

Abstract
The purpose of this study was to determine the effects and mechanisms of sCD40L on endothelial dysfunction in both human coronary artery endothelial cells (HCAECs) and porcine coronary artery rings. HCAECs treated with sCD40L showed significant reductions of endothelial nitric oxide synthase (eNOS) mRNA and protein levels, eNOS mRNA stability, eNOS enzyme activity, and cellular NO levels, whereas superoxide anion (O2−) production was significantly increased. sCD40L enhanced eNOS mRNA 3′UTR binding to cytoplasmic molecules and induced a unique expression pattern of 95 microRNAs. sCD40L significantly decreased mitochondrial membrane potential, and catalase and SOD activities, whereas it increased NADPH oxidase (NOX) activity. sCD40L increased phosphorylation of MAPKs p38 and ERK1/2 as well as IκBα and enhanced NF-κB nuclear translocation. In porcine coronary arteries, sCD40L significantly decreased endothelium-dependent vasorelaxation and eNOS mRNA levels, whereas it increased O2− levels. Antioxidant seleno-l-methionine; chemical inhibitors of p38, ERK1/2, and mitochondrial complex II; as well as dominant negative mutant forms of IκBα and NOX4 effectively blocked sCD40L-induced eNOS down-regulation in HCAECs. Thus, sCD40L reduces eNOS levels, whereas it increases oxidative stress through the unique molecular mechanisms involving eNOS mRNA stability, 3′UTR-binding molecules, microRNAs, mitochondrial function, ROS-related enzymes, p38, ERK1/2, and NF-κB signal pathways in endothelial cells.
Introduction
Endothelial cells play a crucial role in regulating vascular tone, inflammatory response, permeability, and coagulation. Many risk factors including dyslipidemia, hypertension, and diabetes are associated with endothelial dysfunction, which likely explains why they promote atherogenesis.1 Endothelial dysfunction has largely been assessed as alterations of endothelium-dependent vasorelaxation and gene expression. Endothelium-derived nitric oxide (NO), synthesized by the endothelial nitric oxide synthase (eNOS), is a major mediator of endothelium-dependent vasorelaxation, and it is also critically involved in the regulation of other protective properties of the healthy endothelium.1
Recently, increasing evidence supports a central role of the interaction between CD40 ligand (CD40L) and CD40 (its membrane receptor) in the pathogenesis of atherosclerosis.2,3 CD40L is a trimeric transmembrane protein, which is a member of the tumor necrosis factor family.4 CD40L can be found in plasma as a soluble protein,3 which is often elevated in patients with a variety of chronic inflammatory and autoimmune conditions as well as acute coronary syndromes.5,6 Thus, elevated plasma levels of soluble CD40L (sCD40L) are considered as a new independent cardiovascular risk factor.
Several studies have demonstrated that CD40L affects endothelial functions. CD40L-CD40 interaction could trigger the production of several proinflammatory cytokines and chemokines,7–9 and the expression of matrix metalloproteinase, tissue factor, and vascular endothelial growth factor, whereas it decreases thrombomodulin in endothelial cells.10–12 In addition, sCD40L is able to inhibit endothelial cell migration,13 activate platelet aggregation,14 and enhance monocyte tissue factor expression and thrombin generation15 through a mechanism related to the overproduction of reactive oxygen species (ROS). Inhibition of CD40 signaling can effectively reduce atherosclerosis in mice.16
Although previous studies have linked CD40L-CD40 interaction resulting in alternations of gene expression in the vascular system, little is known about whether the sCD40L could affect the expression of eNOS and endothelium-dependent vasorelaxation in coronary arteries. The objective of this study was to determine the effect of sCD40L on eNOS levels and endothelium-dependent vasorelaxation as well as the underlying mechanisms including oxidative stress, MAPK activation, and transcription factor NF-κB involvement. This study may provide new insights into the mechanisms of sCD40L interacting with endothelial cells that may contribute to the vascular lesion formation.
Methods
HCAECs were cultured in endothelial growth medium-2 (EGM-2) with or without sCD40L in both monomer and trimer forms. eNOS mRNA and protein levels were determined by real-time polymerase chain reaction (PCR) and Western blot analysis, respectively. PCR primers are shown in Table S1 (available on the Blood website; see the Supplemental Materials link at the top of the online article). The eNOS mRNA stability was studied with actinomycin D treatment and real-time PCR analysis. Cell proliferation was assessed by a commercial MTS kit (Promega, Madison, WI). Vasomotor reactivity of porcine coronary artery rings was studied with a myograph device. Endothelium-dependent vasorelaxation of porcine coronary artery rings was studied with vasodilator bradykinin. MicroRNA (miRNA) profiling was performed with a commercial 95-miRNA assay kit (SBI Systems Biosciences, Mountain View, CA) and real-time PCR analysis. eNOS mRNA 3′UTR-binding molecules were studied with gel shift assay. Recombinant adenoviruses expressing human Cu/Zn SOD (Ad-SOD), human catalase (Ad-CAT), a dominant negative mutant form of human NADPH oxidase NOX4 (Ad-NOX4 DN), the Escherichia coli β-galactosidase gene (Ad-LacZ), a dominant negative mutant form of human IκBα (Ad-IκB DN), and green florescence protein (Ad-GFP) were included in analyses. Superoxide anion (O2−) levels were studied with dihydroethidium (DHE) staining and flow cytometric analysis for HCAECs as well as the lucigenin-enhanced chemiluminescence method for porcine coronary artery rings. Cellular NO levels and eNOS enzyme activity were studied. Mitochondrial membrane potential was determined with JC-1 staining and flow cytometric analysis. ATP levels were measured with a commercial ATPLite kit (PerkinElmer, Waltham, MA). Activities of NADPH oxidase, CAT, and SOD were determined. Activation of MAPKs and IκBα was determined by Bio-Plex luminex immunoassay (Bio-Rad, Hercules, CA). Chemical inhibitors for p38, ERK1/2, and mitochondrial complex II were included. NF-κB nuclear translocation was studied by Western blot. The CD40L and CD40 immunoreactivity in human atherosclerotic tissues was studied by immunohistochemical staining. Data were reported as mean plus or minus SEM. Mann-Whitney U test and Student t test were used to analyze nonparametric and parametric data, respectively. Detailed materials and methods are available in Document S1.
