

Abstract
Protection of salvinorin B as standard alkoxyalkyl ethers yielded highly potent κ opioid receptor agonists. Ethoxymethyl ether 6 is among the most potent and selective κ agonists reported to date. Fluoroethoxymethyl ether 11 is the first potent, selective fluorinated κ ligand, with potential use in MRI and PET studies. Further enlargement of the alkoxy group, alkylation of the acetal carbon, or heteroatom substitution all reduced activity. These protecting groups may prove useful in related work not only by enabling the use of harsher synthetic conditions, but potentially by optimizing the potency of the products.
Keywords: Salvinorin A, Kappa opioid receptor, Methoxymethyl ether, Protecting groups
1. Introduction
Salvinorin A (1) is a potent and selective naturally-occurring κ (kappa) opioid.1 As one of very few reported non-nitrogenous opioids,2 salvinorin A has created new opportunities for understanding the mechanisms of ligand binding at opioid receptors, which might facilitate drug discovery. One objective has been the development of selective antagonists or partial agonists at κ opioid receptors; such agents have potential utility in the treatment of depression or mania,3 debilitating conditions for which all current treatments have significant limitations (e.g., poor efficacy, delayed onset, marked side effects). Many derivatives of 1 have now been tested at opioid receptors.4 Binding affinity and potency are almost invariably reduced; very few derivatives exhibit potency comparable to 1. Most active derivatives, like the parent compound, are full agonists, but recently partial agonists and antagonists have been reported.4,5 This is potentially significant, since the few available selective κ antagonists exhibit extremely slow onset (~24 hr) and long duration of action (>3 weeks),6 which complicates their use in the study or treatment of psychiatric conditions such as depression.7 However, the modifications which appear to confer partial agonist and antagonist activities on salvinorin derivatives also dramatically reduce binding affinity and selectivity;4,5 methods to optimize these parameters would therefore be useful.
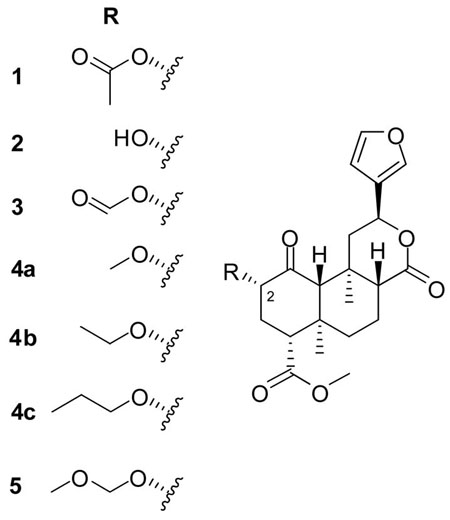
To date the most thoroughly studied functional group of 1 is the C-2 acetate. Interestingly, while deacetylation (giving 2) dramatically lowers affinity and potency,8 demethyl and deoxy analogues 39 and 4b8 each show only modestly reduced affinity. This suggests that these two portions of the acetate may be involved in separate, synergistic interactions with the receptor. Only one derivative with greater potency than 1 has been reported to date: methoxymethyl (MOM) ether 5.10 The increased affinity of 5 may be due to the additional sp3-hybridized oxygen, especially considering the lower affinity of its closest sp2-hybridized analogue, 3. Alternatively, the terminal methyl group of 5 might create an additional interaction, which could be explored by the substitution of other alkoxy groups. A related question is whether methylation of the acetal carbon would further increase potency, as with formate 3. In hopes of optimizing the C-2 substituent, we explored these questions using related protecting groups.
2. Chemistry
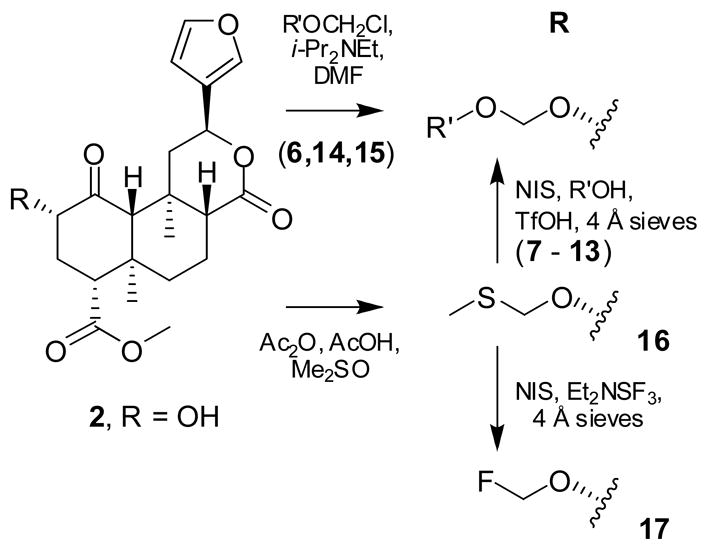
Deacetylation11 of 1, isolated from dried Salvia divinorum leaves as previously described,12 gave 2. MOM ether 5 was prepared from 2 and CH3OCH2Cl as previously described.13 The published 1H NMR data13 required amendment. The molecular formula, not previously established, was confirmed by HRMS. The ethoxymethyl (EOM) ether 6 and several other standard alkoxymethyl ethers were prepared similarly (Scheme 1; see Table 1 for individual structures).14
Scheme 1.
Synthesis of alkoxymethyl ethers and fluoromethyl ether.
Table 1.
Affinities (Ki) and potencies (EC50) at the κ opioid receptor.
| R | Ki ± SEM | EC50 ± SEM | |
|---|---|---|---|
| (nM)a,b | (nM)b,c | ||
| 1 |
|
2.4 ± 0.4 | 1.8 ± 0.5 |
| 5 |
|
0.60 ± 0.07 | 0.40 ± 0.04 |
| 6 |
|
0.32 ± 0.02 | 0.14 ± 0.01 |
| 7 |
|
2.2 ± 0.6 | 5.2 ± 0.4 |
| 8 |
|
5.3 ± 1.7 | 20 ± 3.5 |
| 9 |
|
1.6 ± 0.5 | 4.2 ± 0.7 |
| 10 |
|
35 ± 15 | 108 ± 18 |
| 11 |
|
1.9 ± 0.5 | 3.8 ± 0.3 |
| 12 |
|
31 ± 8 | 75 ± 7 |
| 13 |
|
141 ± 29 | 320 ± 13 |
| 14 |
|
> 1,000 | 1,660 ± 60 |
| 15 |
|
147 ± 26 | 274 ± 16 |
| 16 |
|
13 ± 3 | 31 ± 8 |
| 17 |
|
50 ± 9 | 26 ± 6 |
| 18a |
|
11 ± 1 | 10 ± 1 |
| 18b | 6.6 ± 0.3 | 5.7 ± 0.7 | |
| 19 |

|
72 ± 13 | 72 ± 5 |
| 20 |
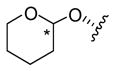
|
4.0 ± 0.4 | 2.8 ± 0.3 |
| U50,488H | 2.2 ± 0.3 | 1.4 ± 0.3 |
Inhibition of [3H]diprenorphine binding to CHO-hKOR.
Mean of three independent experiments performed in duplicate.
Enhancement of [35S]GTPγS binding to CHO-hKOR. All compounds were full agonists (Emax = 81–106% relative to U50,488H).
configuration unknown.
