

Abstract
Checkpoints are evolutionarily conserved signaling mechanisms that arrest cell division and alter cellular stress resistance in response to DNA damage or stalled replication forks. To study the consequences of loss of checkpoint functions in whole animals, checkpoint genes were inactivated in the nematode C. elegans. We show that checkpoint proteins are not only essential for normal development but also determine adult somatic maintenance. Checkpoint proteins play a role in the survival of postmitotic adult cells.
Many DNA damage checkpoint and repair proteins are essential for development. We identified a checkpoint gene in a genetic screen for whole-organism stress resistance. The predicted protein, CID-1, has homology to poly(A)+ polymerase (PAP) domain proteins (fig. S1), including the fission yeast Schizosaccharomyces pombe S-M checkpoint protein caffeine-induced death (Cid) protein–1 (1–3). In C. elegans, we observed that worms carrying the cid-1(rf34::Tc4) mutation or worms with reduced expression of cid-1 by RNA interference (RNAi) were highly thermotolerant (Fig. 1, A and B, and table S1). It was not obvious why checkpoint proteins would affect organismal stress resistance in C. elegans where the soma is composed of nondividing cells. Consequently, we further investigated the extent to which cid-1 determined cell survival in worms and whether it was related to checkpoint functions.
Fig. 1.
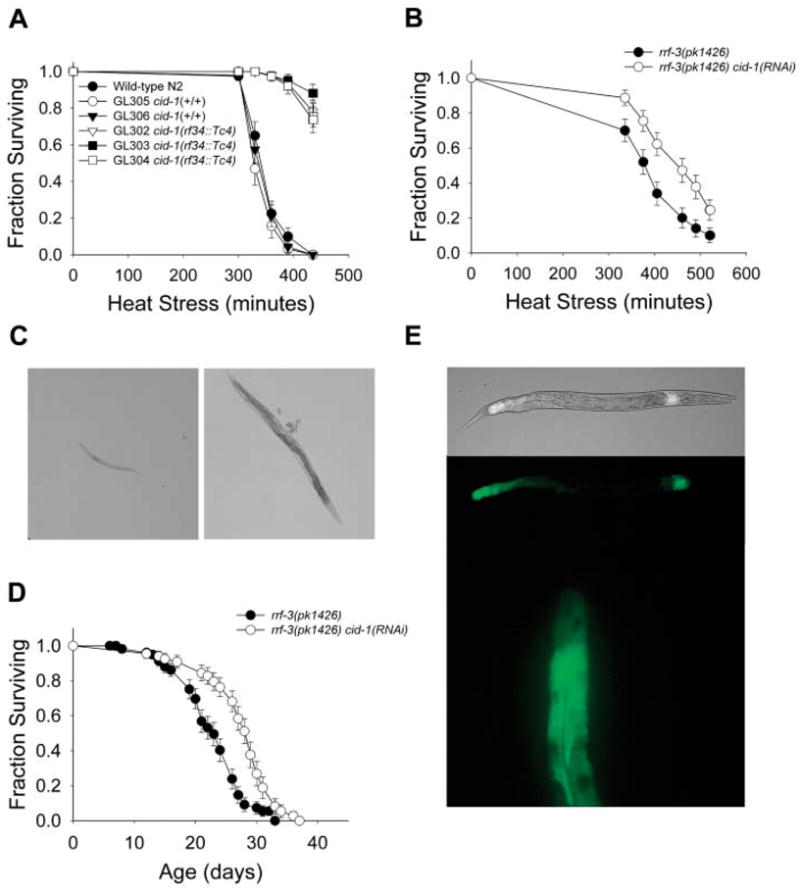
CID-1 regulates stress resistance. (A) Longitudinal thermotolerance assay of cid-1(rf34::Tc4) mutant lines. (+/+) indicates wild-type cid-1 gene. (B) Longitudinal thermotolerance assay of rrf-3(pk1426) cid-1(RNAi) worms. The rrf-3(pk1426) mutation renders worms more sensitive to RNAi. The complete thermotolerance data can be found in table S1. (C) HU-resistance assay. (Left) Wild-type N2 worms showed arrested development at the L1/L2 stage when eggs were placed on HU plates. (Right) cid-1(rf34::Tc4) mutants develop into fertile adults at the same concentration of HU. (D) Life span of rrf-3(pk1426) cid-1(RNAi) worms at 20°C. (E) Transgenic worms expressing a CID-1::GFP gene fusion. (Top) Composite fluorescent and bright-field micrographs and corresponding fluorescent micrographs of the fusion protein localized to the first layer of intestinal cells (Int 1) and to posterior intestinal cells. (Bottom) A high-resolution fluorescent micrograph of the fusion protein localized to the cytoplasm of the posterior intestinal cells.
