

Abstract
1,2,3,4-diepoxybutane (DEB), an in vivo metabolite of 1,3-butadiene (BD), is a carcinogen and mutagen. The strong carcinogenicity/mutagenicity of DEB has been attributed to its high DNA reactivity and cross-linking ability. Recently we have demonstrated that under in vitro physiological conditions (pH 7.4, 37 °C), the reaction of DEB with 2′-deoxyguanosine (dG) produced two diastereomeric pairs of the major nucleoside adducts resulting from alkylation at the N1- and N7-positions of dG, i.e., 2′-deoxy-1-(2-hydroxy-2-oxiranylethyl)-guanosine and 2′-deoxy-7-(2-hydroxy-2-oxiranylethyl)-guanosine, respectively [Zhang, X.-Y., and Elfarra, A. A. (2005) Chem. Res. Toxicol. 18, 1316]. As each of these adducts contains an oxirane ring, the abilities of these adducts to form cross-linking products with dG under physiological conditions were investigated. Incubation of the N7 nucleoside adducts and their corresponding guanine product with dG led to formation of 7,7′-(2,3-dihydroxy-1,4-butanediyl)bis[2-amino-1,7-dihydro-6H-purin-6-one] (bis-N7G-BD), a known DEB cross-linking product. Incubation of the N1 nucleoside adducts with dG led to formation of a pair of diastereomers of 2′-deoxy-1-[4-(2-amino-1,7-dihydro-6H-purin-6-on-7-yl)-2,3-dihydroxybutyl]-guanosine (N7G-N1dG-BD), which are novel cross-linking products. Interestingly, the reaction of DEB with dG in glacial acetic acid at 60 °C yielded different cross-linking products, which were characterized as 2-amino-9-hydroxymethyl-4-{4-[2-amino-9- or 7-(4-acetyloxy-2,3-dihydroxybutyl)-1,7-dihydro-6H-purin-6-on-7- or 9-yl]-2,3-dihydroxybutyl}-8,9-dihydro-7H-[1,4]oxazepino[4,3,2-gh]purin-8-ol (PA2) and 9,9′-bis(4-acetyloxy-2,3-dihydroxybutyl)-7,7′-(2,3-dihydroxy-1,4-butanediyl)bis[2-amino-1,7-dihydro-6H-purin-6-one] (PA4). Collectively, these results increase our understanding of the chemical reactivity and cross-linking ability of DEB under both physiological and non-physiological conditions.
Introduction
1,3-Butadiene (BD)1 is a major industrial chemical used to manufacture synthetic rubber and plastic. Besides occupational exposure, human exposure to BD can result from environmental sources, such as automobile exhaust (1) and cigarette smoke (2-4). Whereas occupational exposure to BD has only been more recently associated with excess mortality from lymphatic and/or hematopoietic cancers (5-8), BD has long been recognized as a rodent mutagen and carcinogen (9-10).
The molecular mechanism of mutagenicity/carcinogenicity of BD has not been fully understood. Metabolism of BD produces 3,4-epoxy-1-butene (EB), 1,2,3,4-diepoxybutane (DEB), and 3,4-epoxy-1,2-butanediol (EBD) (11-18), which are direct-acting mutagens and can form DNA and protein adducts (11, 16, 19). Among these metabolites, DEB seems to play a major role in BD-induced carcinogenicity, because it is ∼100-fold more mutagenic than EB and EBD (20-23). DEB is also ∼50-fold more effective than EB at inducing sister chromatid exchanges in human lymphocytes (24). Whereas both EB and DEB induce DNA base substitutions at GC and AT base pairs and frameshift mutations, only DEB causes large numbers of partial deletions (23, 25, 26).
An important structural feature of DEB is its cross-linking ability, as this feature likely makes crucial contribution to its high mutagenicity. In this regard, DEB-induced interstrand cross-links have long been demonstrated both in vitro (DEB-treated DNA) and in vivo (in mice and rats) (27-33). Stereochemistry of DEB may also play a role in its mutagenicity/carcinogenicity; among the three optical isomers of DEB (R,R-, S,S-, and meso-), S,S-DEB almost exclusively leads to cross-linking of interstrand DNA, whereas meso-DEB forms equal amounts of interstrand and intrastrand cross-links (34). Recent studies concerning DEB-induced N2-N2 guanine intrastrand DNA cross-links have suggested that intrastrand DNA cross-links arising from BD exposure might be inefficiently repaired by the nucleotide excision repair system (35).
Brookes and Lawley were the first to study alkylation of guanosine by DEB in glacial acetic acid (36). After subjecting the reaction mixture to acid hydrolysis (1 M HCl, 100 °C, 1 h), they isolated a DEB–guanine cross-linking product, 7,7′-(2,3-dihydroxy-1,4-butanediyl)bis[2-amino-1,7-dihydro-6H-purin-6-one] (bis-N7G-BD2). This cross-linking product was later detected in DEB-treated salmon sperm DNA (28). Recently, bis-N7G-BD was fully characterized by modern spectroscopic techniques and was also identified from DEB-treated calf thymus DNA (37). Four isomeric guanine–adenine cross-linking products involving the N7 position of guanine and the N1, N7, N3 and N6 positions of adenine have also been characterized from the reaction of DEB with calf thymus DNA (38).
Previous studies in our laboratory demonstrated that racemic DEB readily reacted with 2′-deoxyguanosine (dG) under in vitro physiological conditions (pH 7.4, 37 °C) to produce two major pairs of diastereomers: 2′-deoxy-7-(2-hydroxy-2-oxiranylethyl)-guanosine (P5 and P5′; Scheme 1)3 and 2′-deoxy-1-(2-hydroxy-2-oxiranylethyl)-guanosine (P8 and P9; Scheme 2)3 (39, 40). The four products were labile; their half-lives at pH 7.4 and 37 °C were 2.6, 2.7, 16, and 16 h, respectively (40). P5 and P5′ first decomposed to 2-amino-7-(2-hydroxy-2-oxiranylethyl)-1,7-dihydro-6H-purin-6-one (P5D)3 after cleavage of the glycosidic bond (Scheme 1), and P5D further decomposed to final guanine products through opening of the oxirane ring by water, or chloride or dihydrogen phosphate ion present in the solution (40). In contrast, no cleavage of the glycosidic bond was observed during the decomposition of P8 and P9; all of the identified decomposition products of P8 and P9 contained the deoxyribose moiety (40). Because each of P5, P5′, P5D, P8, and P9 contains an oxirane ring, these adducts were expected to be capable of further reacting with dG to form cross-linking products. Therefore, in the present study, purified P5, P5′, P5D, P8, and P9 were incubated with dG under physiological conditions to investigate formation of cross-linking products. In addition, the reaction of DEB with dG in glacial acetic acid was further investigated in an attempt to prepare cross-linking standards.
Scheme 1.

Formation of Bis-N7G-BD by the Reaction of P5, P5′, or P5D with dG under Physiological Conditions
Scheme 2.
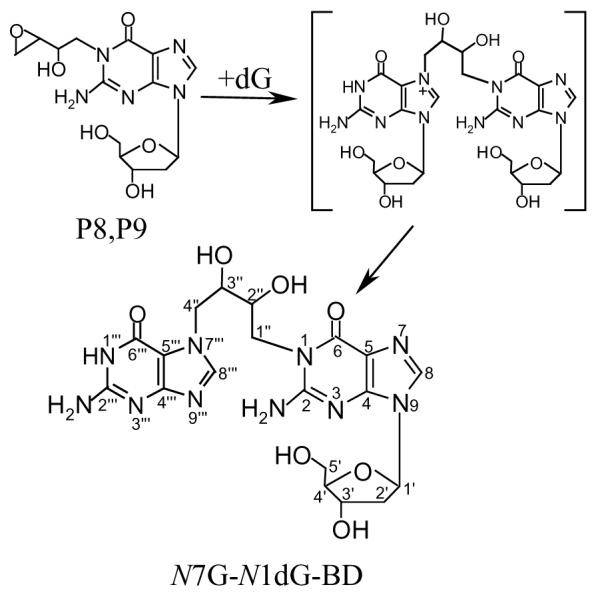
Formation of N7G-N1dG-BD by the Reaction of P8 or P9 with dG under Physiological Conditions
Experimental Procedures
Caution
DEB is a known mutagen and carcinogen and must be handled using proper safety measures.
