

Abstract
Purpose
The purpose of the present study was to investigate the roles of caspase 1 and extracellular signal-regulated kinase (ERK) in inflammation-induced inhibition of lacrimal gland secretion.
Methods
Lacrimal gland inflammation was induced by injection of lipopolysaccharide (LPS; to study the role of caspase 1) or IL-1β (to study the role of ERK). Lacrimal gland protein secretion was measured using a spectrofluorometric assay. Caspase 1 and ERK activities in the lacrimal gland were measured by immunohistochemistry, Western blotting, or both. Aqueous tear production was measured using phenol red-impregnated cotton threads.
Results
Injection of LPS into the lacrimal gland inhibited neurally and adrenergic agonist–induced protein secretion by 77% and 54%, respectively, and activated caspase 1. The degree of inhibition achieved by LPS was similar to that obtained with injection of IL-1β. Inhibition of caspase 1 alleviated the inhibitory effect of LPS on lacrimal gland secretion. IL-1β activated ERK in the lacrimal gland in vitro and in vivo, and this effect was blocked by UO126, an inhibitor of MEK, the ERK-activating enzyme. IL-1β injection into the lacrimal gland inhibited aqueous tear production by 52% and inhibited neurally and adrenergic agonist-induced protein secretion by 80% and 55%, respectively. UO126 alleviated the inhibitory effect of IL-1β on aqueous tear production and lacrimal gland protein secretion.
Conclusions
LPS inhibits lacrimal gland secretion by activating caspase 1, and IL-1β activates the ERK pathway to inhibit lacrimal gland protein secretion and aqueous tear production.
Sjögren syndrome, a systemic inflammatory autoimmune disease, is the leading cause of the aqueous tear- deficient type of dry eye.1,2 Sjögren syndrome may be a primary disorder (primary Sjögren syndrome), or it may be associated with other autoimmune diseases (secondary Sjögren syndrome) such as rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), and systemic sclerosis. Lacrimal gland inflammation is also common in other autoimmune diseases such as diabetes, thyroid disease, and sarcoidosis.3 Other examples of pathologic conditions resulting in lacrimal gland inflammation (for a review, see Zoukhri3) include chronic graft-versus-host disease (cGVHD), acquired immunodeficiency syndrome, hepatitis C, and aging. Increased production of autoantibodies and proinflammatory cytokines, both of which are believed to interfere with tear production, are common in inflammatory disease of the lacrimal gland.
Our previous research showed that IL-1 plays a major role in impairing lacrimal gland secretion.4 IL-1 is a potent proinflammatory cytokine and consists of two isoforms, α and β, and a specific receptor antagonist, IL-1ra.5,6 Both IL-1α and IL-1β are synthesized as 31-kDa precursors without leader sequence.5,6 In contrast to IL-1α, IL-1β is only active in its secreted mature form (17.5 kDa). The IL-1β– converting enzyme (ICE), better known as caspase 1, cleaves the precursor protein at Asp116 to generate the mature active form of IL-1β.5,6 Caspase 1 is also synthesized as an inactive 45-kDa precursor, consisting of p20 and p10 subunits, and is proteolytically cleaved to generate an active enzyme comprising the p20 and p10 polypeptides.5,6 Various microbial agents such as lipopolysaccharide (LPS) are known to induce the activation of caspase 1.7–9
A study by Hirai et al.10 showed that injection of LPS into the lacrimal gland induced an inflammatory response that started 9 hours after injection and became severe at 24 hours. Because LPS is known to activate caspase 1,7–9 we hypothesized that LPS injection will inhibit lacrimal gland secretion by inducing the production of IL-1β through the activation of caspase 1. To test this hypothesis, the effect of LPS on lacrimal gland secretion and caspase 1 activity was investigated, as was the effect of a caspase 1 inhibitor.
IL-1 has two receptors, a signaling receptor named IL-1RI and a decoy receptor named IL-1 RII that does not signal.5 Activation of the IL-1RI triggers a signaling cascade leading to the transcription of several genes involved in acute and chronic inflammation and connective tissue diseases.6 The effect of IL-1 on gene transcription can be mediated by the activation of one or more members of the mitogen-activated protein kinases (MAPK) family.11–13 In mammalian systems, at least six independent MAPK signaling units appear to function.14,15 Among them, the biochemical properties of three MAPKs, the extracellular signal-regulated kinases (ERK1/2), the c-Jun NH2-terminal kinases (JNK1/2), and the p38 MAPKs, have been characterized in some detail.14,15 Once ERK, JNK, and p38 MAPK are activated, they lead to the activation of two major transcription factors, activator protein 1 (AP-1) and nuclear factor-κB (NF-κB).14,15
In a recent study, we showed that JNK inhibition activated by exogenous addition of IL-1β alleviated the inhibitory effect exerted by this cytokine on lacrimal gland secretion and restored tear production in a murine model of aqueous tear- deficient dry eye.16 Hence, a second purpose of the present study was to investigate whether ERK signaling is involved in IL-1β–induced inhibition of lacrimal gland secretion.
Materials and Methods
Chemicals and Antibodies
Recombinant human IL-1β was a generous gift from Craig W. Reynolds (Biological Resources Branch, National Cancer Institute Preclinical Repository, Rockville, MD). Phenylephrine, hydrogen peroxide, bacterial LPS, and a monoclonal antibody against β-actin were from Sigma (St. Louis, MO). Reagent (Amplex Red) was from Molecular Probes (Eugene, OR). Polyclonal antibodies against phosphorylated ERK and JNK and UO126, a dual MEK1/MEK2 inhibitor, were from Cell Signaling Technology (Danvers, MA). Polyclonal antibodies against total ERK were from Cell Signaling Technology or Santa Cruz Biotechnology (Santa Cruz, CA). A polyclonal antibody against the p10 fragment of ICE was from Santa Cruz Biotechnology.