The ribonucleotide reductase inhibitor hydroxyurea (HU) causes stalled replication forks. Wild-type worms had arrested development in the presence of HU, but cid-1(rf34::Tc4) mutants and cid-1(RNAi) worms developed into adults (Fig. 1C). This is consistent with C. elegans CID-1 having a checkpoint function. It is also required for normal development, as cid-1 mutants had shorter, thicker gonads with fewer proliferating and developing germ cells. The worms contained disorganized embryos, produced fewer offspring, and took 24 hours longer to develop into reproductive adults compared with wild-type worms (fig. S2 and table S2). Of cid-1(rf34::Tc4) adult worms examined, 20 to 40% developed a protruding vulval defect that blocked egg laying and resulted in premature death.
Because a number of genes that limit C. elegans’s life span also regulate stress resistance (4), CID-1 could play a role in somatic maintenance during normal aging. Although most cid-1(rf 34::Tc4) mutant worms exhibited premature nonsenescent deaths resulting from their developmental abnormalities, we observed a significant increase in the life span of cid-1(RNAi) worms (Fig. 1D and table S3).
In C. elegans, heat shock proteins (HSPs) influence survival (5, 6). Therefore, we tested whether cid-1(RNAi) worms had altered hsp gene expression. cid-1 RNAi induced expression of hsp-4, which encodes a predicted homolog of the mammalian endoplasmic reticulum chaperone BiP and also increases expression of the Mn superoxide dismutase sod-3 specifically in vulval tissues (Fig. 2). Although the hsp-4 chaperone is induced by the unfolded protein response (UPR), expression of another UPR-associated chaperone, HSP-16 (7, 8), was not up-regulated (Fig. 2). In addition, RNAi of hsp-4 resulted in sick, short-lived adults. Thus, hsp-4 is required for normal survival and is regulated by cid-1, independent of the UPR (table S3). Both hsp-4 and cid-1 are expressed in the postmitotic intestinal cells (Fig. 1E and Fig. 2) during development and adulthood, consistent with a role on postmitotic cell survival.
Fig. 2.