Materials
Racemic DEB, dG (monohydrate), trifluoroacetic acid (TFA), dimethyl sulfoxide-d6 (DMSO-d6), TMS, deuterium oxide, and sodium 3-(trimethylsilyl)propionate-2,2,3,3-d4 were obtained from Sigma-Aldrich Chemical Co. (Milwaukee, WI). HPLC grade acetonitrile was purchased from EM Science (Gibbstown, NJ). HPLC mobile phases were prepared using acetonitrile and water, and the pH was adjusted to 2.5 with 20% (v/v) TFA for acidic mobile phases. Phosphate buffers (100 mM, pH 7.4) containing 100 mM KCl were prepared with KH2PO4 and KCl using KOH to adjust the pH. The DEB–dG adducts P5, P5′, P8, and P9 were prepared according to the procedures described before (39). The reference bis-N7G-BD was synthesized by the reaction of guanosine with DEB in glacial acetic acid (37).
Instruments and Methods
Mass spectra were obtained on an Applied Biosystems MDS Sciex API 365 LC/MS/MS triple quadrupole electrospray ionization mass spectrometer (Foster City, CA). The purified compounds were dissolved in 1:1 acetonitrile–water (v:v) for mass spectrometric analyses using electrospray ionization (ESI). Tandem mass spectrometry (MS-MS) data of molecular ions were also obtained to determine fragmentation patterns. High-resolution mass spectrometric data were obtained using a Micromass Q-TOF2 Hybrid Quadrupole/Orthogonal Time of Flight Mass Spectrometer (Manchester, UK). NMR data were recorded on a Bruker Instruments DMX-400 Avance console, 9.4 T wide-bore magnet NMR spectrometer except the data for the P8–dG and P9–dG cross-linking products, which were obtained on a Bruker Instruments DMX-600 Avance console, 14.1 T standard bore magnet NMR spectrometer. The solvent for NMR samples was DMSO-d6 with TMS as the internal standard. For PA1 and PA2, the NMR spectra in deuterium oxide with sodium 3-(trimethylsilyl)propionate-2,2,3,3-d4 as the internal standard were also obtained. HPLC separation was performed on a Beckman gradient-controlled HPLC system (Irvine, CA) equipped with a Beckman diode array detector (model 168). Acidic mobile phases were used except for the separation concerning P8 and P9, in which pH-unadjusted mobile phases were used. The UV absorption spectra of the products were extracted from data collected by the diode array detector. Analysis of samples was carried out on a Beckman Ultrasphere 5 μm ODS reverse-phase analytical column (250 mm × 4.6 mm) with a flow rate of 1 mL/min. A linear gradient program was used, starting at 2 min from 0% pump B to 10% pump B over 1 min [pump A, 1% (v/v) acetonitrile; pump B, 10% (v/v) acetonitrile], then at 10 min from 10 to 20% pump B over 1 min, at 11 min from 20 to 30% pump B over 9 min, at 20 min from 30 to 65% pump B over 1 min, at 25 min from 65 to 100% pump B over 12 min, finally at 44 min from 100 to 0% pump B over 1 min and stopping at 45 min. Preparative isolation and analysis of products, except for those prepared in glacial acetic acid, were carried out on a Beckman Ultrasphere 5 μm ODS reverse-phase semipreparative column (250 mm × 10 mm) at a flow rate of 3 mL/min (1% acetonitrile at pump A and 10% acetonitrile at pump B). The gradient program was identical to those described above with the analytical column. Analysis and isolation of DEB–dG adducts formed in glacial acetic acid were carried out on a Beckman Ultrasphere 10 μm ODS reverse-phase preparative column (150 mm × 21.2 mm) at a flow rate of 5 mL/min. A linear gradient program was used, which started at 10 min from 20% pump B to 50% pump B over 20 min [pump A, 1% (v/v) acetonitrile; pump B, 25% (v/v) acetonitrile], then at 35 min from 50 to 100% pump B over 12.5 min, at 59 min from 100 to 20% pump B over 1 min and stopped at 60 min. Fractions were collected using a Gilson fraction collector (model 202) with all collection beakers kept on ice. The collected fractions were lyophilized on a Labconco lyophilizer (Kansas City, MO). Centrifugation of the samples was carried out on an Eppendorf 5417R centrifuge (Hamburg, Germany) at 20,200g.
Reactions of P5 and P5′ with dG at Physiological Conditions
A solution (1.4 mL) containing 1.2 mg of P5 or P5′ (3 μmol) and 3 mg of dG (11 μmol) in pH 7.4 phosphate buffer and P5, P5′, and dG controls were incubated at 37 °C for 426 h and then were centrifuged for 15 min. The supernatants were analyzed and products were isolated by HPLC. The precipitates were dissolved in dilute HCl (0.1 M, 0.2 mL) and analyzed by HPLC. To serve as a standard, the synthesized bis-N7G-BD was dissolved in 0.1 M HCl and analyzed by HPLC.
Preparation of P5D and Its Reaction with dG at Physiological Conditions
The crude fraction containing both P5 and P5′, which was obtained from the initial separation of DEB–dG reaction mixture on the Sephadex LH-20 column (39), was concentrated to ∼1 mL by lyophilization. The concentrated solution was then incubated at 37 °C with gentle shaking for 3.7 h to allow P5 and P5′ to fully convert to P5D, which was isolated by HPLC. The solution (3 mL) containing 1.5 mg of P5D (6.3 μmol) and 4.8 mg of dG (17 μmol) in pH 7.4 phosphate buffer, and P5D and dG controls were incubated at 37 °C for 426 h and then centrifuged for 15 min. The supernatants were analyzed by HPLC. The precipitates were dissolved in dilute HCl (0.1 M, 0.2 mL) and analyzed by HPLC. The results were compared with the HPLC results obtained with the standard bis-N7G-BD.
Reactions of P8 and P9 with dG at Physiological Conditions and Isolation of Cross-Linking Products
A solution (0.79 mL) containing 7–10 mg of P8 or P9 (20–28 μmol) and 1.8 mg of dG (6 μmol) in pH 7.4 phosphate buffer, together with P8/P9 and dG controls, were incubated at 37 °C for 160 h. These solutions were analyzed and the cross-linking products (retention times 35.3 min for P8–dG and 36.0 min for P9–dG) were isolated by HPLC.
Determination of Half-Lives of P5, P5′, P5D, P8, and P9 at Physiological Conditions in the Presence of dG
A solution (0.2 mL) containing 1–2 mg of each purified compound and 0.3 mg of dG (1 μmol) in pH 7.4 phosphate buffer was incubated in a water bath at 37 °C with gentle shaking. Aliquots (1 μL) were taken periodically (for P5 and P5′, every hour; for P5D, P8, and P9, every 7–15 h) and analyzed by HPLC. At least six aliquots were taken for HPLC analysis before the adducts decomposed completely. Natural logarithm of the peak areas of the adducts obtained from the chromatograms were plotted against incubation time. The data were regressed and half-lives of the adducts were calculated accordingly.
Reaction of DEB with dG in Glacial Acetic Acid
In an attempt to prepare DEB–dG cross-linking standards, the reaction of DEB with dG in glacial acetic acid was carried out. DEB (97%, 91 μL, 1.14 mmol) was added to a dG (58 mg, 0.20 mmol) solution in glacial acetic acid (2 mL). The reaction mixture was incubated in a water bath at 60 °C for 4 h. After cooling to room temperature, acetone (2 mL) and then ethyl ether (8 mL) were added to the reaction mixture, which was immediately centrifuged for 1 min. The pellet was dissolved in 1 mL of water. The solution was analyzed and the major products were isolated by HPLC.
Effects of HCl Treatment on PA1, PA2, PA3, and PA4
To characterize PA2, PA3, and PA4, concentrated HCl (20 μL) was added to the solutions of purified PA2, PA3, and PA4 (0.1–0.5 mg of compounds was dissolved in 200 μL of water). The solutions were heated in a boiling water bath for 1 h, cooled to room temperature, neutralized to pH ∼6 using ammonium hydroxide, and analyzed by HPLC. The hydrolysis products were isolated by HPLC. For PA1, the procedure was the same except 20 μL of 0.11 M HCl was used instead of concentrated HCl4.