Animals and Treatment
Female BALB/c mice (10–12 weeks old) were purchased from Taconic (Germantown, NY). Animals were maintained in rooms at constant temperature with fixed 12-hour light/12-hour dark intervals and were fed ad libitum. All experiments were conducted in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research and were approved by the Tufts-New England Medical Center Animal Care and Use Committee.
Animals were anesthetized with a mixture of ketamine (60 mg/kg) and xylazine (12 mg/kg), given intraperitoneally. The fur between the ears and the cheeks (where the exorbital lacrimal gland is located) was shaved and cleaned with betadine (10% solution), and a small incision (<1 cm) was made. After the lacrimal glands were located, they were left untreated (control) or were injected with saline (vehicle), rhIL-1β (1 μg), or LPS (25 μg) in a total volume of 2 μL. After injection, the wound was closed using surgical glue. A local anesthetic (proparacaine, 0.5%) plus triple antibiotic ointment (bacitracin ophthalmic ointment, 3.5 g) was applied topically to minimize pain and scratching. Additionally, buprenorphine was given subcutaneously (0.05– 0.1 mg/kg). The entire surgical procedure lasted between 15 and 20 minutes Twenty-four hours after injection, animals were humanely killed by CO2 inhalation; cervical dislocation followed, and the lacrimal glands were removed. Lobules were then prepared and used to measure protein secretion or were processed for Western blotting and immunohistochemistry, as described below.
Measurement of Peroxidase Secretion
Peroxidase secretion was measured as previously described.3,17 Lacrimal glands were cut into small lobules (approximately 2 mm diameter), placed in cell strainers, and incubated at 37°C in Krebs-Ringer bicarbonate buffer (KRB; containing 120 mM NaCl, 5 mM KCl, 1 mM CaCl2, 1.2 mM MgSO4, 1.2 mM KH2PO4, and 25 mM NaHCO3) supplemented with 10 mM HEPES and 5.5 mM glucose (pH 7.4). Cell strainers containing lobules were transferred into fresh KRB solution every 20 minutes for a total of 60 minutes The lobules were incubated for 20 minutes in a total volume of 0.8 mL in normal KRB (referred to as spontaneous secretion), then in depolarizing KRB (evoked secretion) solution, where the concentration of KCl was increased to 75 mM and that of NaCl was decreased to 50 mM to maintain isotonicity. Lacrimal gland lobules were further incubated for 20 minutes in 0.8 mL normal KRB containing phenylephrine (an α1-adrenergic agonist; 10−4 M). After incubation, the lobules were homogenized in 10 mM Tris-HCl (pH 7.5). The amount of peroxidase in the media and tissue homogenate was determined using reagent (Amplex Red; Invitrogen, Carlsbad, CA). For the measurement of peroxidase, 0.1 mL media and 0.01 mL tissue homogenate were spotted in duplicate onto 96-well microplates. To each well was added 0.1 mL assay buffer (50 mM Tris-HCl, pH 7.5) containing 0.2 M reagent (Amplex Red; Invitrogen) and 0.2 M hydrogen peroxide. After incubation, fluorescence was determined in a fluorescence microplate reader using 530 nm excitation wavelength and 590 nm emission wavelength. The amount of secreted peroxidase was expressed as a percentage of the total: [peroxidase in media/(peroxidase in media + peroxidase in tissue)] × 100.
Immunohistochemistry
Lacrimal glands pieces were fixed overnight at 4°C in 4% formaldehyde made in phosphate-buffered saline (PBS; containing 145 mM NaCl, 7.3 mM Na2HPO4, and 2.7 mM NaH2PO4 at pH 7.2). After cryopreservation overnight in 30% sucrose in PBS, the tissue was frozen in optimum cutting temperature embedding medium. Cryostat sections (6 μm) were placed on gelatin-coated slides and dried overnight at 37°C. Sections were incubated for 30 minutes at room temperature in PBS containing 1% bovine serum albumin (BSA) and 0.1% Triton X-100. After blocking for 30 minutes in 5% donkey serum and 1% BSA in PBS, sections were incubated overnight at 4°C with the primary antibody diluted in PBS containing 1% BSA. A secondary antibody conjugated to FITC (1:100 in PBS + 1% BSA) was applied for 30 minutes at room temperature. Coverslips were mounted with a mounting medium (Vectashield; Vector Laboratories, Burlingame, CA). Sections were viewed under a microscope (UFXII; Nikon, Tokyo, Japan) equipped for epi-illumination. Negative control experiments were performed by omitting the primary antibody.
Western Blotting
Total amount of protein in the cell lysate was determined using the method of Bradford, and equal amounts of protein (20 μg) were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE; 4%–15% gradient gels) and transferred to nitrocellulose membranes. The membranes were first blotted with polyclonal antibodies against phosphorylated (active) ERK or JNK (1:1000 dilution). After removal of the antibodies by incubation of the membranes for 40 minutes at 50°C in stripping buffer (62.5 mM Tris-HCl [pH 6.8], 2% SDS, 100 mM β-mercaptoethanol), the membranes were blotted with nondiscriminatory polyclonal antibodies that recognized the whole pools of these enzymes (all at 1:1000 dilution) or a monoclonal antibody against β-actin (1:5000; Sigma). Immunoreactive bands were visualized using the enhanced chemiluminescence method and were quantitated using National Institutes of Health ImageJ software (developed by Wayne Rasband, National Institutes of Health, Bethesda, MD; available at http://rsb.info.nih.gov/ij/index.html). The amount of phosphorylated ERK was normalized to that of total ERK (or β-actin) to control for gel loading and transfer efficiency.
