

Abstract
Olfactory sensory neurons (OSNs) represent a unique population of neurons in which death and regeneration are ongoing throughout adulthood, a feature that makes them an attractive model cell type for the investigation of neuronal death. However, the mechanism by which OSNs die remains elusive. Therefore, we developed a culture system for studying pathways involved in OSN death. Here, we show that inhibition of transcription or translation, by actinomycin D or cycloheximide, respectively, suppresses pathways leading to death, prolonging the survival of OSNs in culture. We discovered that caspase activity and jun N-terminal kinase (JNK) signaling both play a role in OSN death, and inhibition of JNK activity suppresses effector caspase (caspase-3) activation. Results from studies in culture were confirmed in vivo, in a mouse bulbectomy-induced OSN death model. These findings provide new insights into the nature of OSN death and a means of studying OSNs in vitro.
Keywords: Olfactory sensory neuron, neuronal death, culture, bulbectomy, jun N-terminal kinase
Introduction
Olfactory sensory neurons (OSNs) reside in the olfactory epithelium (OE) and serve to detect odors in the environment. These neurons appear to have a limited life span and are continually replaced throughout adulthood (Magrassi and Graziadei, 1995; Paternostro and Meisami, 1994). The mechanism of OSN death is intriguing, as few neuronal populations in the mammalian brain are replenished in this manner; granule and periglomerular cells in the olfactory bulb, and granule cells in the dentate gyrus of the hippocampus are the only other known examples (Kaplan and Hinds, 1977). Of these regions where neuronal turnover persists at high levels into adulthood, the olfactory epithelium is unique in that it is a stratified epithelium containing both progenitor cells and the neurons they give rise to. Therefore, homeostasis of this tissue requires tight control over both death and regeneration. While adult olfactory neurogenesis seems to share mechanisms with olfactory development and with the development of other neurons (Shetty et al., 2005a; Shetty et al., 2005b), adult OSN death appears distinct from developmental neuronal death, suggesting an alternative pathway to those described in development. Here, we seek to understand mechanisms governing the death of this population of neurons; our approach was to screen for factors that block signaling pathways that could lead to OSN death, thus enabling survival.
Programmed cell death occurs at high levels throughout the organism, including the brain, during development; in the nervous system, this death occurs as neuronal connections are established and circuits form. An initial overproduction of neurons is culled as much as half in some brain tissues, as neurons compete for trophic support from their targets; those that fail to form connections die due to a lack of survival signals (Gordon, 1995; Levi-Montalcini and Booker, 1960; Wang and Tessier-Lavigne, 1999). Immature olfactory sensory neurons, which are at a stage in the neuronal lineage prior to functional olfactory receptor expression, mostly die within two to four weeks of birth (Hinds et al., 1984). However, a steady level of death and replacement of mature sensory neurons persists into adulthood after the growth rate of the epithelium has decreased substantially (Fung et al., 1997; Weiler and Farbman, 1997).
While several growth factors and their receptors are expressed throughout the OE (Carter and Roskams, 2002), their effects on survival seem to be primarily during development. Neonatal mice show increased OSN death when neurotrophin-3 (NT3) is deleted, and on postnatal day 0.5, NT3 is protective of cultures of rat OSNs. However, neither NT3 nor other growth factors, including nerve growth factor (NGF) and brain derived neurotrophic factor (BDNF), are effective at preventing OSN death in adult murine olfactory epithelial cultures (Simpson et al., 2002). Thus, the mechanisms governing the ongoing elimination of OSNs in the adult brain appear to be distinct from those governing the elimination of neurons during development.
Multiple hypotheses have been proposed to explain the ongoing death of these sensory neurons. First, due to their position in the nasal cavity, where they are in contact with the environment, OSNs are susceptible to injury. Naris occlusion, which protects OSNs from the environment, does appear to result in an extended lifespan. However, naris occlusion does not inhibit death altogether (Benson et al., 1984; Chess et al., 1992), suggesting an intrinsic process may also govern OSN death. Second, OSN death may be regulated by odorant exposure; under the theory that neuronal turnover provides a mechanism for plasticity, selective death would allow an organism to adapt to changes in a complex and dynamic olfactory environment. Unfortunately, effects of odorant stimulation on survival have been mixed: while one group found a selective enhancement of survival of sensory neurons expressing an activated OR (Watt et al., 2004), a more recent study demonstrated that prolonged odorant exposure induces excitotoxic OSN death in culture after only 3 hours (Brauchi et al., 2006). Thus, it seems likely that the ongoing death of OSNs in the adult brain is intrinsically programmed; defining the mechanism of this form of programmed cell death would therefore be of interest.
A technique that has been useful in elucidating mechanisms of OSN death in vivo involves the removal of their targets in the rostral forebrain – the olfactory bulb. This procedure, known as olfactory bulbectomy (OBX), leads to a retrograde wave of death of all OSNs from which the ablated bulb received input (Costanzo and Graziadei, 1983). OSN death following OBX is accelerated and synchronized, and followed temporally by increased progenitor cell division to re-populate the epithelium, thus providing a model for studying widespread changes that accompany death and regeneration in the olfactory epithelium (Calof et al., 1998; Carr and Farbman, 1992).
Apoptosis is a prevalent form of cell death, and one that is frequently genetically and developmentally programmed. In general, there are two main pathways for apoptosis: the extrinsic death receptor pathway, and the intrinsic mitochondrial pathway. In the former, a death receptor ligand binds to and activates a death receptor on the cell surface leading to the cleavage and activation of caspase-8, the initiator caspase of the extrinsic death pathway. Caspase-8 goes on to cleave and activate caspase-3, the executioner of apoptosis. In the intrinsic form of apoptosis, pro-apoptotic proteins of the Bcl-2 family including Bax and Bak accumulate on the mitochondrial surface and form pores in the outer membrane. The mitochondrial outer membrane is disrupted releasing proteins from the mitochondrial intermembrane space. Cytochrome c is among the proteins released and it interacts with apoptotic protease activating factor 1 (Apaf-1) and caspase-9 to form the apoptosome. When in the apoptosome caspase-9 is active and it cleaves and activates executioner caspase-3 (Hengartner, 2000). While OSN death demonstrates some characteristics of apoptotic cell death, it appears that additional undefined pathways are in operation. Sequential activation of caspases-3 and 9 has been documented in the olfactory neuronal layer following OBX, and the extent of OSN death following OBX decreases in caspase-3-null mice (Cowan et al., 2001), however pan-caspase inhibitors are ineffective at protecting OSNs from death in culture (personal communication, Dr. Glennis Matthews). Caspase 3 activation and OE thinning, due to OSN loss following OBX, was reduced in the absence of the pro-apoptotic protein, Bax. However, even in these bax knock-out mice, initial death of OSNs occurs within the first 24 hours post-OBX (Robinson et al., 2003).