Results
Identification of the Cross-Linking Products of P5 or P5′ with dG at Physiological Conditions
Direct HPLC analysis of the reaction mixture of P5 or P5′ with dG using regular injection volume (≤ 20 μL), after incubation at pH 7.4 and 37 °C for over 400 h, did not show any new peaks compared with chromatograms of P5, P5′, and dG controls. Since bis-N7G-BD was insoluble in water but soluble in HCl, the reaction mixture was centrifuged. The precipitate on the wall of the tube, which was marginally visible, was dissolved in 0.1 M HCl and analyzed by HPLC. The chromatogram showed an overwhelmingly predominant peak at 23.2 min, whose retention time and UV absorption spectrum matched those of the standard, confirming the presence of bis-N7G-BD in the precipitate (Scheme 1).
When a large volume of supernatant (250 μL) was loaded onto the analytical HPLC column, a minor peak at 33.2 or 33.6 min for P5 or P5′, respectively, was observed. These minor peaks did not appear in the chromatograms of P5, P5′, and dG controls. The compounds that the two minor peaks represent had the same UV absorption spectra (λmax 254 nm) and their ESI-mass spectra showed the same molecular ion at m/z 505. The molecular weight is consistent with a guanine moiety cross-linked to dG through a dihydroxybutanediyl bridge chain. The two products were assumed to be a pair of diastereomers because of their close retention times, and their identical UV absorption spectra and molecular weights. The quantities of the two products were too small to allow further characterization by NMR; however, their MS results suggest they were the unstable precursors of bis-N7G-BD (Scheme 1). The possibility that these products were N1-N7 cross-linking products was excluded because the retention times and UV absorption spectra of these products did not match those of the P8–dG and P9–dG cross-linking products (see below).
Identification of the Cross-Linking Products of P5D with dG at Physiological Conditions
Similar to P5 and P5′, incubation of P5D with dG at pH 7.4 and 37 °C for 426 h confirmed the presence of bis-N7G-BD in the precipitate after the reaction mixture was centrifuged (Scheme 1).
The chromatogram of the supernatant on an analytical HPLC column with a large injection volume (250 μL) showed two minor peaks at 33.4 and 33.7 min (Figure 1), which were not observed in the chromatograms of P5D and dG controls. The products that the two minor peaks represent were characterized as the same unstable precursors of bis-N7G-BD described above from the incubation of P5 or P5′ with dG based upon comparison of the retention times and the UV absorption spectra. Because P5D was prepared from a mixture of P5 and P5′, it was actually a mixture of a pair of enantiomers rather than an optically pure isomer like P5 or P5′. As a result, a diastereomeric pair of unstable precursors of bis-N7G-BD was produced in the case of P5D, whereas only one of this pair of diastereomers was yielded with P5 or P5′.
Figure 1.
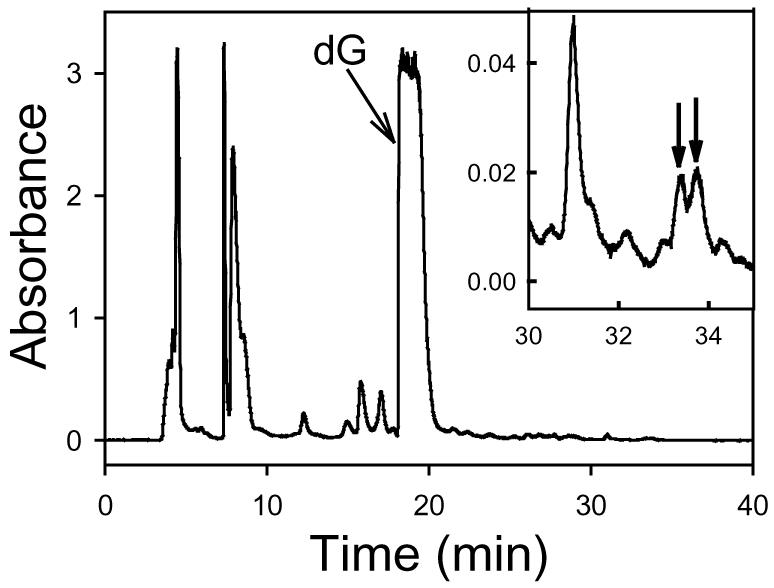
The HPLC chromatogram of the reaction mixture of P5D and dG incubated at pH 7.4 and 37 °C for 426 h (monitoring wavelength: 260 nm). Inset: The magnified chromatogram between 30 and 35 min to show the minor peaks. The two peaks, which were not observed in the chromatograms of P5D and dG controls, are marked with arrows.
Structural Characterization of the Cross-Linking Products of P8 and P9 with dG at Physiological Conditions
The chromatogram of the reaction mixture of P8 and dG incubated under physiological conditions for 118 h showed many peaks (Figure 2A). A comparison of the chromatogram of the P8–dG reaction mixture with that of P8 control (Figure 2B) revealed four new peaks, marked with numbers 1, 2, 3, and 4 (Figure 2A). However, peak 1, 2, and 3 were also observed in the chromatogram of dG control (data not shown). Therefore, only peak 4 (retention time 35.3 min) was new and assumed to represent the reaction product of P8 with dG. Similarly, only one new peak at 36.0 min was observed in the chromatogram of the P9–dG reaction mixture.
Figure 2.
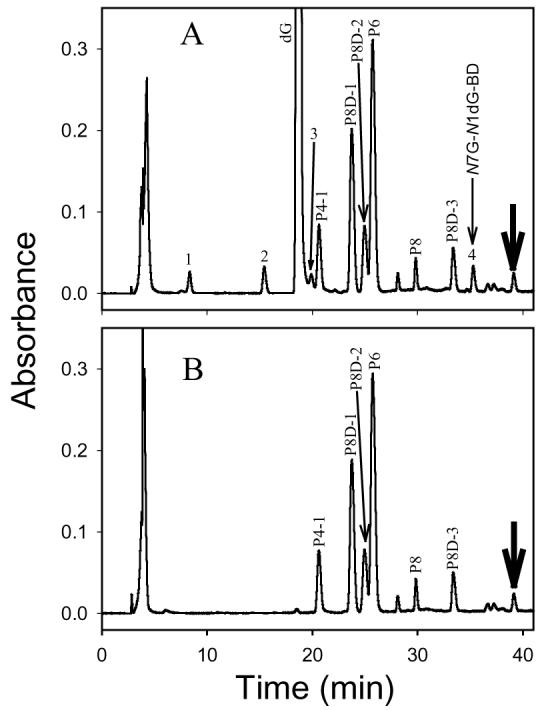
The HPLC chromatogram of P8 incubated at pH 7.4 and 37 °C for 118 h in the presence (A) and absence (B) of dG (monitoring wavelength: 260 nm). P4-1, P8D-1, P8D-2, P6, and P8D-3 are decomposition products of P8 (40). Peaks 1, 2, and 3 are decomposition products of dG. Peak 4 (N7G-N1dG-BD) represents the cross-linking product of P8 and dG. The thick arrow refers to a cross-linking product of two P8 molecules.
The UV absorption spectrum of the compound that peak 4 represents exhibited two peaks at 252 and 276 nm in pH-unadjusted mobile phase (Figure 3), which was different from the UV absorption spectra of dG (λmax at 253 nm) and P8 (λmax at 256 nm) obtained under the same conditions (Figure 3). The molecular weight of this compound was determined to be 504 by ESI-MS (data not shown), consistent with a structure of a dG molecule being linked with a guanine molecule through a dihydroxybutanediyl bridge chain. The MS-MS spectrum of the protonated molecular ion at m/z 505 showed an overwhelmingly predominant fragment at m/z 389, indicating loss of a deoxyribose moiety. These results identified this compound as a P8–dG cross-linking product. The compound that the peak at 36.0 min in the chromatogram of the P9–dG reaction mixture represents exhibited a UV absorption spectrum and molecular weight (data not shown) identical to those observed with the P8–dG cross-linking product. Collectively, the results indicated that the P9–dG cross-linking product was a diastereomer of the P8–dG cross-linking product.
Figure 3.
