

Abstract
Multidisciplinary research from our laboratory shows that disruption at various levels of leptin signaling to the interactive hypothalamic network of NPY and cohorts contributes to the antecedent pathophysiological sequalae of the disease cluster of metabolic syndrome. Disruptions in NPY signaling due to both high or low abundance of NPY and cognate receptors dysregulate the homeostatic milieu to promote hyperinsulinemia, hyperglycemia, fat accrual and overt diabetes. Hyperleptinemia induced by consumption of energy-enriched diets inhibits leptin transport across the blood brain barrier and thereby produces leptin insufficiency in the hypothalamus. Sustained leptin insufficiency results in loss of hypothalamic restraint on pancreatic insulin secretion, diminished glucose metabolism and energy expenditure. This chain of events culminates in hyperinsulinemia, hyperglycemia and diabetes. Our recent studies show that increasing the supply of leptin centrally by gene therapy reinstated the restraint on hypothalamic NPY signaling, ameliorated diabetes and the attendant disease cluster of metabolic syndrome. Thus, newer therapies that would enhance leptin transport across blood brain barrier in a timely manner or reinstate leptin restraint on NPY signaling through either central leptin gene therapy, or pharmacologically with leptin mimetics, are likely to curtail the pathophysiological sequalae of diabetes and related ailments of metabolic syndrome.
Keywords: leptin insufficiency, metabolic syndrome, diabetes, inflammation, hyperinsulinemia, hypothalamic control, neuropeptide Y
Introduction
Convergence of clinical, epidemiological and experimental evidence over the past decade has affirmed a worldwide pandemic of chronic metabolic diseases [1–3]. It is projected that if the upward trend in the incidence of type 2 diabetes and the dependent disease cluster of metabolic syndrome - dyslipidemia, low grade systemic inflammation, atherosclerosis, cardiovascular diseases (CVD), hypertension and certain cancers - is not curtailed, it would adversely impact lifespan [1–3]. Lifestyle modifications in energy intake and expenditure facilitated by recent technological advances disrupt the homeostatic minute-to-minute crosstalk between the afferent hormonal signals from the periphery and the hypothalamic network of neuropeptide Y (NPY) and cohorts, the obligatory central pathway mediating metabolic homeostasis [4–6]. Derangements in the dynamic feedback of fat-derived leptin and the central effector pathways progressively promote fat accrual and obesity. These sequalae, in turn, increase the risks of the disease cluster of metabolic syndrome [2,3,6]. In this context, there is a general consensus that type 2 diabetes and low grade systemic inflammation, the major risk factors for CVD, are etiologically linked in the periphery with abdominal obesity and altered secretion of adipokines [1,5,6–8].However, it has also been reported that a large percentage of diabetics are not obese and, likewise, not all obese patients exhibit type 2 diabetes [1,6–8]. In addition, evidence from studies involving ablation of discrete hypothalamic sites and neural transections to interrupt the relay of efferent neural outflow from the hypothalamus, suggested a centrally mediated link between hyperphagia - obesity and hyperinsulinemia - diabetes [4,5,9].
These observations, however, could not rule out the possibility of a crucial control by the hypothalamus in monitoring insulin-glucose homeostasis, independent of the influence of increased fat accrual. Recent experimental evidence from our laboratory has identified a common causality, the central leptin insufficiency syndrome, independently orchestrating hyperphagia, fat accrual, derangements in glucose-insulin homeostasis that culminate in type 2 diabetes, and low grade systemic inflammation [3,6,10]. This new insight has uncovered new therapeutic avenues for exploration to independently either decelerate the rate of fat accrual, or curtail the development of pathophysiological sequences that confer type 2 diabetes and low grade systemic inflammatory disease.
This article briefly summarizes the dynamic relationship between the adipocyte leptin and the hypothalamic network of NPY and cohorts in regulation of (i) pancreatic insulin secretion and insulin resistance, (ii) glucose metabolism, glucose intolerance and hyperglycemia, and (iii) systemic low grade inflammation.
(1) HYPOTHALAMIC NETWORK OF NEUROPEPTIDE Y (NPY) AND COHORTS
Since the first demonstration of the potent appetite stimulating effects of NPY in 1984 [11], a wealth of evidence documents that NPY is, indeed, a physiological central appetite transducing signal molecule produced by two clusters of neurons, one located in the arcuate nucleus (ARC) of the hypothalamus that co-express orexigenic agouti-related peptide (AgrP), and gamma-aminobutyric acid (GABA) and the other in the brainstem that co-express adrenergic neurotransmitter [Fig.1; 4,5,11]. The timely release of NPY and co-expressed transmitters in the paraventricular nucleus (PVN) of the hypothalamus evokes appetite [4,5,12]. In addition, the release of NPY and cohorts in the ARC-PVN axis restrain anorexigenic melanocortin signaling. Thus, a two-prong action of NPY, stimulation of appetite mediated by Y1 and Y5 receptors in the PVN, and repression of the inhibitory melanocortin signaling mediated by Y1 receptors in the ARC pro-opiomelanocortin (POMC) and cocaine and amphetamine-regulated transcript (CART) co-expressing neurons, coordinates the daily feeding pattern. The precise operation of the hypothalamic NPY network in propagation and termination of appetite has been reviewed extensively [Fig.1; 4,5,13]. An unanticipated revelation from the results of various experiments was that both upregulation and downregulation of NPY abundance or NPYergic signaling in the ARC-PVN axis, evoked relentless hyperphagia, and abnormal rate of weight gain to culminate in overt obesity, consistently accompanied by hyperinsulinemia [5,9]. Seemingly, incessant NPY Y1 and Y5 receptor activation in the ARC-PVN axis induced by either overabundance of NPY or development of compensatory NPY receptor supersensitivity in response to low abundance of NPY, promoted unremitting positive energy intake, fat accretion and hyperinsulinemia [5,9].
