogen bonds. The O-1 phosphonate atom makes H-bonds with the main chain NH and the side chain OG atoms of Ser149. The O-3 phosphonate oxygen participates in hydrogen bonding with OG of Thr210 and OG of Ser148 and with a water of the Mg2+ coordination sphere. The O-2 phosphonate atom makes a hydrogen bond with NH of Gly53 (Fig. 5B). Additional stabilization factors include hydrophobic interactions between C-1 and C-2 of fosfomycin and hydrophobic parts of Thr210, Ile61, and Gly57. The primary role of Ser148, Ser149, and Thr210 in catalysis could be in proper positioning and orientation of fosfomycin for the enzymatic reaction. There are no direct interactions between fosfomycin and the nucleotide molecule. However, water-mediated hydrogen bonds between the phosphonate group of fosfomycin and the γ-phosphate of AMPPNP are observed (Fig. 5B).
Insights into Phosphoryl Transfer—The best studied enzyme of the AAKF is NAGK, for which kinetic data as well as crystal structures of different complexes and reaction state intermediates are available. The enzymatic reaction proceeds via associative in-line phosphoryl transfer with the assistance of the catalytical residues Lys8, Lys217, and Asp162 and with no need for general acid-base catalysis (33). The crystal structures of two FomA complexes presented here reveal a great degree of similarity between FomA and NAGK in the overall molecular fold as well as in the conservation of amino acid residues participating in nucleotide binding (Figs. 3B and 5C). The residues coordinating the adenine moiety of ATP appear to be less well conserved. However, several key residues coordinating the phosphate groups are conserved in the enzymes. These include Lys9, Asp150, and Lys216 corresponding to Lys8, Asp162, and Lys217 in NAGK, respectively (Fig. 5C). Asp150 makes hydrogen bonds with Lys9 and Lys216 in both FomA complexes, and its likely role is in the proper orientation of Lys9 and Lys216 for catalysis, similarly as in case of NAGK. Lys216, which hydrogen bonds to a nonbridging oxygen of the β-phosphate, should activate the departure of the leaving phosphate group. Lys9 in the FomA·DPO complex, which is H-bonded to the γ-phosphate group, is likely to participate in the stabilization of the transition state intermediate, in a similar manner as observed in the NAGK. On the other hand, the conformation of the triphosphate moiety of the AMPPNP molecule in the ternary complex of FomA does not allow hydrogen bond interactions between Lys9 and the γ-phosphate of AMPPNP (Fig. 5C). However, in this case Lys18 (there is no homologous residue in NAGK) is H-bonded to a nonbridging oxygen of the γ-phosphate (Fig. 5, B and C). Such a “switch” from one lysine to another allows Lys18 to be considered the candidate for a catalytic residue, which neutralizes the charge and polarizes the leaving γ-phosphoryl group. Magnesium is coordinated with the α-, β-, and γ-phosphate groups of the nucleotide and together with Lys18 and Lys216 is presumably involved in orientation of the nucleotide and charge neutralization and polarization of the γ-phosphate group.
As expected, the biggest differences between FomA and NAGK are observed in the substrate-binding site. The amino acid residues Ser148, Ser149, and Thr210 participate in binding with phosphonate oxygens of fosfomycin via H-bonds. Their likely role in the enzymatic reaction is in proper positioning and orientation of the antibiotic for the catalysis.