Conversely, in transgenic mice in which mature OSNs overexpress the anti-apoptotic protein Bcl-2, OSNs proceed to die following OBX. However, OSNs generated post-OBX experience an extended lifespan in the absence of the bulb when compared to non-transgenic animals, perhaps due to increased Bcl-2 levels (Hayward et al., 2004). Microarray analyses also show upregulation of several pro-apoptotic mRNAs one-day post OBX, when death of OSNs is initiated, accompanied by down-regulation of anti-apoptotic factors (Shetty et al., 2005a). These changes shift the intracellular balance in favor of apoptosis.
Taken together, these studies point to a multi-faceted program regulating the death of olfactory sensory neurons in the adult mouse. While classic apoptotic death appears to be a component of the OSN death program, other elements remain to be discovered. By probing the mechanism of death operating within this unique population of neurons, we may discover new mechanisms of programmed neuronal death with potential relevance to neurodegeneration. Here, we show that OSNs die through an active process, involving caspase activity and mediated by JNK signaling.
Results
Neuronal Survival Assay
In order to assess the viability of olfactory sensory neurons (OSNs) following dissociation, we utilized OMP-GFP mice (Potter et al., 2001). In this knock-in mouse line, enhanced GFP-1 (green fluorescent protein) was inserted into an OMP (olfactory marker protein) -targeting vector and is transcribed under the control of the OMP promoter. A confocal image of a 16-µm section through the intact olfactory epithelium (OE) shows GFP expression in the mature neuronal layer, and in the dendritic knobs of these neurons (Fig 1A). However, when the olfactory epithelium is dissociated for culturing, individual cell types are no longer discernable by their position in the structured epithelium. Therefore, we took advantage of the fact that OMP expression is confined to mature OSNs (Sydor et al., 1986), so OSNs can be identified as the only cells in culture producing a GFP signal. We verified that GFP expression is in fact confined to mature OSNs, by labeling with an antibody against OMP (data not shown). We saw that 100% of GFP-positive cells are OMP-positive (n=438), while an additional 27% of OMP-positive cells are GFP-negative. This discrepancy could be due to some non-specific labeling by the OMP antibody, or quenching of some of the GFP signal in the staining procedure; nonetheless, the number of GFP-positive cells provides a useful estimate of the number of mature OSNs.
Figure 1. GFP is expressed in mature olfactory sensory neurons of OMP-GFP knock-in mice.
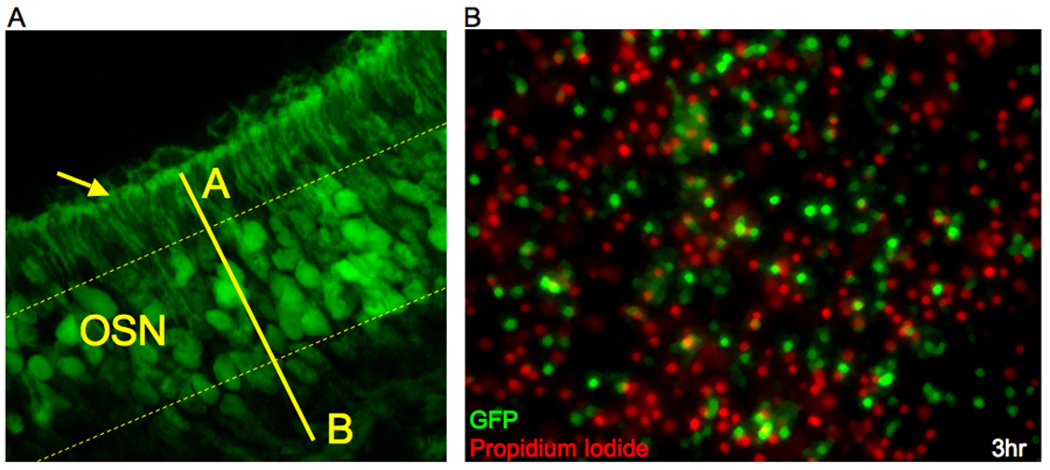
(a) Coronal section though the olfactory epithelium of a transgenic adult mouse in which the transcription of GFP has been placed under the control of the OMP promoter. GFP expression is visible in the soma of neurons within the mature neuronal layer, labeled OSN between dashed yellow lines, and in their dendritic knobs (arrow) that protrude from the apical surface of the epithelia (A = apical; B = basal). (b) Dissociated cells from the olfactory epithelium of an adult OMP-GFP mouse. Mature sensory neurons retain GFP expression (green) while they are alive, and dead or dying cells within the culture stain positive for propidium iodide (PI, red) and no longer express GFP. While there are high numbers of both PI+ cells and GFP+ cells, there is very little overlap of the two labels, indicating that OSNs no longer express GFP as they are dying: of 310 GFP+ cells counted, only 1.6% of those GFP+ cells stained positive for PI. Therefore the GFP signal can be used as an indicator of live mature OSNs.
Because we were interested in monitoring only live mature OSNs, we verified that the GFP signal is no longer produced from OSNs once they have died by performing a test for propidium iodide (PI) exclusion. PI is a nuclear stain that is only able to enter a cell once the plasma membrane has been permeabilized; therefore PI staining may be used as an assay for dead or dying cells (Darzynkiewicz et al., 1992). We found that cells expressing GFP do not also stain positive for PI: only 1.6% of GFP+ cells (n=310) were also PI+, while the vast majority of GFP+ cells, 98.4%, excluded PI (Fig 1B). Therefore, counts of GFP+ cells remaining under each treatment condition provide accurate estimates of live OSNs. No differences in rate of death were detected between OSNs from homozygous and heterozygous OMP-GFP mice, so homozygotes were used due to their increased production of GFP signal and ease of breeding.
Active synthesis of new proteins is required for OSN death
While untreated neurons from dissociated olfactory epithelium proceeded to die within 48 hours in culture, treatment with RNA and protein synthesis inhibitors prolonged their survival. Actinomycin D (ActD), an inhibitor of transcription, and cycloheximide (CHX), an inhibitor of translation, significantly prolonged the survival of OSNs at extended time points in culture (Fig 2A). GFP+ cells remained dense at 48 hours of treatment with either inhibitor, while few GFP+ cells remained when the same initial number of total cells plated was untreated (Fig 2B). This shift in the time course of death demonstrates that active expression of newly synthesized proteins contributes to the execution of death following OE dissociation. If this increased protein expression is prevented, OSNs survive longer, and then eventually die in the absence of protein synthesis.
Figure 2. Suppressors of protein synthesis prolong survival of OSNs in culture.

(a) The majority of olfactory neurons die within 48 hours in culture when untreated (control). Cells treated with transcriptional inhibitor, actinomycin D (80 ng/ml) or translational inhibitor, cycloheximide (3 µM) remain alive in culture significantly longer in comparison to no treatment (* p<.05; **p<.01; ***p<.001; N=4). (b) Images of GFP signal produced from live OSNs in 48 hour cultures of from left to right, untreated, actinomycin D, and cycloheximide treated cultures.