Fig. 1.
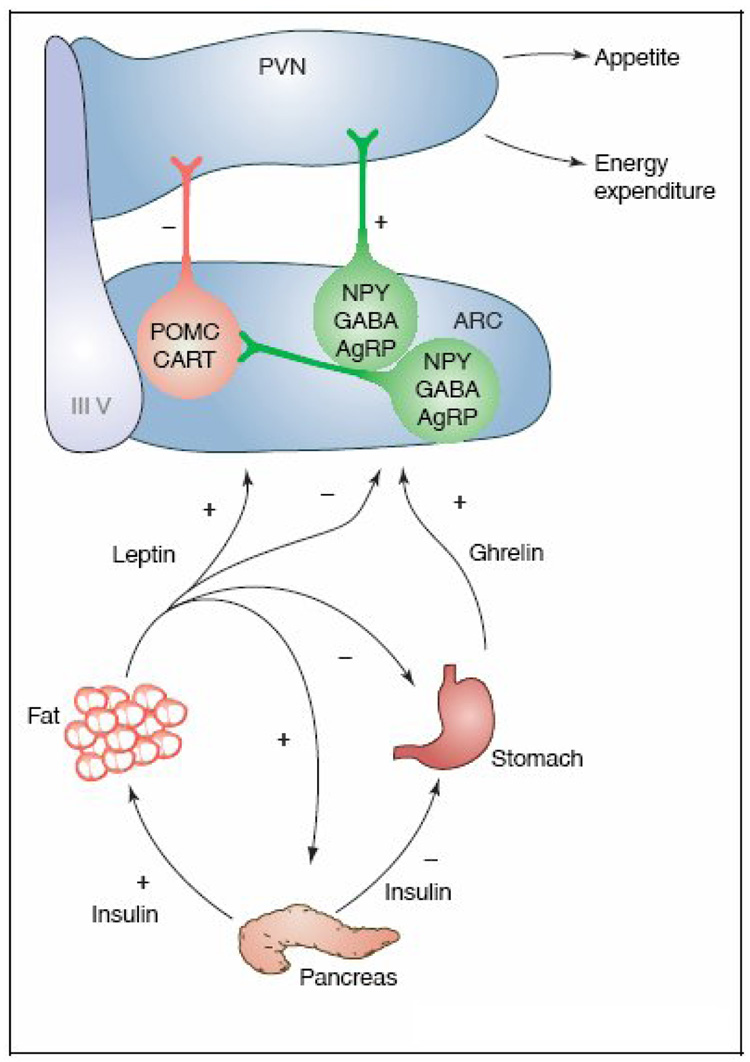
Depicts the dynamic crosstalk between hypothalamic network of NPY and cohorts in the ARC-PVN axis and afferent hormonal signals, adipocyte leptin and gastric ghrelin. A feedback link in the periphery, the adipoinsular axis is also shown. Leptin regulates appetite and energy expenditure by a control on the NPY signaling in the hypothalamus, repressing the orexigenic effects of ghrelin centrally and gastric ghrelin secretion in the periphery. For detail, see text. (With permission from Trends in Pharmacological Sciences 26:488–495; 2005).
PVN = paraventricular nucleus; ARC = arcuate nucleus; NPY = neuropeptide Y; GABA = gamma aminobutyric acid; AgrP = agouti-related peptide; POMC = pro-opiomelanocortin; CART = cocaine and amphetamine regulating transcript; (+) = stimulatory; (−) = inhibitory; IIIV - third ventricle.
(2) LEPTIN: A PRIMARY AFFERENT SIGNAL REGULATING NPY SIGNALING IN THE ARC-PVN AXIS
A host of afferent hormonal signals of diverse chemical composition secreted by white adipose tissue (WAT), pancreas and gastrointestinal tract inhibit energy intake by repressing NPY signaling in the ARC-PVN axis [Fig.1; 4,5,14]. In marked contrast, gastric ghrelin stimulates appetite by upregulating hypothalamic NPY signaling [Fig.1; 4,15]. However, the inverse temporal relationship between the episodic circulating patterns of inhibitory leptin and excitatory ghrelin, and the existence of a similar inverse relationship between these two hormones in obese, hyperleptinmic rodents and human subjects, implied a new role of leptin in energy homeostasis [15–19]. We found that leptin inhibited ghrelin efflux from the stomach and reduced ghrelin-induced feeding mediated by the hypothalamic NPY network [16]. These newer findings strengthened our hypothesis that leptin is the primary afferent hormonal signal that plays a prominent role in maintaining energy homeostasis by a dual action [Fig.1]. Centrally, leptin directly restrains the release of NPY and cohorts from the hypothalamic NPY neuronal network and opposes the orexigenic action of ghrelin, both of these effects mediated by the long isoform of leptin receptor, OB-Rb. Peripherally, leptin represses the release of gastric ghrelin to secondarily reduce its central appetite stimulating effects [16].