Results
sCD40L decreases the level and activity of eNOS in HCAECs
HCAECs were treated with sCD40L in a concentration- and time-dependent manner. eNOS mRNA and protein levels were detected using real-time PCR (n = 3) and Western blot (n = 3), respectively. When cells were treated with sCD40L (1 or 5 μg/mL) for 24 hours, eNOS mRNA levels were decreased by 22% and 61%, respectively, compared with controls (P < .01, t test; Figure 1A). Heat-inactivated (HI)–sCD40L showed no effect on eNOS mRNA levels (Figure 1A). When cells were cultured with sCD40L (5 μg/mL) for different times, 12-, 24-, and 48-hour treatment groups showed significant reductions of eNOS mRNA levels (P < .05 or P < .01, t test; Figure 1B). Anti-CD40L antibody or isotype IgG was used to determine the specificity of sCD40L. When cells were treated with anti-CD40L antibody (5 μg/mL), the sCD40L-induced eNOS decrease was significantly blocked (P < .01, t test; Figure 1C). To investigate the role of sCD40L receptor CD40 on endothelial cells, anti-CD40 antibody was used and also effectively blocked sCD40L-induced eNOS down-regulation in HCAECs (P < .05 or P < .01, t test; Figure 1D). To determine whether sCD40L-induced decrease of eNOS mRNA levels could be due to increased mRNA degradation, actinomycin D, a direct inhibitor of RNA polymerase II, was used to treat HCACEs for different times in the presence or absence sCD40L, and eNOS mRNA levels were determined by real-time PCR. Control HCAECs had a half-life of eNOS mRNA around 12 hours, whereas sCD40L-treated cells had a half-life of eNOS mRNA less than 3 hours (P < .05, U test; Figure 1E).
Figure 1.

Effects of sCD40L on eNOS mRNA levels in HCAECs. (A) Concentration-dependent study. HCAECs were treated with different concentrations of sCD40L for 24 hours. The eNOS levels were determined by real-time PCR. Heat-inactivated (HI) sCD40L was included as a negative control. (B) Time course study. HCAECs were treated with sCD40L (5 μg/mL) for different times. The eNOS levels were determined by real-time PCR. (C) Effect of anti-CD40L antibody and (D) effect of anti-CD40 antibody. HCAECs were pretreated with different concentrations of anti-CD40L antibody or anti-CD40 antibody for 30 minutes and followed with sCD40L treatment for 24 hours. The eNOS mRNA levels were determined by real-time PCR. Isotype IgG was used for a negative control. (E) eNOS mRNA stability. HCAECs were treated with actinomycin D (2.5 μg/mL) in the presence or absence of sCD40L (5 μg/mL) for different time points, and eNOS mRNA levels were determined by real-time PCR. (F) eNOS mRNA 3′UTR-binding molecules. Biotin-labeled human eNOS mRNA 3′UTR probe was incubated with cytoplasmic extracts of HCAECs treated with or without sCD40L for 24 hours. The binding reaction was electrophoresed on a native polyacrylamide gel. For the competition experiment, excess unlabeled RNA probe was preincubated with cytoplasmic protein prior to the addition of biotin-labeled RNA probe. *P < .05 and **P < .01, compared with the control. #P < .05 and ##P < .01, compared with sCD40L treatment. n = 3. Data are means and SE of multiple experiments (n).
Since TNF-α could reduce eNOS mRNA stability through potential mechanisms of unknown cytoplasmic proteins binding to eNOS mRNA 3′UTR sequences,17–19 we performed a similar experiment with some modifications. Biotin-labeled human eNOS mRNA 3′UTR probe was incubated with cytoplasmic extracts of HCAECs treated with or without sCD40L for 24 hours. Gel shift assay demonstrated that there was an enhanced molecular binding to the eNOS mRNA 3′UTR in sCD40L-treated cells compared with control cells (Figure 1F). This binding activity was specific because excess unlabeled RNA probe completely blocked biotin-labeled RNA probe. Furthermore, we performed a 95-miRNA profiling experiment in HCAECs treated with sCD40L or TNF-α for 24 hours. sCD40L treatment induced a specific expression pattern of 95 miRNAs, which was also similar to that in TNF-α–treated cells (Figure S1; Table S2). For example, we found sCD40L and TNF-α could increase miR-221 levels by 38% and 47%, respectively, whereas they decreased miR-222 levels by about 10%. miR-221 and miR-222 may have functions to regulate eNOS levels.20 However, we performed predictive algorithm analyses (Sanger miRBase Targets21 and TargetScan22) and found that miR-221/222 might be not able to bind to the eNOS mRNA 3′UTR. Furthermore, we used Sanger miRBase Targets predictive algorithm and found that 5 miRNAs (miR-125a, miR-214, miR-155, miR-185, and miR-150) are potential molecules to be able to bind to eNOS mRNA 3′UTR. Expression levels of these 5 miRNAs were changed in response to sCD40L and TNF-α treatments in HCAECs. sCD40L and TNF-α increased the expression of miR-125a, miR-214, and miR-155, whereas they decreased the levels of miR-185 and miR-150 (Table 1). Thus, sCD40L and TNF-α may share similar mechanisms to down-regulate eNOS mRNA in human endothelial cells. These effects of sCD40L were not related to cytotoxicity because the treatment of sCD40L (5 μg/mL) did not affect cell proliferation for up to 4 days compared with untreated cells (Figure S2).
Table 1.
The expression of 5 miRNAs in response to sCD40L or TNF-α treatment in HCAECs
| miRNA | miRBase no. | miRNA sequence | RT-PCR primer sequence | sCD40L vs control | TNF-α vs control |
|---|---|---|---|---|---|
| miR-125a | MIMAT0000443 | ucccugagacccuuuaaccugug | tccctgagaccctttaacctgtg | 1.0353 | 1.1892 |
| miR-150 | MIMAT0000451 | ucucccaacccuuguaccagug | tctcccaacccttgtaccagtg | 0.6598 | 0.7579 |
| miR-155 | MIMAT0000646 | uuaaugcuaaucgugauagggg | ttaatgctaatcgtgatagggg | 1.0210 | 1.2570 |
| miR-185 | MIMAT0000455 | uggagagaaaggcaguuc | tggagagaaaggcagttc | 0.7526 | 0.8066 |
| miR-214 | MIMAT0000271 | acagcaggcacagacaggcag | acagcaggcacagacaggcag | 1.7901 | 1.1810 |
Sanger miRBase Targets predictive algorithm search indicates that these 5 miRNAs may potentially target eNOS mRNA 3′UTR.
The eNOS protein levels were determined by Western blot analysis. HCAECs treated with sCD40L (5 μg/mL) for 24 hours showed a significant decrease in eNOS protein levels compared with untreated controls (P < .05, U test; Figure 2A). The eNOS enzyme activity was studied using an eNOS detection kit (n = 3). sCD40L (5 μg/mL) significantly decreased eNOS enzyme activity by 34% compared with controls (P < .05, U test; Figure 2B). Diaminofluoroescein-FM diacetate (DAF-FM DA) staining is a unique method to measure NO levels in living cells or solutions.23 HCAECs treated with sCD40L (5 μg/mL) had a substantial decrease of cellular NO levels by 77% compared with untreated controls (Figure 2C).
Figure 2.