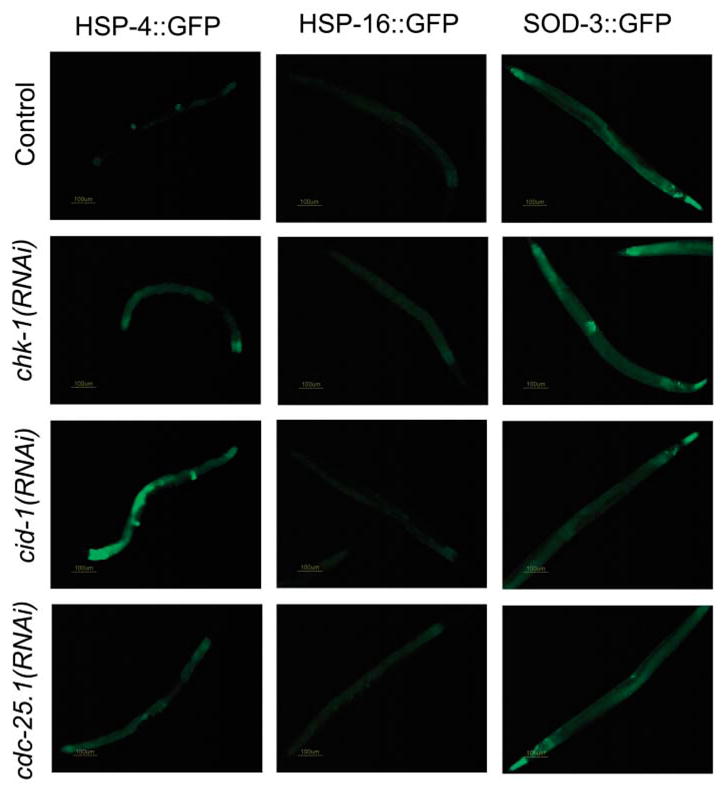
Checkpoint protein regulation of stress genes in postmitotic cells. (Left) Expression pattern of hsp-4::GFP in control, chk-1(RNAi), cdc-25.1(RNAi), and cid-1(RNAi) worms. Elevated levels of hsp-4::GFP were observed in the intestine, particularly in the first layer of intestinal cells, when either component of the pathway was inactivated. (Middle) Expression pattern of hsp-16::GFP in control, chk-1(RNAi), cdc-25.1(RNAi), and cid-1(RNAi) worms. (Right) Expression pattern of sod-3::GFP in control, chk-1(RNAi), cdc-25.1(RNAi), and cid-1(RNAi) worms. Inactivation of cid-1/chk-1/cdc-25.1 results in elevated levels of sod-3::GFP in the vulva. The pictures are representative of results observed in three independent experiments.
We wished to determine whether other checkpoint proteins affect organismal survival. In S. pombe, cell-cycle progression requires activation of a cyclin-dependent kinase (Cdk) complex by the phosphatase Cdc25, which is regulated by several kinases including the serine-threonine kinase Chk1 (9). In C. elegans, cid-1 interacts genetically with chk-1 and members of the cdc-25 gene family. chk-1 is active in the S-M and DNA repair checkpoints (10) and is a DNA-replication checkpoint coupling cell-cycle length to asymmetric cell division of the two-cell embryo (11). chk-1 and cid-1 interacted genetically to determine normal embryonic development, and cid-1 and cdc-25.1 interacted to determine normal hermaphrodite self-fertility (fig. S3).
The offspring of worms treated with chk-1 RNAi are viable but sterile (fig. S3). In characterizing the somatic maintenance of these offspring, we observed that chk-1 RNAi caused large increases in organismal resistance to thermal stress (Fig. 3A and table S1). In addition, the life span of chk-1(RNAi) worms increased by 15 to 25% (Fig. 4, A to C, and table S3). chk-1 RNAi also induced hsp-4 expression (Fig. 2).
Fig. 3.