In the complete absence of either leptin or hypothalamic leptin receptors, NPY signaling is upregulated to promote unabated hyperphagia and fat storage [3,5,20,21]. Under these very conditions, the disease cluster of metabolic syndrome, namely hyperinsulinemia, glucose intolerance, hyperglycemia and diabetes is the norm in rodents and humans [3,5,20,21]. Severe cardiovascular anomalies and early mortality have also been reported in leptin- deficient rodents [10,22]. Remarkably, markedly reduced leptin levels in the CNS and hypothalamic sites have also been observed in obese but not in lean aging rodents, in obese rodents consuming high fat diets (HFD), as well as in obese human subjects [3,6,23,24]. This central leptin insufficiency associated with obesity is also accompanied by a breakdown in downstream signaling in the network of NPY and cohorts, concomitant with hyperinsulinemia, insulin resistance, alterations in glucose metabolism in the brown adipose tissue (BAT), liver, and skeletal muscles, and reduced non-thermogenic energy expenditure [2,3,5,6,20,21]. Seemingly, besides an essential role in regulation of energy intake and expenditure, leptin-NPYergic signaling is engaged also in the central regulation of insulin secretion and glucose metabolism. Manifestation of similar metabolic responses in the Y1, Y2 and Y4 knockout mice lend credence to the notion that leptin plays a prominent part [25], not recognized previously [26], in the hypothalamic regulation of insulin secretion and glucose metabolism.
(3) CENTRAL LEPTIN INSUFFICIENCY SYNDROME AND PANDEMIC OF METABOLIC SYNDROME
Leptin, secreted by adipocytes and stomach in pulsatile fashion, is transported across the blood brain barrier (BBB) by an active process mediated by the short isoform of leptin receptor (OB-Ra) located in the endothelium of the choroid plexus and circumventricular organs [3,6,23,24]. Leptin concentrations in the cerebrospinal fluid (CSF) and parenchyma vary in accordance with the quantity and pattern of circulating leptin concentrations. A host of circulating metabolic factors, in addition, modify the dynamics of leptin entry across the BBB. A noteworthy observation relevant in the leptin-NPY crosstalk is that when leptin levels fall in the pre-prandial interval, and in calorie restricted and fasting rodents, brain leptin concentrations are reduced apparently to stimulate appetite. Also, the capacity of the BBB to transport leptin into the brain gradually diminishes with age, but more so in those aging rodents that exhibit increased adiposity and hyperinsulinemia [Fig.2; 6,23,24,27]. Even in young, severely hyperleptinemic, obesity-prone rodents and human subjects consuming energy-enriched diets, leptin levels in the CSF are reduced contemporaneously with hyperinsulinemia, glucose intolerance, elevated blood glucose concentrations and diminished non-shivering thermogenesis [6,23,24,28]. In general, leptin insufficiency in the brain produced by enlarging abdominal adiposity and hyperleptinemia is coincident with disruptions in glucose-insulin homeostasis, dyslipidemia, the chain of pathophysiological events if sustained for extended period, eventuate into type 2 diabetes and CVD [Fig.2; 6]. This tight etiological association between central leptin insufficiency and disease cluster of metabolic syndrome raised a paramount question. What is the precise role of leptin-NPY signaling in the regulation of pancreatic insulin secretion and glucose metabolism?
Fig. 2.
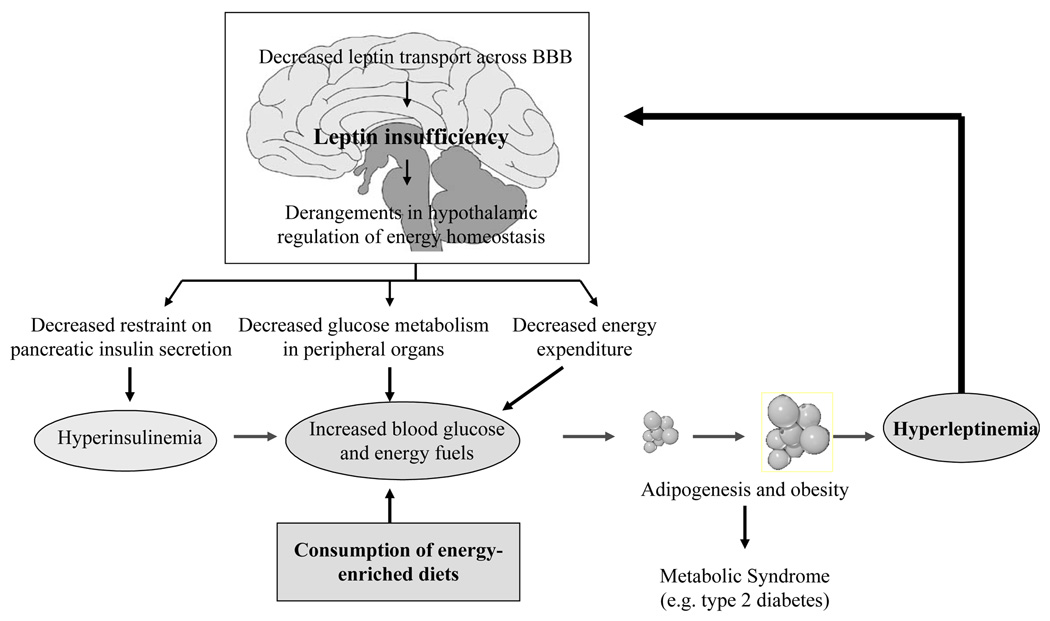
A flow chart of sequential metabolic and neural events initiated by consumption of energy-enriched diets leading to leptin insufficiency in the brain and derangements in the hypothalamic regulation of insulin secretion, glucose metabolism and energy expenditure that together promote fat accrual and metabolic disorders including type 2 diabetes.