The shortest distance between oxygen atoms of the phosphonate group of fosfomycin and phosphorus atom of AMPPNP is 5.19 Å, a value that is long for the optimal in-line phosphoryl transfer reaction. In addition, the triphosphate tail of AMPPNP is not aligned toward the phosphonate group of fosfomycin. Such observations could suggest that the catalytic conformation of ATP may be significantly different from the one observed for AMPPNP in the FomA·MgAMPPNP·fosfomycin crystal structure. The nucleotide molecule could undergo a conformational change that places the γ-phosphate closer to the fosfomycin phosphonate group to allow the in-line phosphoryl transfer to proceed similar to what is seen for NAGK. Indeed, there is plenty of room to accommodate such a conformational change of the AMPPNP molecule without the need for any restructuring of the active site of the enzyme. The preservation of architecture of the catalytical residues of NAGK (Lys9, Lys216, Asp150 in FomA) in the nucleotide-binding site, as well as the position of the diphosphate molecule in the FomA·DPO complex that superimposes well to the β- and γ-phosphates of AMPPNP in NAGK ternary complex with MgAMPPNP and NAG, suggests that the mechanisms of the enzymatic reaction could be very similar and adds additional ground for a possible conformational change of the nucleotide molecule in the FomA catalyzed reaction. On the other hand, water-mediated catalysis is another possibility that cannot yet be ruled out because interactions between the nucleotide molecule and fosfomycin are mediated by two water molecules. One of them is coordinated to the Mg2+ ion and is hydrogen-bonded to Asp150 (equivalent to the key organizing residue Asp162 in NAGK) (Fig. 5C). Such coordination could suggest general base catalysis. Asp150 is positioned to deprotonate the water and could function as a general base. There is also another water molecule that mediates interactions between AMPPNP and the antibiotic via hydrogen bonds of 2.82 Å to the phosphonate group of fosfomycin and 2.73 Åtothe γ-phosphate of AMPPNP. The water molecule is 3.05 Å from His58 (Fig. 5B). His58 is in close vicinity to both the fosfomycin and AMPPNP molecules (3.83 and 3.45 Å to fosfomycin and AMPPNP, respectively) and is positioned approximately at the midpoint of the phosphoryl transfer site. It should be noted that His58 belongs to the completely disordered part in the fosfomycin-free FomA·DPO structure. In contrast, in the FomA·MgAMPPNP·fosfomycin complex His58 is a part of the ordered α2-helix. The ordering of the helix clearly depends on the binding of fosfomycin in the active site of the enzyme. Hence, His58 could also play an important role in the enzymatic reaction. Mutational and kinetic studies will be necessary to confirm the possible catalytical role of His58 and Asp150.
It is not clear from the present structures whether the FomA-catalyzed phosphoryl transfer proceeds via the associative or the dissociative mechanism. The later mechanism, however, is generally not favorable for enzymatic reactions assisted by ATP (46). Crystal structures of additional complexes, such as complexes with ATP (FomA·ATP and FomA·MgATP), the ternary complex with ADP, Mg(II), and the product of the reaction fosfomycin monophosphate, and a reaction intermediate or a transition state analog will be needed to determine the precise mechanism of the enzymatic reaction.
Supplementary Material
Acknowledgments
We thank Dr. Henry Bellamy and Dr. David Neau for assistance with data collection.
The atomic coordinates and structure factors (codes 3D40 and 3D41) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
This work was supported, in whole or in part, by National Institutes of Health Grant R03AI074770. This work was also supported in part by the Louisiana Governor's Biotechnology Initiative. The Gulf Coast Protein Crystallography Beamline was supported by National Science Foundation Grant DBI-9871464 with co-funding from the National Institute for General Medical Sciences. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Table S1.
Footnotes
The abbreviations used are: AMPPNP, adenosine 5′-(β,γ-imido)-triphosphate; AAKF, amino acid kinase family of enzymes; NAG, N-acetyl-l-glutamate; NAGK, N-acetyl-l-glutamate kinase; UMPK, uridylate kinase; GK, glutamate-5 kinase; DPO, diphosphate moiety; SeMet, seleno-l-methionine; MES, 4-morpholineethanesulfonic acid.