Effects of sCD40L on eNOS protein levels and NO production in HCAECs. (A) Western blot analysis. HCAECs were treated with sCD40L for 24 hours and eNOS protein levels were determined by Western blot. (B) eNOS enzyme activity. HCAECs were treated with sCD50L for 24 hours. The eNOS enzyme activity was determined by a commercial eNOS fluorimetric assay kit. (C) Cellular NO levels. HCAECs were treated with sCD40L for 24 hours and cellular NO levels were determined by DAF-FM DA staining and flow cytometric analysis. (D) Effect of sCD40L trimer on eNOS mRNA and (E) protein levels. HCAECs were treated with sCD40L monomer form or sCD40L trimer form for 24 hours, and eNOS mRNA and protein levels were determined by real-time PCR analysis and Western blot, respectively. *P < .05 and **P < .01, compared with the control. n = 3. Data are means and SE of multiple experiments (n).
Since sCD40L trimer is the major physiological form of sCD40L in human plasma,24 we obtained commercially available recombinant sCD40L trimer form from R&D Systems (Minneapolis, MN). We performed additional experiments and confirmed that both sCD40L trimer and monomer forms at the same concentration (5 μg/mL) substantially reduced eNOS expression at both mRNA (P < .01, t test; Figure 2D) and protein (P < .05, U test; Figure 2E) levels.
sCD40L decreases eNOS levels and endothelium-dependent vasorelaxation in porcine coronary arteries
Porcine coronary artery rings were cultured with sCD40L (5 μg/mL) for 24 hours. The eNOS mRNA levels were determined by real-time PCR analysis (n = 3). The eNOS mRNA levels were significantly reduced by 49% in the sCD40L-treated group compared with controls (P < .05, t test; Figure 3A). The endothelial function was also studied using a well-characterized myograph system (n = 8). No significant difference was observed in all experimental groups in response to thromboxane A2 analog U46619 (contraction) or sodium nitroprusside (endothelium-independent vasorelaxation) compared with controls (Figure S3). However, in response to a series of concentrations of bradykinin, the endothelium-dependent vasorelaxation was significantly decreased in the sCD40L-treated vessel rings compared with control vessels. For example, sCD40L (5 μg/mL) significantly reduced vasorelaxation by 23% in response to bradykinin at 10−6 M concentration (P < .05, U test; Figure 3B).
Figure 3.

Effects of sCD40L on eNOS mRNA levels and endothelium-dependent vasorelaxation in porcine coronary arteries. (A) Porcine eNOS mRNA levels. Porcine coronary artery rings were treated with sCD40L (5 μg/mL) for 24 hours, and porcine eNOS mRNA levels were determined by real-time PCR. *P < .05 compared with the control. t test. n = 4. (B) Endothelium-dependent vasorelaxation. Porcine coronary artery rings were treated with sCD40L for 24 hours. Vasomotor reactivity was analyzed with a myograph device. The vessel ring was initially contracted with thromboxane A2 analog U46619, and then a relaxation concentration-response curve was generated by 4 cumulative additions of the endothelium-dependent vasodilator bradykinin. *P < .05 compared with the control. U test. n = 8. Data are means and SE of multiple experiments (n).
sCD40L increases O2− production in HCAECs and porcine coronary arteries
To study whether ROSs could play a role in sCD40L-induced endothelial dysfunction and eNOS down-regulation, O2− production in HCAECs was measured using a fluorescence dye DHE staining and flow cytometry analysis. When HCAECs were cultured with sCD40L (5 μg/mL) for 24 hours, the O2− production was substantially increased by 76% compared with controls (P < .05, t test; Figure 4A). Similarly, lucigenin-enhanced chemiluminescence assay showed that the O2− production was significantly increased by 31% in sCD40L-treated porcine coronary artery rings compared with control vessels (P < .05, t test; Figure 4B).
Figure 4.

Effects of sCD40L on O2− production in HCAECs and porcine coronary arteries. (A) DHE staining in HCAECs. Cells were treated with sCD40L for 24 hours, and intracellular O2− levels were determined by DHE staining and flow cytometric analysis. U test. n = 3. (B) Porcine coronary artery rings. The vessel rings were treated with sCD40L for 24 hours and O2− levels in the endothelial layer of porcine coronary arteries were tested with lucigenin-enhanced chemiluminescence assay. *P < .05 compared with the control. t test. n = 6. (C) Mitochondrial membrane potential (representative histograms of flow cytometric analysis). HCAECs were treated with sCD40L for 24 hours, and mitochondrial membrane potential was determined by JC-1 staining and flow cytometric analysis. (D) Quantitative data of JC-1 staining showed a significant decrease in mitochondrial membrane potential after sCD40L treatment. Antioxidant SeMet effectively reversed these changes. U test. n = 3. (E) ATP content. HCAECs were treated with sCD40L for 24 hours, and ATP production was determined by a commercial ATPLite kit. Antioxidant SeMet was included. t test. n = 3. (F) Effect of mitochondrial complex II inhibitor TTFA on eNOS protein levels. HCAECs were treated with sCD40L in the presence or absence of mitochondrial complex II inhibitor TTFA (10 μM) for 24 hours, and eNOS protein levels were determined by Western blot analysis. *P < .05 and **P < .01, compared with the control. #P < .05 compared with sCD40L treatment. U test. n = 3. Data are means and SE of multiple experiments (n).
sCD40L decreases mitochondrial membrane potential (Δψm) and ATP production in HCAECs
Mitochondria are known to be one of the major sources of ROSs during the mitochondrial respiration and ATP production, and Δψm can serve as an indicator for the function of mitochondrial respiration chain.25 HCAECs were cultured with or without sCD40L (5 μg/mL) for 24 hours. sCD40L treatment substantially reduced Δψm by 58% compared with controls (P < .05, U test, Figure 4C,D). When cocultured with antioxidant SeMet, the Δψm was reversed to control levels. In addition, sCD40L (5 μg/mL) treatment significantly reduced ATP levels by 28% (P < .05, t test, Figure 4E), whereas antioxidant SeMet effectively blocked sCD40L-induced inhibition in ATP production. SeMet treatment did not affect cell numbers and viability for up to 4 days (Figure S2). In addition, to study the functional link between mitochondrial function and sCD40L action, we determined the effect of mitochondrial complex II inhibitor TTFA on sCD40L-induced eNOS down-regulation. Indeed, HCAECs treated with sCD40L in the presence of TTFA for 24 hours showed a near complete blocking effect of TTFA on sCD40L-induced eNOS down-regulation at the protein level (P < .01, U test, Figure 4F).
sCD40L increases NOX activity, whereas it decreases CAT and SOD activities in HCAECs
In addition to mitochondrial dysfunction, ROSs can be generated from other enzymatic sources such as increased levels and/or activities of NADPH oxidase (NOX)26 as well as decreased levels and/or activities of internal antioxidant enzymes including superoxide dismutase (SOD) and catalase (CAT).26,27 In the current study, we determined NOX activity by chemiluminescence assay (n = 3). HCAECs were treated with sCD40L (5 μg/mL) and/or SeMet (20 μM) for 24 hours. With the presence of β-NADPH, the NOX activity in sCD40L-treated cells showed a 53% increase compared with controls (P < .05, t test, Figure 5A). O2− scavenger Tiron or flavoprotein inhibitor DPI coculture abolished this sCD40L-induced increase of NOX activity, suggesting the specificity of the assay system for NOX activity. Antioxidant SeMet coculture also significantly blocked this sCD40L-induced change to the control levels (P < .05, t test, Figure 5A).