Measurement of Aqueous Tear Production
Tear production was measured on anesthetized mice using phenol red–impregnated cotton threads (Zone-Quick; Lacrimedics, Eastsound, WA), as previously described.16 The threads were held with jeweler forceps and applied to the ocular surface, on both eyes, in the lateral canthus for 10 seconds. Wetting of the thread (which turns red in contact with tears) was measured in millimeters under a dissecting microscope.
Data Presentation and Statistical Analysis
Data are expressed as mean ± SEM and were statistically analyzed using one- or two-way analysis of variance (ANOVA) followed by post hoc t-test. P < 0.05 was considered significant.
Results
Effect of LPS Lacrimal Gland Protein Secretion and Caspase 1 Activity
Injection of LPS into the lacrimal gland resulted in significant inhibition of KCl- and phenylephrine-induced peroxidase secretion by 80% and 60%, respectively. Interestingly, the degree of inhibition of lacrimal gland secretion induced by LPS was comparable to that induced by IL-1β (Fig. 1), suggesting that the effect of LPS on lacrimal gland secretion might be mediated through caspase 1–induced production of IL-1β.7–9
Figure 1.
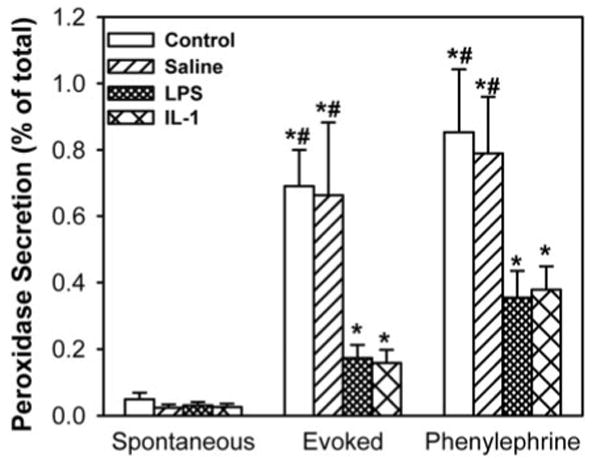
Effect of LPS on lacrimal gland secretion. The lacrimal glands of female BALB/c mice were left untreated or were injected with saline (vehicle), LPS (25 μg), or IL-1β (as a positive control, 1 μg). Twenty-four hours later, lacrimal glands were removed, and lobules were prepared and processed for peroxidase secretion experiments. Lobules were incubated for 20 minutes each in normal KRB buffer (spontaneous), in depolarizing KRB (evoked) solution in which the concentration of KCl was increased to 75 mM, and then in KRB containing phenylephrine (an α1D-adrenergic agonist; 10−4 M). After incubation, the amount of peroxidase in the media and tissue homogenate was determined using reagent. Data are the mean ± SEM from three to six independent experiments. *Statistically significant difference from spontaneous secretion. #Statistically significant difference from LPS or IL-1β.
To test this hypothesis, lacrimal glands removed from control animals and saline- and LPS-injected mice were processed for immunohistochemistry using an antibody that specifically recognizes the cleaved (active) p10 fragment of caspase 1. Caspase 1 immunoreactivity was scarce in control and saline-injected lacrimal glands (Fig. 2). The arrows in Figure 2 indicate caspase 1 immunoreactivity associated with two resident plasma cells. In contrast, strong caspase 1 immunoreactivity was detected in LPS-injected lacrimal glands (Fig. 2). Omission of the primary antibody resulted in a loss of immunoreactivity (Fig. 2, Negative). These results suggested that LPS-induced inflammation in the lacrimal gland is accompanied by activation of caspase 1, which would result in increased production of IL-1β.
Figure 2.
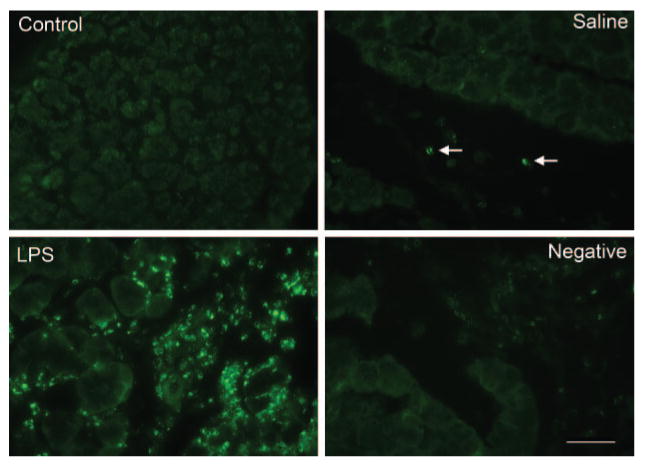
Effect of LPS on caspase 1 activity. Female BALB/c mice were anesthetized, and their exorbital lacrimal glands were left untreated (control) or were injected with saline (vehicle, control) or LPS (25 μg). Lacrimal glands were removed 24 hours after injection, fixed, and processed for immunocytochemistry using a rabbit polyclonal antibody against the p10 subunit of caspase 1. Results depicted in the images were replicated at least three times on lacrimal glands from separate animals. Scale bar, 50 μm.
To further investigate the role of caspase 1, we tested the effect of a Z-YVAD-FMK, an inhibitor of caspase 1, on LPS-induced inhibition of lacrimal gland protein secretion. Injection of LPS inhibited KCl- and phenylephrine-induced lacrimal gland secretion by 77% and 54%, respectively (Fig. 3). Z-YVAD-FMK significantly alleviated the inhibitory effect of LPS on KCl- and phenylephrine-induced protein secretion by 64% and 34%, respectively (Fig. 3). These results suggest that caspase 1, and indirectly IL-1β, plays a central role in inflammation-induced inhibition of lacrimal gland secretion.