OSN death in culture – temperature dependence
We found that survival of OSNs is prolonged when cells are cultured at 33 °C, the temperature of the nasal mucosa, rather than 37 °C, the typical cell culture temperature (Fig 3). When cultured at 37°C, most OSNs died within 48 hours; few GFP+ cells were observed at that time. However, when the incubation temperature was lowered to 33°C, survival of OSNs was extended significantly by 24 hours, continuing to 72 hours. This effect was observed at 30°C as well, with a significant additional benefit to survival over 33°C at 48 and 72 hours. Separate studies in which the time course of death in other cell types is observed over the same temperature range have not revealed such a shift in survival with decreasing temperature (data not shown). The temperature-dependence of OSN death seems to be a distinct characteristic of these cells, as opposed to a universal slowing of metabolic processes and cell death with decreased temperature. This finding aided our search for regulators of OSN death, because at 37°C protein synthesis inhibitors, which block active mechanisms, were the only agents that were effective at protecting OSNs from death. However, when screening for rescue at 33°C, we were able to detect a further enhancement of survival with specific inhibitors.
Figure 3. Survival of OSNs in culture is prolonged by decreased incubation temperature.

While the majority of OSNs die within 48 hours of incubation at 37°C, significantly more OSNs survive at extended time points when cultured at 33°C or 30°C. A difference in survival between neurons cultured at 33°C versus 30°C becomes significant at 48 and 72 hours (* p<.01; **p<.001; N=5).
A role for JNK signaling and caspase activity
With this modified cell culture system, we tested inhibitors of known mechanisms related to cell death, including apoptosis, necrosis, and autophagy, as well as trophic factors and inhibitors of numerous kinases, to identify components of the OSN death machinery (Sup. Table 1). While none of the trophic factors or inhibitors of various death pathways were effective, one kinase inhibitor, that of jun N-terminal kinase (JNK), SP600125, showed significant extension of OSN survival (Fig 4A). In order to verify this mechanism, we tested another JNK inhibitor: JNK inhibitor VIII (JIVIII). This inhibitor was also protective (Fig 4A). The fact that both JNK inhibitors prolong OSN survival decreases the likelihood that one or the other is protecting through an off-target effect.
Figure 4. Pan-caspase and JNK inhibitors delay death of OSNs.

(a) Survival of OSNs is extended when treated with pan-caspase inhibitors Boc-D-FMK (boc, 45 µM) and QVD-OPH (qvd, 3 µM) or JNK inhibitors, JNK Inhibitor VIII (j8, 5 µM) or SP600125 (sp, 5 µM) when compared to the control no treatment condition (nt). (b) Combinatorial treatments of each caspase inhibitor paired with each JNK inhibitor lead to further protection over either caspase or JNK inhibitor. Combinatorial effects were most significant by 96 hours in culture (* p<.05; **p<.01; ***p<.001; N=4).
To further validate the role of JNK signaling in OSN death, we monitored JNK activity by probing for the phosphorylation of a downstream target, the transcription factor c-jun. With an antibody directed against phosphorylated c-jun, we found that the level of phosphorylation was low initially. However, by 18 hours in culture, phospho-c-jun levels increased 3-fold; this increase was prevented with treatment by JIVIII (Fig 5A). Because caspase activity has been shown to accompany OSN death in vivo following olfactory bulbectomy (Cowan et al., 2001), we probed for activation of caspase-3 and cleavage of poly(ADP-Ribose) polymerase (PARP1), a target of caspase-3. We found that inhibition of JNK activity inhibited caspase-3 activation and the subsequent cleavage of PARP1 (Fig 5C). The levels of both the 46 and 54kD JNK family members, all of which were identified for their ability to phosphorylate c-jun, were unchanged (Fig 5B). Hence, protection from death can be attributed to inhibition of kinase activity of JNK already present in OSNs. JNK inhibition prevents the phosphorylation of c-jun, thereby suppressing its downstream transcriptional activity.
Figure 5. JNK inhibition prevents both c-jun phosphorylation and caspase-3 activation in cultured OSNs.
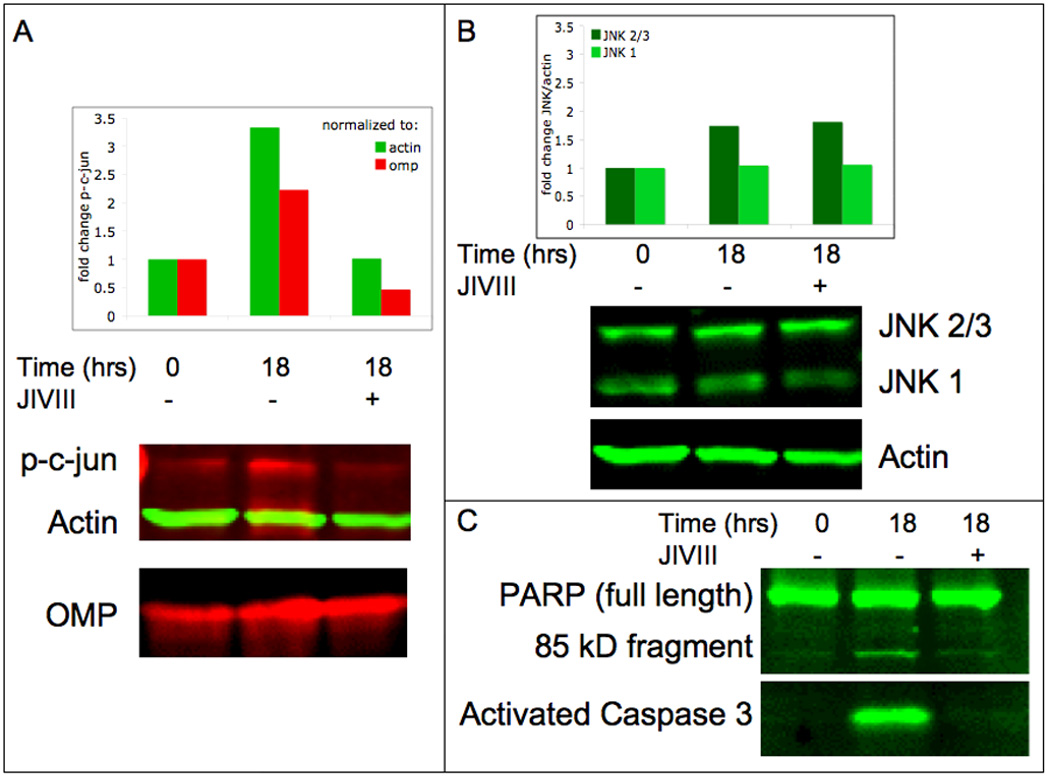
(a) In comparison to OSN cultures at time 0, OSNs cultured for 18 hours show a 3-fold increase in phosphorylation of c-jun as indicated by western blotting with an antibody to the phosphorylated form of c-jun (p-c-jun) when normalized to actin signal (2-fold when normalized to OMP signal). This increased c-jun phosphorylation is prevented when the same 18-hour culture is treated with JNK inhibitor VIII (JIVIII, 10 µM). (b) The levels of the kinase itself – both the 46kD (JNK1) and 54kD (JNK2/3) JNK family members – remain unchanged. (c) Caspase-3 activation is also evident after 18 hours in culture as indicated by an antibody to the cleaved fragment of caspase-3 and the downstream cleavage of PARP1. Both indicators of caspase activity are suppressed by treatment with JIVIII.