With permission from (Peptides 29:127–138, 2008).
a. Hyperinsulinemia
Hyperleptinemia and hyperinsulinemia in association with enlarging abdominal adiposity, and the recently revealed coincidence of these metabolic hormones with attenuated brain leptin levels, as discussed above, when considered together with the evidence that ablation of discrete hypothalamic sites that decrease NPY signaling and abolish central leptin feedback, also elicit hyperinsulinemia and insulin resistance, unraveled a novel role of central leptin feedback [1,2,5,6,9,15]. We proposed that normally leptin exerts a restraint on episodic pancreatic secretion [Fig.2; 3,15,17,18,29–32]. The fact that selective activation of leptin receptors by leptin delivered directly into the hypothalamus, to circumvent impedance of leptin entry by BBB, readily suppressed insulin hypersecretion and abrogated insulin resistance, reinforces the notion that the hypothalamus regulates pancreatic insulin secretion independent of its role in governing energy intake [33,34].
Furthermore, we employed central leptin gene therapy technology to dissect out the precise role of leptin-NPY signaling in the control of episodic insulin secretion [2,3,17,29–32,35]. A single injection either intracerebroventricularly or into discrete hypothalamic sites of the replicative-deficient, non-pathogenic and non-immunogenic recombinant adeno-associated viral vector encoding leptin gene, readily transduced leptin in the hypothalamus. We observed that increased bioactive leptin transduced by leptin transgene expression in the hypothalamus, suppressed hypothalamic NPY gene expression and insulin efflux from the pancreas for extended periods [29,30,35–37]. Central leptin gene expression initiated at prepubertal and pubertal stages in rodents also abolished the gradual age-related rise in circulating insulin and maintained normoinsulinemia for the lifetime of rodents [35–37]. In fact, increased leptin transgene expression in the hypothalamus suppressed the amount of insulin discharged per episode, abolished the onset of high fat diet (HFD)-induced hyperinsulinemia that normally precedes fat accrual and attenuated the rapid post-prandial spurt in insulin secretion [17,18,20,31,38,39].
The strongest evidence in support of our proposal that central leptin normally restrains pancreatic insulin secretion, was obtained in leptin mutant ob/ob mice [3,10,22,28,31,38]. In these hyperinsulinemic mice, consuming either normal chow or HFD, leptin availability selectively in the hypothalamus, without leakage to the periphery , maintained normo-insulinemia for the life time. Remarkably, these mice exhibited a loss of insulin resistance and their lifespan doubled as compared to mutant mice without experimentally-induced central leptin restraint on pancreatic insulin secretion [22]. Thus, prevention of central leptin insufficiency by providing leptin in optimal amounts with the aid of gene therapy can restrain hyperinsulinemia and impose normoinsulinemia presumably by activating descending hypothalamic pathways that traverse caudally through the brainstem to innervate pancreatic β-cells [Fig.3, 3,5,9,13,17,40]. Consequently, optimal leptin-NPY signaling is a physiological regulatory mechanism that restrains insulin secretion and prevents the occurrence of hyperinsulinemia, insulin resistance and loss of pancreatic β-cells, normally encountered in severely hyperleptinemic obese subjects exhibiting central leptin insufficiency [Fig.2].
Fig. 3.