Figure 3.
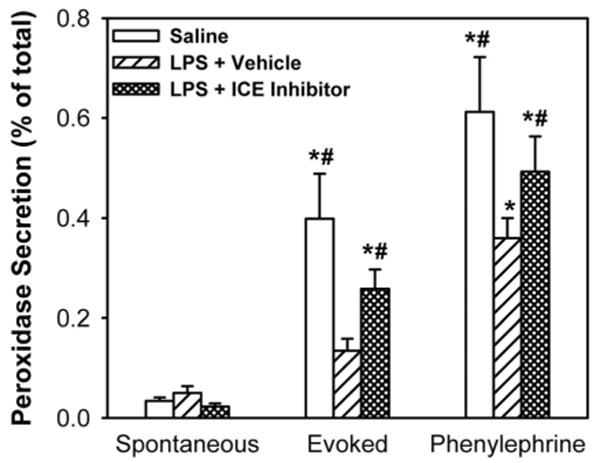
Effect of a caspase 1 inhibitor on LPS-induced inhibition of lacrimal gland secretion. Lightly anesthetized female BALB/c mice received subcutaneous injections (100 μL) of vehicle (DMSO+PEG400) or Z-YVAD-FMK (0.1 mg). Twenty-four hours later, the mice received second subcutaneous injections of Z-YVAD-FMK (or vehicle) followed by injection (2 μL) of LPS (25 μg) or saline (vehicle for LPS) into the exorbital lacrimal glands. Twenty-four hours later, lacrimal glands were removed and lobules were prepared to measure protein secretion. Data are the mean ± SEM from three independent experiments. *Statistically significant difference from spontaneous secretion. #Statistically significant difference from LPS + vehicle.
Effect of IL-1β on ERK Activity In Vitro
We showed in a recent study that concomitant with IL-1β–induced inhibition of lacrimal gland secretion, JNK activity was increased and JNK inhibition alleviated the inhibitory effect of IL-1β and restored aqueous tear production in a murine model of aqueous-deficient dry eye.16 We hypothesized that IL-1β activates the ERK signaling pathway to inhibit lacrimal gland secretion and aqueous tear production. In a first series of experiments, we tested the effects of IL-1β on ERK activity in an in vitro assay in which lacrimal gland lobules were incubated in the presence or absence of IL-1β for 1 to 120 minutes. IL-1β induced a time-dependent activation of p42ERK and p44ERK with a maximum 3.0-fold at 10 minutes (Fig. 4). ERK activity declined with time and returned to basal level between 60 and 120 minutes. These results show that IL-1β activated p42ERK and p44ERK in the lacrimal gland in a time-dependent manner.
Figure 4.
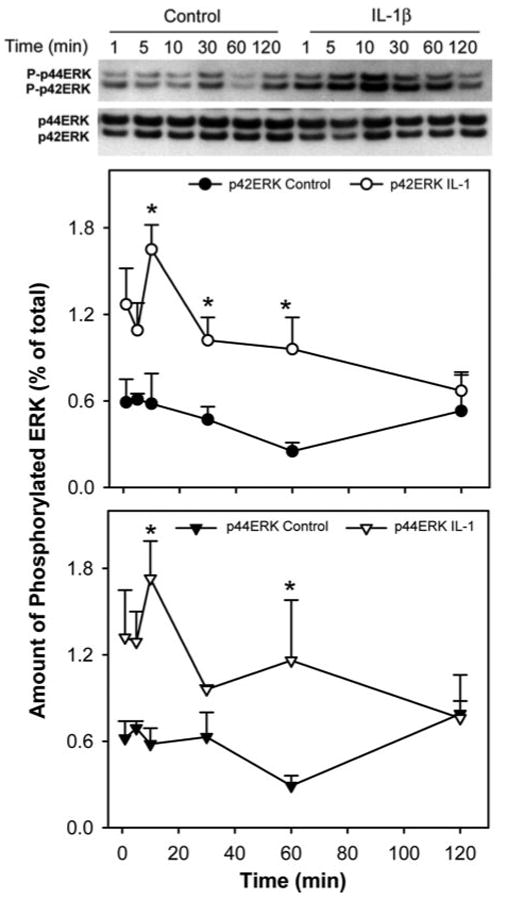
Kinetics of IL-1β–induced activation of ERK in the lacrimal gland. Lacrimal gland lobules were incubated in the presence or absence of IL-1β (10 ng/mL) for 0 to 120 minutes and were homogenized, and the proteins in the cell lysate were separated by SDS-PAGE followed by Western blotting using an antibody against phosphorylated ERK or an antibody against the whole pool of ERK. Data are the mean ± SEM of three independent experiments. *Statistically significant difference from control.
Effect of IL-1β on ERK Activity In Vivo
In a second series of experiments, we tested the effect of IL-1β on ERK in an in vivo setting in which the cytokine or saline was injected directly into the lacrimal glands of female BALB/c mice. Another set of animals whose lacrimal glands were untouched served as controls. Compared with control and saline-injected animals, the amount of phosphorylated p42ERK was increased 4.8-fold and that of p44ERK was increased 5.4-fold in IL-1β–injected lacrimal glands (Fig. 5A).
Figure 5.