To determine whether the protection by cycloheximide that we had observed was due to the inhibition of downstream transcriptional activity, or due to inhibition upstream of JNK, prior to c-jun phosphorylation, we performed a similar experiment with CHX treatment in place of the JNK inhibitor. When comparing levels of phosphorylated c-jun in lysate from untreated OSNs prepared immediately upon dissociation, to OSNs collected after 18 hours in culture with and without CHX treatment, we saw an increase in phospho-c-jun by 18 hours in the untreated cells, but not in those treated with CHX. Cycloheximide treatment also prevented caspase-3 activation and suppressed PARP1 cleavage (Sup. Fig. 2). Therefore, it appears that protection derived from CHX treatment is due to its prevention of the synthesis of an upstream regulator of JNK. As JNK is a member of the MAPK family of kinases, this regulation could be due to inhibition of the synthesis of an MKK or MKKK that activates JNK. Candidate kinases in the JNK pathway include MKK4 and MKK7, and 13 known MKKKs, according to the STKE JNK Pathway Connections Map.
Because JNK inhibition additionally prevented caspase-3 activation, we tested to see if pan-caspase inhibitors also offered protection of OSNs in culture. Although initially the pan-caspase inhibitors, z-VAD-fmk and BOC-D-fmk, were ineffective at protecting OSNs from death at 37°C, we tested them, as well as a more potent caspase inhibitor, Q-VD-OPH, while incubating cultures at 33°C. We found that the protective effect of caspase inhibition was detectable at 33°C, most notably by BOC-D-FMK (50 uM) and Q-VD-OPH (3 uM) (Fig 4A). The rescue by caspase inhibition was even more effective than that by JNK inhibitors. Furthermore, co-treatment with JNK and caspase inhibitors yielded protection modestly greater than either type of inhibitor on its own, according to the Bliss Independence model of synergy. The supra-additive effects of these combinations support the notion that JNK signaling and caspase activity are wired in series (Lehar et al., 2007). This effect is not due to an increase in total concentration of inhibitor, but the combination of inhibitors; 2-fold dose response analyses for both JNK inhibitors, SP600125 and JIVIII, show no further enhancement of survival by either inhibitor at 10 µM versus 5 µM, the dose selected for combinatorial treatments (Sup. Fig. 1).
In vivo JNK activity
In order to validate that the changes we observed during OSN death in culture accompany OSN death in vivo, we examined the olfactory epithelium of mice following unilateral olfactory bulbectomy (OBX). Because axons from each side of the OE connect to the ipsilateral bulb, we performed unilateral OBXs, leaving the opposite bulbs and corresponding OEs intact to serve as an internal control. In sections of OE from mice perfused 5 days post OBX, when most neurons have died and the OE is thinnest, the bulbectomized side shows higher levels of phospho-c-jun compared to controls (Fig 6). This indicates that c-jun is phosphorylated during OSN death in vivo and suggests that the JNK pathway is active during death in vivo post-OBX. Furthermore it supports our culture model, demonstrating that an inhibitor that shows protective activity in culture holds relevance to OSN death occurring in vivo.
Figure 6. Phosphorylation of c-jun increases in vivo following removal of the olfactory bulb.
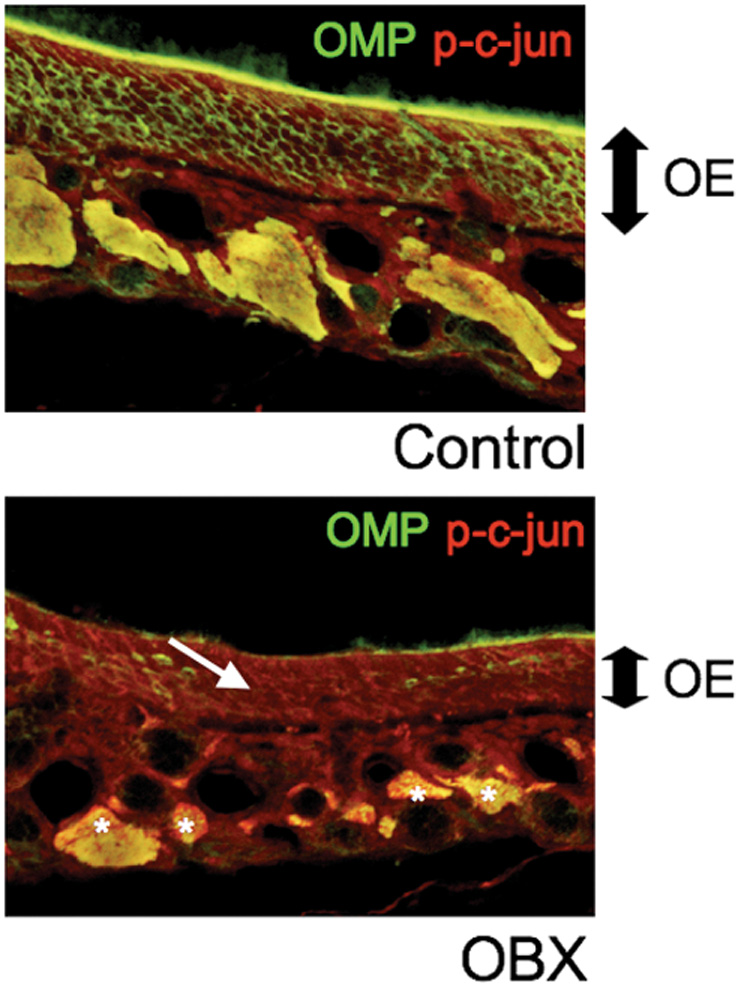
Tissue from the olfactory epithelium of an adult mouse was fixed after a 5-day survival period following unilateral olfactory bulbectomy (OBX) in which one lobe of the olfactory bulb was removed, and the opposite side was left in tact as an internal control. On the unoperated side (top panel), the neuronal layer of the epithelium remains thick as demonstrated by OMP labeling, while the operated side (lower panel) has decreased in thickness as sensory neurons have died. Concurrently, the level of c-jun phosphorylation has increased on the operated side in the neuronal layer (arrow) and in the axons bundles (asterix); p-c-jun levels remain low on the unoperated side.