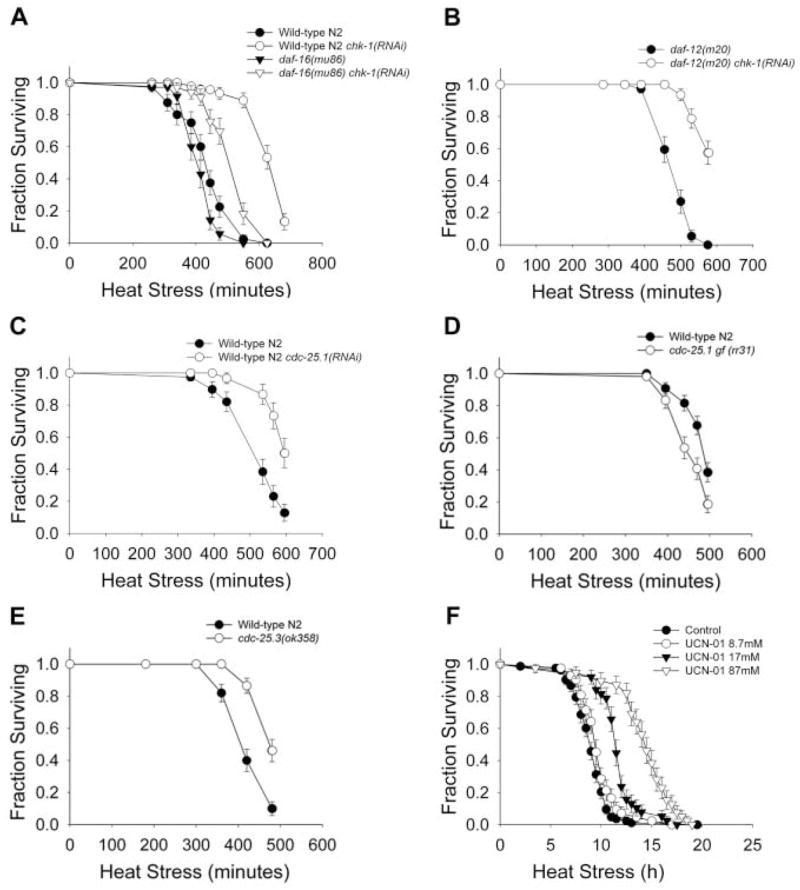
Inactivation of chk-1 and cdc-25 confers organismal stress resistance. Longitudinal thermotolerance assays. (A) Thermotolerance of wild-type N2 and daf-16(mu86) mutant worms treated with chk-1 RNAi. (B) Thermotolerance of daf-12(m20) chk-1(RNAi) worms. (C) Thermotolerance of cdc-25.1(RNAi) worms. (D) Thermosensitivity of cdc-25.1(rr31) I; rrIs1 gain-of-function mutant worms. (E) Thermotolerance of cdc-25.3(ok358) mutant worms. (F) Thermotolerance of wild-type N2 worms treated with UCN-01 during adulthood. Our thermotolerance data can be found in table S1.
Fig. 4.
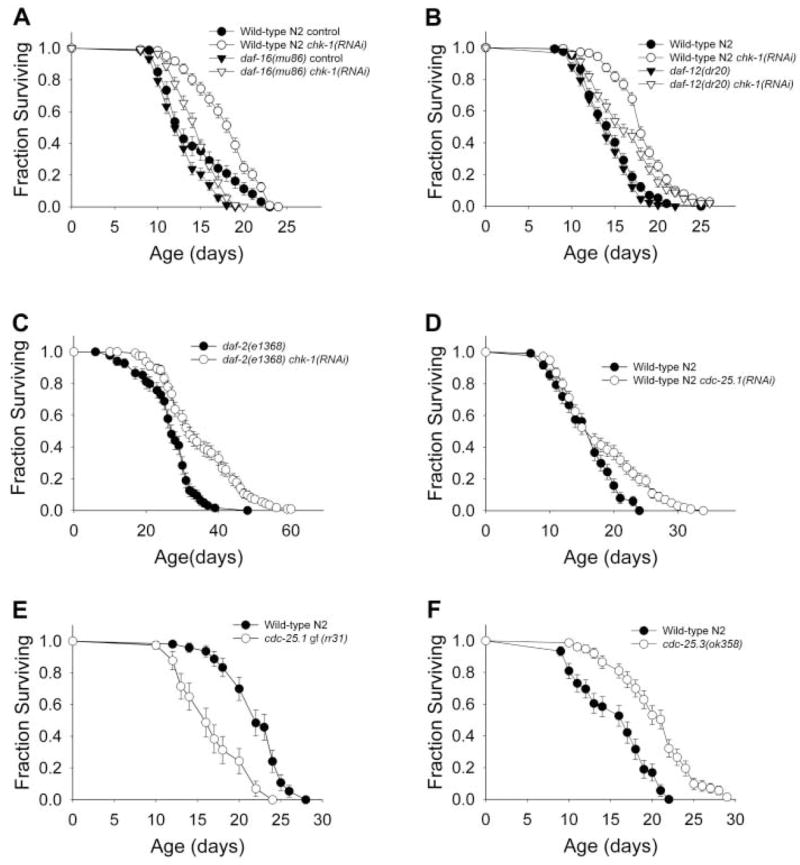
Inactivation of chk-1 and cdc-25 confers increased life span independent of germline signal. (A) Life span of wild-type N2 and daf-16(mu86) mutant worms treated with chk-1 RNAi at 25°C. (B) Life span of chk-1 RNAi treated wild-type N2 and daf-12(m20) mutant worms at 25°C. (C) Life span of chk-1 RNAi treated daf-2(e1368) mutant worms at 20°C. (D) Life span of cdc-25.1(RNAi) worms at 25°C. (E) Life span of cdc-25.1(rr31) I; rrIs1 gf mutant worms at 20°C. (F) Life span of cdc-25.3(ok358) mutant worms at 25°C. All the survival data can be found in table S3.