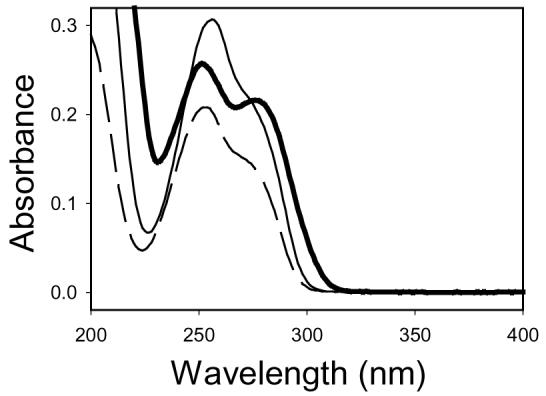
The UV absorption spectra of the P8–dG cross-linking product (thick solid line), P8 (thin solid line), and dG (dashed line) obtained from the HPLC chromatogram data with pH-unadjusted mobile phase.
The dG control showed no decomposition to guanine. Therefore the possibility that the P8–dG and P9–dG cross-linking products were formed through the reaction of P8 and P9 with guanine was excluded. The two cross-linking products were considered to be either N1-N7 or N1-N1 cross-linking products based upon the known reactivity of the N7 and N1 positions of dG towards DEB and EB (11, 39, 40). A cross-linking product of P8 or P9 with dG, which contains two deoxyribose moieties, would have a molecular weight of 619 or 620 for N1-N1 or N1-N7 cross-links, respectively. The observed molecular weights of the two cross-linking products indicated that these molecules contained only one deoxyribose moiety; i.e., one deoxyribose moiety was already cleaved during incubation. Since N7-alkylated dG adducts are known to lose their deoxyribose moieties under physiological conditions whereas N1-alkylated dG adducts will not undergo cleavage of glycosidic bond at the same conditions (40, 41), the P8–dG and P9–dG cross-linking products were determined to be N1-N7 cross-linking products and thus designated as N7G-N1dG-BD5 (Scheme 2).
The 1H NMR spectra of the P8–dG and P9–dG cross-linking products appeared nearly identical (Table 1), confirming that they are indeed a pair of diastereomers. In the 1H NMR spectrum of the P8–dG cross-linking product in DMSO-d6 (Figure 4A), the broad signal at 10.74 ppm with one proton (Figure 4A, inset) was assigned to the N1 proton. The presence of an N1 proton indicated that this cross-linking product was indeed an N1-N7 cross-linking product as suggested above.
Table 1.
The 1H NMR Spectroscopic Data Obtained with the P8–dG and P9–dG Cross-Linking Products (diastereomers of N7G-N1dG-BD; Scheme 2) in DMSO-d6
| P8–dG cross- linking product |
P9–dG cross- linking product |
assignments |
|---|---|---|
| 10.74 (br s, 1H) | 10.69 (s, 1H) | H-1‴ |
| 7.95 (s, 1H) | 7.94 (s, 1H) | H-8 |
| 7.82 (s, 1H) | 7.81 (s, 1H) | H-8‴ |
| 6.77 (s, 2H) | 6.74 (s, 2H) | 2-NH2 |
| 6.13 (t, 1H) | 6.13 (dd, 1H) | H-1′ |
| 6.08 (s, 2H) | 6.04 (s, 2H) | 2‴-NH2 |
| 5.48 (d, 1H) | 5.45 (d, 1H) | 2″-OH or 3″-OH |
| 5.27 (d, 2H) | 5.22 (d, 2H) | 3′-OH and 2″-OH or 3″-OH |
| 4.94 (t, 1H) | 4.90 (t, 1H) | 5′-OH |
| 4.34 (m, 1H) | 4.34 (m, 1H) | H-3′ |
| 4.33 (m, 1H) | 4.33 (m, 1H) | H-4″ |
| 4.18 (dd, 1H) | 4.19 (dd, 1H) | H-4″ |
| 4.06 (m, 1H) | 4.09 (m, 1H) | H-1″ |
| 3.97 (m, 1H) | 3.96 (m, 1H) | H-1″ |
| 3.81 (m, 2H) | 3.82 (m, 2H) | H-4′ and H-2″ or H-3″ |
| 3.72 (m, 1H) | 3.73 (m, 1H) | H-2″ or H-3″ |
| 3.55 (m, 1H) | 3.55 (m, 1H) | H-5′ |
| 3.49 (m, 1H) | 3.50 (m, 1H) | H-5′ |
| 2.5 (m, 1H)a | 2.51 (m, 1H)a | H-2′ |
| 2.19 (m, 1H) | 2.20 (m, 1H) | H-2′ |
These values of the chemical shifts are approximate because the solvent peak (DMSO) appeared at 2.50 ppm.
Figure 4.
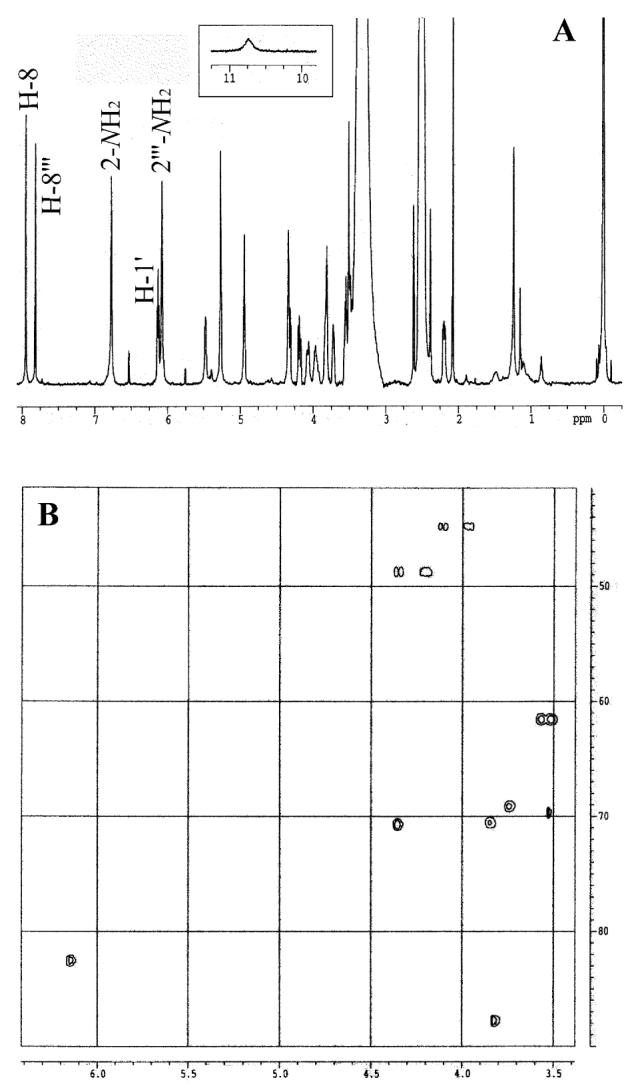
A. The 1H NMR spectrum of the P8–dG cross-linking product in DMSO-d6. Inset: The signal between 10 and 11 ppm; B. The HMQC spectrum of the P9–dG cross-linking product in DMSO-d6.
The HMQC spectrum of the P9–dG cross-linking product (Figure 4B) showed two methylene carbons at high field (45.1 and 49.0 ppm) and two methine carbons at low field (69.5 and 70.4 ppm), indicating that the two ends of the bridge chain were attached to N1 and N7. Therefore, N7G-N1dG-BD were structurally characterized as a pair of diastereomers of 2′-deoxy-1-[4-(2-amino-1,7-dihydro-6H-purin-6-on-7-yl)-2,3-dihydroxybutyl]-guanosine (Scheme 2).
P8–P8 and P9–P9 Cross-Linking Products
A minor peak at 39.1 min was detected in the chromatograms of both P8 control and P8–dG reaction mixture (Figure 2, marked with a thick arrow). The compound that this peak represents had a molecular weight of 590 as determined by ESI-MS, consistent with a P8–P8 cross-linking product after losing one deoxyribose moiety. Because the N1 position in P8 was already alkylated, the reaction was expected to occur at N7. Similar to the P8–dG cross-linking product, the initial product was expected to lose one deoxyribose moiety during incubation. A minor peak with a similar retention time was also observed in the chromatograms of P9 control and P9–dG reaction mixture (data not shown). However, the latter minor products were not further characterized.
The Half-Lives of P5, P5′, P5D, P8, and P9 in the Presence of dG
In an effort to assess the relative rates of the cross-linking reactions in comparison with overall decomposition reactions, the half-lives of P5, P5′, P5D, P8, and P9 in the presence of dG were determined to be 2.6, 2.7, 33, 16, and 16 h, respectively, which are identical to the half-lives of these adducts in the absence of dG (40). These results, which indicate that the rates of the cross-linking reactions were low relative to the overall decomposition reactions, are consistent with the observation that the peak areas of N7G-N1dG-BD accounted for less than 1% of the total peak areas in the HPLC chromatograms of the P8–dG and P9–dG reaction mixtures (Figure 2A).