For the more unusual alkoxymethyl ethers 7–12, the corresponding chloromethyl ethers were expensive or hard to obtain. Rather than prepare and purify these volatile carcinogens, an alternative route was used. Methylthiomethyl ether 16 was prepared from 2 using Ac2O and AcOH in Me2SO (Scheme 1).15 When following the published procedure, side reactions resulted in poor yields, but we found that these were suppressed by a large excess of AcOH. The alkoxymethyl compounds 7–12 were then produced by alcoholysis of 16, using N-iodosuccinimide (NIS) and catalytic TfOH with the appropriate alcohol.16 Although low-yielding (< 40%), this route offers access to a wide range of products from a common intermediate and readily available alcohols. The NIS route was also employed for the 2-methoxyethoxymethyl (MEM) ether 13. Although the corresponding chloromethyl ether is readily available, 13 is difficult to separate from residual starting material 2, making purification difficult; the NIS route from 16 circumvented this difficulty, since 13 and 16 are easily separated. Due to the low yields of the NIS/TfOH route, other conditions were tested (AgNO3/2,6-lutidine17 and HgCl2);18 unfortunately, neither proved effective. Fluoromethyl ether 17 was prepared using a closely related method: NIS and Et2NSF3.19 Typical 1JCF and 2JHF couplings were observed in the 1H, 13C and 19F NMR spectra of all fluorinated compounds (11, 12 and 17).
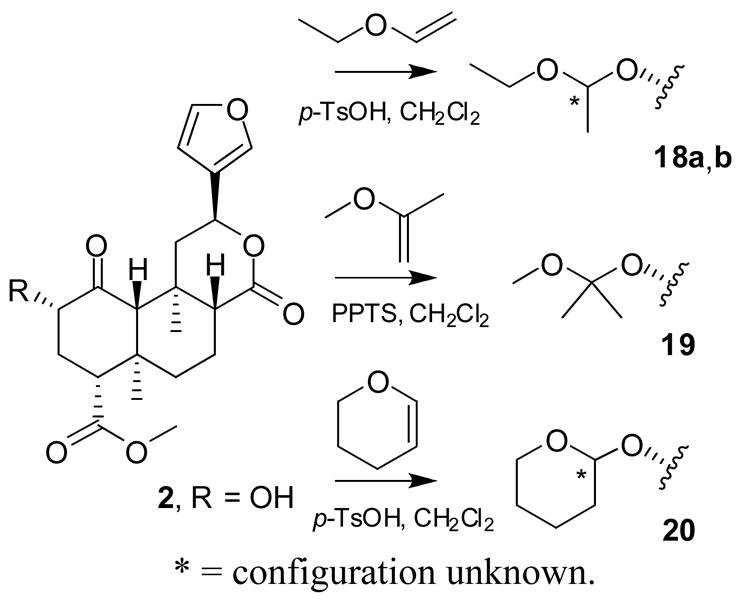
Compounds 18, 19, and 20 were prepared using ethoxyethene, 2-methoxypropene, and 3,4-dihydro-2H-pyran, respectively, with catalytic p-TsOH or PPTS (Scheme 2).14 The two epimers of 18 were separated with difficulty by repeated flash chromatography; complete separation was not achieved. Epimerization also occurred in CDCl3, so NMR data were collected in C6D6. The relative configurations of 18a and b were not determined. Tetrahydropyranyl ether 20 was also formed as an epimeric mixture, but only one epimer could be isolated after chromatography on silica gel, contaminated with a small amount of the other. Again, the configuration at the acetal carbon is unknown. For NMR, CDCl3 filtered through basic Al2O3 was satisfactory for brief exposures, but for longer experiments C6D6 was required.
Scheme 2.
Synthesis of other alkoxyalkyl ethers.
3. Results and Discussion
Binding affinities and potencies at the κ receptor are shown in Table 1. All compounds were full agonists, with efficacy approximately equal to that of U50,488H. All compounds except 14 showed submicromolar affinity and potency, generally in the low nanomolar range. None of the compounds bound to μ or δ receptors (Ki > 1 μM). This series as a whole shows markedly higher affinity and selectivity than previously reported series of derivatives of 1.
Among the n-alkoxymethyl ethers 5–8, the ethyl substituent was optimal (6), conferring extreme (subnanomolar) affinity and potency. Given the lack of μ and δ affinity, this compound is therefore also extremely selective (μ/κ and δ/κ > 3,000). There have been very few reports of compounds with μ/κ selectivity over 1,000.20 Ethoxymethyl ether 6 is thus among the most potent and selective κ opioids reported to date. While the naltrexone derivative nalfurafine (TRK-820) is more potent still (EC50 = 0.025 nM under the same conditions), this compound shows much lower selectivity (μ/κ < 100).21
The potency of the MOM ether 5, while lower than 6, was nonetheless higher than 1 or U50,488H, as previously reported.10 The selectivity of this compound, which has not previously been quantified, is thus also extremely high (μ/κ and δ/κ > 1,600). Further extension of the alkyl chain reduced activity, but the propoxymethyl and butoxymethyl ethers 7 and 8 retained activity comparable to 1. The same trend was previously reported in the alkyl ether8 and ester22 series, but with strikingly inferior absolute values. This provides further confirmation that substitution of oxygen in this position dramatically increases binding affinity. However, the increase in binding affinity from 5 to 6 suggests that an additional interaction involving the terminal alkyl group also contributes to the extremely high affinities of these compounds. For instance, the receptor may possess a hydrophobic pocket just deep enough to accommodate an ethyl group.
Branching of the alkyl chain was not as well tolerated as elongation; the isopropoxy and tert-butoxy compounds 9 and 10 showed a steep, progressive loss of activity relative to 6. MEM ether 13, with the same chain length as butoxymethyl ether 8, showed lower affinity than that compound. Larger protecting groups were also poorly tolerated: the 2-(trimethylsilyl)ethoxymethyl (SEM, 14) and benzyloxymethyl (BOM, 15) ethers were the least active compounds in the series.
2-Fluoroethoxymethyl ether 11 showed potency and affinity approximately equal to salvinorin A (1) itself. To our knowledge, 11 is the first potent, selective fluorinated ligand at the κ opioid receptor to be reported in the peer-reviewed literature. This is potentially significant, since the presence of fluorine permits in vivo imaging through 19F magnetic resonance imaging (MRI), or (with 18F labeling) positron emission tomography (PET).23 Should labeling prove feasible, compound 11 has important potential advantages over the existing [11C]-labelled PET ligand, GR103545.24 Although often described as κ-selective, that compound and its racemate in fact display higher affinity for μ than κ receptors (μ/κ = 0.5).25 By contrast, compound 11 is highly selective (μ/κ > 500). Another potential advantage of 11 is that the half-life of 18F is five times longer than that of 11C, which increases sensitivity and allows a wider range of experiments.
Heteroatom substitution at the alkoxy group reduced activity. Methylthiomethyl ether 16 showed much lower potency than 5, comparable to the previously-reported propyl ether 4c of equal chain length (EC50 = 67 nM under the same conditions).8 The potency of fluoromethyl ether 17 was also greatly reduced relative to 5. Although fluorine is widely regarded as isosteric with hydrogen, it is in fact much closer in size to oxygen, and can serve as an effective bioisostere for hydroxy and methoxy groups.26 Thus, while many would regard 17 as a bioisostere for the methyl ether 4a, it is in fact closer to 5. Direct comparison of fluorine and oxygen is confounded, however, by the terminal methyl substituent in 5, which as discussed above appears to contribute to that compound’s potency. An indirect comparison is nonetheless possible. The potency of 17 was similar to that of ethyl ether 4b (EC50 = 18 nM).8 Thus, whereas substitution of oxygen for C-2 of the propyl and butyl ethers (giving 5 and 6) dramatically increases activity, substitution of fluorine for C-2 of the ethyl ether (giving 17) has little effect. These results for 16 and 17 suggest that the alkoxymethyl group may act as an H-bond acceptor, since organic fluorine is a very poor H-bond acceptor,27 and sulfur is not an acceptor.
Contrary to the trend observed with the formate 3, methyl substitution of the acetal carbon dramatically lowered affinity. Compound 18 (the methyl analogue of 6), had at least 20-fold lower affinity, while 19 (the dimethyl analogue of 5) exhibited a greater than 100-fold reduction. This implies that this series of compounds may bind in a different manner than the ester series. The tetrahydropyranyl ether 20 showed comparable affinity to 1; thus, monosubstitution of the acetal carbon is better tolerated than disubstitution. There was not a dramatic difference in potency between the epimers of 18; unfortunately, only one epimer of 20 could be isolated, preventing any additional investigation of this question.