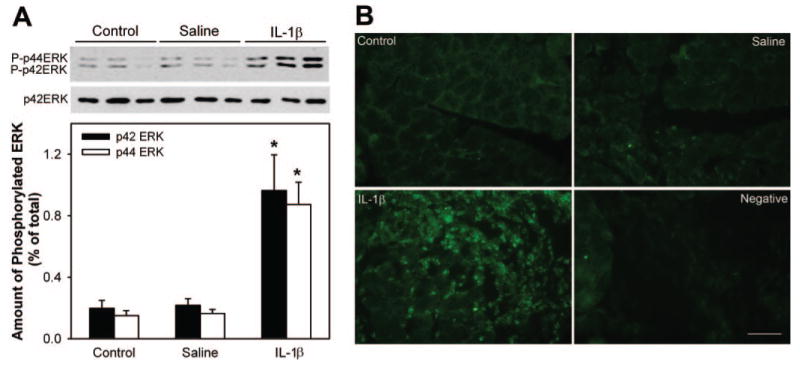
Effect of IL-1β on lacrimal gland ERK activity in vivo. Female BALB/c mice were anesthetized, and their exorbital lacrimal glands were left untreated (control) or were injected with saline (vehicle) or IL-1β (1 μg). Lacrimal gland lobules were removed 24 hours after injection and were homogenized or fixed in 4% buffered formaldehyde. (A) Proteins in the cell lysate separated by SDS-PAGE followed by Western blotting using an antibody against phosphorylated ERK or an antibody against the whole pool of ERK. Data in the plot are the mean ± SEM from three independent experiments. *Statistically significant difference from control and saline. (B) Lacrimal gland sections were processed for immunofluorescence studies using an antibody against phosphorylated ERK. Results depicted in the images were replicated on lacrimal glands from at least three separate animals. Scale bar, 50 μm.
Because the lacrimal gland is composed of several cell types—acini, duct, nerve, myoepithelial, mast, and plasma—it was important to determine the cell type in which IL-1 induced its effects. To that purpose, we performed immunohistochemical studies. Phospho-ERK immunoreactivity was scarce in control and saline-injected lacrimal glands (Fig. 5B). In contrast, strong immunoreactivity for phospho-ERK was detected in cell nuclei of IL-1-injected lacrimal glands (Fig. 5B). Phospho-ERK immunoreactivity seems to be detected only in the infiltrating immune cells. Omission of the primary antibody led to a loss of immunoreactivity (Fig. 5B). These results show that injection of IL-1β into the lacrimal gland activated p42ERK and p44ERK isoforms.
Effect of UO126 on IL-1–Induced Inhibition of Lacrimal Gland Secretion
In a recent study, we showed that JNK activation played an important role in IL-1–induced inhibition of lacrimal gland secretion. The data presented thus far suggest that ERK activity might also be implicated in IL-1–induced inhibition of lacrimal gland secretion. To test this hypothesis, we used UO126, an inhibitor of MEK (the kinase upstream of ERK). In a first series of experiments, we determined the efficiency of this inhibitor, in vitro, on IL-1β–induced activation of ERK. As control for the specificity of UO126, the activity of JNK was also analyzed by Western blotting. UO126 inhibited ERK activity in a concentration-dependent manner with maximum or 100% inhibition achieved at 10 μM (Fig. 6). In contrast, JNK activity was not affected by UO126 at all the concentrations tested (Fig. 6).
Figure 6.
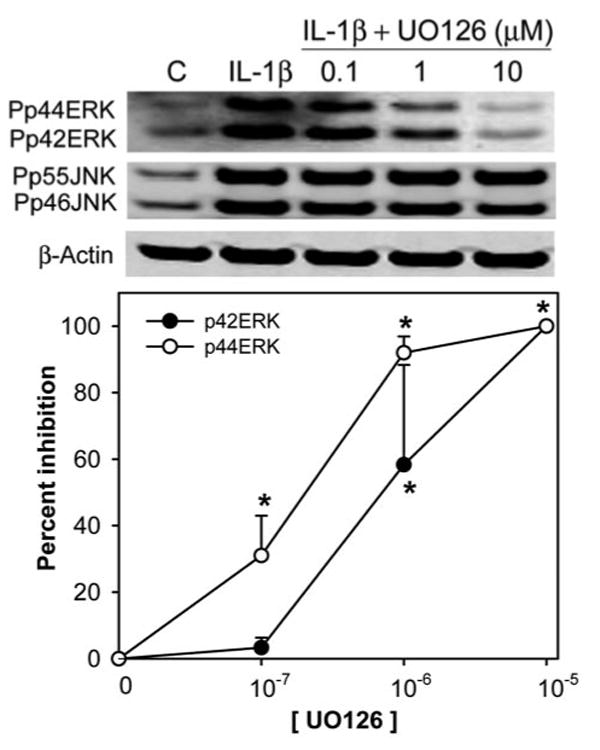
Effect of UO126 on IL-1β–induced activation of ERK. Lacrimal glands lobules were preincubated for 30 minutes in the absence or presence of increasing concentrations of UO126 (0.1, 1, 10 μM). Lobules were then incubated with or without IL-1β (10 ng/mL) for 10 minutes (C, control). After incubation, the lobules were homogenized, and the proteins in the homogenate were separated by SDS-PAGE followed by Western blotting. Data in the plot are the mean ± SEM from three independent experiments. *Statistically significant difference from IL-1β.
In another series of experiments, we tested the effect of UO126 in vivo (2 mg, as described by Li et al.18) on IL-1β–induced inhibition of lacrimal gland secretion. As shown in Figure 7A and in accordance with our previous studies,19 compared with saline-treated animals, aqueous tear production was inhibited 52% after injection of IL-1β into vehicle-treated animals. Data analysis using one-way ANOVA showed a statistically significant difference among the three groups (P = 0.03). The post hoc t-test found a statistically significant difference between IL-1 + vehicle and control (showing inhibition of tear production by IL-1; P = 0.029) and IL-1 + vehicle and IL-1 + UO126 (showing reversal of IL-1–induced inhibition of tear production by subcutaneous treatment with UO126; P = 0.026; Fig. 7A). These results suggest that UO126 reversed the inhibitory effect of IL-1β on lacrimal gland secretion. This hypothesis was directly tested using lobules prepared from vehicle- or UO126-treated animals. As shown in Figure 7B and in agreement with our published findings,4,16,19 IL-1β inhibited KCl- and phenylephrine-induced protein secretion by 80% and 55%, respectively. Importantly, although UO126 only partially alleviated the inhibitory effect of IL-1β on KCl-induced protein secretion, it completely alleviated the inhibitory effect of IL-1β on phenylephrine-induced protein secretion (Fig. 7B). These results show that ERK inhibition with UO126 alleviated the inhibitory effect of IL-1β on aqueous tear production and lacrimal gland protein secretion.