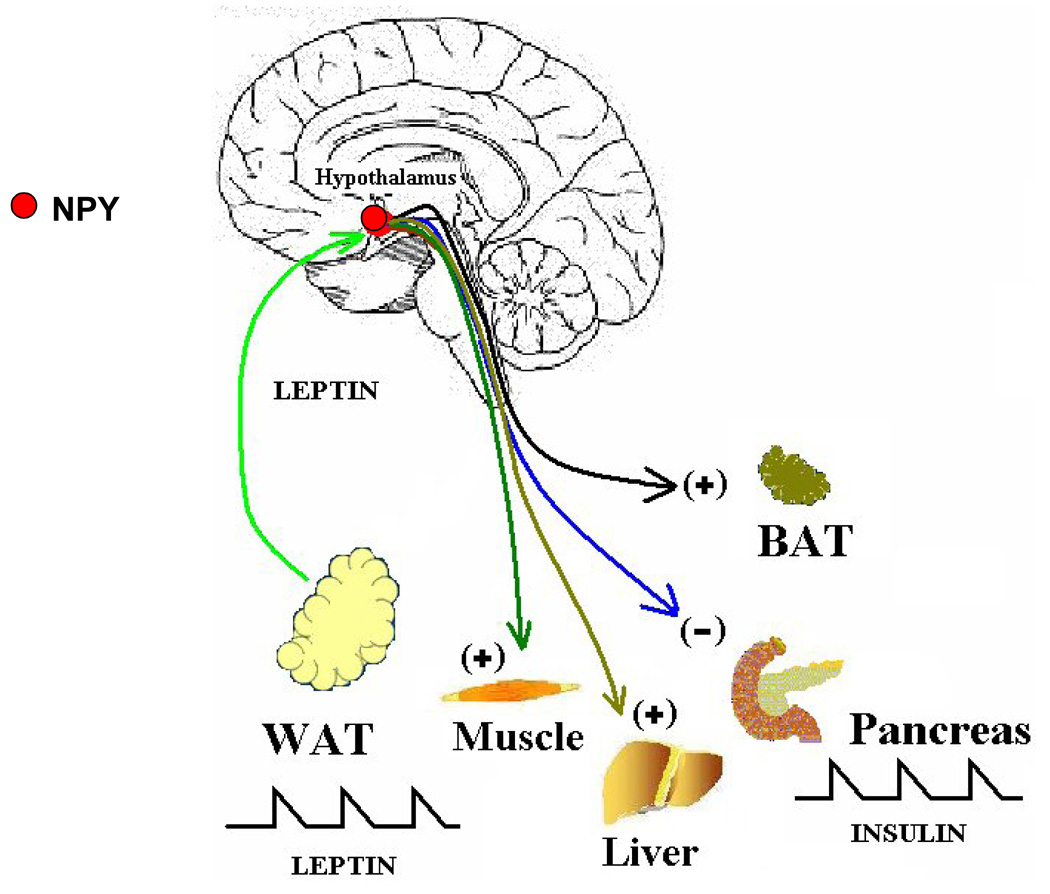
Depicts the independent course traversed by leptin-responsive pathways emanating from hypothalamus and descending caudally through brainstem to innervate WAT (white adipose tissue), skeletal muscle, liver, pancreas and BAT (brown adipose tissue) in the periphery. Information relay along these tracts regulate episodic adipocyte leptin and pancreatic insulin secretion and glucose metabolism in skeletal muscle, BAT and liver is detailed in the text.
b. Hyperglycemia and glucose metabolism
It is generally held that the age-related or HFD-induced progressive antecedent hyperinsulinemia, decrease in insulin sensitivity and downregulation of insulin receptors coalesce into insulin resistance [1,26]. This disruption in insulin signaling in the peripheral targets gradually impels glucose intolerance, hyperglycemia and type 2 diabetes. However, contrary to this established mode for the genesis of type 2 diabetes, evidence also shows that direct activation of hypothalamic leptin receptor by intraventricular injections of leptin lowers blood glucose levels and glucose intolerance [33,34].
Additionally, we observed that enhanced leptin availability produced by hypothalamic expression of leptin transgene, enforced euglycemia through the extended duration of the experiments despite concurrent suppression of insulin secretion and adiposity [3,17,18,29–32,35–39]. This sustenance of euglycemia contemporaneous with insulinopenia implied a novel role of leptin in the hypothalamic regulation of glucose metabolism in the periphery. Indeed, alleviation of central leptin insufficiency conferred euglycemia in rodents of all ages, as well as in extremely hyperglycemic HFD-consuming rodents and leptin-deficient ob/ob mice [3,17,18,29–32,35–39]. In aggregate, these findings uncovered the existence of an independent leptin responsive pathway that can impose euglycemia. Furthermore, similar perseverance of euglycemia was observed in streptozotocin-treated, insulin -deficient diabetic mice and insulinopenic, non-obese diabetic Akita mice [Kojima,S., Amitani,H., Asakawa,M., Inui,A., and Kalra,S. (Unpublished), 38]. Thus, neural signaling propagated by leptin from the hypothalamus can impose euglycemia for the lifetime of rodents, independent of the varied macronutrient composition of the diet, the existing level of adiposity and insulin concentrations. Evidently, euglycemia in rodents receiving central leptin gene therapy results from sustained activation of those leptin responsive hypothalamic pathways that accelerate glucose metabolism in BAT, liver and skeletal muscles, independent of the circulating leptin and insulin levels [Fig.3; 2,3,33,34,40].
Anatomical evidence and results of neural transection to interrupt information outflow also suggested that efferent signals generated by leptin-responsive targets in the ventromedial hypothalamus, ARC-PVN axis and medial preoptic area descend caudally to the BAT, liver and skeletal muscles to upregulate glucose metabolism [Fig.3, 4,5,9,10,13,33,34,38,40]. The paramount question of how optimal leptin signaling transduced by leptin transgene expression selectively in the hypothalamus optimizes the rate of glucose metabolism at cellular and molecular levels to confer long-lasting euglycemia remain to be elucidated.
c. Chronic low grade systemic inflammation and cardiovascular diseases
A large body of clinical evidence suggests a strong causal relationship between obesity and chronic low grade systemic inflammation which itself is considered an underlying causal factor in the development of diabetes and CVD [1,7,8]. A rise in proinflammatory cytokines, especially interleukin-6 (IL-6) and acute phase protein, C-reactive protein (CRP), are reliable biomarkers of systemic low grade inflammation and predictor of CVD [7,8].