To examine the temporal requirements for CHK-1 regulation of stress responses, we used the CHK-1 chemical inhibitor UCN-01. When worms were treated throughout development, their growth was arrested before they reached adulthood. However, treatment of young adults resulted in a dose-dependent increase in thermotolerance (Fig. 3F). Therefore, inhibition of CHK-1 function during adulthood is sufficient to confer increased somatic maintenance.
After the molt into an adult worm, cell division is confined to the germ line (12). Therefore, CHK-1 could influence somatic maintenance through its action in the germline stem cells. Ablation of germline precursor cells leads to organismal stress resistance and life-span extension through a mechanism requiring the forkhead (FOXO) transcription factor DAF-16 and also the nuclear hormone receptor DAF-12 (13–16). Thermotolerance of chk-1(RNAi) worms was not dependent on DAF-16 or DAF-12 (Fig. 3, A and B, and table S1). Likewise, life-span extension caused by RNAi of chk-1 was observed in both daf-16 and daf-12 mutant backgrounds (Fig. 4, A and B, and table S3). The magnitude of life-span extension was smaller in the daf-12 mutant worms, which suggests that the effect of the checkpoint proteins may be partially dependent on cycling germ cells. Exposure to stress or germline ablation causes DAF-16 translocation from the cytoplasm to the nucleus (16–18). We examined the localization of a green fluorescent protein reporter, DAF-16::GFP, in chk-1(RNAi) worms and observed DAF-16 in the cytoplasm of intestinal cells (fig. S4). However, after a heat stress, RNAi of chk-1 did not block translocation of DAF-16 to the nucleus, which suggested that CHK-1 activity is independent of DAF-16. chk-1 RNAi also extended the life span of an already long-lived insulin receptor–like mutant daf-2(e1368) (19, 20) (Fig. 4C). Taken together, these results suggest CHK-1 regulates somatic maintenance through processes different from the previously described germline signal and the insulin signaling pathway.
Because cid-1 genetically interacts with cdc-25.1, the generality of checkpoint proteins influencing survival was examined. In mammalian cells, CHK-1 targets CDC25A for degradation by phosphorylation in response to DNA damage (21). There are four Cdc25 homologs in C. elegans, with the cdc-25.1 gene predominantly expressed in the germ line (22), where it is required for proliferation (23). Inactivation of cdc-25.1 causes embryonic lethality, with some surviving embryos developing into sterile adults with abnormal germ lines. Sterile chk-1(RNAi) worms were similar in appearance to worms with inactivated cdc-25.1 (24) (fig. S3). RNAi of cdc-25.1 resulted in increased thermotolerance and life-span extension (Figs. 3C and 4D). In addition, worms carrying a cdc-25.1 gain-of-function mutation were thermosensitive (Fig. 3D and table S1) and short-lived (Fig. 4E and table S3). Inactivation of isoforms cdc-25.2 and cdc-25.3 by RNAi also conferred increased thermotolerance, whereas loss of cdc-25.4 had no effect (table S1). cdc-25.3(RNAi) worms had significantly increased life span (table S3) and, like cid-1(RNAi) worms, increased expression of hsp-4 in the intestine and sod-3 in the vulva (Fig. 4). A cdc-25.3 knockout (KO) mutant displayed both significantly increased thermotolerance (Fig. 3E) and 40% extended life span (Fig. 4F and table S3). The cdc-25.3 KO mutants also exhibited many of the phenotypes resulting from lack of cid-1 and chk-1 gene products, including delayed development and reduced fertility, which suggests that these genes act in either a single or functionally related pathway. Thus, at least three cdc-25 genes, chk-1, and cid-1 affect survival, consistent with the general notion that checkpoint proteins have a role of somatic maintenance.