Figure 5.

Effects of sCD40L and SeMet on activities of NOX, CAT, and SOD and eNOS mRNA levels in HCAECs. (A) NADPH oxidase (NOX) activity. HCAECs were treated with sCD40L for 24 hours, and NOX activities were determined by lucigenin-enhanced chemiluminescence with the presence of its substrate β-NADPH. O2− scavenger Tiron or flavoprotein inhibitor DPI was included in the assay to confirm the specificity of NOX activity. (B) CAT activity. HCAECs were treated with sCD40L for 24 hours and CAT activity was determined with a commercial kit. Antioxidant SeMet was included. (C) SOD activity. HCAECs were treated with sCD40L for 24 hours and SOD activity was determined with a commercial kit. Antioxidant SeMet was included. (D) eNOS mRNA levels. HCAECs were treated with sCD40L and/or SeMet for 24 hours, and eNOS mRNA levels were determined by real-time PCR analysis. *P < .05 and **P < .01, compared with the control. #P < .05 compared with sCD40L treatment. t test. n = 3. Data are means and SE of multiple experiments (n).
The enzyme activities of CAT and SOD were studied with the commercial assay kits (n = 3 for each). sCD40L (5 μg/mL) treatment significantly reduced CAT activities by 28% (P < .05, t test, Figure 5B). Similarly, SOD activities were also reduced by 36% (P < .05, t test, Figure 5C). Coculture with SeMet (20 μM) effectively blocked sCD40L-induced decrease of activities of both enzymes (Figure 5B,C). Thus, sCD40L increases O2− production through the dysfunction of mitochondria, increase of NADPH oxidase activity, and decrease of internal antioxidant enzyme activities. SeMet has been used as a potent antioxidant in our previous studies.28 It effectively blocked sCD40L-induced decrease in eNOS mRNA levels in HCAECs compared with those of sCD40L-treated cells without SeMet (P < .01, t test, Figure 5D). SeMet alone showed no effect on eNOS mRNA levels.
sCD40L induces the activation of MAPK p38 and ERK1/2 in HCAECs
Phosphorylation of MAPKs was investigated with a Bio-Plex luminex immunoassay. p38 and ERK2 phosphorylation showed an early peak at 20 minutes after sCD40L (5 μg/mL) treatment in HCACEs (P < .001, U test, Figure 6A). JNK did not show any significant changes in all time points (data not known). To confirm the role of p38 and ERK1/2 activation in sCD40L-induced endothelial dysfunction, specific p38 inhibitor (SB239036, 1 μM) or ERK1/2 inhibitor (PD98059, 40 μM) was used to pretreat HCAECs for 30 minutes before cells were cultured with sCD40L (5 μg/mL) for 24 hours. After culture, the eNOS mRNA levels were determined by real-time PCR analysis. SB239036 or PD98059 effectively blocked sCD40L-induced eNOS mRNA decrease (P < .05 or P < .01, t test, Figure 6B). SB239036 or PD98059 alone did not show any effect on eNOS mRNA levels. In addition, effects of SB239036 and PD98059 on eNOS protein levels were determined by Western blot. As shown in Figure 6C,D, SB239036 (1, 5, and 10 μM) and PD98059 (10, 30, and 50 μM) concentration-dependently blocked sCD40L-induced decrease in eNOS protein levels. Effects of MAPK inhibitors and mitochondrial complex II inhibitor TTFA on sCD40L-induced oxidative stress were also determined with DHE staining and flow cytometry analysis. SB239036, PD98059, and TTFA effectively blocked sCD40L-induced increase in O2− production (Figure 6E).
Figure 6.

Effects of sCD40L on activation of MAPKs in HCAECs. (A) MAPK p38 and ERK2 phosphorylation. HCAECs were treated with sCD40L for different time points and the phosphorylation levels of MAPK p38 and ERK2 were determined by Bio-Plex luminex immunoassay with a commercial kit. U test. (B) Effects of MAPK inhibitors on eNOS mRNA levels. HCAECs were treated with sCD40L in the presence or absence of p38 inhibitor (SB239036) or ERK1/2 inhibitor (PD98059) for 24 hours, and eNOS mRNA levels were determined by real-time PCR analysis. t test. (C) Effect of p38 inhibitor (SB239036) on eNOS protein levels. HCAECs were treated with sCD40L in the presence or absence of different concentrations of p38 inhibitor (SB239036) for 24 hours, and eNOS protein levels were determined by Western blot. (D) Effect of ERK1/2 inhibitor (PD98059) on eNOS protein levels. HCAECs were treated with sCD40L in the presence or absence of different concentrations of ERK1/2 inhibitor (PD98059) for 24 hours, and eNOS protein levels were determined by Western blot. (E) Effects of sCD40L, MAPK inhibitors, and mitochondrial complex II inhibitor TTFA on O2− production (DHE staining). HCAECs were treated with sCD40L monomer form or sCD40L trimer form in the presence or absence of MAPK inhibitors (p38 and ERK2) or TTFA for 24 hours. O2− production was determined by DHE staining and flow cytometric analysis. (F) Effects of recombinant adenovirus–based gene delivery on eNOS protein levels. HCAECs were infected with different recombinant adenoviruses for 48 hours, and followed by sCD40L treatment for 24 hours. Ad-LacZ was included as a negative control. eNOS mRNA levels were determined by Western blot analysis. **P < .01 compared with the control. #P < .05 compared with sCD40L treatment. n = 3. Data are means and SE of multiple experiments (n).