Figure 7.
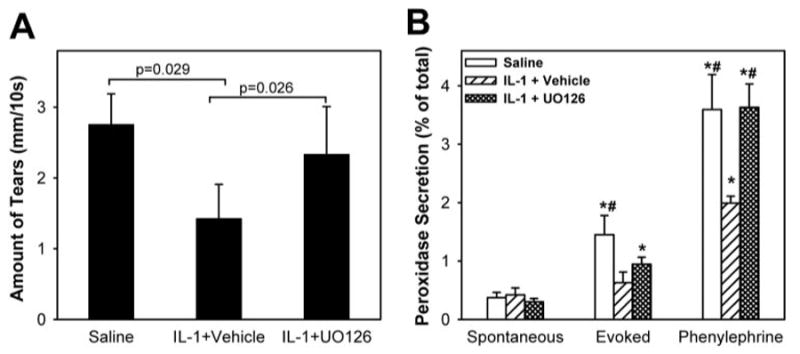
Effect of UO126 on IL-1β–induced inhibition of lacrimal gland secretion. Animals were left untreated or received subcutaneous injections of vehicle or UO126 (2 mg). Twenty-four hours after injection of saline or IL-1β (1 μg), the lacrimal glands were removed, and lobules were prepared and used to measure protein secretion. Immediately before kill, animals were lightly anesthetized, and aqueous tear production was measured. (A) Aqueous tear production was measured using phenol red–impregnated cotton threads. Wetting of the thread was measured in millimeters under a dissecting microscope. Data are the mean ± SEM from three independent experiments. (B) Lobules were incubated for 20 minutes each in normal KRB buffer (spontaneous), in depolarizing KRB (evoked) solution in which the concentration of KCl was increased to 75 mM, and in KRB containing phenylephrine (an α1D-adrenergic agonist; 10−4 M). After incubation, the amount of peroxidase in the media and tissue homogenate was determined using reagent. Data are the mean ± SEM from three independent experiments. *Statistically significant difference from spontaneous secretion. #Statistically significant difference from IL-1 + vehicle.
Discussion
In previous reports we showed that the injection of IL-1 (α or β) into murine exorbital lacrimal glands led to a severe inflammatory response and impaired protein secretion in response to nerve and exogenous agonist stimulation.4,16,19 In a recent study, we showed that concomitant with IL-1β–induced inhibition of lacrimal gland secretion, JNK activity was increased and JNK inhibition alleviated the inhibitory effect of IL-1β and restored aqueous tear production in a murine model of aqueous-deficient dry eye.16 In the present study, we present evidence suggesting that IL-1β plays a pivotal role in inflammation-induced impairment of lacrimal gland secretion because we found that LPS-induced inhibition of lacrimal gland secretion was significantly alleviated when caspase 1 activity was inhibited. We also present evidence showing that the ERK signaling pathway is activated by IL-1β to inhibit lacrimal gland secretion and to diminish aqueous tear production.
LPS-induced inflammation of the lacrimal gland was previously described and was shown to induce the leakage of aquaporin 5 into tears.10 Our data show that LPS injection inhibited neurally induced and agonist-induced lacrimal gland secretion concomitant with the activation of caspase 1. Furthermore, we show that the degree of inhibition induced by LPS was similar to that induced by IL-1β and that the effect of LPS was significantly inhibited by a caspase 1 inhibitor. Together, these data suggest that the effect of LPS on lacrimal gland secretion is largely mediated through caspase 1–induced production of IL-1β.
Activation of caspase 1 by LPS to increase IL-1β production is well documented and has been associated with several inflammatory diseases.7–9,20–23 Furthermore, caspase 1–deficient mice were shown to have a major defect in the production of mature IL-1β production on LPS stimulation and were resistant to LPS-induced endotoxic shock.24,25 Consequently, strategies aimed at reducing IL-1β levels based on the inhibition of caspase 1 activity have been established, and some caspase 1 inhibitors are being tested in clinical trials.26–30
The role of the MAPK signaling pathways in ocular surface inflammation and dry eye has been recently documented.31–33 It was shown that all three MAPK pathways are activated in response to inflammatory triggers, including high osmolarity and desiccating stress.32,33 Furthermore, activation of the MAPK pathway has been linked to production of the inflammatory cytokines IL-1β and TNFα and of matrix metalloproteinase 9 (MMP9) by ocular surface epithelia and their secretion into tears.31–33
Our findings that the inhibition of JNK16 or ERK (present study) alleviated the inhibitory effect of IL-1 on lacrimal gland secretion and restored tear production suggest that these pathways are redundant. This is in line with findings from the study by Li et al.,33 in which it was shown that the inhibition of JNK or ERK activity resulted in diminished production of IL-1β, TNFα, and MMP9 by the ocular surface. It is well documented that intracellular signaling in general and the MAPK pathway in particular are often redundant in eukaryotic cells, with one agonist activating multiple intracellular effectors.34,35 Consequently, inhibition of either of these activated signaling pathways would theoretically abrogate the measured biological response.35 Because of the redundancy in signaling and the lack of selectivity of the available chemical inhibitors, however, it is often necessary to inhibit more than one pathway to completely terminate a given biological response, especially in pathologic conditions. Nevertheless our data are in line with those published and suggest that strategies to inhibit or modulate the MAPK signaling pathway to alleviate dry eye syndromes caused by ocular surface inflammation are a viable option.16,31–33 Investigating the effect of direct delivery of such inhibitors to the lacrimal gland, however, to minimize potential side effects on other systems is warranted.