The high affinities of the alkoxymethyl ethers are of particular interest because these protecting groups are more stable than acetates under many synthetic conditions. For instance, 1 is readily deacetylated by weak bases, and the α-hydroxy ketone function thus exposed is highly sensitive to strong bases,12 while MOM ethers are virtually inert under strongly basic conditions.14 Similarly, MOM ethers are generally more stable to nucleophiles, organometallic and hydride reagents than acetates.14 A very pertinent illustration of these advantages is the first total synthesis of 1, which employs a BOM protecting group at C-2 in place of the acetate.28
4. Conclusions
It is fortuitous that standard protecting groups, stable and unreactive under a wide range of harsh conditions, should also confer high potency. For synthetic transformations elsewhere in the salvinorin scaffold, this permits the use of conditions incompatible with an acetate or hydroxyl. The resulting derivatives may also possess higher affinity and selectivity than the corresponding acetate, which would be valuable in ameliorating the effects of transformations elsewhere in the parent compound. For instance, some salvinorin derivatives appear to exhibit antagonism or partial agonism,4,5 but this is accompanied by severe reductions in affinity and selectivity. In such cases, it would be interesting to explore whether a C-2 alkoxymethyl ether substituent attenuated these reductions in affinity and selectivity. Alkoxymethyl ethers are also likely to be more stable in vivo, which would be desirable given salvinorin A’s brief duration of action. In a recent report, a MOM ether showed equal in vitro activity against HIV to the corresponding acetate, and greater stability in plasma.29
5. Experimental
5.1. General Experimental Conditions
1H NMR (300 MHz) and 13C NMR (75.5 MHz) chemical shifts are referenced to residual solvent peaks as internal standards: CDCl3 (7.26 and 77 ppm), C6D6 (7.16 and 128 ppm), and C6D5N (135.5 ppm). 19F NMR chemical shifts are referenced to CCl3F (0 ppm). Where the coupling constants of a discrete multiplet could not be determined, the separation of the outermost peaks (Δν) is given in Hz. Flash column chromatography (FCC) was performed on silica gel (230–400 mesh, 60 Å), eluting with a stepped gradient over the specified range. Where compound 2 was present due to incomplete reaction or hydrolysis of products, the column was stripped with 20% MeOH/CH2Cl2 to maximize recovery.
5.2. Binding Assays
Binding affinities at μ, δ, and κ opioid receptors were determined, as previously described,21 by competitive inhibition of [3H]diprenorphine binding to membranes prepared from Chinese hamster ovary (CHO) cells stably transfected with the human κ (hKOR), rat μ, or mouse δ receptors. Compounds were initially screened at 3 μM; those compounds causing > 50% displacement of [3H]diprenorphine (equivalent under the assay conditions to Ki < 1 μM) were tested further for determination of binding affinity (Ki), potency (EC50) and efficacy (Emax). Positive controls were U50,488H (κ), DAMGO (μ), SNC80 (δ), and etorphine (μ/δ), which all caused > 90% displacement of [3H]diprenorphine at 3 μM. Potencies and efficacies were determined by [35S]GTPγS binding to membranes of CHO-hKOR cells, as previously described.21 Testing was blinded: neither identity nor molecular mass were known to the testers.
5.3. Salvinorin B methoxymethyl ether (5)
Prepared as previously described.13 TLC (50% EtOAc/hexanes): hRf = 40 (5), 30 (2); 1H NMR (CDCl3): δ 7.41 (1H, dt, J = 1.8, 0.9 Hz), 7.40 (1H, t, J = 1.8 Hz), 6.38 (1H, dd, J = 1.8, 0.9 Hz), 5.54 (1H, dd, J = 11.8, 5.2 Hz), 4.72 (1H, d, J = 7.0 Hz), 4.70 (1H, d, J = 7.0 Hz), 4.14 (1H, dd, J = 12.2, 7.4 Hz), 3.71 (3H, s), 3.38 (3H, s), 2.68 (1H, dd, J = 13.5, 3.5 Hz), 2.53 (1H, dd, J = 13.5, 5.3 Hz), 2.36 (1H, ddd, J = 13.5, 7.6, 3.6 Hz), 2.19 (1H, td, J = 13.4, 12.2 Hz), 2.15 (1H, dq, J = 13.5, 3.4 Hz), 2.06 (1H, br s), 2.05 (1H, dd, J = 11.3, 3.2 Hz), 1.78 (1H, m, Δν = 18.6 Hz), 1.71–1.45 (3H, m), 1.46 (3H, s), 1.11 (3H, s); 13C NMR (CDCl3): δ 205.8, 171.8, 171.2, 143.7, 139.4, 125.3, 108.3, 95.7, 77.8, 71.9, 64.3, 55.8, 53.8, 51.9, 51.5, 43.5, 41.9, 38.1, 35.5, 32.6, 18.1, 16.4, 15.2; HRMS(ESI): [M+NH4]+ m/z 452.2295 (calcd for C23H30O8, 452.2284).
5.4. Salvinorin B ethoxymethyl ether (6)
Salvinorin B (2) (49.7 mg, 127 μmol) was added to dry DMF (1 mL) under Ar with warming. i-Pr2NEt (110 μL, 631 μmol) and EtOCH2Cl (60 μL, 646 μmol) were added. The white slurry was stirred at r.t. for 24 h. The solution was diluted in EtOAc and washed with 0.1 M aq. HCl (× 3), H2O, sat. aq. NaHCO3, and brine and dried (MgSO4). FCC (25–50% EtOAc/hexanes, then 20% MeOH/CH2Cl2) gave 6 as an amorphous white solid (45.6 mg, 80%). TLC (50% EtOAc/hexanes): hRf = 47 (6), 30 (2); 1H NMR (CDCl3): δ 7.41 (1H, dt, J = 1.6, 0.9 Hz), 7.40 (1H, t, J = 1.8 Hz), 6.37 (1H, dd, J = 1.8, 0.9 Hz), 5.54 (1H, dd, J = 11.6, 5.1 Hz), 4.77 (1H, d, J = 7.0 Hz), 4.74 (1H, d, J = 7.0 Hz), 4.16 (1H, dd, J = 12.1, 7.4 Hz), 3.71 (3H, s), 3.69 (1H, dq, J = 9.7, 7.0 Hz), 3.58 (1H, dq, J = 9.7, 7.0 Hz), 2.69 (1H, dd, J = 13.3, 3.4 Hz), 2.53 (1H, dd, J = 13.3, 5.1 Hz), 2.34 (1H, ddd, J = 13.5, 7.4, 3.5 Hz), 2.18 (1H, td, J = 13.4, 12.1 Hz), 2.15 (1H, dq, J = 13.7, 3.5 Hz), 2.06 (1H, br s), 2.05 (1H, m, Δν = 15 Hz), 1.77 (1H, m, Δν = 18.3 Hz), 1.71–1.44 (3H, m), 1.46 (3H, s), 1.18 (3H, t, J = 6.9 Hz), 1.11 (3H, s); 13C NMR (CDCl3): δ 206.0, 171.8, 171.2, 143.7, 139.3, 125.3, 108.3, 94.3, 77.7, 71.9, 64.2, 63.8, 53.8, 51.8, 51.4, 43.4, 41.9, 38.1, 35.4, 32.6, 18.1, 16.4, 15.2, 15.1; HRMS(ESI): [M+H]+ m/z 449.2193 (calcd for C24H32O8, 449.2175).