Characterization of the DEB–dG Adducts Formed in Glacial Acetic Acid
The HPLC chromatogram of the DEB–dG reaction mixture in glacial acetic acid at 60 °C for 4 h showed four major peaks (designated PA1, PA2, PA3, and PA46) and several minor peaks (Figure 5). The 1H NMR data indicated that the four major products did not contain the deoxyribose moiety (Table 2). Their accurate molecular weights were determined by high-resolution ESI-MS and the corresponding molecular formulas were calculated accordingly (Table 3). Characterization of each of the four adducts is briefly described below, and a full description can be found in the Supporting Information.
Figure 5.

The HPLC chromatogram of the reaction mixture of DEB and dG in glacial acetic acid after incubation at 60 °C for 4 h (monitoring wavelength: 260 nm).
Table 2.
The 1H NMR Spectroscopic Data Obtained with PA1, PA2, PA3, and PA4 in DMSO-d6
| PA1 | PA2 | PA3 | PA4 | ||||
|---|---|---|---|---|---|---|---|
| δ (ppm) | Assignment | δ (ppm) | Assignment | δ (ppm) | Assignment | δ (ppm) | Assignment |
| 11.88 (s, 1H) | H-1″ | 11.94 (s, 1H) | H-1 | 11.97 (d, 2H) | H-1 | ||
| 9.22 (d, 1H) | H-5 | 9.26 (s, 1H) 9.24 (s, 1H) |
H-5, H- 8″ |
9.20 (d, 1H) | H-8 | 9.23 (s, 2H) | H-8 |
| 7.74 (s, 2H) | NH2 | 7.77 (br s, 2H) | 2-NH2 | ||||
| 7.34 (br s, 2H) | 2″-NH2 | 7.38 (br s, 2H) | NH2 | 7.41 (br s, 4H) | NH2 | ||
| 5.91 (br s, 1H) 5.84 (br s, 1H) |
8-OH, 9a-OH |
5.96 (m, 1H) 5.86 (m, 1H) |
8-OH, 9a- OH |
||||
| 5.58 (m, 1H) 5.52 (m, 1H) |
2′,3′-OH | 5.59 (s, 1H) 5.54 (s, 1H) |
2′,3′- OH |
||||
| 5.30 (br s, 2H) | 2′,3′-OH | 5.32 (m, 1H) 5.25 (m, 1H) |
2‴,3‴- OH |
5.31 (s, 2H) 5.26 (s, 2H) |
2′,3′,2″, 3″-OH |
5.33 (br s, 2H) 5.27 (br s, 2H) |
2″,3″- OH |
| 4.60-4.68 (m, 2H) |
H-8, H-9 | 4.66 (m, 2H) | H-8, H-9 | ||||
| 4.50 (dd, 1H) | H-7 | 4.52 (m, 2H) | H-7, H-4′ or 1‴ |
4.51 (m, 1H) | H-1′ | 4.55 (m, 2H) | H-1′/4′ |
| 4.26-4.37 (m, 2H) |
H-7, H-1′ | 4.38 (m, 2H) | H-7, H-4′ or 1‴ |
4.35 (m, 1H) | H-1′ | 4.40 (m, 2H) | H-1′/4′ |
| 4.14-4.30 (m, 4H) |
H-1′, H- 1‴ or 4′ |
4.24 (dd, 1H) | H-1″ | 4.24 (m, 2H) | H-1″ | ||
| 4.18 (dd, 1H) | H-1′ | 4.18 (m, 1H) | H-1″ | 4.18 (m, 2H) | H-1″ | ||
| 4.04 (m, 4H) | H-9a, H- 4′ |
4.05 (m, 6H) | H-9a, H- 4‴, H-2′, H-3′ |
4.06 (m, 4H) | H-4′, H- 4″ |
4.05 (m, 6H) | H-4″, H-2′, H- 3′ |
| 3.88 (m, 1H) | H-2′ | 3.96 (m, 1H) | H-2‴ | 3.93 (br, 2H) | H-2′, H- 2″ |
3.94 (m, 2H) | H-2″ |
| 3.73 (m, 1H) | H-3′ | 3.76 (s, 1H) | H-3‴ | 3.75 (s, 2H) | H-3′, H- 3″ |
3.76 (s, 2H) | H-3″ |
| 2.02 (s, 3H) | CH3CO | 2.04 (s, 3H) | CH3CO | 2.03 (s, 6H) | CH3CO | 2.01-2.03 (m, 6H) | CH3CO |
Table 3.
The High Resolution ESI-MS Data Obtained with PA1, PA2, PA3, and PA4 and Their Molecular Formula as Determined from the Resultsa
| molecular ion charge |
m/z of molecular ions (low-resolution) |
formula weight | molecular formula |
||
|---|---|---|---|---|---|
| measured | theoretical | ||||
| PA1 | 1+ | 384 | 384.1540 | 384.1519 | C15H22N5O7 |
| PA2 | 2+ | 311 | 621.2350 | 621.2381 | C24H34N10O10 |
| PA3 | 1+ | 444 | 444.1700 | 444.1731 | C17H26N5O9 |
| PA4 | 2+ | 341 | 681.2580 | 681.2592 | C26H38N10O12 |
It was found that the molecular ions of PA1 and PA3 (appeared at m/z 384 and 444, respectively) were single charged, whereas those of PA2 and PA4 (appeared at m/z 311 and 341, respectively) carried two positive charges. These molecular ions were not protonated because they were quaternary ammonium ions. As a result, the formula weights of PA1, PA2, PA3, and PA4 were 384, 622, 444, and 682, respectively. When measuring accurate formula weights of the four compounds by high-resolution ESI-MS, single-charged ions were observed. Namely, for PA1 and PA3, the molecular ions at m/z 384 and 444 were observed; however, for PA2 and PA4, the ions at m/z 621 and 681, rather than the molecular ions at m/z 311 and 341, were observed. The ions at m/z 621 and 681 were apparently formed after the molecular ions of PA2 and PA4 each lost a proton.
PA1
The 1H NMR spectrum of PA1 did not show any signal beyond 10 ppm (Table 2), indicating absence of the guanine N1 proton. This observation was attributed to formation of a fused seven-membered ring by the reaction of the guanine O6 with the remaining oxirane moiety in the DEB-derived side chain at the guanine N7 position (see Supporting Information for details; Scheme 3). The signal of the C8 proton appeared at 9.22 ppm, a remarkable downfield shift from the C8 proton of dG (7.93 ppm, 42), and disappeared in D2O (data not shown), clearly indicating that the imidazole ring of guanine was positively-charged (41, 43). That is, PA1 is a quaternary ammonium salt. Thus, PA1 must be a 7,9-dialkylated guanine derivative. Hydrolysis of PA1 in 0.01 M HCl4 (100 °C, 1 h) yielded a predominant product with a molecular ion at m/z 342 as determined by ESI-MS, indicating that the acid hydrolysis merely caused loss of an acetyl moiety and excluding presence of an oxirane ring in this molecule. Based on these observations and the molecular formula, it is easy to establish the structure of PA1 as 2-amino-9-hydroxymethyl-4-(4-acetyloxy-2,3-dihydroxybutyl)-8,9-dihydro-7H-[1,4]oxazepino[4,3,2-gh]purin-8-ol (Scheme 3).
Scheme 3.

The Chemical Structures of PA1, PA2, PA3, and PA4
PA2
The ESI-mass spectrum of PA2 showed that the molecular ion, appearing at m/z 311, carried two positive charges. This indicated that the actual mass of the molecular ion is 622 and suggested the guanine moieties in this adduct are both 7,9-dialkylated. The 1H NMR spectrum showed a single peak representing one proton at 11.87 ppm, which was apparently the signal of the N1 proton. Because PA2 contains two guanine moieties but only has one N1 proton, one of the two guanine moieties must have lost its N1 proton. Similar to PA1, this observation was attributed to formation of a fused seven-membered ring involving both the N7 and O6 positions. The 1H NMR spectrum also showed two overlapping single peaks corresponding to two protons at 9.24 and 9.26 ppm, which could only be assigned to the two C8 protons. Like PA1, the high chemical shifts and disappearance of the signals of the two C8 protons in D2O (data not shown) indicated that the guanine moieties were both 7,9-dialkylated, which was consistent with the MS results. Thus, PA2 was considered a quaternary ammonium salt. The acid hydrolysis experiment excluded the possibility of presence of an oxirane ring in this molecule. Therefore the structure of PA2 was characterized as 2-amino-9-hydroxymethyl-4-{4-[2-amino-9- or 7-(4-acetyloxy-2,3-dihydroxybutyl)-1,7-dihydro-6H-purin-6-on-7- or 9-yl]-2,3,-dihydroxybutyl}-8,9-dihydro-7H-[1,4]oxazepino[4,3,2-gh]purin-8-ol (Scheme 3).