To further confirm the functional role of oxidative stress in sCD40L-induced eNOS down-regulation, we used a genetic approach by infecting HCAECs with several recombinant adenoviruses delivering a dominant negative mutant (Ad-NOX4 DN) form of NOX4 gene and a wild-type form of CAT and SOD gene (Ad-CAT and Ad-SOD). Ad-LacZ was included as a negative control. HCAECs were infected with these adenoviruses for 48 hours, and were treated with sCD40L for 24 hours. The overexpression of these genes delivered by recombinant adenoviruses was confirmed by real-time PCR analysis (Figure S4). Ad-NOX4 DN substantially blocked sCD40L-induced decrease in eNOS protein levels, and Ad-CAT or Ad-SOD showed a partial blocking effect (Figure 6F). These effects were specific because Ad-LacZ did not change eNOS protein levels. Thus, both mitochondrial dysfunction and NADPH oxidase activation are prominent for sCD40L's effects on eNOS levels.
sCD40L effects are involved in the activation of transcription factor NF-κB in HCAECs
Transcription factor NF-κB activation is mediated by phosphorylation of IκBα, an inhibitor of NF-κB, and nuclear translocation of NF-κB p65.29 In the present study, we observed an early peak of IκBα phosphorylation at 10 minutes and a second peak at 60 minutes after sCD40L treatment (Figure 7A). Nuclear translocation of NF-κB p65 was substantially increased in response to sCD40L treatment at 10 and 45 minutes (P < .01, U test, Figure 7B). Recombinant adenovirus expressing a dominant negative mutant form of IκBα (Ad-IκB DN) was used to infect HCAECs, which were no longer responsible for sCD40L increasing NF-κB p65 nuclear translocation (Figure 7B). To confirm the functional role of NF-κB activation, HCAECs were infected with Ad-IκB DN and treated with sCD40L; and eNOS mRNA and protein levels were studied. As shown in Figure 7C,D, Ad-IκB DN infection effectively blocked sCD40L-induced eNOS down-regulation at both mRNA and protein levels. These effects were specific because Ad-GFP infection had no effect on eNOS levels. Thus, NF-κB activity induced by sCD40L is responsible for the decreased eNOS levels.
Figure 7.

Effects of sCD40L on NF-κB activation in HCAECs. (A) IκBα phosphorylation. HCAECs were treated with sCD40L for different time points, and phosphorylation levels of IκBα were determined by a Bio-Plex Luminex Immunoassay with a commercial kit. U test. (B) NF-κB p65 protein nuclear translocations. HCAECs were infected with recombinant adenovirus Ad-IκB DN (inhibition of IκBα) or Ad-GFP (a negative control). The cells were then treated with sCD40L for different time frames, and nuclear extract was prepared. Nuclear NF-κB protein levels were determined by Western blot analysis. Nuclear protein laminin was included as a loading control. (C) Effect of dominant negative mutant form of IκBα on eNOS mRNA levels. HCAECs were infected with recombinant adenovirus Ad-IκB DN or Ad-GFP for 72 hours, and then cells were treated with sCD40L for 24 hours. The NOS mRNA levels of eNOS were determined by real-time PCR analysis. U test. (D) Effect of dominant negative mutant form of IκBα on eNOS protein levels. HCAECs were infected with recombinant adenovirus Ad-IκB DN or Ad-GFP for 72 hours, and then cells were treated with sCD40L for 24 hours. The eNOS protein levels of eNOS were determined by Western blot analysis. *P < .05; **P < .01 compared with the control. n = 3. Data are means and SE of multiple experiments (n).
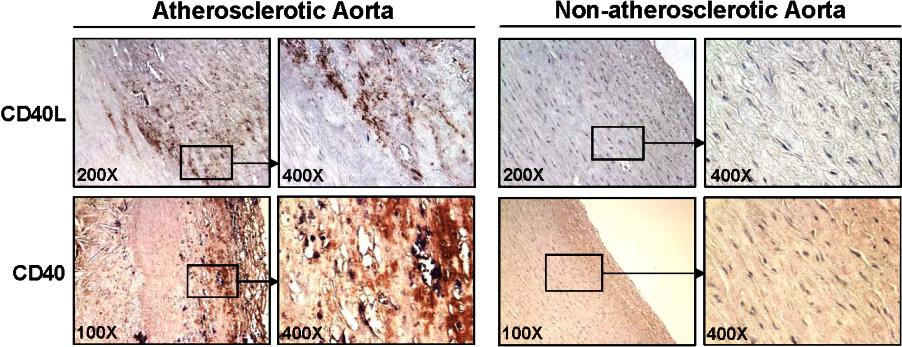
Expression of human CD40L and CD40 is increased in human atherosclerotic vessels
We performed immunostaining to detect the expression of CD40L and CD40 in human aortic tissues. Nonatherosclerotic aortic tissues showed no or a very limited immunoreactivity of CD40L and CD40. By contrast, atherosclerotic regions of these vessels showed a strong CD40L and CD40 immunoreactivity. The increased signal of CD40L and CD40 was located mainly in the intima and media areas of atherosclerotic plaques (Figure S5). These results were observed in 3 atherosclerosis cases and 2 nonatherosclerosis cases.
Discussion
The present study demonstrates that recombinant human sCD40L induces endothelial dysfunction in both HCAECs and porcine coronary artery rings. Specifically, sCD40L significantly reduces eNOS mRNA and protein levels, eNOS enzyme activity, and NO bioavailability in HCAECs. In addition, sCD40L significantly increases production of O2− and activation of ERK1/2, p38, and NF-κB in HCAECs. sCD40L also significantly decreased endothelium-dependent vasorelaxation in porcine coronary artery rings. This study provides several novel discoveries of the biologic functions and related mechanisms of sCD40L in the vascular system.
Homology of CD40L and CD40 amino acid sequences between human and porcine was analyzed from reported genes in the GenBank30 (Table S3). Human CD40L shares 86% protein sequence homology with porcine CD40L. Human CD40 variant 1 or variant 2 shares 79% or 82% protein sequence homology with porcine CD40. Wienhold et al reported that the deduced amino acid sequence of the porcine CD40L shared 82%, 88%, and 93% similarity with the CD40L protein of mouse, human, and cattle.31 Furthermore, experimental evidence shows human CD40L can interact with porcine CD40 on porcine endothelial cells and induce biologic changes including gene expression.32 Thus, we also studied effects of human sCD40L on porcine coronary arteries.