5.5. Salvinorin B propoxymethyl ether (7)
Propanol was stored over freshly activated 4 Å sieves for 1 h. To a mixture of methylthiomethyl ether 16 (37.6 mg, 83.5 μmol), N-iodosuccinimide (28.6 mg, 127 μmol, 1.5 eq), and 4 Å molecular sieves (beads) under Ar was added CH2Cl2 (0.5 mL). The flask was cooled to 0 °C, and dry propanol (1.5 mL, excess) was added, followed by TfOH (1 μL), and the solution was stirred for 5 min. NaHCO3 (~100 mg) was added, then the solution was diluted in EtOAc and washed with sat. aq. NaHCO3, 10% aq. NaS2O3, and brine. The organic layer was dried over MgSO4, filtered, and concentrated under reduced pressure. FCC (5–10% EtOAc/CH2Cl2 gradient) gave 7 as a clear resin (21%); TLC (10% EtOAc/CH2Cl2): hRf = 27 (7), 35 (16); 1H NMR (CDCl3): δ 7.41–7.40 (2H, m), 6.37 (1H, dd, J = 1.6, 0.9 Hz), 5.55 (1H, dd, J = 11.7, 5.1 Hz), 4.77 (1H, d, J = 7.2 Hz), 4.76 (1H, d, J = 7.2 Hz), 4.17 (1H, dd, J = 12.1, 7.6 Hz), 3.71 (3H, s), 3.59 (1H, dt, J = 9.4, 6.6 Hz), 3.47 (1H, dt, J = 9.4, 6.6 Hz), 2.69 (1H, dd, J = 13.4, 3.5 Hz), 2.53 (1H, dd, J = 13.4, 5.2 Hz), 2.34 (1H, ddd, J = 13.5, 7.6, 3.5 Hz), 2.18 (1H, td, J = 13.2, 12.1 Hz), 2.15 (1H, dq, J = 13.5, 3.5 Hz), 2.06 (1H, br s), 2.05 (1H, dd, J = 11.7, 3.2 Hz), 1.77 (1H, m, Δν = 18.3 Hz), 1.71–1.46 (5H, m), 1.46 (3H, s), 1.10 (3H, s), 0.90 (3H, t, J = 7.4 Hz); 13C NMR (CDCl3): δ 206.0, 171.8, 171.2, 143.7, 139.3, 125.3, 108.3, 94.4, 77.6, 72.0, 70.2, 64.2, 53.9, 51.9, 51.4, 43.4, 42.0, 38.1, 35.5, 32.6, 22.8, 18.1, 16.4, 15.2, 10.6; HRMS(ESI): [M+H]+ m/z 463.2339 (calcd for C25H34O8, 463.2332).
5.6. Salvinorin B butoxymethyl ether (8)
Procedure as for 7, using butanol. FCC (3–6% EtOAc/CH2Cl2 gradient) gave 8 as a clear resin (24%); TLC (5% EtOAc/CH2Cl2): hRf = 17 (8), 22 (16); 1H NMR (CDCl3): δ 7.41–7.39 (2H, m), 6.37 (1H, dd, J = 1.9, 1.0 Hz), 5.55 (1H, dd, J = 11.7, 5.1 Hz), 4.76 (1H, d, J = 7.1 Hz), 4.74 (1H, d, J = 7.1 Hz), 4.17 (1H, dd, J = 12.1, 7.4 Hz), 3.71 (3H, s), 3.63 (1H, dt, J = 9.4, 6.5 Hz), 3.51 (1H, dt, J = 9.4, 6.5 Hz), 2.69 (1H, dd, J = 13.3, 3.4 Hz), 2.53 (1H, dd, J = 13.3, 5.2 Hz), 2.34 (1H, ddd, J = 13.5, 7.5, 3.5 Hz), 2.18 (1H, td, J = 13.4, 12.2 Hz), 2.19–2.12 (1H, m), 2.06 (1H, br s), 2.05 (1H, dd, J = 11.5, 3.1 Hz), 1.78 (1H, m, Δν = 18.8 Hz), 1.71–1.45 (5H, m), 1.47 (3H, s), 1.34 (2H, m, Δν = 37 Hz), 1.11 (3H, s), 0.89 (3H, t, J = 7.3 Hz); 13C NMR (CDCl3): δ 206.0, 171.8, 171.2, 143.7, 139.3, 125.2, 108.3, 94.4, 77.6, 72.0, 68.3, 64.2, 53.9, 51.9, 51.5, 43.5, 42.0, 38.2, 35.5, 32.5, 31.7, 19.3, 18.1, 16.4, 15.2, 13.8; HRMS(ESI): [M+H]+ m/z 477.2506 (calcd for C26H36O8, 477.2488).
5.7. Salvinorin B isopropoxymethyl ether (9)
Procedure as for 7, using 2-propanol. FCC (0–10% EtOAc/CH2Cl2 gradient) gave 9 as a clear resin (18%); TLC (10% EtOAc/CH2Cl2): hRf = 27(9), 41 (16); 1H NMR (CDCl3): δ 7.41–7.39 (2H, m), 6.37 (1H, dd, J = 1.6, 0.9 Hz), 5.54 (1H, dd, J = 11.7, 5.1 Hz), 4.83 (1H, d, J = 7.4 Hz), 4.74 (1H, d, J = 7.4 Hz), 4.21 (1H, dd, J = 12.2, 7.5 Hz), 3.93 (1H, sept, J = 6.1 Hz), 3.72 (3H, s), 2.70 (1H, dd, J = 13.5, 3.5 Hz), 2.53 (1H, dd, J = 13.2, 5.2 Hz), 2.33 (1H, ddd, J = 13.5, 7.5, 3.7 Hz), 2.18 (1H, dt, J = 13.4, 12.0 Hz), 2.15 (1H, dq, J = 13.5, 3.5 Hz), 2.07 (1H, br s), 2.05 (1H, dd, J = 11.4, 3.1 Hz), 1.78 (1H, m, Δν = 18.3 Hz), 1.71–1.45 (3H, m), 1.47 (3H, s), 1.18 (3H, d, J = 6.2 Hz), 1.13 (3H, d, J = 6.2 Hz), 1.10 (3H, s); 13C NMR (CDCl3): δ 206.2, 171.9, 171.2, 143.7, 139.3, 125.3, 108.3, 92.2, 77.3, 72.0, 69.7, 64.2, 53.9, 51.9, 51.5, 43.4, 42.0, 38.2, 35.5, 32.5, 23.0, 22.0, 18.1, 16.4, 15.2; HRMS(ESI): [M+NH4]+ m/z 480.2618 (calcd for C25H34O8, 480.2597).
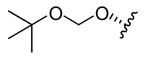
5.8. Salvinorin B tert-butoxymethyl ether (10)
Procedure as for 7, using 2-methyl- 2-propanol. FCC (0–5% EtOAc/CH2Cl2 gradient) gave 10 as a clear resin (40%); TLC (10% EtOAc/CH2Cl2): hRf = 32 (10), 34 (16); 1H NMR (CDCl3): δ 7.41–7.39 (2H, m), 6.36 (1H, dd, J = 1.7, 1.0 Hz), 5.53 (1H, dd, J = 11.7, 5.0 Hz), 4.99 (1H, d, J = 8.2 Hz), 4.71 (1H, d, J = 8.2 Hz), 4.26 (1H, dd, J = 12.2, 7.3 Hz), 3.71 (3H, s), 2.70 (1H, dd, J = 13.3, 3.4 Hz), 2.51 (1H, dd, J = 13.3, 5.1 Hz), 2.32 (1H, ddd, J = 13.5, 7.3, 3.5 Hz), 2.20–2.10 (2H, m), 2.07 (1H, br s), 2.04 (1H, dd, J = 11.7, 3.1 Hz), 1.76 (1H, m, Δν = 17.9 Hz), 1.70–1.43 (3H, m), 1.45 (3H, s), 1.22 (9H, s), 1.09 (3H, s); 13C NMR (CDCl3): δ 206.5, 171.9, 171.2, 143.7, 139.3, 125.2, 108.3, 88.7, 77.1, 75.0, 71.9, 64.1, 53.9, 51.9, 51.4, 43.4, 42.0, 38.1, 35.4, 32.5, 28.7, 18.1, 16.4, 15.2; HRMS(ESI): [M+ NH4]+ m/z 494.2773 (calcd for C26H36O8, 494.2754).