The separation between postmitotic soma and mitotic germ line in C. elegans has provided an opportunity to study the roles of checkpoint functions in postmitotic cells in a whole organism. Because postmitotic tissues do not form tumors in C. elegans, the effects of checkpoint proteins on acute stress survival and normal life span have been uncovered. The notion that checkpoint proteins function in postmitotic cells suggests a checkpoint surveillance function that is unrelated to the decision to undergo cell division but still regulates cell survival. Checkpoints act as tumor suppressor pathways in mammals in part by promoting cellular senescence. A role in normal aging has been hypothesized; mice with hyperactive p53 exhibit signs of accelerated aging consistent with a trade-off between checkpoint fidelity and tissue regeneration (25, 26), and decreasing p53 activity in fruit fly neurons extends life span (27). This is consistent with the prediction that reduced checkpoint competence leads to reduced rates of tissue aging (28). Our observation indicates that postmitotic cell survival is also regulated by checkpoint proteins, and thus, a trade-off between checkpoint function and cellular maintenance is likely in both mitotic and postmitotic tissues. This prompts the question whether specific alleles of checkpoint proteins in humans may place individuals at risk for some cancers but protect them against other age-associated diseases. It also appears critical that neurons are maintained in a noncycling, terminally differentiated state to avoid apoptosis. Checkpoint proteins are likely to have a role in maintaining neurons in a postmitotic state; they may also influence intrinsic resistance to stress.
Supplementary Material
www.sciencemag.org/cgi/content/full/312/5778/1381/DC1 Materials and Methods
References and Notes
- 1.Wang SW, Toda T, MacCallum R, Harris AL, Norbury C. Mol Cell Biol. 2000;20:3234. doi: 10.1128/mcb.20.9.3234-3244.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Saitoh S, et al. Cell. 2002;109:563. doi: 10.1016/s0092-8674(02)00753-5. [DOI] [PubMed] [Google Scholar]
- 3.Wang L, Eckmann CR, Kadyk LC, Wickens M, Kimble J. Nature. 2002;419:312. doi: 10.1038/nature01039. [DOI] [PubMed] [Google Scholar]
- 4.Kenyon C. Cell. 2005;120:449. doi: 10.1016/j.cell.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 5.Walker GA, Lithgow GJ. Aging Cell. 2003;2:131. doi: 10.1046/j.1474-9728.2003.00045.x. [DOI] [PubMed] [Google Scholar]
- 6.Yokoyama K, et al. FEBS Lett. 2002;516:53. doi: 10.1016/s0014-5793(02)02470-5. [DOI] [PubMed] [Google Scholar]
- 7.Calfon M, et al. Nature. 2002;415:92. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- 8.Urano F, et al. J Cell Biol. 2002;158:639. doi: 10.1083/jcb.200203086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Melo J, Toczyski D. Curr Opin Cell Biol. 2002;14:237. doi: 10.1016/s0955-0674(02)00312-5. [DOI] [PubMed] [Google Scholar]
- 10.Kalogeropoulos N, Christoforou C, Green AJ, Gill S, Ashcroft NR. Cell Cycle. 2004;3:1196. [PubMed] [Google Scholar]