References
- 1.Shoji, J., Kato, T., Hinoo, H., Hattori, T., Hirooka, K., Matsumoto, K., Tanimoto, T., and Kondo, E. (1986) J. Antibiot. (Tokyo) 39 1011-1012 [DOI] [PubMed] [Google Scholar]
- 2.Hendlin, D., Stapley, E. O., Jackson, M., Wallick, H., Miller, A. K., Wolf, F. J., Miller, T. W., Chaiet, L., Kahan, F. M., Foltz, E. L., Woodruff, H. B., Mata, J. M., Hernandez, S., and Mochales, S. (1969) Science 166 122-123 [DOI] [PubMed] [Google Scholar]
- 3.Christensen, B. G., Leanza, W. J., Beattie, T. R., Patchett, A. A., Arison, B. H., Ormond, R. E., Kuehl, F. A., Jr., Albers-Schonberg, G., and Jardetzky, O. (1969) Science 166 123-125 [DOI] [PubMed] [Google Scholar]
- 4.Lobel, B. (2003) Int. J. Antimicrob. Agents 22 (Suppl. 2) 85-87 [DOI] [PubMed] [Google Scholar]
- 5.Cassone, M., Campanile, F., Pantosti, A., Venditti, M., and Stefani, S. (2004) Microb. Drug Resist. 10 43-49 [DOI] [PubMed] [Google Scholar]
- 6.Nakazawa, H., Kikuchi, Y., Honda, T., Isago, T., and Nozaki, M. (2003) J. Infect. Chemother. 9 304-309 [DOI] [PubMed] [Google Scholar]
- 7.Gobernado, M. (2003) Rev. Esp. Quimioter. 16 15-40 [PubMed] [Google Scholar]
- 8.Galatti, L., Sessa, A., Mazzaglia, G., Pecchioli, S., Rossi, A., Cricelli, C., Schito, G. C., Nicoletti, G., and Caputi, A. P. (2006) J. Antimicrob. Chemother. 57 551-556 [DOI] [PubMed] [Google Scholar]
- 9.Stein, G. E. (1998) Ann. Pharmacother. 32 215-219 [DOI] [PubMed] [Google Scholar]
- 10.Kahan, F. M., Kahan, J. S., Cassidy, P. J., and Kropp, H. (1974) Ann. N. Y. Acad. Sci. 235 364-386 [DOI] [PubMed] [Google Scholar]
- 11.Marquardt, J. L., Brown, E. D., Lane, W. S., Haley, T. M., Ichikawa, Y., Wong, C. H., and Walsh, C. T. (1994) Biochemistry 33 10646-10651 [DOI] [PubMed] [Google Scholar]
- 12.Kadner, R. J., and Winkler, H. H. (1973) J. Bacteriol. 113 895-900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsuruoka, T., and Yamada, Y. (1975) J. Antibiot. (Tokyo) 28 906-911 [DOI] [PubMed] [Google Scholar]
- 14.Venkateswaran, P. S., and Wu, H. C. (1972) J. Bacteriol. 110 935-944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garcia-Lobo, J. M., and Ortiz, J. M. (1982) J. Bacteriol. 151 477-479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Suarez, J. E., and Mendoza, M. C. (1991) Antimicrob. Agents Chemother. 35 791-795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bernat, B. A., Laughlin, L. T., and Armstrong, R. N. (1997) Biochemistry 36 3050-3055 [DOI] [PubMed] [Google Scholar]
- 18.Cao, M., Bernat, B. A., Wang, Z., Armstrong, R. N., and Helmann, J. D. (2001) J. Bacteriol. 183 2380-2383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Etienne, J., Gerbaud, G., Courvalin, P., and Fleurette, J. (1989) FEMS Microbiol. Lett. 52 133-137 [DOI] [PubMed] [Google Scholar]
- 20.Kobayashi, S., Kuzuyama, T., and Seto, H. (2000) Antimicrob. Agents Chemother. 44 647-650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Woodyer, R. D., Shao, Z., Thomas, P. M., Kelleher, N. L., Blodgett, J. A., Metcalf, W. W., van der Donk, W. A., and Zhao, H. (2006) Chem. Biol. 13 1171-1182 [DOI] [PubMed] [Google Scholar]
- 22.Shimizu, M., Shigeobu, F., Miyakozawa, I., Nakamura, A., Suzuki, M., Mizukoshi, S., O'Hara, K., and Sawai, T. (2000) Antimicrob. Agents Chemother. 44 2007-2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Studier, F. W. (2005) Protein Expr. Purif. 41 207-234 [DOI] [PubMed] [Google Scholar]