5.9. Salvinorin B 2-fluoroethoxymethyl ether (11)
Procedure as for 7, using 2- fluoroethanol. FCC (0–5% EtOAc/CH2Cl2) gave 11 as a clear resin (18%). TLC (10% EtOAc/CH2Cl2): hRf = 26 (11), 40 (16); 1H NMR (CDCl3): δ 7.42 (1H, dt, J = 1.6, 0.9 Hz), 7.40 (1H, t, J = 1.9 Hz), 6.38 (1H, dd, J = 1.9, 1.0 Hz), 5.55 (1H, dd, J = 11.6, 5.1 Hz), 4.82 (2H, s), 4.55 (2H, ~dt, J = 47.8, 4.1 Hz), 4.22 (1H, dd, J = 12.2, 7.5 Hz), 3.85 (2H, ~dt, J = 30.2, 4.1 Hz), 3.72 (3H, s), 2.70 (1H, dd, J = 13.2, 3.1 Hz), 2.53 (1H, dd, J = 13.1, 4.8 Hz), 2.35 (1H, ddd, J = 13.5, 7.5, 3.5 Hz), 2.18 (1H, dt, J = 13.2, 12.0 Hz), 2.18–2.12 (1H, m), 2.08 (1H, br s), 2.05 (1H, dd, J = 11.7, 2.9 Hz), 1.78 (1H, m, Δν = 18.2 Hz), 1.72–1.45 (3H, m), 1.47 (3H, s), 1.11 (3H, s); 13C NMR (CDCl3): δ 205.8, 171.8, 171.2, 143.7, 139.3, 125.3, 108.3, 94.4, 82.7 (d, J = 168 Hz), 77.7, 72.0, 67.2 (d, J = 22 Hz), 64.3, 53.8, 51.9, 51.4, 43.4, 42.0, 38.2, 35.5, 32.4, 18.1, 16.4, 15.2; 19F NMR (CDCl3): δ 5.6 (tt, J = 47.8, 30.3 Hz); HRMS(ESI): [M+H]+ m/z 467.2096 (calcd for C24H31FO8, 467.2081).
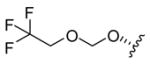
5.10. Salvinorin B 2,2,2-trifluoroethoxymethyl ether (12)
Procedure as for 7, using 2,2,2-trifluoroethanol. FCC (0–5% EtOAc/CH2Cl2) gave 12 as a clear resin (11%). TLC (10% EtOAc/CH2Cl2): hRf = 37 (12), 35 (16); 1H NMR (CDCl3): δ 7.42 (1H, dt, J = 1.8, 0.9 Hz), 7.40 (1H, t, J = 1.8 Hz), 6.38 (1H, dd, J = 1.9, 0.9 Hz), 5.55 (1H, dd, J = 11.6, 5.0 Hz), 4.84 (1H, d, J = 7.5 Hz), 4.82 (1H, d, J = 7.5 Hz), 4.17 (1H, dd, J = 12.2, 7.5 Hz), 4.05 (1H, dq, J = 12.0, 8.7 Hz), 3.95 (1H, dq, J = 12.0, 8.7 Hz), 3.72 (3H, s), 2.70 (1H, dd, J = 13.3, 3.5 Hz), 2.52 (1H, dd, J = 13.3, 5.1 Hz), 2.33 (1H, ddd, J = 13.6, 7.5, 3.7 Hz), 2.20 (1H, td, J = 13.3, 12.2 Hz), 2.16 (1H, dq, J = 13.6, 3.4 Hz), 2.08 (1H, br s), 2.05 (1H, dd, J = 11.9, 3.1 Hz), 1.79 (1H, m, Δν = 18.3 Hz), 1.72–1.49 (3H, m), 1.47 (3H, s), 1.10 (3H, s); 13C NMR (CDCl3): δ 205.4, 171.6, 171.1, 143.7, 139.3, 125.3, 123.7 (q, J = 278 Hz), 108.3, 94.5, 78.5, 72.0, 64.9 (q, J = 34.6 Hz), 64.3, 53.7, 51.9, 51.4, 43.4, 42.0, 38.2, 35.5, 32.3, 18.1, 16.4, 15.2; 19F NMR (CDCl3): δ −74.8 (t, J = 8.9 Hz); HRMS(ESI): [M+H]+ m/z 503.1870 (calcd for C24H29F3O8, 503.1893).
5.11. Salvinorin B 2-methoxyethoxymethyl ether (13)
Procedure as for 7, using 2- methoxyethanol. FCC (33–50% EtOAc/hexanes) gave 13 as a clear resin (15%). TLC (10% EtOAc/CH2Cl2): hRf = 14 (13), 35 (16); 1H NMR (CDCl3): δ 7.42 (1H, dt, J = 1.6, 0.9 Hz), 7.40 (1H, t, J = 1.9 Hz), 6.38 (1H, dd, J = 1.8, 0.9 Hz), 5.54 (1H, dd, J = 11.7, 5.1 Hz), 4.82 (1H, d, J = 7.3 Hz), 4.80 (1H, d, J = 7.3 Hz), 4.22 (1H, dd, J = 12.2, 7.2 Hz), 3.79 (1H, m, Δν = 20.1 Hz), 3.71 (3H, s), 3.70 (1H, m, Δν = 20.1 Hz), 3.52 (2H, t, J = 4.5 Hz), 3.35 (3H, s), 2.68 (1H, dd, J = 13.5, 3.5 Hz), 2.52 (1H, dd, J = 13.3, 5.1 Hz), 2.36 (1H, ddd, J = 13.6, 7.2, 3.4 Hz), 2.18 (1H, td, J = 13.5, 12.3 Hz), 2.20–2.11 (1H, m), 2.06 (1H, br s), 2.04 (1H, dd, J = 11.6, 3.1 Hz), 1.78 (1H, m, Δν = 18.5 Hz), 1.71–1.44 (3H, m), 1.47 (3H, s), 1.11 (3H, s); 13C NMR (CDCl3): δ 205.9, 171.8, 171.2, 143.7, 139.4, 125.3, 108.3, 94.6, 77.6, 71.9, 71.6, 67.3, 64.3, 59.0, 53.8, 51.9, 51.5, 43.5, 42.0, 38.2, 35.5, 32.5, 18.1, 16.4, 15.2; HRMS(ESI): [M+NH4]+ m/z 496.2528 (calcd for C25H34O9, 496.2547).
5.12. Salvinorin B 2-(trimethylsilyl)ethoxymethyl ether (14)
Procedure as for 6, using 2-(trimethylsilyl)ethyl chloromethyl ether for 23 h. FCC (33–50% EtOAc/hexanes) gave 12 as an amorphous white solid (53%). TLC (50% EtOAc/hexanes): hRf = 72 (14), 24 (2); 1H NMR (CDCl3): δ 7.41 (1H, dt, J = 1.6,0.9 Hz), 7.40 (1H, t, J = 1.7 Hz), 6.37 (1H, dd, J = 1.7, 0.9 Hz), 5.54 (1H, dd, J = 11.7, 5.0 Hz), 4.77 (1H, d, J = 7.2 Hz), 4.73 (1H, d, J = 7.2 Hz), 4.16 (1H, dd, J = 12.2, 7.5 Hz), 3.69 (1H, m, Δν = 26.8 Hz), 3.71 (3H, s), 3.59 (1H, m, Δν = 26.8 Hz), 2.69 (1H, dd, J = 13.5, 3.4 Hz), 2.52 (1H, dd, J = 13.3, 5.1 Hz), 2.33 (1H, ddd, J = 13.3, 7.2, 3.4 Hz), 2.18 (1H, td, J = 13.3, 12.2 Hz), 2.19–2.11 (1H, m), 2.06 (1H, br s), 2.05 (1H, dd, J = 11.3, 3.1 Hz), 1.77 (1H, m, Δν = 18.0 Hz), 1.71–1.48 (3H, m), 1.46 (3H, s), 1.10 (3H, s), 0.88 (2H, m, Δν = 16.8 Hz), 0.00 (9H, s); 13C NMR (CDCl3): δ 206.0, 171.8, 171.2, 143.7, 139.3, 125.2, 108.3, 93.9, 77.7, 71.9, 65.7, 64.2, 53.9, 51.9, 51.4, 43.5, 41.9, 38.1, 35.5, 32.5, 18.1, 18.1, 16.4, 15.2, −1.4; HRMS(ESI): [M+NH4]+ m/z 538.2855 (calcd for C27H40O8Si, 538.2836).