The endothelium plays a crucial role in the control of vascular tone by releasing endothelium-derived vessel-relaxing factors including mainly NO generated by eNOS.33 However, under various pathological conditions, eNOS expression is decreased or becomes dysfunctional.34 These changes of eNOS not only impair endothelium-dependent vasorelaxation but also accelerate the atherosclerotic lesion formation.35 In the present study, we demonstrated a direct effect of sCD40L on eNOS levels in cultured HCAECs and porcine coronary artery endothelial cells. Using real-time PCR analysis, we showed that sCD40L in both monomer and trimer forms can decrease eNOS mRNA levels. Western blot analysis also confirmed that eNOS protein levels were decreased after sCD40L treatment. These effects are the result of the specific interaction between sCD40L and CD40 on endothelial cells because anti-CD40L or anti-CD40 antibody effectively blocked sCD40L-induced eNOS down-regulation. We have confirmed that one of mechanisms of sCD40L-induced eNOS down-regulation is the decrease of eNOS mRNA stability in sCD40L-treated human endothelial cells. Interestingly, sCD40L treatment substantially enhances eNOS mRNA 3′UTR sequence binding to cytoplasmic molecules in HCAECs. This observation was similar to that in TNF-α–treated endothelial cells.17–19 Properties of these binding molecules are unknown. Since miRNA can regulate protein translation from mRNA and mRNA degradation, we performed a 95-miRNA profiling experiment in HCAECs treated with sCD40L or TNF-α for 24 hours. All 95 miRNAs are selected based on their potential roles in cell regulations. sCD40L and TNF-α induced a similar expression pattern of 95 miRNAs, indicating sCD40L and TNF-α may share similar mechanisms to down-regulate eNOS mRNA in human endothelial cells. A recent study demonstrated that the knockdown of Dicer increased eNOS expression, whereas transfection of miR-221 and miR-222 mimics partially restored the elevated eNOS protein levels.20 In our study, we found sCD40L and TNF-α could increase miR-221 levels by 38% and 47%, respectively; whereas they decreased miR-222 levels by about 10%. These data indicate that miR-221 or miR-222 may be involved in the regulation of eNOS levels. However, using different predictive algorithms (Sanger miRBase Targets and TargetScan), miR-221/222 might be not able to bind to eNOS mRNA 3′UTR. We used Sanger miRBase Targets predictive algorithm and found that 5 miRNAs in our profiling list may have a potential binding capability to eNOS mRNA 3′UTR (miR-125a, miR-214, miR-155, miR-185, and miR-150). In response to sCD40L and TNF-α treatment, the expression of miR-125a, miR-214, and miR-155 was increased, whereas the level of miR-185 and miR-150 was decreased. However, whether these miRNAs regulate eNOS mRNA translation and degradation is not known.
Our previous study demonstrates that sCD40L can increase the expression of its receptor CD40 on HCAECs through which sCD40L could enhance its biologic functions in the vascular system.36 Cellular NO levels in sCD40L-treated HCAECs showed a 77% decrease, and the eNOS enzyme activity was also decreased. In addition, sCD40L significantly reduced endothelium-dependent vasorelaxation, suggesting a decrease in NO bioavailability. There has only been one study showing the relationship between NO and sCD40L in platelet activation.37 Acute inhibition of NO production increased platelet surface expression of sCD40L. There are no previous reports showing CD40L could regulate eNOS expression. Our novel data obtained from endothelial cells directly demonstrate the causal relationship between sCD40L and eNOS/NO levels. sCD40L directly inhibits the NO bioavailability by reducing the eNOS levels and/or activity in endothelial cells. Since sCD40L trimer is the major physiological form of sCD40L in human plasma,24 we obtained commercially available recombinant sCD40L trimer from R&D Systems. This recombinant sCD40L has a predicted molecular mass of 16 911 Da. SDS–polyacrylamide gel electrophoresis (PAGE) analysis showed a single band at approximately 17 kDa under both reducing and nonreducing conditions. Upon size exclusion chromatography, the recombinant sCD40L eluted with an apparent molecular mass of 45 kDa, suggesting that the protein is a noncovalent linked homotrimer. We performed additional experiments and confirmed that both sCD40L trimer and monomer forms at the same concentration (5 μg/mL) substantially reduced eNOS mRNA and protein levels. Since sCD40L levels are increased in the progression of atherosclerosis and other inflammation conditions,2–6 our new discoveries provide a better understanding of the functional roles of sCD40L in these clinical disorders.
Atherosclerosis is a multifactorial disease for which the molecular etiology of many related risk factors is still not fully understood.38 Among them, ROSs have been shown to be the key mediators for vascular inflammation and atherogenesis.26,39 In the current study, we demonstrate that sCD40L in both monomer and trimer forms induces a significant increase of O2− in HCAECs. Similar change is also found in sCD40L-treated porcine coronary rings. These data demonstrate that sCD40L has strong impact on O2− production, which could be one of the mechanisms for sCD40L-induced eNOS dysfunction. In fact, O2− could deplete NO to form peroxynitrite (ONOO−), resulting in NO being unavailable for vasorelaxation and other biologic functions.40 We further investigated the cause of ROS overproduction. ROSs can be produced by a variety of sources including mitochondrial respiration chain,25 NADPH oxidase (NOX),41 xanthine oxidase, and lipoxygenases.42 Accumulating evidence has suggested a predominant role of NOX in generating ROSs in endothelial cells.43 On the other hand, several cellular mechanisms have been shown to counterbalance the production of ROSs, including enzymatic and nonenzymatic pathways.44 Among them, the best-characterized enzymatic pathways are SOD, CAT, and GPX. To study potential sources of increased O2− in sCD40L-treated cells, we detected a significant decrease in both mitochondrial membrane potential and ATP production in sCD40L-treated cells. To confirm the functional role of mitochondrial dysfunction in sCD40L treatment, we treated HCAECs with mitochondrial complex II inhibitor TTFA and showed an effective blocking effect of TTFA on sCD40L-induced increase in O2− production and decrease in eNOS levels. In addition, we tested activities of NOX, CAT, and SOD in HCAECs. Our data show that sCD40L substantially increases NOX activity, whereas it decreases CAT and SOD activities. The impact of these ROS enzymes on sCD40L-induced eNOS levels was determined by recombinant adenovirus–based gene delivery experiments. A dominant negative mutant form of NOX4 (Ad-NOX4 DN) effectively blocked sCD40L-induced decrease in eNOS protein levels. Increase expression of CAT and SOD through adenovirus infection (Ad-SOD and Ad-CAT) also effectively blocked sCD40L-induced decrease in eNOS protein levels. Taken together, these data indicate that increased O2− production in sCD40L-treated cells may result from mitochondrial dysfunction and the unbalance of cellular redox enzymes. Recently, Chakrabarti et al showed that sCD40L significantly enhanced ROS generation in platelets as measured by DCFHDA oxidation and confocal microscopy, and NADPH inhibitor (apocynin) was able to inhibit sCD40L-induced ROS generation.14
Since O2− was overproduced in sCD40L-treated endothelial cells, we then tested the blocking effect of antioxidant SeMet on sCD40L-induced decrease of eNOS levels. Indeed, SeMet effectively blocked sCD40L-induced eNOS down-regulation. SeMet is a relatively nontoxic compound and is especially interesting in terms of its antioxidant properties. It is the major component of dietary selenium, and undergoes an intramolecular transsulfuration reaction to form selenocysteine.45 Recently, we have demonstrated that SeMet can effectively block lysophosphatidylcholine-induced oxidative stress and endothelial dysfunction in porcine coronary arteries.46 Chakrabarti et al showed that sCD40L induced release of CD40L and MCP1 in human umbilical vein endothelial cells (HUVECs) by a redox-sensitive mechanism because ROS scavenger N-acetylcysteine effectively blocked this effect of sCD40L.9 These findings suggest a possible role of antioxidants in treating vascular diseases related to risk factors such as sCD40L.