Additionally, we administered SP600125 to 2-month old OMP-GFP mice by intraperitoneal injection prior to performing unilateral OBXs. 24 hours after removal of the olfactory bulb, animals were perfused, and tissues from the OE of SP600125-treated and untreated mice were sectioned and examined for two hallmarks of apoptotic death: caspase-3 activation, and TUNEL reactivity. While there was no detectable loss of OSNs by 24 hours on either side of either treatment group, as evidenced by the constant thickness of the GFP-expressing layer of mature olfactory neurons in each image, the OE from untreated mice on the operated side showed both caspase activity (cleaved caspase-3 antigen detection) and DNA fragmentation (TUNEL positivity) within the mature neuronal layer, indicating initiation of OSN death. Both indicators of death onset were undetectable in OE sections when SP600125 was administered 30 minutes prior to OBX, suggesting that JNK inhibition can delay the onset of death following target removal in vivo (Fig 7). This suppression of death by SP600125 treatment remained at 32 hours, but was no longer observed by 48 hours. Failure of SP600125 to suppress death at 48 hours could be due to its inability to provide permanent protection against death inducing stimuli, or to the metabolism of the drug in vivo, in which case further protection may be possible with a more rigorous delivery schedule. These results further validate the use of cultured olfactory neurons to study OSN death occurring in mice.
Figure 7. Inhibition of JNK activity suppresses OSN death following OBX.
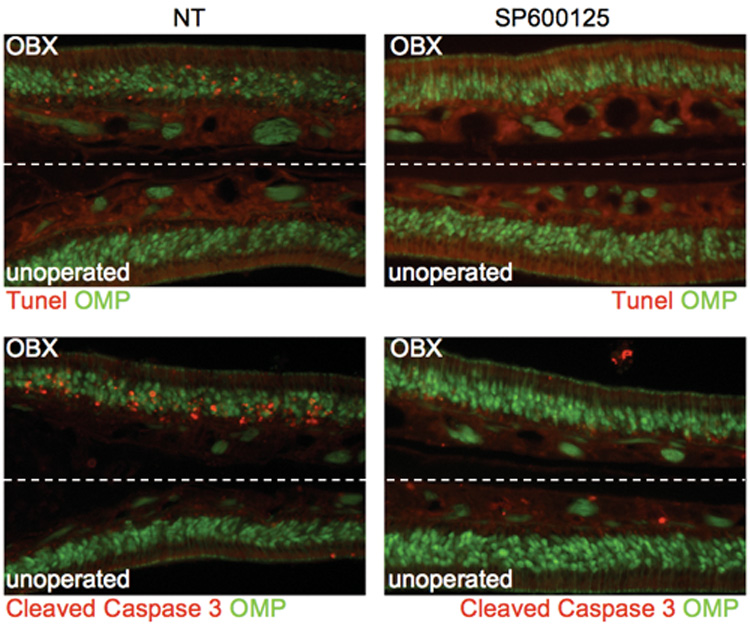
Olfactory epithelia from adult OMP-GFP mice were fixed 24 hours after unilateral OBX, and stained for both TUNEL reactivity and the presence of cleaved caspase-3. Both indicators of cellular death are present on the operated sides (OBX) of non-treated mice (NT). When mice were injected intraperitoneally with SP600125 (10 mg/kg) preceding OBX, both TUNEL and caspase-3 staining were absent indicating a delayed onset of death.
Discussion
Jun kinases have been previously characterized as stress-activated protein kinases in the mitogen-activated protein kinase (MAPK) family. This group of MAPKs regulates transcription by binding and subsequently phosphorylating the transcription factor c-Jun (Johnson and Lapadat, 2002). JNK activity has been implicated in multiple forms of cell death, including apoptosis (Tournier et al., 2000) and necrosis (Wu et al., 2008), in response to many and varied stimuli. Here we show that the death of olfactory sensory neurons is mediated by JNK signaling; JNK inhibitors prolong the survival of OSNs in vitro (Fig 4) and in vivo (Fig 7), and JNK signaling is high following induction of olfactory sensory neuronal death in culture (Fig 5) or in vivo (Fig 6). Previous studies investigating the death of olfactory sensory neurons collectively implicate a complex and multifaceted program of intracellular events regulating the execution of this unique neuronal death. We now identify jun kinases as regulators of OSN death; these kinases play a key role at a focal point of the death program, as caspase activation is also prevented in the presence of JNK inhibitors (Fig 5).
A separate study used microarray profiling to predict that Jun kinases Mapk9, Mapk10, and Jun kinase inhibitory protein Map8ip, were expressed in OSNs (Shetty et al., 2005a). These members of the JNK family may be responsible for the coordination of the death of sensory neurons in the olfactory epithelium. Further studies would be necessary to confirm the precise roles of each isoform, but nonetheless this suggests the presence of candidate JNKs in OSNs with potential to regulate their death program. This source of regulation, while commonly occurring in developmental death, and in adult death in disease models, has not been observed previously in ongoing neuronal death within healthy adult tissue (Repici and Borsello, 2006). In fact, in dorsal root ganglia sensory neurons, although the JNK pathway is induced by apoptotic stimuli in both developing and adult neuronal cultures, only neonatal cultures are susceptible to death by c-jun phosphorylation; adult neurons are protected downstream of JNK activity (Walsh et al., 2004). Thus it appears that JNK signaling in non-pathological, adult, neuronal death is unique to OSNs.
In a microarray study using Rat-1a cells to monitor c-jun-responsive genes following c-jun induction by tetracycline, it was found that most changes in expression levels triggered by c-jun activation occurred in cytoskeletal and adhesion-related genes (Kinoshita et al., 2003). Cell adhesion genes, as characterized by Gene Ontology annotation, are also the most predominantly upregulated group in dying OSNs following OBX (data not shown). Additionally, another group, using a lac-Z reporter to follow c-jun induction in the OE following OBX, discovered increased expression of the reporter in response to bulb removal (Hayward and Morgan, 1995). Because both c-jun and a shared set of genes are upregulated following OBX, it is possible that c-jun activation following OBX is responsible for the transcriptional regulation of these shared genes upregulated in the rat-1a model. These previous observations are in line with our current finding that c-jun activation is responsible for the changes in the olfactory epithelium that lead to neuronal death. Furthermore, loss of substrate adhesion is a common cause of cellular death (i.e. anoikis), and we have observed death of OSNs that are unable to adhere to the surface of the culture well. Genes induced by c-jun-regulated transcription may regulate changes in adhesion, leading to neuronal death.
While we have focused on the role of JNK signaling and c-jun activation in OSN death, a role for c-jun as a regulator of both neuronal death and regeneration has been previously described (Herdegen et al., 1997). Both of these processes occur in the OE following OBX, although they are separated temporally, with death occurring early on, followed later by regeneration. We observe c-jun phosphorylation coinciding with the time course of OSN death (Fig. 5). Therefore it is more likely that the increased numbers of live, mature OSNs, as detected by GFP expression, that we observed in cultures treated with JNK inhibitors (Fig 4) represent neurons that have been protected from dying, rather than neurons regenerated from progenitor cells that were induced to divide and differentiate.
Here, we have elucidated the role of JNK signaling in the execution of olfactory sensory neuronal death. In the mammalian olfactory epithelium, where mature neurons die and are replaced throughout adult life, tight control over the mechanism of neuronal death is required to avoid tumor generation or untimely depletion of sensory neurons. The JNK signaling pathway offers a mode of such precise control with multiple levels of upstream regulation by numerous MAPK kinase kinases (MKKKs), and downstream via the many target genes that are transcriptionally regulated by c-jun once phosphorylated.