That dysregulation of leptin-NPY signaling resulting from central leptin insufficiency may play a role in inducing chronic low grade inflammation, was suggested by our recent findings [10]. Increased leptin supply selectively in the hypothalamus with the aid of gene therapy, suppressed the elevated plasma levels of CRP and IL-6 in obese and diabetic ob/ob mice [10]. This suppression of the two biomarkers of CVD prevailed, even if the mice were maintained on either regular chow or HFD [10]. Consequently, it is obvious that leptin insufficiency in the hypothalamus raises the circulating levels of proinflammatory biomarkers and alleviation of this central deficiency has the potential to prevent CVD that invariably follows the low grade systemic inflammation and obesity. Indeed, we recently reported that these benefits may contribute to the recently reported life-prolonging impact of reinstatement of central leptin sufficiency in ob/ob obese mice [22].
CONCLUSION
New insight into the varied central feedback effects of leptin mediated by the hypothalamic network of NPY and cohorts has emerged. This includes a powerful restraint on pancreatic insulin secretion and maintenance of euglycemia by upregulation of glucose metabolism in the periphery, independent of the status of circulating insulin. A new road map can now be drawn that delineates the sequential etiological chain of events in the genesis of the coexistence of environmentally -induced obesity and the disease cluster of metabolic syndrome. Increased release of insulin in response to rises in blood glucose and other energy fuels derived from consumption of energy-rich diets, promote adipogenesis and conversion of excess energy into fat in adipocytes [Fig.4]. In the periphery, the sustained hyperleptinemia inflicts leptin insensitivity by downregulating OB-Rb receptors to, thereby, curb the restraint on insulin secretion. Also, centrally this increased energy intake and the resultant enhanced rate of fat accrual and severe hyperleptinemia over extended intervals, in turn, engender leptin insufficiency in the leptin-responsive target pathways in the hypothalamus. Hypothalamic leptin insufficiency curtails the restraint on insulin secretion that promotes hyperinsulinemia, and simultaneously attenuates peripheral glucose metabolism and energy expenditure and, thereby, raise blood glucose levels [Fig.2 & 4]. Hyperinsulinemia and increased circulating fuels together augment fat accretion and morbid obesity. The adverse outcome of these metabolic derangements , such as insulin receptor downregulation, insulin resistance and progressive loss of pancreatic β-cells, in concert instill hyperglycemia, glucose intolerance eventuating into type 2 diabetes, CVD and other co-morbidities of metabolic syndrome.
Fig. 4.

Peripheral and central sequence of events mediating the obesity-induced type 2 diabetes. For details, see text.
Consequently, to prevent the onset of disease cluster of metabolic syndrome, it is necessary that the NPY-leptin link operates at the optimal range. This new insight into the neurobiology of leptin is endorsed by a series of findings documenting that sustenance of leptin sufficiency experimentally in the hypothalamus blocks the age-related and HFD-induced obesity and co-morbidities of metabolic syndrome and prevents early mortality. Thus, as proposed earlier [2,3,5,6,13,35], newer therapies that augment leptin transport across the blood brain barrier in a timely manner or reinstate leptin restraint on NPY signaling through either central leptin gene therapy or pharmacologically with leptin mimetics, are likely to curtail pathophysiological sequalae of diabetes and related ailments of metabolic syndrome.
Acknowledgements
The research embodied in this paper was supported by grants from National Institute of Health (DK 37273 and NS 32727). Secretarial assistance of Mr. Nicholas Cross is acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Grundy SM. Obesity, metabolic syndrome, and cardiovascular disease. J Clin Endocrinol Metab. 2004;89:2595–2600. doi: 10.1210/jc.2004-0372. [DOI] [PubMed] [Google Scholar]
- 2.Kalra PS, Kalra SP. Obesity and Metabolic Syndrome: Long-term benefits of central leptin gene therapy. In: Prous JR, editor. Drugs of Today. Barcelona, Spain: Prous Science; 2002. pp. 745–757. [DOI] [PubMed] [Google Scholar]
- 3.Kalra SP, Kalra PS. Gene transfer technology: a preventive neurotherapy to curb obesity, ameliorate metabolic syndrome and extend life-expectancy. Trends Pharmacol Sci. 2005;26:488–495. doi: 10.1016/j.tips.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 4.Kalra SP, Dube MG, Pu S, Xu B, Horvath TL, Kalra PS. Interacting appetite-regulating pathways in the hypothalamic regulation of body weight. Endocr Rev. 1999;20:68–100. doi: 10.1210/edrv.20.1.0357. [DOI] [PubMed] [Google Scholar]