To connect sCD40L-induced overexpression of O2− with down-regulation of eNOS in sCD40L-treated HCACEs, we investigated oxidation-sensitive signal transduction pathways such as MAPKs and transcription factors such as NF-κB. There is increasing evidence that MAPKs are involved in various cardiovascular disorders such as cardiac hypertrophy or atherosclerosis.47 Three major subfamilies of structurally related MAPKs have been identified in mammalian cells, which are termed p44/42 MAPK (extracellular-signal regulated kinase 1/2 [ERK1/2]), p38 MAPK, and Jun N-terminal kinase (JNK). p38 and ERK pathways have been linked to sCD40L-mediated mature dendritic cell survival.47 In the present study, we demonstrate that p38 and ERK2 are activated in response to sCD40L stimulation in human endothelial cells, and p38 inhibitor SB239063 and ERK1/2 inhibitor PD98059 effectively block sCD40L-induced eNOS down-regulation at both mRNA and protein levels. These new data are consistent with our previous report that sCD40L-induced up-regulation of CD40 in HCAECs is mediated by enhanced O2− production and ERK1/2 activation.36 However, a recent study showed that sCD40L was also able to activate JNK in HUVECs. This result is different from our current data that sCD40L did not activate JNK in HCAECs. The possible reasons for this difference may include different cell types and culture conditions. For example, we did not treat HCAECs with serum starvation, whereas HUVECs were treated with serum starvation before incubation with sCD40L.9
NF-κB is a crucial transcription factor that regulates many genes. Prior to cytokine stimulation, NF-κB is restricted to the cytosol as an inactive complex with its inhibitors such as IκBα. Upon activation by cytokines, IκBα is phosphorylated, dissociates from the NF-κB, and is subsequently ubiquitinated and degraded. This allows active NF-κB to enter the nucleus and bind to κB consensus regulatory elements in the promoters of many responsive genes. Consequently, NF-κB could increase or decrease the expression of certain specific genes via direct or indirect mechanisms. In the present study, IκBα phosphorylation levels are significantly increased 20 minutes after treatment, which suggests the activation of NF-κB in sCD40L-treated HCAECs. Indeed, we showed that nuclear translocation of NF-κB p65 was substantially increased in response to sCD40L treatment at 10 and 45 minutes. When we blocked IκBα function in HCAECs by infection of recombinant adenovirus expressing a dominant negative mutant form of IκBα (Ad-IκB DN), HCAECs were no longer responsible for sCD40L increasing NF-κB p65 nuclear translocation and decreasing eNOS levels. These data are consistent with previous report that sCD40 activates NF-κB activation in HUVECs.9 Therefore, NF-κB may play an important role in the interaction between sCD40L and endothelial cells.
Although we showed that sCD40L treatment increased O2− production in both HCAECs (DHE staining) and porcine coronary artery rings (lucigenin-enhanced chemiluminescence), we did not perform any experiment using SOD or other antioxidants to block O2− production. Instead, we clearly demonstrated that overexpression of SOD through adenovirus gene delivery and antioxidant SeMet effectively blocked sCD40L-induced eNOS down-regulation in HCAECs. In addition, a dominant negative mutant form of NOX4, CAT overexpression, and mitochondrial complex II inhibitor TTFA effectively blocked sCD40L-induced decrease in eNOS levels. These data indicate that sCD40L indeed induces O2− production. However, it cannot be excluded that other types of ROSs may be involved in the effect of sCD40L. DHE staining and lucigenin-enhanced chemiluminescence may not exclusively detect O2− levels.
Although different clinical studies reported different serum or plasma levels of sCD40L due to different measurement procedures and conditions, human sCD40L levels usually range from picogram per milliliter to nanogram per milliliter in both healthy individuals and patients with cardiovascular diseases.6,47–50 In the current study, we have used a much higher concentration (1 or 5 μg/mL) for in vitro experiments than those in human serum or plasma levels. The major reason for this choice is considering that similar concentrations of sCD40L are used in the previous discoveries of sCD40L functions and underlying mechanisms from in vitro experiments in endothelial cells.12,13,15,51 Thus, the data generated from the current study are comparable with those in the previous publications. In addition, local concentrations of CD40L at the vascular lesion site may be much higher than those in patients' serum or plasma. Our immunohistologic staining showed that immunoreactivity of CD40L and CD40 was substantially increased in atherosclerotic plaque regions compared with that in nonatherosclerotic tissues. These data are consistent with previous reports.52,53
In summary, our study demonstrates that sCD40L significantly decreases eNOS expression and NO production through unique molecular mechanisms (Figure S6). sCD40L specifically interacts with its membrane receptor CD40 on endothelial cells and increases O2− production through reduction of mitochondrial membrane potential, activation of NADPH oxidase, and inhibition of CAT and SOD activities. Dysfunctional mitochondria are also demonstrated by reduction of ATP production. O2− could trigger activation of p38 and ERK1/2, which may lead to the phosphorylation of IκBα and transcriptional factor NF-κB activation. O2− may be able to directly activate the IκBα and NF-κB system. O2− and activation of p38, ERK1/2, and NF-κB could decrease eNOS mRNA stability and/or eNOS transcriptional activity, resulting in decrease in eNOS levels and NO bioavailability. O2− could directly interact with NO and reduce NO bioavailability, which causes endothelial dysfunction. Atherosclerotic tissues showed increased immunoreactivity of CD40L and CD40, indicating that the high local concentrations of CD40L and CD40 may have strong impact on endothelial dysfunction at the regions of vascular lesions. Antioxidant SeMet may have potential clinical applications in blocking sCD40L-induced endothelial dysfunction, thereby preventing vascular disease.
Supplementary Material
Acknowledgments
The authors thank Dr Barry Goldstein (Thomas Jefferson University, Philadelphia, PA) for his kind gifts of recombinant adenoviruses expressing a dominant negative mutant form of human NADPH oxidase NOX4 (Ad-NOX4 DN) and the Escherichia coli β-galactosidase gene (Ad-LacZ), and Dr Toren Finkel (National Institutes of Health, Bethesda, MD) for his generous gifts of recombinant adenoviruses expressing human Cu/Zn SOD (Ad-SOD), and human catalase (Ad-CAT). We also thank Dr Jie Du (Baylor College of Medicine) for his critical comments for revision and technical assistance.
This work is partially supported by research grants from the National Institutes of Health (DE15543 and AT003094 [Q.Y.]; and EB-002436 and HL083471 [C.C.]) and by the Michael E. DeBakey Department of Surgery, Baylor College of Medicine.