With this understanding of OSN death, and a means to slow death in culture, we can maintain a longer-term culture system for OSNs, in which we can study not only their mode of demise, but their functional activity. Each OSN expresses one of over 1,000 olfactory receptor (OR) genes in rodents (Zhang and Firestein, 2002), and although this receptor superfamily was first identified over 15 years ago (Buck and Axel, 1991), few ORs have characterized ligands. The lack of a viable culture system has hindered studies of OR function and regulation. A reliable primary culture system would facilitate further investigation into olfactory detection and signaling.
Experimental Methods
Animals
OMP-GFP mice were provided as a generous gift by Dr. Peter Mombaerts of the Rockefeller University (New York, NY) (Potter et al., 2001). Experiments were performed in compliance with the Columbia University Institutional Animal Care and Use Committee.
Olfactory epithelium dissection, dissociation, and culture
Olfactory sensory neurons were isolated from epithelia as follows, modified from Araneda 2000 (Araneda et al., 2000). Following overdose with anaesthetics (ketamine 210 mg/kg, xylazine 25 mg/kg), mice were decapitated. The head was cut open sagitally and the septum was removed to expose the medial surface of the olfactory turbinates. The epithelium was quickly dissected out in divalent cation-free Ringer’s (145 mM NaCl, 5.6 mM KCl, 10 mM HEPES, 10 mM glucose, 4 mM EGTA, pH 7.4). The tissue was then incubated at 37°C for 45 min in 5 ml of divalent-free Ringer containing 5 mg/ml bovine serum albumin (Sigma, St Louis, Missouri), 25 ug bovine serum albumin (Sigma, St Louis, Missouri), 2.5 ug Collagenase Type I (Gibco BRL, Grand Island, New York), 22 U/ml dispase (Invitrogen, 10295825001) and 50 ug deoxyribonuclease II (Sigma). The tissue was then transferred to a fresh vial and washed 3 times with media, DMEM/F12 (Gibco BRL). Media was then replaced with culture media consisting of DMEM/F12 supplemented with 10% FBS, 100 uM ascorbic acid, 1% insulin-transferrin-selenium-X (Gibco BRL), 100 U/ml penicillin and 100 ug/ml streptomycin (Gibco BRL), and cells were dissociated by banging the tube containing the tissue firmly atop a lab bench. Cells were passed through a 40 uM cell strainer to remove undissociated clusters. Media volume was adjusted for a final concentration of 105 cells/180uL. Cells were then homogenized by gentle pipetting and 180uL of cell solution was plated per well in 16 well chamber slides (Nunc, Rochester, NY) that had been coated with poly-D-ornithine (Sigma). Cultures were incubated at 33°C or 37°C as designated and maintained in the presence of 5% CO2.
Drug treatments
In culture, following plating, cells were treated with 20 uL of each drug prepared in culture media at 10-fold the treatment concentration. Final concentrations are as follows: cycloheximide, 3uM (Sigma); Actinomycin D, 64nM (Sigma); Z-VAD-FMK, 45uM (Biomol, Plymouth Meeting, PA); BOC-D-FMK, 45uM (MP Biomedicals, Solon, OH); Q-VD-OPH, 3uM (MP Biomedicals); SP600125, 5uM (Axxora, San Diego, CA); JNK Inhibitor VIII, 5uM (Calbiochem, Gibbstown, NJ). In vivo drug treatments were delivered via intraperitoneal injections of SP600125 in PBS solution (10mg/kg) or PBS alone (vehicle control). Injections were administered 30 minutes before bulbectomies were performed.
Cell counting
GFP+ cells were counted daily following dissociation and drug treatments. Cell counts were made by taking the total cell number from 5 non-overlapping fields of view visualized using a 20X-objective on an inverted fluorescence microscope. Each drug treatment was applied to three separate wells, and the average cell count from each treatment group was used for comparisons.
Propidium Iodide Staining
20 uL of propidium iodide (Sigma, 287075) solution, 400 ug/ml in PBS, was added to 200 uL media and cells in each well and incubated for 30 minutes. Cultures were then imaged on an inverted fluorescence microscope to determine the extent of overlap of propidium iodide staining with GFP label.
Western blotting
Cells from olfactory epithelial cultures were trypsinized, collected, and washed with cold PBS. Cell lysates were prepared, denatured, separated by SDS-polyacrylamide gel electrophosesis on 10–20% gradient Criterion Tris-HCl gels (Biorad, Hercules, CA), transferred to a polyvinylidene difluoride membrane, blocked and probed by the necessary antibodies, and scanned using the Licor Odyssey Imaging System as described by Yagoda et. al., 2007 (Yagoda et al., 2007). Primary antibodies included mouse anti-phospho-c-jun (1:500; Santa Cruz Biotechnology, Santa Cruz, CA, sc-822), rabbit anti-JNK/SAPK (1:500; Cell Signaling Technology, Danvers, MA, 9258), rabbit anti-actin (1:1000, Santa Cruz Biotechnology, sc-1616R), goat anti-OMP (1:1000; Wako Chemicals, Richmond, VA, 544–10001), rabbit anti-PARP1 (Santa Cruz Biotechnology, sc-7150), and rabbit anti- activated caspase-3 (1:500; Cell Signaling Technology, 9661). Secondary antibodies included IRDye 800 goat anti-rabbit antibody (Rockland Immunochemicals, Gilbertsville, PA, 611-132-122), Alexa Fluor 680 goat anti-mouse (Molecular Probes, Eugene, OR, A21058) and Alexa Fluor 680 donkey anti- goat (Molecular Probes, A21084).
Bulbectomy and Perfusion
2-month old mice were given a survival dose of anesthetic (ketamine 60 mg/kg, xylazine 20 mg/kg) by intraperitoneal injection. A rostral to caudal incision was made above the nose to behind the ears. With the skin held open, a small hole was made in the frontal bone over the right olfactory bulb using a dremmel drill and the bulb was removed by aspiration using a p10 tip. The cavity was filled with Gelfoam to control bleeding and keep the left bulb in position, and the skin was closed using veterinary glue. Animals were housed singly during recovery. Following a 24- to 48-hour delay period, animals were overdosed with anesthetic (ketamine 210 mg/kg, xylazine 25 mg/kg), perfused with PBS till blood was cleared, and fixed by perfusion with 4% paraformaldehyde in 0.1 M phosphate buffer, pH 7.0. Heads were removed and post-fixed overnight at 4°C.
Decalcification, embedding, and sectioning
Following fixation, heads were transferred to 0.5M EDTA, pH 8.0, for 5 days of decalcification. Solutions were changed daily. On the fifth day, EDTA was replaced by 30% sucrose for cryoprotection. The following day, excess tissue was removed, and bulb and OE were mounted in Tissue-Tek (Sakura Finetek USA, Torrance, CA), frozen on dry ice, and stored at −80°C before sectioning. 14µm sections were made using a cryostat (Leica CM1850) and immediately mounted on SuperFrost Plus Gold microscope slides (Fisher Scientific). Slides were stored at −20°C for staining.