PA3
The molecular formula of PA3 (Table 3) showed that this molecule contains one guanine moiety and two acetyloxydihydroxybutyl side chains. The signals at 11.94 (1 proton), 9.20 (1 proton), and 7.38 ppm (broad, 2 protons) in the 1H NMR spectrum clearly were those of the protons at the N1, C8 and N2 positions, respectively. The high chemical shift of the C8 proton indicated 7,9-dialkylation. The two side chains were easily determined as 4-acetyloxy-2,3-dihydroxybutyl by analyzing 13C, 13C DEPT 135, COSY, and HMQC data. Therefore the structure of PA3 is established as 2-amino-7,9-bis(4-acetyloxy-2,3-dihydroxybutyl)-1,7-dihydro-6H-purin-6-one (Scheme 3).
PA4
ESI-mass spectrum showed that the molecular ion of PA4, which appeared at m/z 341, was doubly-charged. Thus the actual mass of the molecular ion was 682. According to the molecular formula (Table 3), PA4 contained two guanine moieties, one dihydroxybutanediyl bridge chain and two acetyloxydihydroxybutyl side chains. The doubly-charged molecular ion indicated that the guanine moieties were both 7,9-dialkylated, which was further supported by the 1H NMR data. The signals at 11.97 (2 protons), 9.23 (2 protons) and 7.41 ppm (broad, 4 protons) (Table 2) were assigned as the signals of the protons at the N1, C8 and N2 positions, respectively. Unlike PA2, the presence of two N1 protons ruled out formation of fused rings involving O6. Further analysis of NMR data revealed that the structure of PA4 was 9,9′-bis(4-acetyloxy-2,3-dihydroxybutyl)-7,7′-(2,3-dihydroxy-1,4-butanediyl)bis[2-amino-1,7-dihydro-6H-purin-6-one] (Scheme 3). Interestingly, a comparison of the HPLC chromatogram of the HCl hydrolysis mixture of PA4 with that of the standard bis-N7G-BD showed the acid hydrolysis mixture did not contain bis-N7G-BD.
Discussion
The results obtained from the present study demonstrated that P5, P5′, P5D, P8, and P9 were all able to form cross-linking products with dG at physiological conditions. Based upon the major products characterized from the reactions of DEB and EB with dG (11, 39, 40, 41), it was expected that the cross-linking reactions of the oxirane moieties in the adducts with dG would occur at either the N7 or N1 position of dG. However, no cross-linking reaction involving the N1 position of dG was detected, suggesting an N7 selectivity in the reactions of these adducts with dG to form cross-linking products.
The reactions of P5 and P5′ with dG at physiological conditions can occur along two different pathways (Scheme 1). In the first pathway, P5 and P5′ can directly react with dG to produce an unstable intermediate with two positive charges, which subsequently may lose the two deoxyribose moieties in one or two steps to yield the final cross-linking product, bis-N7G-BD. In the second pathway, P5 and P5′ may first decompose to the less labile P5D, which then reacts with dG to produce an unstable intermediate with a positive charge. This intermediate is expected to be labile, and would convert to bis-N7G-BD after cleavage of its glycosidic bond.
The cross-linking products were minor products under the used reaction conditions. This fact, together with the unchanged half-lives of P5, P5′, P5D, P8, and P9 in the presence of dG, indicated low rates for the cross-linking reactions. Modest reactivity of dG towards the remaining oxirane rings in the monoadducts and/or low dG concentrations in the reaction mixtures may have contributed to these results. It should be noted, however, that the stacking and close proximity of guanine bases in DNA could greatly facilitate the cross-linking reaction. Analysis of DEB-treated DNA indicated that the molar ratio of bis-N7G-BD and the monoadduct, 2-amino-7-(2,3,4-trihydroxybutyl)-1,7-dihydro-6H-purin-6-one, was close to 1 at low DEB concentration (0.05–0.1 mM) (37).
To our knowledge, this is the first study to identify guanine N1-N7 cross-linking products that could be formed from DEB. We have previously demonstrated that in addition to the N7-alkylated dG adducts (P5 and P5′), the N1-alkylated dG adducts (P8 and P9) were also major products from the reaction of DEB and dG at in vitro physiological conditions (39, 40). Because P8 and P9 are much less labile than P5 and P5′ (40), it is expected that the N1 adducts and their derived cross-linking products may preferentially accumulate in target tissues, suggesting a more prominent role for the N1 adducts and their derived cross-linking products in mutagenicity and carcinogenicity.
The initial purpose of carrying out the reaction of DEB with dG in glacial acetic acid was to prepare cross-linking standards that could be used to characterize the reaction products of P5, P5′, P5D, P8, and P9 with dG under physiological conditions. Interestingly, the four major compounds isolated were all 7,9-dialkylated guanine derivatives. These results could be explained by instability of P5 and P5′, the likely major initial products of the DEB–dG reaction in acetic acid. P5 and P5′ are short-lived; their half-lives at pH 7.4 and 37 °C are 2.6 and 2.7 h, respectively (40). They are even much more short-lived at weakly acidic conditions; their half-lives at pH 4.0 and 37 °C are only 44 min7. Thus, it is reasonable to speculate that P5 and P5′ could be more short-lived in glacial acetic acid at 60 °C than at pH 4.0 and 37 °C. Decomposition of P5 and P5′, as our previous work has demonstrated (40), predominantly yields a pair of enantiomers of the corresponding guanine adduct P5D (Scheme 4). P5D can then undergo further reactions: the oxirane ring of the side chain can form a fused ring with the purine O6 or be opened by a molecule of acetic acid. Alternatively, the oxirane ring of P5D can react with dG to form cross-linking products. Because each of these products and P5D has a free N9 position, these molecules can further react with DEB to form the various 7,9-dialkylated adducts (Scheme 4).
Scheme 4.
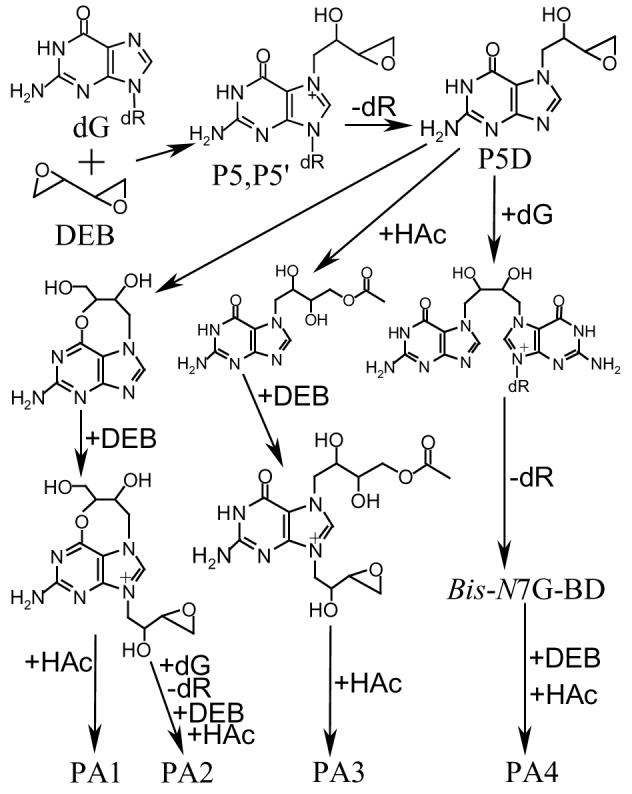
Formation of DEB–dG Products in Glacial Acetic Acid; dR: deoxyribose; HAc: acetic acid
Unexpectedly, bis-N7G-BD was not among the major products detected from the reaction of DEB and dG in glacial acetic acid at 60 °C for 4 h despite the fact that bis-N7G-BD was prepared through the reaction of guanosine with DEB in glacial acetic acid at 80 °C (37). This is probably caused by the large excess of DEB in our experiment (DEB:dG = 5.7:1) in comparison with the conditions used to prepare bis-N7G-BD (DEB:guanosine = 1.1:1). The finding that heating PA2 and PA4 in 1 M HCl at 100 °C for 1 h did not lead to formation of bis-N7G-BD is consistent with a previous report that alkyl substituents at N9 were not cleaved by heating in HCl (36).