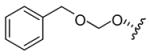
5.13. Salvinorin B benzyloxymethyl ether (15)
Procedure as for 6, using BnOCH2Cl with NaI (1 eq) for 96 h. FCC (33% EtOAc/hexanes, then 5% MeOH/CH2Cl2) gave 15 as an amorphous white solid (34% [47% borsm]). TLC (50% EtOAc/hexanes): hRf = 52 (15), 27 (2); 1H NMR (CDCl3): δ 7.41–7.40 (2H, m), 7.32– 7.28 (5H, m), 6.37 (1H, m, Δν = 2.8 Hz), 5.54 (1H, dd, J = 12.2, 5.3 Hz), 4.86 (1H, d, J = 7.2 Hz), 4.84 (1H, d, J = 7.2 Hz), 4.65 (2H, s), 4.19 (1H, dd, J = 12.0 ,7.5 Hz), 3.71 (3H, s), 2.66 (1H, dd, J = 13.0, 3.7 Hz), 2.50 (1H, dd, J = 13.2, 4.8 Hz), 2.33–2.11 (3H, m), 2.05 (1H, br s), 2.04 (1H, dd, J = 11.3, 2.9 Hz), 1.78 (1H, m, Δν = 18.6 Hz), 1.71–1.45 (3H, m), 1.47 (3H, s), 1.11 (3H, s); 13C NMR (CDCl3): δ 205.8, 171.8, 171.2, 143.7, 139.4, 137.4, 128.5, 127.94, 127.90, 125.2, 108.4, 93.9, 77.9, 71.9, 70.1, 64.2, 53.8, 51.9, 51.5, 43.5, 42.0, 38.1, 35.5, 32.4, 18.1, 16.4, 15.2; HRMS(ESI): [M+NH4]+ m/z 528.2606 (calcd for C29H34O8, 528.2597).
5.14. Salvinorin B methylthiomethyl ether (16)
Salvinorin B (2) (32.4 mg, 83 μmol) was dissolved in Me2SO (1 mL). AcOH was added (1 mL), followed by Ac2O (0.5 mL). The resulting white suspension was stirred at r.t. for 65 h, giving a clear yellow solution. Aq. NaOH (5.0 M, 5 mL) was added drop-wise, then the solution was diluted in EtOAc and washed with sat. aq. NaHCO3 (× 3) and brine and dried (MgSO4). Evaporation in vacuo gave 16 (33.7 mg, 90%) as an amorphous white solid of adequate purity for synthetic use. The receptor binding sample was purified by FCC (25% EtOAc/hexanes, stripped in 20% MeOH/CH2Cl2); TLC (50% EtOAc/hexanes): hRf = 54 (16), 30 (2); 1H NMR (CDCl3): δ 7.42 (1H, dt, J = 1.7, 0.8 Hz), 7.39 (1H, t, J = 1.7 Hz), 6.38 (1H, dd, J = 1.9, 0.9 Hz), 5.54 (1H, dd, J = 11.7, 5.1 Hz), 4.84 (1H, d, J = 11.9 Hz), 4.66 (1H, d, J = 11.9 Hz), 4.26 (1H, dd, J = 12.0, 7.4 Hz), 3.70 (3H, s), 2.72 (1H, dd, J = 13.4, 3.7 Hz), 2.54 (1H, dd, J = 13.4, 5.2 Hz), 2.28 (1H, ddd, J = 13.4, 7.5, 3.7 Hz), 2.19–2.10 (2H, m), 2.13 (3H, s), 2.09 (1H, br s), 2.05 (1H, dd, J = 12.2, 2.8 Hz), 1.76 (1H, m, Δν = 19 Hz), 1.70–1.48 (3H, m), 1.45 (3H, s), 1.10 (3H, s); 13C NMR (CDCl3): δ 206.1, 171.8, 171.4, 143.7, 139.4, 125.4, 108.4, 74.5, 71.9, 64.4, 53.8, 51.8, 51.5, 43.6, 42.0, 38.2, 35.5, 32.3, 18.2, 16.4, 15.2, 13.7 (one signal not observed); 13C NMR (C5D5N): δ 207.0, 172.4, 171.4, 144.3, 140.4, 126.6, 109.4, 78.0, 74.8, 71.9, 63.5, 53.6, 51.6, 51.3, 43.4, 42.1, 38.3, 35.9, 33.0, 18.8, 16.5, 15.3, 13.7; HRMS(ESI): [M+H]+ m/z 451.1792 (calcd for C23H30O7S, 451.1790).
5.15. Salvinorin B fluoromethyl ether (17)
Procedure as for 7, substituting Et2NSF3 (1.5 eq) for PrOH and omitting TfOH. FCC (66 – 100% Et2O/hexanes, then 20% MeOH/CH2Cl2) gave 17 as an amorphous white solid (64% [81% based on recovered 2]). A high-Rf byproduct was also recovered, along with 2. These were pooled and briefly refluxed in MeOH/CH2Cl2/AcOH. Evaporation under reduced pressure and rinsing with minimal MeOH gave 2. TLC (Et2O): hRf = 65 (byproduct), 54 (16), 48 (17), 32 (2); 1H NMR (CDCl3): δ 7.42 (1H, dt, J = 1.7, 0.9 Hz), 7.40 (1H, t, J = 1.8 Hz), 6.38 (1H, dd, J = 1.8, 0.9 Hz), 5.55 (1H, dd, J = 11.7, 5.1 Hz), 5.40 (1H, dd, J = 53.2, 3.0 Hz), 5.26 (1H, dd, J = 58.7, 3.0 Hz), 4.24 (1H, dd, J = 12.1, 7.6 Hz), 3.72 (3H, s), 2.70 (1H, dd, J = 13.8, 3.4 Hz), 2.53 (1H, dd, J = 13.3, 5.2 Hz), 2.42 (1H, ddd, J = 13.5, 7.4, 3.5 Hz), 2.24 (1H, td, J = 13.5, 12.3 Hz), 2.16 (1H, dq, J = 13.5, 3.2 Hz), 2.08 (1H, br s), 2.05 (1H, dd, J = 11.0, 3.2 Hz), 1.79 (1H, m, Δν = 18.3 Hz), 1.71–1.42 (3H, m), 1.46 (3H, s), 1.12 (3H, s); 13C NMR (CDCl3): δ 204.5, 171.5, 171.0, 143.8, 139.4, 125.2, 108.3, 102.4 (d, J = 214 Hz), 79.8, 71.9, 64.3, 53.6, 51.9, 51.4, 43.5, 42.0, 38.1, 35.5, 32.3, 18.1, 16.4, 15.2; 19F NMR (CDCl3): δ −152.2 (dd, J = 58.7, 53.8 Hz); HRMS(ESI): [M+H]+ m/z 423.1813 (calcd for C22H27FO7, 423.1819).