- 24.Otwinowski, Z., and Minor, W. (1997) Methods Enzymol. 276 307-326 [DOI] [PubMed] [Google Scholar]
- 25.Miller, R., Gallo, S. M., Khalak, H. G., and Weeks, C. M. (1994) J. Appl. Crystallogr. 27 613-621 [Google Scholar]
- 26.Brunger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T., and Warren, G. L. (1998) Acta Crystallogr. D Biol. Crystallogr. 54 905-921 [DOI] [PubMed] [Google Scholar]
- 27.Jones, T. A., Zou, J. Y., Cowan, S. W., and Kjeldgaard, M. (1991) Acta Crystallogr. A 47 110-119 [DOI] [PubMed] [Google Scholar]
- 28.Bailey, S. (1994) Acta Crystallogr. D Biol. Crystallogr. 50 760-763 [DOI] [PubMed] [Google Scholar]
- 29.Painter, J., and Merritt, E. A. (2006) J. Appl. Crystallogr. 39 109-111 [Google Scholar]
- 30.Howlin, B., Butler, S. A., Moss, D. S., Harris, G. W., and Driessen, H. P. C. (1993) J. Appl. Crystallogr. 26 622-624 [Google Scholar]
- 31.DeLano, W. L. (2002) The PyMOL Molecular Graphics System
- 32.Ramon-Maiques, S., Fernandez-Murga, M. L., Gil-Ortiz, F., Vagin, A., Fita, I., and Rubio, V. (2006) J. Mol. Biol. 356 695-713 [DOI] [PubMed] [Google Scholar]
- 33.Ramon-Maiques, S., Marina, A., Gil-Ortiz, F., Fita, I., and Rubio, V. (2002) Structure 10 329-342 [DOI] [PubMed] [Google Scholar]
- 34.Gil-Ortiz, F., Ramon-Maiques, S., Fita, I., and Rubio, V. (2003) J. Mol. Biol. 331 231-244 [DOI] [PubMed] [Google Scholar]
- 35.Marco-Marin, C., Gil-Ortiz, F., Perez-Arellano, I., Cervera, J., Fita, I., and Rubio, V. (2007) J. Mol. Biol. 367 1431-1446 [DOI] [PubMed] [Google Scholar]
- 36.Marina, A., Alzari, P. M., Bravo, J., Uriarte, M., Barcelona, B., Fita, I., and Rubio, V. (1999) Protein Sci. 8 934-940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mas-Droux, C., Curien, G., Robert-Genthon, M., Laurencin, M., Ferrer, J. L., and Dumas, R. (2006) Plant Cell 18 1681-1692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kotaka, M., Ren, J., Lockyer, M., Hawkins, A. R., and Stammers, D. K. (2006) J. Biol. Chem. 281 31544-31552 [DOI] [PubMed] [Google Scholar]
- 39.Faehnle, C. R., Liu, X. Y., Pavlovsky, A., and Viola, R. E. (2006) Acta Crystallogr. F Struct. Biol. Crystallization Commun. 62 962-966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jensen, K. S., Johansson, E., and Jensen, K. F. (2007) Biochemistry 46 2745-2757 [DOI] [PubMed] [Google Scholar]
- 41.Marco-Marin, C., Gil-Ortiz, F., and Rubio, V. (2005) J. Mol. Biol. 352 438-454 [DOI] [PubMed] [Google Scholar]
- 42.Egeblad-Welin, L., Welin, M., Wang, L., and Eriksson, S. (2007) FEBS J. 274 6403-6414 [DOI] [PubMed] [Google Scholar]
- 43.Briozzo, P., Evrin, C., Meyer, P., Assairi, L., Joly, N., Barzu, O., and Gilles, A. M. (2005) J. Biol. Chem. 280 25533-25540 [DOI] [PubMed] [Google Scholar]
- 44.Krissinel, E., and Henrick, K. (2004) Acta Crystallogr. D Biol. Crystallogr. 60 2256-2268 [DOI] [PubMed] [Google Scholar]
- 45.Krissinel, E., and Henrick, K. (2007) J. Mol. Biol. 372 774-797 [DOI] [PubMed] [Google Scholar]
- 46.Cleland, W. W., and Hengge, A. C. (2006) Chem. Rev. 106 3252-3278 [DOI] [PubMed] [Google Scholar]
- 47.Kabsch, W., and Sander, C. (1983) Biopolymers 22 2577-2637 [DOI] [PubMed] [Google Scholar]
- 48.Gouet, P., Courcelle, E., Stuart, D. I., and Metoz, F. (1999) Bioinformatics 15 305-308 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.