In summary, our results show that the inhibition of caspase 1 activity alleviates LPS-induced inhibition of lacrimal gland secretion. They also show that inhibition of IL-1–induced activation of ERK signaling alleviates IL-1β–induced aqueous tear deficiency and inhibition of lacrimal gland secretion. It remains to be tested whether the inhibition of caspase 1 or of ERK would restore normal tear production in animal models of aqueous tear– deficient dry eye.
Acknowledgments
The authors thank Craig Reynolds for the generous gift of recombinant human cytokines, Darlene Dartt and Robin Hodges for critical reading of the manuscript, and Fara Sourie for her invaluable contribution to this work.
Supported by National Institutes of Health/National Eye Institute Grant RO1 12383.
Footnotes
Disclosure: D. Zoukhri, None; S. Ko, None; P.C. Stark, None; C.L. Kublin, None
References
- 1.Talal N, Moutsopoulos HM, Kassan SS. Sjögren's Syndrome: Clinical and Immunological Aspects. Berlin: Springer Verlag; 1987. [Google Scholar]
- 2.Sullivan DA, Wickham LA, Krenzer KL, Rocha EM, Toda I. Aqueous tear deficiency in Sjögren's syndrome: possible causes and potential treatment. In: Pleyer U, Hartmenn C, editors. Oculodermal Diseases. Buren, Netherlands: Aeolus Press; 1997. pp. 95–152. [Google Scholar]
- 3.Zoukhri D. Effect of inflammation on lacrimal gland function. Exp Eye Res. 2006;82:885–898. doi: 10.1016/j.exer.2005.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zoukhri D, Hodges RR, Byon D, Kublin CL. Role of proinflammatory cytokines in the impaired lacrimation associated with autoimmune xerophthalmia. Invest Ophthalmol Vis Sci. 2002;43:1429–1436. [PMC free article] [PubMed] [Google Scholar]
- 5.Dinarello CA. Biologic basis for interleukin-1 in disease. Blood. 1996;87:2095–2174. [PubMed] [Google Scholar]
- 6.Dinarello CA. Proinflammatory cytokines. Chest. 2000;118:503–508. doi: 10.1378/chest.118.2.503. [DOI] [PubMed] [Google Scholar]
- 7.Schumann RR, Belka C, Reuter D, et al. Lipopolysaccharide activates caspase-1 (interleukin-1-converting enzyme) in cultured monocytic and endothelial cells. Blood. 1998;91:577–584. [PubMed] [Google Scholar]
- 8.Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10:417–426. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 9.Yamamoto M, Yaginuma K, Tsutsui H, et al. ASC is essential for LPS-induced activation of procaspase-1 independently of TLR-associated signal adaptor molecules. Genes Cells. 2004;9:1055–1067. doi: 10.1111/j.1365-2443.2004.00789.x. [DOI] [PubMed] [Google Scholar]
- 10.Hirai SI, Ishida N, Watanabe K, Mita S. Leakage of aquaporin 5 in the tear of dacryoadenitis mice. Invest Ophthalmol Vis Sci. 2000;41:2432–2437. [PubMed] [Google Scholar]
- 11.Auron PE. The interleukin 1 receptor: ligand interactions and signal transduction. Cytokine Growth Factor Rev. 1998;9:221–237. doi: 10.1016/s1359-6101(98)00018-5. [DOI] [PubMed] [Google Scholar]
- 12.O'Neill LA, Dinarello CA. The IL-1 receptor/toll-like receptor superfamily: crucial receptors for inflammation and host defense. Immunol Today. 2000;21:206–209. doi: 10.1016/s0167-5699(00)01611-x. [DOI] [PubMed] [Google Scholar]
- 13.Baud V, Karin M. Signal transduction by tumor necrosis factor and its relatives. Trends Cell Biol. 2001;11:372–377. doi: 10.1016/s0962-8924(01)02064-5. [DOI] [PubMed] [Google Scholar]
- 14.Ichijo H. From receptors to stress-activated MAP kinases. Oncogene. 1999;18:6087–6093. doi: 10.1038/sj.onc.1203129. [DOI] [PubMed] [Google Scholar]
- 15.Paul A, Wilson S, Belham CM, et al. Stress-activated protein kinases: activation, regulation and function. Cell Signal. 1997;9:403–410. doi: 10.1016/s0898-6568(97)00042-9. [DOI] [PubMed] [Google Scholar]
- 16.Zoukhri D, Macari E, Choi SH, Kublin CL. c-Jun NH2-terminal kinase mediates interleukin-1β-induced inhibition of lacrimal gland secretion. J Neurochem. 2006;96:126–135. doi: 10.1111/j.1471-4159.2005.03529.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zoukhri D, Kublin CL. Impaired neurotransmitter release from lacrimal and salivary gland nerves of a murine model of Sjogren's syndrome. Invest Ophthalmol Vis Sci. 2001;42:925–932. [PMC free article] [PubMed] [Google Scholar]
- 18.Li Y, Je HD, Malek S, Morgan KG. Role of ERK1/2 in uterine contractility and preterm labor in rats. Am J Physiol Regul Integr Comp Physiol. 2004;287:R328–R335. doi: 10.1152/ajpregu.00042.2004. [DOI] [PubMed] [Google Scholar]