Immunohistochemistry
Immunohistochemistry was performed as previously described by Zou et al., 2004 (Zou et al., 2004) in serial coronal sections through the olfactory epithelium and the olfactory bulb of homozygous OMP-GFP mice. The primary antibodies used were goat anti-OMP (1:1000; Wako Chemicals), mouse anti-phospho-c-jun (1:200; Santa Cruz Biotechnology), and rabbit anti-cleaved caspase-3 (1:200; Cell Signaling Technology). The secondary antibodies were Alexa Fluor 488-conjugated donkey anti-goat (1:750; Invitrogen), Alexa Fluor 594-conjugated donkey anti-mouse (1:750; Invitrogen), and Alexa Fluor 594-conjugated donkey anti-rabbit (1:750; Invitrogen).
TUNEL assay
Terminal dUTP Nick End Labeling (TUNEL) was performed following instructions for fixed tissue sections provided with In Situ Cell Death Detection Kit, TMR Red (Roche 12 156 792 910).
Supplementary Material
Acknowledgements
This research of Brent R. Stockwell was supported in part by a Beckman Young Investigator Award from the Arnold and Mabel Beckman Foundation, by a Career Award at the Scientific Interface from the Burroughs Wellcome Fund, and by the NIH (1R01GM085081). The laboratory of Stuart Firestein was supported by the grants from the National Institutes of Health, National Institute on Deafness and Other Communication Disorders (NIDCD). We thank Dr. Peter Mombaerts of the Rockefeller University (New York, NY) for the OMP-GFP mice.
Abbreviations
- OSN
Olfactory Sensory Neuron
- OE
Olfactory Epithelium
- OR
Olfactory Receptor
- NT3
Neurotrophin-3
- NGF
Nerve Growth Factor
- BDNF
Brain Derived Neurotrophic Factor
- OBX
Olfactory Bulbectomy
- APAF-1
Apoptotic protease activating factor 1
- GFP
Green Fluorescent Protein
- OMP
Olfactory Marker Protein
- PI
Propidium Iodide
- TUNEL
Terminal transferase-mediated deoxyUridine triphosphate Nick End Labeling
- ActD
Actinomycin D
- CHX
Cycloheximide
- JNK
jun N-terminal Kinase
- JIVIII
JNK inhibitor VIII
- PARP1
Poly(ADP-Ribose) Polymerase
- MAPK
Mitogen-Activated Protein Kinase
- MKKK
MAPK Kinase Kinase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Araneda RC, Kini AD, Firestein S. The molecular receptive range of an odorant receptor. Nat Neurosci. 2000;3:1248–1255. doi: 10.1038/81774. [DOI] [PubMed] [Google Scholar]
- Benson TE, Ryugo DK, Hinds JW. Effects of sensory deprivation on the developing mouse olfactory system: a light and electron microscopic, morphometric analysis. J Neurosci. 1984;4:638–653. doi: 10.1523/JNEUROSCI.04-03-00638.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brauchi S, Cea C, Farias JG, Bacigalupo J, Reyes JG. Apoptosis induced by prolonged exposure to odorants in cultured cells from rat olfactory epithelium. Brain Res. 2006;1103:114–122. doi: 10.1016/j.brainres.2006.05.072. [DOI] [PubMed] [Google Scholar]
- Buck L, Axel R. A novel multigene family may encode odorant receptors: a molecular basis for odor recognition. Cell. 1991;65:175–187. doi: 10.1016/0092-8674(91)90418-x. [DOI] [PubMed] [Google Scholar]
- Calof AL, Rim PC, Askins KJ, Mumm JS, Gordon MK, Iannuzzelli P, Shou J. Factors regulating neurogenesis and programmed cell death in mouse olfactory epithelium. Ann N Y Acad Sci. 1998;855:226–229. doi: 10.1111/j.1749-6632.1998.tb10571.x. [DOI] [PubMed] [Google Scholar]
- Carr VM, Farbman AI. Ablation of the olfactory bulb up-regulates the rate of neurogenesis and induces precocious cell death in olfactory epithelium. Exp Neurol. 1992;115:55–59. doi: 10.1016/0014-4886(92)90221-b. [DOI] [PubMed] [Google Scholar]
- Carter LA, Roskams AJ. Neurotrophins and their receptors in the primary olfactory neuraxis. Microsc Res Tech. 2002;58:189–196. doi: 10.1002/jemt.10148. [DOI] [PubMed] [Google Scholar]
- Chess A, Buck L, Dowling MM, Axel R, Ngai J. Molecular biology of smell: expression of the multigene family encoding putative odorant receptors. Cold Spring Harb Symp Quant Biol. 1992;57:505–516. doi: 10.1101/sqb.1992.057.01.056. [DOI] [PubMed] [Google Scholar]
- Costanzo RM, Graziadei PP. A quantitative analysis of changes in the olfactory epithelium following bulbectomy in hamster. J Comp Neurol. 1983;215:370–381. doi: 10.1002/cne.902150403. [DOI] [PubMed] [Google Scholar]
- Cowan CM, Thai J, Krajewski S, Reed JC, Nicholson DW, Kaufmann SH, Roskams AJ. Caspases 3 and 9 send a pro-apoptotic signal from synapse to cell body in olfactory receptor neurons. J Neurosci. 2001;21:7099–7109. doi: 10.1523/JNEUROSCI.21-18-07099.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darzynkiewicz Z, Bruno S, Del Bino G, Gorczyca W, Hotz MA, Lassota P, Traganos F. Features of apoptotic cells measured by flow cytometry. Cytometry. 1992;13:795–808. doi: 10.1002/cyto.990130802. [DOI] [PubMed] [Google Scholar]
- Fung KM, Peringa J, Venkatachalam S, Lee VM, Trojanowski JQ. Coordinate reduction in cell proliferation and cell death in mouse olfactory epithelium from birth to maturity. Brain Res. 1997;761:347–351. doi: 10.1016/s0006-8993(97)00467-8. [DOI] [PubMed] [Google Scholar]
- Gordon N. Apoptosis (programmed cell death) and other reasons for elimination of neurons and axons. Brain Dev. 1995;17:73–77. doi: 10.1016/0387-7604(94)00109-b. [DOI] [PubMed] [Google Scholar]
- Hayward MD, Morgan JI. The olfactory system as a model for the analysis of the contribution of gene expression to programmed cell death. Chem Senses. 1995;20:261–269. doi: 10.1093/chemse/20.2.261. [DOI] [PubMed] [Google Scholar]
- Hayward MD, Bocchiaro CM, Morgan JI. Expression of Bcl-2 extends the survival of olfactory receptor neurons in the absence of an olfactory bulb. Brain Res Mol Brain Res. 2004;132:221–234. doi: 10.1016/j.molbrainres.2004.08.003. [DOI] [PubMed] [Google Scholar]
- Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770–776. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- Herdegen T, Skene P, Bahr M. The c-Jun transcription factor--bipotential mediator of neuronal death, survival and regeneration. Trends Neurosci. 1997;20:227–231. doi: 10.1016/s0166-2236(96)01000-4. [DOI] [PubMed] [Google Scholar]