- 5.Kalra SP, Kalra PS. NPY and Cohorts in Regulating Appetite, Obesity and Metabolic Syndrome: Beneficial Effects of Gene Therapy. Neuropeptides. 2004;38:201–211. doi: 10.1016/j.npep.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 6.Kalra SP. Central leptin insufficiency syndrome: An interactive etiology for obesity, metabolic and neural diseases and for designing new therapeutic interventions. Peptides. 2008;29:127–138. doi: 10.1016/j.peptides.2007.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carr MC, Brunzell JD. Abdominal obesity and dyslipidemia in the metabolic syndrome: importance of type 2 diabetes and familial combined hyperlipidemia in coronary artery disease risk. J Clin Endocrinol Metab. 2004;89:2601–2607. doi: 10.1210/jc.2004-0432. [DOI] [PubMed] [Google Scholar]
- 8.Pearson TA, Mensah GA, Alexander RW, Anderson JL, Cannon RO, 3rd, Criqui M, et al. Markers of inflammation and cardiovascular disease: application to clinical and public health practice: A statement for healthcare professionals from the Centers for Disease Control and Prevention and the American Heart Association. Circulation. 2003;107:499–511. doi: 10.1161/01.cir.0000052939.59093.45. [DOI] [PubMed] [Google Scholar]
- 9.Dube MG, Kalra SP, Kalra PS. Low abundance of NPY in the hypothalamus can produce hyperphagia and obesity. Peptides. 2007;28:475–479. doi: 10.1016/j.peptides.2006.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dube MG, Torto R, Kalra SP. Increased leptin expression selectively in the hypothalamus suppresses inflammatory markers CRP and IL-6 in leptin-deficient diabetic obese mice. Peptides. 2008;29:593–598. doi: 10.1016/j.peptides.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clark JT, Kalra PS, Crowley WR, Kalra SP. Neuropeptide Y and human pancreatic polypeptide stimulate feeding behavior in rats. Endocrinology. 1984;115:427–429. doi: 10.1210/endo-115-1-427. [DOI] [PubMed] [Google Scholar]
- 12.Kalra SP, Dube MG, Sahu A, Phelps CP, Kalra PS. Neuropeptide Y secretion increases in the paraventricular nucleus in association with increased appetite for food. Proc Natl Acad Sci U S A. 1991;88:10931–10935. doi: 10.1073/pnas.88.23.10931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kalra SP, Kalra PS. To subjugate NPY is to improve the quality of life and live longer. Peptides. 2007;28:413–418. doi: 10.1016/j.peptides.2006.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wynne K, Stanley S, McGowan B, Bloom S. Appetite control. J Endocrinol. 2005;184:291–318. doi: 10.1677/joe.1.05866. [DOI] [PubMed] [Google Scholar]
- 15.Kalra SP, Bagnasco M, Otukonyong EE, Dube MG, Kalra PS. Rhythmic, reciprocal ghrelin and leptin signaling: new insight in the development of obesity. Regul Pept. 2003;111:1–11. doi: 10.1016/s0167-0115(02)00305-1. [DOI] [PubMed] [Google Scholar]
- 16.Kalra SP, Ueno N, Kalra PS. Stimulation of appetite by ghrelin is regulated by leptin restraint: peripheral and central sites of action. J Nutr. 2005;135:1331–1335. doi: 10.1093/jn/135.5.1331. [DOI] [PubMed] [Google Scholar]
- 17.Otukonyong EE, Dube MG, Torto R, Kalra PS, Kalra SP. Central Leptin Differentially Modulates Ultradian Secretory Patterns of Insulin, Leptin and Ghrelin Independent of Effects on Food Intake and Body Weight. Peptides. 2005;26:2559–2566. doi: 10.1016/j.peptides.2005.04.015. [DOI] [PubMed] [Google Scholar]
- 18.Otukonyong EE, Dube MG, Torto R, Kalra PS, Kalra SP. High fat diet-induced ultradian leptin and insulin hypersecretion and ghrelin is absent in obesity-resistant rats. Obesity Research. 2005;13:991–999. doi: 10.1038/oby.2005.116. [DOI] [PubMed] [Google Scholar]
- 19.Tschop M, Weyer C, Tataranni PA, Devanarayan V, Ravussin E, Heiman ML. Circulating ghrelin levels are decreased in human obesity. Diabetes. 2001;50:707–709. doi: 10.2337/diabetes.50.4.707. [DOI] [PubMed] [Google Scholar]
- 20.O'Rahilly S, Farooqi IS, Yeo GS, Challis BG. Minireview: human obesity-lessons from monogenic disorders. Endocrinology. 2003;144:3757–3764. doi: 10.1210/en.2003-0373. [DOI] [PubMed] [Google Scholar]
- 21.Cohen P, Zhao C, Cai X, Montez JM, Rohani SC, Feinstein P, Mombaerts P, Friedman JM. Selective deletion of leptin receptor in neurons leads to obesity. J Clin Invest. 2001;108:1113–1121. doi: 10.1172/JCI13914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boghossian S, Ueno N, Dube MG, Kalra P, Kalra S. Leptin gene transfer in the hypothalamus enhances longevity in adult monogenic mutant mice in the absence of circulating leptin. Neurobiol Aging. 2007;28:1594–1604. doi: 10.1016/j.neurobiolaging.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 23.Banks WA, Farr SA, Morley JE. The effects of high fat diets on the blood-brain barrier transport of leptin: failure or adaptation? Physiol Behav. 2006;88:244–248. doi: 10.1016/j.physbeh.2006.05.037. [DOI] [PubMed] [Google Scholar]