Acknowledgments
This research was supported by NIH grant ES06841 from the National Institute of Environmental Health Sciences. We thank Dr. Mark Anderson for his assistance in measuring NMR spectra. This study made use of the National Magnetic Resonance Facility at Madison, which is supported by National Institutes of Health grants P41RR02301 (Biomedical Research Technology Program, National Center for Research Resources) and P41GM66326 (National Institute of General Medical Sciences). Equipment in the facility was purchased with funds from the University of Wisconsin, the National Institutes of Health (P41GM66326, P41RR02301, RR02781, RR08438), the National Science Foundation (DMB-8415048, OIA-9977486, BIR-9214394), and the U.S. Department of Agriculture.
Footnotes
Abbreviations: BD: 1,3-butadiene; COSY: correlation spectroscopy; DEB: 1,2,3,4-diepoxybutane; DEPT: distortionless enhancement by polarization transfer; dG: 2′-deoxyguanosine; DMSO: dimethyl sulfoxide; EB: 3,4-epoxy-1-butene; EBD: 3,4-epoxy-1,2-butanediol; ESI: electrospray ionization; HMQC: heteronuclear multiple quantum coherence; MS: mass spectrometry; MS-MS: tandem mass spectrometry; TFA: trifluoroacetic acid; λmax: the wavelength with maximum absorption.
This compound is named according to the Chemical Abstracts nomenclature rules. However, to be consistent with the literature, the designation bis-N7G-BD, which is based on another name for this compound, i.e., 1,4-bis-(guanin-7-yl)-2,3-butanediol, is also used.
The designations P5, P5′, P8, and P9 were used previously to describe products isolated from the reaction of DEB with dG based upon HPLC retention times (39). The designation P5D was used to describe the major decomposition product of P5 and P5′ (40). These designations are used in the present article to be consistent with our previous publications (39, 40).
Hydrolysis of PA1 in higher concentrations of HCl (0.1 and 1 M) produced complex results that are beyond the scope of this article.
These products are named according to the Chemical Abstracts nomenclature rules. However, to simplify the reference to them, the designation N7G-N1dG-BD, which is based on another name for these compounds, i.e., 4-(guanin-7-yl)-1-(2′-deoxyguanosin-1-yl)-2,3-butanediol, is also used.
PA refers to product in acetic acid.
Zhang, X.-Y., and Elfarra, A. A., unpublished data.
References
- 1.Pelz N, Dempster NM, Shore PR. Analysis of low molecular weight hydrocarbons including 1,3-butadiene in engine exhaust gases using an aluminum oxide porous-layer open-tubular fused-silica column. J. Chromatogr. Sci. 1990;28:230–235. doi: 10.1093/chromsci/28.5.230. [DOI] [PubMed] [Google Scholar]
- 2.Hecht SS. Tobacco smoke carcinogens and lung cancer. J. Natl. Cancer Inst. 1999;91:1194–1210. doi: 10.1093/jnci/91.14.1194. [DOI] [PubMed] [Google Scholar]
- 3.Fowles J, Dybing E. Application of toxicological risk assessment principles to the chemical constituents of cigarette smoke. Tobacco Control. 2003;12:424–430. doi: 10.1136/tc.12.4.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nazaroff WM, Singer B. Inhalation of hazardous air pollutants from environmental tobacco smoke in US residences. J. Expo. Anal. Environ. Epidemiol. 2004;14:571–577. doi: 10.1038/sj.jea.7500361. [DOI] [PubMed] [Google Scholar]
- 5.U.S. Environmental Protection Agency (EPA) Health assessment of 1,3-butadiene. National Center for Environmental Assessment; Washington, DC: 2002. EPA/600/P-98/001F. [Google Scholar]
- 6.Santos-Burgoa C, Matanoski GM, Zeger S, Schwartz L. Lymphohematopoietic cancer in styrene-butadiene polymerization workers. Am. J. Epidemiol. 1992;136:843–854. doi: 10.1093/aje/136.7.843. [DOI] [PubMed] [Google Scholar]
- 7.Divine BJ, Wendt JK, Hartman CM. Cancer mortality among workers at a butadiene production facility. In: Sorsa M, Peltonen K, Vainio H, Hemminki K, editors. Butadiene and Styrene: Assessment of Health Hazards. IARC; Lyon: 1993. pp. 345–362. (IARC Scientific Publications no. 127). [PubMed] [Google Scholar]
- 8.Delzell E, Sathiakumar N, Hovinga M, Macaluso M, Julian J, Larson R, Cole P, Muir DCF. A follow-up study of synthetic rubber workers. Toxicology. 1996;113:182–189. doi: 10.1016/0300-483x(96)03443-9. [DOI] [PubMed] [Google Scholar]
- 9.Owen PE, Glaister JR, Gaunt IF, Pullinger DH. Inhalation toxicity studies with 1,3-butadiene: 3. Two year toxicity/carcinogenicity study in rats. Am. Ind. Hyg. Assoc. J. 1987;48:407–413. doi: 10.1080/15298668791384959. [DOI] [PubMed] [Google Scholar]
- 10.Melnick RL, Huff J. 1,3-Butadiene: toxicity and carcinogenicity in laboratory animals and in humans. Rev. Environm. Contamination Toxicol. 1992;124:111–144. doi: 10.1007/978-1-4612-2864-6_5. [DOI] [PubMed] [Google Scholar]
- 11.Elfarra A, Moll T, Krause R, Kemper R, Selzer R. Reactive metabolites of 1,3-butadiene: DNA and hemoglobin adduct formation and potential roles in carcinogenicity. In: Dansette, et al., editors. Biological Reactive Intermediates VI. Kluwer Academic/Plenum Publishers; 2001. pp. 93–103. [DOI] [PubMed] [Google Scholar]
- 12.Csanády GA, Guengerich FP, Bond JA. Comparison of the biotransformation of 1,3-butadiene and its metabolite, butadiene monoepoxide, by hepatic and pulmonary tissues from humans, rats and mice. Carcinogenesis. 1992;13:1143–1153. doi: 10.1093/carcin/13.7.1143. [DOI] [PubMed] [Google Scholar]
- 13.Duescher RJ, Elfarra AA. 1,3-Butadiene oxidation by human myeloperoxidase. Role of chloride ion in catalysis of divergent pathways. J. Biol. Chem. 1992;267:19859–19865. [PubMed] [Google Scholar]
- 14.Duescher RJ, Elfarra AA. Human liver-microsomes are efficient catalysts of 1,3-butadiene oxidation. Evidence for major roles by cytochromes P450 2A6 and 2E1. Arch. Biochem. Biophys. 1994;311:342–349. doi: 10.1006/abbi.1994.1246. [DOI] [PubMed] [Google Scholar]
- 15.Krause RJ, Elfarra AA. Oxidation of butadiene monoxide to meso- and (±)-diepoxybutane by cDNA-expressed human cytochrome P450s and by mouse, rat, and human liver microsomes: evidence for preferential hydration of meso-diepoxybutane in rat and human microsomes. Arch. Biochem. Biophys. 1997;337:176–184. doi: 10.1006/abbi.1996.9781. [DOI] [PubMed] [Google Scholar]
- 16.Henderson RF, Thornton-Manning JR, Bechtold WE, Dahl AR. Metabolism of 1,3-butadiene: species differences. Toxicology. 1996;113:17–22. doi: 10.1016/0300-483x(96)03422-1. [DOI] [PubMed] [Google Scholar]