- Hinds JW, Hinds PL, McNelly NA. An autoradiographic study of the mouse olfactory epithelium: evidence for long-lived receptors. Anat Rec. 1984;210:375–383. doi: 10.1002/ar.1092100213. [DOI] [PubMed] [Google Scholar]
- Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–1912. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- Kaplan MS, Hinds JW. Neurogenesis in the adult rat: electron microscopic analysis of light radioautographs. Science. 1977;197:1092–1094. doi: 10.1126/science.887941. [DOI] [PubMed] [Google Scholar]
- Kinoshita I, Leaner V, Katabami M, Manzano RG, Dent P, Sabichi A, Birrer MJ. Identification of cJun-responsive genes in Rat-1a cells using multiple techniques: increased expression of stathmin is necessary for cJun-mediated anchorage-independent growth. Oncogene. 2003;22:2710–2722. doi: 10.1038/sj.onc.1206371. [DOI] [PubMed] [Google Scholar]
- Lehar J, Zimmermann GR, Krueger AS, Molnar RA, Ledell JT, Heilbut AM, Short GF, 3rd, Giusti LC, Nolan GP, Magid OA, Lee MS, Borisy AA, Stockwell BR, Keith CT. Chemical combination effects predict connectivity in biological systems. Mol Syst Biol. 2007;3:80. doi: 10.1038/msb4100116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levi-Montalcini R, Booker B. Destruction of the Sympathetic Ganglia in Mammals by an Antiserum to a Nerve-Growth Protein. Proc Natl Acad Sci U S A. 1960;46:384–391. doi: 10.1073/pnas.46.3.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magrassi L, Graziadei PP. Cell death in the olfactory epithelium. Anat Embryol (Berl) 1995;192:77–87. doi: 10.1007/BF00186993. [DOI] [PubMed] [Google Scholar]
- Paternostro MA, Meisami E. Quantitative [3H]thymidine autoradiography of neurogenesis in the olfactory epithelium of developing normal, hypothyroid and hypothyroid-rehabilitated rats. Brain Res Dev Brain Res. 1994;83:151–162. doi: 10.1016/0165-3806(94)00811-6. [DOI] [PubMed] [Google Scholar]
- Potter SM, Zheng C, Koos DS, Feinstein P, Fraser SE, Mombaerts P. Structure and emergence of specific olfactory glomeruli in the mouse. J Neurosci. 2001;21:9713–9723. doi: 10.1523/JNEUROSCI.21-24-09713.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repici M, Borsello T. JNK pathway as therapeutic target to prevent degeneration in the central nervous system. Adv Exp Med Biol. 2006;588:145–155. doi: 10.1007/978-0-387-34817-9_13. [DOI] [PubMed] [Google Scholar]
- Robinson AM, Conley DB, Kern RC. Olfactory neurons in bax knockout mice are protected from bulbectomy-induced apoptosis. Neuroreport. 2003;14:1891–1894. doi: 10.1097/00001756-200310270-00002. [DOI] [PubMed] [Google Scholar]
- Shetty RS, Bose SC, Nickell MD, McIntyre JC, Hardin DH, Harris AM, McClintock TS. Transcriptional changes during neuronal death and replacement in the olfactory epithelium. Mol Cell Neurosci. 2005a;30:583–600. [PubMed] [Google Scholar]
- Shetty RS, Bose SC, Nickell MD, McIntyre JC, Hardin DH, Harris AM, McClintock TS. Transcriptional changes during neuronal death and replacement in the olfactory epithelium. Mol Cell Neurosci. 2005b;30:90–107. doi: 10.1016/j.mcn.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Simpson PJ, Miller I, Moon C, Hanlon AL, Liebl DJ, Ronnett GV. Atrial natriuretic peptide type C induces a cell-cycle switch from proliferation to differentiation in brain-derived neurotrophic factor- or nerve growth factor-primed olfactory receptor neurons. J Neurosci. 2002;22:5536–5551. doi: 10.1523/JNEUROSCI.22-13-05536.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sydor W, Teitelbaum Z, Blacher R, Sun S, Benz W, Margolis FL. Amino acid sequence of a unique neuronal protein: rat olfactory marker protein. Arch Biochem Biophys. 1986;249:351–362. doi: 10.1016/0003-9861(86)90011-1. [DOI] [PubMed] [Google Scholar]
- Tournier C, Hess P, Yang DD, Xu J, Turner TK, Nimnual A, Bar-Sagi D, Jones SN, Flavell RA, Davis RJ. Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science. 2000;288:870–874. doi: 10.1126/science.288.5467.870. [DOI] [PubMed] [Google Scholar]
- Walsh GS, Orike N, Kaplan DR, Miller FD. The invulnerability of adult neurons: a critical role for p73. J Neurosci. 2004;24:9638–9647. doi: 10.1523/JNEUROSCI.1299-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Tessier-Lavigne M. En passant neurotrophic action of an intermediate axonal target in the developing mammalian CNS. Nature. 1999;401:765–769. doi: 10.1038/44521. [DOI] [PubMed] [Google Scholar]
- Watt WC, Sakano H, Lee ZY, Reusch JE, Trinh K, Storm DR. Odorant stimulation enhances survival of olfactory sensory neurons via MAPK and CREB. Neuron. 2004;41:955–967. doi: 10.1016/s0896-6273(04)00075-3. [DOI] [PubMed] [Google Scholar]
- Weiler E, Farbman AI. Proliferation in the rat olfactory epithelium: age-dependent changes. J Neurosci. 1997;17:3610–3622. doi: 10.1523/JNEUROSCI.17-10-03610.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YT, Zhang S, Kim YS, Tan HL, Whiteman M, Ong CN, Liu ZG, Ichijo H, Shen HM. Signaling pathways from membrane lipid rafts to JNK1 activation in reactive nitrogen species-induced non-apoptotic cell death. Cell Death Differ. 2008;15:386–397. doi: 10.1038/sj.cdd.4402273. [DOI] [PubMed] [Google Scholar]
- Yagoda N, von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, Wolpaw AJ, Smukste I, Peltier JM, Boniface JJ, Smith R, Lessnick SL, Sahasrabudhe S, Stockwell BR. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature. 2007;447:864–868. doi: 10.1038/nature05859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Firestein S. The olfactory receptor gene superfamily of the mouse. Nat Neurosci. 2002;5:124–133. doi: 10.1038/nn800. [DOI] [PubMed] [Google Scholar]
- Zou DJ, Feinstein P, Rivers AL, Mathews GA, Kim A, Greer CA, Mombaerts P, Firestein S. Postnatal refinement of peripheral olfactory projections. Science. 2004;304:1976–1979. doi: 10.1126/science.1093468. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.