- 24.Kastin AJ, Pan W, Maness LM, Koletsky RJ, Ernsberger P. Decreased transport of leptin across the blood-brain barrier in rats lacking the short form of the leptin receptor. Peptides. 1999;20:1449–1453. doi: 10.1016/s0196-9781(99)00156-4. [DOI] [PubMed] [Google Scholar]
- 25.Lin EJ, Sainsbury A, Lee NJ, Boey D, Couzens M, Enriquez R, Slack K, Bland R, During MJ, Herzog H. Combined deletion of Y1, Y2, and Y4 receptors prevents hypothalamic neuropeptide Y overexpression-induced hyperinsulinemia despite persistence of hyperphagia and obesity. Endocrinology. 2006;147:5094–5101. doi: 10.1210/en.2006-0097. [DOI] [PubMed] [Google Scholar]
- 26.Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006;444:840–846. doi: 10.1038/nature05482. [DOI] [PubMed] [Google Scholar]
- 27.Caro JF, Kolaczynski JW, Nyce MR, Ohannesian JP, Opentanova I, Goldman WH, et al. Decreased cerebrospinal-fluid/serum leptin ratio in obesity: a possible mechanism for leptin resistance. Lancet. 1996;348:159–161. doi: 10.1016/s0140-6736(96)03173-x. [DOI] [PubMed] [Google Scholar]
- 28.Yokosuka M, Xu B, Pu S, Kalra PS, Kalra SP. Neural substrates for leptin and neuropeptide Y (NPY) interaction: hypothalamic sites associated with inhibition of NPY-induced food intake. Physiol Behav. 1998;64:331–338. doi: 10.1016/s0031-9384(98)00065-1. [DOI] [PubMed] [Google Scholar]
- 29.Dhillon H, Kalra SP, Prima V, Zolotukhin S, Scarpace PJ, Moldawer LL, Muzyczka N, Kalra PS. Central Leptin Gene Therapy Suppresses Body Weight Gain, Adiposity and Serum Insulin Without Affecting Food Consumption in Normal Rats: A Long-Term Study. Regul Pept. 2001;99:69–77. doi: 10.1016/s0167-0115(01)00237-3. [DOI] [PubMed] [Google Scholar]
- 30.Bagnasco M, Dube MG, Katz A, Kalra PS, Kalra SP. Leptin expression in hypothalamic PVN reverses dietary obesity and hyperinsulinemia but stimulates ghrelin. Obesity Research. 2003;11:1463–1470. doi: 10.1038/oby.2003.196. [DOI] [PubMed] [Google Scholar]
- 31.Boghossian S, Dube MG, Torto R, Kalra PS, Kalra SP. Hypothalamic clamp on insulin release by leptin-transgene expression. Peptides. 2006;27:3245–3254. doi: 10.1016/j.peptides.2006.07.022. [DOI] [PubMed] [Google Scholar]
- 32.Dube MG, Beretta E, Dhillon H, Ueno N, Kalra PS, Kalra SP. Central leptin gene therapy blocks high fat diet-induced weight gain, hyperleptinemia and hyperinsulinemia: effects on serum ghrelin levels. Diabetes. 2002;51:1729–1736. doi: 10.2337/diabetes.51.6.1729. [DOI] [PubMed] [Google Scholar]
- 33.Chinookoswong N, Wang JL, Shi ZQ. Leptin restores euglycemia and normalizes glucose turnover in insulin- deficient diabetes in the rat. Diabetes. 1999;48:1487–1492. doi: 10.2337/diabetes.48.7.1487. [DOI] [PubMed] [Google Scholar]
- 34.Miyanaga F, Ogawa Y, Ebihara K, Hidaka S, Tanaka T, Hayashi S, Masuzaki H, Nakao K. Leptin as an adjunct of insulin therapy in insulin-deficient diabetes. Diabetologia. 2003;46:1329–1337. doi: 10.1007/s00125-003-1193-6. [DOI] [PubMed] [Google Scholar]
- 35.Dhillon H, Ge Y, Minter RM, Prima V, Moldawer LL, Muzyczka N, Zolotukhin S, Kalra PS, Kalra SP. Long-term differential modulation of genes encoding orexigenic and anorexigenic peptides by leptin delivered by rAAV vector in ob/ob mice. Relationship with body weight change. Regul Pept. 2000;92:97–105. doi: 10.1016/s0167-0115(00)00155-5. [DOI] [PubMed] [Google Scholar]
- 36.Bagnasco M, Dube MG, Kalra PS, Kalra SP. Evidence for the existence of distinct central appetite and energy expenditure pathways and stimulation of ghrelin as revealed by hypothalamic site-specific leptin gene therapy. Endocrinology. 2002;143:4409–4421. doi: 10.1210/en.2002-220505. [DOI] [PubMed] [Google Scholar]
- 37.Beretta E, Dube MG, Kalra PS, Kalra SP. Long-term suppression of weight gain, adiposity, and serum insulin by central leptin gene therapy in prepubertal rats: Effects on serum ghrelin and appetite-regulating genes. Ped. Res. 2002;52:189–198. doi: 10.1203/00006450-200208000-00010. [DOI] [PubMed] [Google Scholar]
- 38.Ueno N, Inui A, Kalra SP, Kalra PS. Leptin transgene expression in the hypothalamus enforces euglycemia in diabetic, insulin-deficient nonobese Akita mice and leptin-deficient obese ob/ob mice. Peptides. 2006;27:2332–2342. doi: 10.1016/j.peptides.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 39.Boghossian S, Lecklin AH, Torto R, Kalra PS, Kalra SP. Suppression of fat deposition for the life time of rodents with gene therapy. Peptides. 2005;26:1512–1519. doi: 10.1016/j.peptides.2005.03.039. [DOI] [PubMed] [Google Scholar]
- 40.Kreier F, Kap YS, Mettenleiter TC, van Heijningen C, van der Vliet J, Kalsbeek A, Sauerwein HP, Fliers E, Romijn JA, Buijs RM. Tracing from fat tissue, liver, and pancreas: a neuroanatomical framework for the role of the brain in type 2 diabetes. Endocrinology. 2006;147:1140–1147. doi: 10.1210/en.2005-0667. [DOI] [PubMed] [Google Scholar]