- 17.Nieusma JL, Claffey DJ, Maniglier-Poulet C, Imiolczyk T, Ross D, Ruth JA. Stereochemical aspects of 1,3-butadiene metabolism and toxicity in rat and mouse liver microsomes and freshly isolated rat hepatocytes. Chem. Res. Toxicol. 1997;10:450–456. doi: 10.1021/tx960199m. [DOI] [PubMed] [Google Scholar]
- 18.Malvoisin E, Roberfroid M. Hepatic microsomal metabolism of 1,3-butadiene. Xenobiotica. 1982;12:137–144. doi: 10.3109/00498258209046787. [DOI] [PubMed] [Google Scholar]
- 19.Elfarra AA, Krause RJ, Selzer RR. Biochemistry of 1,3-butadiene metabolism and its relevance to 1,3-butadiene-induced carcinogenicity. Toxicology. 1996;113:23–30. doi: 10.1016/0300-483x(96)03423-3. [DOI] [PubMed] [Google Scholar]
- 20.Gervasi PG, Citti L, Del Monte M, Longo V, Benetti D. Mutagenicity and chemical reactivity of epoxidic intermediates of the isoprene metabolism and other structurally related compounds. Mutat. Res. 1985;156:77–82. doi: 10.1016/0165-1218(85)90009-6. [DOI] [PubMed] [Google Scholar]
- 21.Tice RR, Boucher R, Luke CA, Shelby MD. Comparative cytogenetic analysis of bone marrow damage induced in male B6C3F1 mice by multiple exposures to gaseous 1,3-butadiene. Environ. Mutagen. 1987;9:235–250. doi: 10.1002/em.2860090303. [DOI] [PubMed] [Google Scholar]
- 22.Cochrane JE, Skopek TR. Mutagenicity of butadiene and its epoxide metabolites: I. Mutagenic potential of 1,2-epoxybutene, 1,2,3,4-diepoxybutane and 3,4-epoxy-1,2-butanediol in cultured human lymphoblasts. Carcinogenesis. 1994;15:713–717. doi: 10.1093/carcin/15.4.713. [DOI] [PubMed] [Google Scholar]
- 23.Cochrane JE, Skopek TR. Mutagenicity of butadiene and its epoxide metabolites: II. Mutational spectra of butadiene, 1,2-epoxybutene and diepoxybutane at the hprt locus in splenic T cells from exposed B6C3F1 mice. Carcinogenesis. 1994;15:719–723. doi: 10.1093/carcin/15.4.719. [DOI] [PubMed] [Google Scholar]
- 24.Sasiadek M, Norppa H, Forsa M. 1,3-Butadiene and its epoxides induce sister-chromatid exchanges in human lymphocytes in vitro. Mutat. Res. 1991;261:117–121. doi: 10.1016/0165-1218(91)90057-s. [DOI] [PubMed] [Google Scholar]
- 25.Recio L, Steen AM, Pluta LJ, Meyer KG, Saranko CJ. Mutational spectrum of 1,3-butadiene and metabolites 1,2-epoxybutene and 1,2,3,4-diepoxybutane to assess mutagenic mechanisms. Chem.-Biol. Interact. 2001;135-136:325–341. doi: 10.1016/s0009-2797(01)00220-4. [DOI] [PubMed] [Google Scholar]
- 26.Meng Q, Singh N, Heflich RH, Bauer MJ, Walker VE. Comparison of the mutations at Hprt exon 3 of T-lymphocytes from B6C3F1 mice and F344 rats exposed by inhalation to 1,3-butadiene or the racemic mixture of 1,2:3,4-diepoxybutane. Mutat. Res. 2000;464:169–184. doi: 10.1016/s1383-5718(99)00157-6. [DOI] [PubMed] [Google Scholar]
- 27.Lawley PD, Brookes P. Molecular mechanism of the cytotoxic action of difunctional alkylating agents and resistance to this action. Nature. 1965;206:480–483. doi: 10.1038/206480a0. [DOI] [PubMed] [Google Scholar]
- 28.Lawley PD, Brookes P. Interstrand cross-linking of DNA by difunctional alkylating agents. J. Mol. Biol. 1967;25:143–160. doi: 10.1016/0022-2836(67)90285-9. [DOI] [PubMed] [Google Scholar]
- 29.Verly WG, Brakier L. The lethal action of monofunctional and bifunctional alkylating agents on T7 coliphage. Biochim. Biophys. Acta. 1969;174:674–685. doi: 10.1016/0005-2787(69)90296-2. [DOI] [PubMed] [Google Scholar]
- 30.Verly WG, Brakier L, Feit PW. Inactivation of the T7 coliphage by the diepoxybutane stereoisomers. Biochim. Biophys. Acta. 1971;228:400–406. doi: 10.1016/0005-2787(71)90046-3. [DOI] [PubMed] [Google Scholar]
- 31.Jelitto B, Vangala RR, Laib RJ. Species differences in DNA damage by butadiene: role of diepoxybutane. Arch. Toxicol. 1989;13(suppl):246–249. doi: 10.1007/978-3-642-74117-3_42. [DOI] [PubMed] [Google Scholar]
- 32.Vangala RR, Laib RJ, Bolt HM. Evaluation of DNA damage by alkaline elution technique after inhalation exposure of rats and mice to 1,3-butadiene. Arch. Toxicol. 1993;67:34–38. doi: 10.1007/BF02072032. [DOI] [PubMed] [Google Scholar]
- 33.Millard JT, White MM. Diepoxybutane cross-links DNA at 5′-GNC sequence. Biochemistry. 1993;32:2120–2124. doi: 10.1021/bi00059a034. [DOI] [PubMed] [Google Scholar]
- 34.Park S, Anderson C, Loeber R, Seetharaman M, Jones R, Tretyakova N. Interstrand and intrastrand DNA–DNA cross-linking by 1,2,3,4-diepoxybutane: Role of stereochemistry. J. Am. Chem. Soc. 2005;127:14355–14365. doi: 10.1021/ja051979x. [DOI] [PubMed] [Google Scholar]
- 35.Carmical JR, Kowalczyk A, Zou Y, Van Houten B, Nechev LV, Harris CM, Harris TM, Lloyd RS. Butadiene-induced intrastrand DNA cross-links: a possible role in deletion mutagenesis. J. Biol. Chem. 2000;275:19482–19489. doi: 10.1074/jbc.M002037200. [DOI] [PubMed] [Google Scholar]
- 36.Brookes P, Lawley PD. The alkylation of guanosine and guanylic acid. J. Chem. Soc. 1961:3923–3928. [Google Scholar]
- 37.Park S, Tretyakova N. Structural characterization of the major DNA-DNA cross-link of 1,2,3,4-diepoxybutane. Chem. Res. Toxicol. 2004;17:129–136. doi: 10.1021/tx0342058. [DOI] [PubMed] [Google Scholar]
- 38.Park S, Hodge J, Anderson C, Tretyakova N. Guanine-adenine DNA cross-linking by 1,2,3,4-diepoxybutane: potential basis for biological activity. Chem. Res. Toxicol. 2004;17:1638–1651. doi: 10.1021/tx0498206. [DOI] [PubMed] [Google Scholar]
- 39.Zhang X-Y, Elfarra AA. Identification and characterization of a series of nucleoside adducts formed by the reaction of 2′-deoxyguanosine and 1,2,3,4-diepoxybutane under physiological conditions. Chem. Res. Toxicol. 2003;16:1606–1615. doi: 10.1021/tx0341355. [DOI] [PubMed] [Google Scholar]
- 40.Zhang X-Y, Elfarra AA. Reaction of 1,2,3,4-diepoxybutane with 2′-deoxyguanosine: Initial products and their stabilities and decomposition patterns under physiological conditions. Chem. Res. Toxicol. 2005;18:1316–1323. doi: 10.1021/tx0500979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Selzer RR, Elfarra AA. Synthesis and biochemical characterization of N1-, N2-, and N7-guanosine adducts of butadiene monoxide. Chem. Res. Toxicol. 1996;9:126–132. doi: 10.1021/tx950101o. [DOI] [PubMed] [Google Scholar]
- 42.Aldrich Chemical Co., Inc. In: The Aldrich Library of 13C and 1H FT NMR Spectra. 1st ed. Pouchert CJ, Behnke J, editors. Vol. 3. Aldrich Chemical Co., Inc.; Milwaukee, WI: 1993. p. 223A. [Google Scholar]
- 43.Tomasz M. Extreme lability of the C-8 proton: a consequence of 7-methylation of guanine residues in model compounds and in DNA and its analytical application. Biochim. Biophys. Acta. 1970;199:18–28. doi: 10.1016/0005-2787(70)90690-8. [DOI] [PubMed] [Google Scholar]