- 11.Brauchle M, Baumer K, Gonczy P. Curr Biol. 2003;13:819. doi: 10.1016/s0960-9822(03)00295-1. [DOI] [PubMed] [Google Scholar]
- 12.Sulston JE, Horvitz HR. Dev Biol. 1977;56:110. doi: 10.1016/0012-1606(77)90158-0. [DOI] [PubMed] [Google Scholar]
- 13.Ogg S, et al. Nature. 1997;389:994. doi: 10.1038/40194. [DOI] [PubMed] [Google Scholar]
- 14.Lin K, Dorman JB, Rodan A, Kenyon C. Science. 1997;278:1319. doi: 10.1126/science.278.5341.1319. [DOI] [PubMed] [Google Scholar]
- 15.Hsin H, Kenyon C. Nature. 1999;399:362. doi: 10.1038/20694. [DOI] [PubMed] [Google Scholar]
- 16.Lin K, Hsin H, Libina N, Kenyon C. Nat Genet. 2001;28:139. doi: 10.1038/88850. [DOI] [PubMed] [Google Scholar]
- 17.Henderson ST, Johnson TE. Curr Biol. 2001;11:1975. doi: 10.1016/s0960-9822(01)00594-2. [DOI] [PubMed] [Google Scholar]
- 18.Libina N, Berman JR, Kenyon C. Cell. 2003;115:489. doi: 10.1016/s0092-8674(03)00889-4. [DOI] [PubMed] [Google Scholar]
- 19.Kenyon C, Chang J, Gensch E, Rudner A, Tabtiang R. Nature. 1993;366:461. doi: 10.1038/366461a0. [DOI] [PubMed] [Google Scholar]
- 20.Kimura KD, Tissenbaum HA, Liu Y, Ruvkun G. Science. 1997;277:942. doi: 10.1126/science.277.5328.942. [DOI] [PubMed] [Google Scholar]
- 21.Mailand N, et al. Science. 2000;288:1425. doi: 10.1126/science.288.5470.1425. [DOI] [PubMed] [Google Scholar]
- 22.Ashcroft NR, Srayko M, Kosinski ME, Mains PE, Golden A. Dev Biol. 1999;206:15. doi: 10.1006/dbio.1998.9135. [DOI] [PubMed] [Google Scholar]
- 23.Ashcroft N, Golden A. Genesis. 2002;33:1. doi: 10.1002/gene.10083. [DOI] [PubMed] [Google Scholar]
- 24.Clucas C, Cabello J, Bussing I, Schnabel R, Johnstone IL. EMBO J. 2002;21:665. doi: 10.1093/emboj/21.4.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tyner SD, et al. Nature. 2002;415:45. doi: 10.1038/415045a. [DOI] [PubMed] [Google Scholar]
- 26.Maier B, et al. Genes Dev. 2004;18:306. doi: 10.1101/gad.1162404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bauer JH, Poon PC, Glatt-Deeley H, Abrams JM, Helfand SL. Curr Biol. 2005;15:2063. doi: 10.1016/j.cub.2005.10.051. [DOI] [PubMed] [Google Scholar]
- 28.Wodarz D. Oncogene. 2004;23:7799. doi: 10.1038/sj.onc.1207833. [DOI] [PubMed] [Google Scholar]
- 29.We thank M. Benedetti and A. Foster for help with the UCN-01 thermotolerance assays; M. Lund and the members of the Lithgow Laboratory, the Kapahi Laboratory, J. Campisi, C. Benz, and D. Bhaumik for helpful discussions; and J. Ahringer and the Medical Research Council gene service for providing the RNAi library. UCN-01 was made available from the Drug Synthesis and Chemistry Branch, Developmental Therapeutics Program, National Cancer Institute. A.O. was supported by the Danish Research Academy and the Danish Cancer Society. G.J.L. was supported by NIH AG21069, AG22868, and NS050789-01; the Ellison Medical Foundation; the Glenn Foundation for Medical Research; the Biotechnology and Biological Sciences Research Council, Science of Ageing Initiative; and the Herbert Simon Family Medical Foundation.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
www.sciencemag.org/cgi/content/full/312/5778/1381/DC1 Materials and Methods