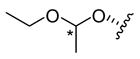
5.16. Salvinorin B 1-ethoxyethyl ether (18)
Salvinorin B (2) (49.1 mg, 126 μmol) was dissolved in dry CH2Cl2 (1.5 mL) under Ar and stirred at 0 °C. Ethoxyethene (100 μL, 1.04 mmol) and a speck of p-TsOH (≪ 1 mg, catalytic) were added. The resulting suspension was removed from the icebath and stirred at room temperature for 10 min, when it had clarified and turned yellow. TLC (50% EtOAc/hexanes) showed minimal starting material. The solution was diluted in EtOAc and washed with sat. aq. NaHCO3 (× 3) and dried (MgSO4). Evaporation under reduced pressure and repeated FCC (25– 50% EtOAc/hexanes, then 20% MeOH/CH2Cl2) gave 18a as an amorphous white solid (20.6 mg, 36%); TLC (50% EtOAc/hexanes): hRf = 48 (18a), 43 (18b), 30 (2); 1H NMR (C6D6): δ 7.10 (1H, dt, J = 1.6, 0.9 Hz), 7.05 (1H, t, J = 1.7 Hz), 6.14 (1H, dd, J = 1.9, 0.9 Hz), 5.18 (1H, dd, J = 11.7, 5.0 Hz), 4.94 (1H, q, J = 5.4 Hz), 4.00 (1H, m, Δν = 19 Hz), 3.44 (1H, dq, J = 9.2, 7.1 Hz), 3.32 (1H, dq, J = 9.2, 7.1 Hz), 3.29 (3H, s), 2.35–2.09 (5H, m), 1.54–1.43 (3H, m), 1.40 (3H, d, J = 5.4 Hz), 1.30 (1H, br s), 1.27 (3H, s), 1.24–1.05 (2H, m), 1.07 (3H, t, J = 7.1 Hz), 0.89 (3H, s); 13C NMR (C6D6): δ 206.3, 171.7, 170.1, 143.7, 139.4, 126.5, 108.7, 99.2, 76.9, 71.5, 63.6, 58.4, 53.7, 51.3, 51.1, 43.6, 41.7, 38.1, 35.5, 33.2, 19.8, 18.6, 16.2, 15.7, 15.1; HRMS(ESI): [M+H]+ m/z 463.2318 (calcd for C25H34O8, 463.2332).
Mixed fractions were pooled with other runs; further chromatography gave 18b as an amorphous white solid (16%); 1H NMR (C6D6): δ 7.11 (1H, dt, J = 1.7, 0.9 Hz), 7.05 (1H, t, J = 1.7 Hz), 6.16 (1H, dd, J = 1.9, 0.9 Hz), 5.18 (1H, dd, J = 11.7, 5.0 Hz), 4.73 (1H, q, J = 5.4 Hz), 3.83 (1H, m, Δν = 19 Hz), 3.64 (1H, dq, J = 9.4, 7.1 Hz), 3.52 (1H, dq, J = 9.4, 7.1 Hz), 3.31 (3H, s), 2.36–2.22 (3H, m), 2.20–2.07 (2H, m), 1.59–1.41 (3H, m), 1.32 (1H, br s), 1.29 (3H, s), 1.25–1.04 (2H, m), 1.21 (3H, d, J = 5.4 Hz), 1.06 (3H, t, J = 7.1 Hz), 0.91 (3H, s); 13C NMR (C6D6): δ 205.5, 171.8, 170.1, 143.7, 139.4, 126.6, 108.6, 98.6, 75.3, 71.5, 63.8, 60.9, 53.8, 51.3, 51.1, 43.5, 41.8, 38.2, 35.5, 33.4, 20.0, 18.7, 16.1, 15.5, 15.1; HRMS(ESI): [M+H]+ m/z 463.2342 (calcd for C25H34O8, 463.2332).
5.17. Salvinorin B 2-methoxy-2-propyl ether (19)
Salvinorin B (2) (40.4 mg, 103 μmol) was dissolved in dry CH2Cl2 (1 mL) under Ar. Pyridinium p-toluenesulfonate in dry CH2Cl2 (10 mM) was added (1 mL, 10 μmol). 2-Methoxypropene (100 μL, 1.04 mmol) was added, and the solution was stirred at room temperature for 35 minutes, monitored by TLC (50% EtOAc/hexanes). The reaction mixture was quenched with excess NEt3 (200 μL), loaded directly onto silica gel and purified by FCC (0.5% NEt3/10 – 50% EtOAc/hexanes, then 20% MeOH/CH2Cl2) to give 19 as an amorphous white solid (13.7 mg, 31%); TLC (50% EtOAc/hexanes): hRf = 59 (byproduct), 43 (19), 30 (2); 1H NMR (CDCl3): δ 7.40–7.39 (2H, m), 6.36 (1H, dd, J = 1.6, 0.9), 5.53 (1H, dd, J = 11.6, 5.1 Hz), 4.22 (1H, ~dd, J = 20.9, 8.4 Hz), 3.71 (3H, s), 3.21 (3H, s), 2.70 (1H, ~dd, J = 10.9, 5.6 Hz), 2.46 (1H, dd, J = 13.2, 5.1 Hz), 2.26–2.15 (2H, m), 2.14 (1H, dq, J = 13.5, 3.5 Hz), 2.09 (1H, br s), 2.04 (1H, dd, J = 11.3, 2.7 Hz), 1.79 (1H, m, Δν = 18.5 Hz), 1.71–1.45 (3H, m), 1.47 (3H, s), 1.40 (3H, s), 1.27 (3H, s), 1.09 (3H, s); 13C NMR (CDCl3): δ 206.6, 171.9, 171.2, 143.7, 139.4, 125.3, 108.3, 101.3, 73.5, 71.8, 64.5, 54.2, 51.8, 51.4, 49.4, 43.3, 42.1, 38.3, 35.4, 33.8, 25.0, 24.7, 18.1, 16.2, 15.1; HRMS(ESI): [M+H]+ m/z 463.2342 (calcd for C25H34O8, 463.2332).
5.18. Salvinorin B tetrahydropyran-2-yl ether (20)
Procedure as for 18, using 3,4- dihydro-2H-pyran. Repeated FCC (33–50% EtOAc/hexanes, then 20% MeOH/CH2Cl2) gave 20 as a clear resin (28.6 mg, 47%); TLC (50% EtOAc/hexanes): hRf = 58 (20), 52 (epimeric acetal), 35 (2); 1H NMR (CDCl3, filtered through basic Al2O3): δ 7.43–7.39 (2H, m), 6.38 (1H, dd, J = 1.9, 0.9 Hz), 5.53 (1H, dd, J = 11.9, 5.3 Hz), 4.74 (1H, t, J = 3.0 Hz), 4.25 (1H, dd, J = 12.2, 7.6 Hz), 3.81 (1H, ddd, J = 11.6, 9.0, 3.0 Hz), 3.71 (3H, s), 3.50 (1H, dt, J = 11.1, 4.5 Hz), 2.71 (1H, dd, J = 13.2, 3.7 Hz), 2.54 (1H, dd, J = 13.2, 5.1 Hz), 2.36 (1H, ddd, J = 13.5, 7.6, 3.8 Hz), 2.24 (1H, td, J = 13.4, 12.2 Hz), 2.15 (1H, dq, J = 13.2, 3.2 Hz), 2.06 (1H, br s), 2.05 (1H, dd, J = 13.3, 3.2 Hz), 1.88–1.41 (10H, m), 1.46 (3H, s), 1.12 (3H, s); 1H NMR (C6D6): δ 7.05–7.04 (2H, m), 6.11 (1H, q, J = 1.5 Hz), 5.16 (1H, dd, J = 11.8, 5.2 Hz), 5.00 (1H, t, J = 2.9 Hz), 4.06 (1H, m, Δν = 18 Hz), 3.70 (1H, td, J = 11.1, 3.0 Hz), 3.36 (1H, m, Δν = 20 Hz), 3.30 (3H, s), 2.43–2.23 (4H, m), 2.11 (1H, m, Δν = 26 Hz), 1.93 (1H, m, Δν = 24 Hz), 1.80–1.62 (2H, m), 1.49–1.00 (8H, m), 1.27 (3H, s), 1.02 (1H, td, J = 7.2, 1.5 Hz), 0.90 (3H, s); 13C NMR (C6D6): δ 206.3, 171.8, 170.1, 143.6, 139.4, 126.5, 108.7, 97.8, 76.9, 71.5, 63.7, 61.5, 53.7, 51.3, 51.1, 43.7, 41.6, 38.1, 35.5, 33.1, 30.3, 25.8, 18.8, 18.6, 16.2, 15.1; HRMS(ESI): [M+H]+ m/z 475.2342 (calcd for C26H34O8, 475.2332).