- 19.Zoukhri D, Macari E, Kublin CL. A single injection of interleukin-1 induces reversible aqueous-tear deficiency, lacrimal gland inflammation, and acinar and ductal cell proliferation. Exp Eye Res. 2007;84:894–904. doi: 10.1016/j.exer.2007.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ghayur T, Banerjee S, Hugunin M, et al. Caspase-1 processes IFN-gamma-inducing factor and regulates LPS-induced IFN-gamma production. Nature. 1997;386:619–623. doi: 10.1038/386619a0. [DOI] [PubMed] [Google Scholar]
- 21.Paszkowski AS, Rau B, Mayer JM, Moller P, Beger HG. Therapeutic application of caspase 1/interleukin-1β-converting enzyme inhibitor decreases the death rate in severe acute experimental pancreatitis. Ann Surg. 2002;235:68–76. doi: 10.1097/00000658-200201000-00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saha N, Moldovan F, Tardif G, Pelletier JP, Cloutier JM, Martel-Pelletier J. Interleukin-1beta-converting enzyme/caspase-1 in human osteoarthritic tissues: localization and role in the maturation of interleukin-1beta and interleukin-18. Arthritis Rheum. 1999;42:1577–1587. doi: 10.1002/1529-0131(199908)42:8<1577::AID-ANR3>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 23.Vereker E, Campbell V, Roche E, McEntee E, Lynch MA. Lipopolysaccharide inhibits long term potentiation in the rat dentate gyrus by activating caspase-1. J Biol Chem. 2000;275:26252–26258. doi: 10.1074/jbc.M002226200. [DOI] [PubMed] [Google Scholar]
- 24.Li P, Allen H, Banerjee S, et al. Mice deficient in IL-1β-converting enzyme are defective in production of mature IL-1β and resistant to endotoxic shock. Cell. 1995;80:401–411. doi: 10.1016/0092-8674(95)90490-5. [DOI] [PubMed] [Google Scholar]
- 25.Li P, Allen H, Banerjee S, Seshadri T. Characterization of mice deficient in interleukin-1β-converting enzyme. J Cell Biochem. 1997;64:27–32. doi: 10.1002/(sici)1097-4644(199701)64:1<27::aid-jcb5>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 26.Dinarello CA. Modalities for reducing interleukin 1 activity in disease. Immunol Today. 1993;14:260–264. doi: 10.1016/0167-5699(93)90042-J. [DOI] [PubMed] [Google Scholar]
- 27.Livingston DJ. In vitro and in vivo studies of ICE inhibitors. J Cell Biochem. 1997;64:19–26. [PubMed] [Google Scholar]
- 28.Randle JC, Harding MW, Ku G, Schonharting M, Kurrle R. ICE/caspase-1 inhibitors as novel anti-inflammatory drugs. Expert Opin Investig Drugs. 2001;10:1207–1209. doi: 10.1517/13543784.10.7.1207. [DOI] [PubMed] [Google Scholar]
- 29.Stack JH, Beaumont K, Larsen PD, et al. IL-converting enzyme/caspase-1 inhibitor VX-765 blocks the hypersensitive response to an inflammatory stimulus in monocytes from familial cold autoinflammatory syndrome patients. J Immunol. 2005;175:2630–2634. doi: 10.4049/jimmunol.175.4.2630. [DOI] [PubMed] [Google Scholar]
- 30.Wannamaker W, Davies R, Namchuk M, et al. (S)-1-((S)-2-{[1-(4-amino-3-chloro-phenyl)-methanoyl]-amino}-3,3-dimethyl-butanoyl)-pyrrolidine-2-carboxylic acid ((2R,3S)-2-ethoxy-5-oxo-tetrahydrofuran-3-yl)-amide (VX-765), an orally available selective interleukin (IL)-converting enzyme/caspase-1 inhibitor, exhibits potent anti-inflammatory activities by inhibiting the release of IL-1beta and IL-18. J Pharmacol Exp Ther. 2007;321:509–516. doi: 10.1124/jpet.106.111344. [DOI] [PubMed] [Google Scholar]
- 31.Luo L, Li DQ, Doshi A, Farley W, Corrales RM, Pflugfelder SC. Experimental dry eye stimulates production of inflammatory cytokines and MMP-9 and activates MAPK signaling pathways on the ocular surface. Invest Ophthalmol Vis Sci. 2004;45:4293–4301. doi: 10.1167/iovs.03-1145. [DOI] [PubMed] [Google Scholar]
- 32.Pflugfelder SC, de Paiva CS, Tong L, Luo L, Stern ME, Li DQ. Stress-activated protein kinase signaling pathways in dry eye and ocular surface disease. Ocul Surf. 2005;3:S154–S157. doi: 10.1016/s1542-0124(12)70244-6. [DOI] [PubMed] [Google Scholar]
- 33.Li DQ, Luo L, Chen Z, Kim HS, Song XJ, Pflugfelder SC. JNK and ERK MAP kinases mediate induction of IL-1beta, TNF-alpha and IL-8 following hyperosmolar stress in human limbal epithelial cells. Exp Eye Res. 2006;82:588–596. doi: 10.1016/j.exer.2005.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dunne A, O'Neill LA. The interleukin-1 receptor/Toll-like receptor superfamily: signal transduction during inflammation and host defense. Sci STKE. 2003;2003:re3. doi: 10.1126/stke.2003.171.re3. [DOI] [PubMed] [Google Scholar]
- 35.Berg EL, Kunkel EJ, Hytopoulos E. Biological complexity and drug discovery: a practical systems biology approach. Syst Biol (Stevenage) 2005;152:201–206. doi: 10.1049/ip-syb:20050036. [DOI] [PubMed] [Google Scholar]