Supplementary Material
Supporting Information available: 1H and 13C NMR spectra for compounds 5–20; IUPAC International Chemical Identifiers (InChIs); statement of author contributions.
Acknowledgments
This work was supported by grants from the Stanley Medical Research Institute, the National Institute of Mental Health (MH63266), the National Alliance for Research on Schizophrenia and Depression (NARSAD) and the Engelhard Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Roth BL, Baner K, Westkaemper R, Siebert D, Rice KC, Steinberg S, Ernsberger P, Rothman RB. Proc Natl Acad Sci U S A. 2002;99:11934. doi: 10.1073/pnas.182234399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McCurdy CR, Scully SS. Life Sci. 2005;78:476. doi: 10.1016/j.lfs.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 3.Carlezon WA, Jr, Béguin C, DiNieri JA, Baumann MH, Richards MR, Todtenkopf MS, Rothman RB, Ma Z, Lee DY, Cohen BM. J Pharmacol Exp Ther. 2006;316:440. doi: 10.1124/jpet.105.092304. [DOI] [PubMed] [Google Scholar]
- 4.Holden KG, Tidgewell K, Marquam A, Rothman RB, Navarro H, Prisinzano TE. Bioorg Med Chem Lett. 2007;17:6111. doi: 10.1016/j.bmcl.2007.09.050. and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Simpson DS, Katavic PL, Lozama A, Harding WW, Parrish D, Deschamps JR, Dersch CM, Partilla JS, Rothman RB, Navarro H, Prisinzano TE. J Med Chem. 2007;50:3596. doi: 10.1021/jm070393d. [DOI] [PubMed] [Google Scholar]
- 6.Carroll I, Thomas JB, Dykstra LA, Granger AL, Allen RM, Howard JL, Pollard GT, Aceto MD, Harris LS. Eur J Pharmacol. 2004;501:111. doi: 10.1016/j.ejphar.2004.08.028. [DOI] [PubMed] [Google Scholar]
- 7.Mague SD, Pliakas AM, Todtenkopf MS, Tomasiewicz HC, Zhang Y, Stevens WC, Jr, Jones RM, Portoghese PS, Carlezon WA., Jr J Pharmacol Exp Ther. 2003;305:323. doi: 10.1124/jpet.102.046433. [DOI] [PubMed] [Google Scholar]
- 8.Béguin C, Richards MR, Wang Y, Chen Y, Liu-Chen L-Y, Ma Z, Lee DYW, Carlezon WA, Jr, Cohen BM. Bioorg Med Chem Lett. 2005;15:2761. doi: 10.1016/j.bmcl.2005.03.113. [DOI] [PubMed] [Google Scholar]
- 9.Munro TA, Rizzacasa MA, Roth BL, Toth BA, Yan F. J Med Chem. 2005;48:345. doi: 10.1021/jm049438q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee DY, Karnati VVR, He M, Liu-Chen L-Y, Kondareti L, Ma Z, Wang Y, Chen Y, Béguin C, Carlezon WA, Jr, Cohen B. Bioorg Med Chem Lett. 2005;15:3744. doi: 10.1016/j.bmcl.2005.05.048. [DOI] [PubMed] [Google Scholar]
- 11.Tidgewell K, Harding WW, Schmidt M, Holden KG, Murry DJ, Prisinzano TE. Bioorg Med Chem Lett. 2004;14:5099. doi: 10.1016/j.bmcl.2004.07.081. [DOI] [PubMed] [Google Scholar]
- 12.Munro TA. PhD Thesis. University of Melbourne; Australia: 2006. http://eprints.infodiv.unimelb.edu.au/archive/00002327. [Google Scholar]
- 13.Béguin C, Carlezon WA, Jr, Cohen BM, He M, Lee DY-W, Richards MR, Liu-Chen L-Y. 20060052439. US Patent Application. 2006
- 14.Wuts PGM, Greene TW. Greene's Protective Groups in Organic Synthesis. 4. Wiley; Hoboken, NJ: 2007. [Google Scholar]
- 15.Pojer PM, Angyal SJ. Aust J Chem. 1978;31:1031. doi: 10.1071/CH9781031. [DOI] [Google Scholar]
- 16.Konradsson P, Udodong UE, Fraser-Reid B. Tetrahedron Lett. 1990;31:4313. doi: 10.1016/S0040-4039(00)97609-3. [DOI] [Google Scholar]
- 17.Breslow R, Pandey PS. J Org Chem. 1980;45:740. doi: 10.1021/jo01292a046. [DOI] [Google Scholar]
- 18.Chowdhury PK, Sarma DN, Sharma RP. Chem Ind. 1984:803. [Google Scholar]
- 19.Iimura S, Uoto K, Ohsuki S, Chiba J, Yoshino T, Iwahana M, Jimbo T, Terasawa H, Soga T. Bioorg Med Chem Lett. 2001;11:407. doi: 10.1016/S0960-894X(00)00682-X. see note 21. [DOI] [PubMed] [Google Scholar]
- 20.Aldrich JV, Vigil-Cruz SC. In: Burger's Medicinal Chemistry and Drug Discovery. 6. Burger A, Abraham DJ, editors. Vol. 6. Wiley; Hoboken, N.J: 2003. pp. 329–481. [Google Scholar]
- 21.Wang Y, Tang K, Inan S, Siebert DJ, Holzgrabe U, Lee DYW, Huang P, Li JG, Cowan A, Liu-Chen LY. J Pharmacol Exp Ther. 2005;312:220. doi: 10.1124/jpet.104.073668. [DOI] [PubMed] [Google Scholar]
- 22.Tidgewell K, Harding WW, Lozama A, Cobb H, Shah K, Kannan P, Dersch CM, Parrish D, Deschamps JR, Rothman RB, Prisinzano TE. J Nat Prod. 2006;69:914. doi: 10.1021/np060094b. [DOI] [PubMed] [Google Scholar]
- 23.Lever JR. Curr Pharm Des. 2007;13:33. doi: 10.2174/138161207779313821. [DOI] [PubMed] [Google Scholar]
- 24.Talbot PS, Narendran R, Butelman ER, Huang Y, Ngo K, Slifstein M, Martinez D, Laruelle M, Hwang DR. J Nucl Med. 2005;46:484. [PubMed] [Google Scholar]
- 25.Butelman ER, Ko MC, Traynor JR, Vivian JA, Kreek MJ, Woods JH. J Pharmacol Exp Ther. 2001;298:1049. [PubMed] [Google Scholar]
- 26.Müller K, Faeh C, Diederich F. Science. 2007;317:1881. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
- 27.Dunitz JD, Taylor R. Chem Eur J. 1997;3:89. doi: 10.1002/chem.19970030115. [DOI] [Google Scholar]
- 28.Scheerer JR, Lawrence JF, Wang GC, Evans DA. J Am Chem Soc. 2007;129:8968. doi: 10.1021/ja073590a. [DOI] [PubMed] [Google Scholar]
- 29.Matsuya Y, Yu Z, Yamamoto N, Mori M, Saito H, Takeuchi M, Ito M, Nemoto H. Bioorg Med Chem. 2005;13:4383. doi: 10.1016/j.bmc.2005.04.056. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information available: 1H and 13C NMR spectra for compounds 5–20; IUPAC International Chemical Identifiers (InChIs); statement of author contributions.