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: C.C. and H.C. designed research, performed research, analyzed data, and wrote the paper; X.W., J.J., M.S.J., D.L., Y.Z., H.W., S.Z., and M.L. designed research, performed research, and analyzed data; U.B. analyzed data; and P.L. and Q.Y. designed research and analyzed data.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Changyi (Johnny) Chen or Michael E. DeBakey, Department of Surgery-MARB 413, Baylor College of Medicine, One Baylor Plaza, Mail stop: BCM390, Houston, TX 77030-3411; e-mail: jchen@bcm.tmc.edu.
References
- 1.Landmesser U, Hornig B, Drexler H. Endothelial function: a critical determinant in atherosclerosis? Circulation. 2004;109:II27–II33. doi: 10.1161/01.CIR.0000129501.88485.1f. [DOI] [PubMed] [Google Scholar]
- 2.Lutgens E, Daemen MJ. CD40-CD40L interactions in atherosclerosis. Trends Cardiovasc Med. 2002;12:27–32. doi: 10.1016/s1050-1738(01)00142-6. [DOI] [PubMed] [Google Scholar]
- 3.André P, Nannizzi-Alaimo L, Prasad SK, Phillips DR. Platelet-derived CD40L: the switch-hitting player of cardiovascular disease. Circulation. 2002;106:896–899. doi: 10.1161/01.cir.0000028962.04520.01. [DOI] [PubMed] [Google Scholar]
- 4.Grewal IS, Flavell RA. The CD40 ligand: at the center of the immune universe? Immunol Res. 1997;16:59–70. doi: 10.1007/BF02786323. [DOI] [PubMed] [Google Scholar]
- 5.Koutroubakis IE, Theodoropoulou A, Xidakis C, et al. Association between enhanced soluble CD40 ligand and prothrombotic state in inflammatory bowel disease. Eur J Gastroenterol Hepatol. 2004;16:1147–1152. doi: 10.1097/00042737-200411000-00011. [DOI] [PubMed] [Google Scholar]
- 6.Heeschen C, Dimmeler S, Hamm CW, et al. Soluble CD40 ligand in acute coronary syndromes. N Engl J Med. 2003;348:1104–1111. doi: 10.1056/NEJMoa022600. [DOI] [PubMed] [Google Scholar]
- 7.Thienel U, Loike J, Yellin MJ. CD154 (CD40L) induces human endothelial cell chemokine production and migration of leukocyte subsets. Cell Immunol. 1999;198:87–95. doi: 10.1006/cimm.1999.1583. [DOI] [PubMed] [Google Scholar]
- 8.Déchanet J, Grosset C, Taupin JL, et al. CD40 ligand stimulates proinflammatory cytokine production by human endothelial cells. J Immunol. 1997;159:5640–5647. [PubMed] [Google Scholar]
- 9.Chakrabarti S, Blair P, Freedman JE. CD40-40L signaling in vascular inflammation. J Biol Chem. 2007;282:18307–18317. doi: 10.1074/jbc.M700211200. [DOI] [PubMed] [Google Scholar]
- 10.Mach F, Schonbeck U, Fabunmi RP, et al. T lymphocytes induce endothelial cell matrix metalloproteinase expression by a CD40L-dependent mechanism: implications for tubule formation. Am J Pathol. 1999;154:229–238. doi: 10.1016/S0002-9440(10)65269-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller DL, Yaron R, Yellin MJ. CD40L-CD40 interactions regulate endothelial cell surface tissue factor and thrombomodulin expression. J Leukoc Biol. 1998;63:373–379. doi: 10.1002/jlb.63.3.373. [DOI] [PubMed] [Google Scholar]
- 12.Flaxenburg JA, Melter M, Lapchak PH, Briscoe DM, Pal S. The CD40-induced signaling pathway in endothelial cells resulting in the overexpression of vascular endothelial growth factor involves Ras and phosphatidylinositol 3-kinase. J Immunol. 2004;172:7503–7509. doi: 10.4049/jimmunol.172.12.7503. [DOI] [PubMed] [Google Scholar]
- 13.Urbich C, Dernbach E, Aicher A, Zeiher AM, Dimmeler S. CD40 ligand inhibits endothelial cell migration by increasing production of endothelial reactive oxygen species. Circulation. 2002;106:981–986. doi: 10.1161/01.cir.0000027107.54614.1a. [DOI] [PubMed] [Google Scholar]
- 14.Chakrabarti S, Varghese S, Vitseva O, Tanriverdi K, Freedman JE. CD40 ligand influences platelet release of reactive oxygen intermediates. Arterioscler Thromb Vasc Biol. 2005;25:2428–2434. doi: 10.1161/01.ATV.0000184765.59207.f3. [DOI] [PubMed] [Google Scholar]
- 15.Sanguigni V, Ferro D, Pignatelli P, et al. CD40 ligand enhances monocyte tissue factor expression and thrombin generation via oxidative stress in patients with hypercholesterolemia. J Am Coll Cardiol. 2005;45:35–42. doi: 10.1016/j.jacc.2004.09.047. [DOI] [PubMed] [Google Scholar]
- 16.Mach F, Schonbeck U, Sukhova GK, Atkinson E, Libby P. Reduction of atherosclerosis in mice by inhibition of CD40 signaling. Nature. 1998;394:200–203. doi: 10.1038/28204. [DOI] [PubMed] [Google Scholar]
- 17.Lai PF, Mohamed F, Monge JC, Stewart DJ. Downregulation of eNOS mRNA expression by TNF-α: identification and functional characterization of RNA-protein interactions in the 3′UTR. Cardiovasc Res. 2003;59:160–168. doi: 10.1016/s0008-6363(03)00296-7. [DOI] [PubMed] [Google Scholar]
- 18.Alonso J, Sánchez de Miguel L, Montón M, Casado S, López-Farré A. Endothelial cytosolic proteins bind to the 3′ untranslated region of endothelial nitric oxide synthase mRNA: regulation by tumor necrosis factor alpha. Mol Cell Biol. 1997;17:5719–5726. doi: 10.1128/mcb.17.10.5719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Searles CD, Miwa Y, Harrison DG, Ramasamy S. Posttranscriptional regulation of endothelial nitric oxide synthase during cell growth. Circ Res. 1999;85:588–595. doi: 10.1161/01.res.85.7.588. [DOI] [PubMed] [Google Scholar]
- 20.Suárez Y, Fernández-Hernando C, Pober JS, Sessa WC. Dicer dependent microRNAs regulate gene expression and functions in human endothelial cells. Circ Res. 2007;100:1164–1173. doi: 10.1161/01.RES.0000265065.26744.17. [DOI] [PubMed] [Google Scholar]
- 21.Sanger Institute. [Accessed June 4, 2008];miRBase Targets. http://microrna.sanger.ac.uk/targets/v5.
- 22.Whitehead Institute for Biomedical Research. [Accessed June 4, 2008];Target Scan. http://www.targetscan.org.
- 23.Kojima H, Urano Y, Kikuchi K, Higuchi T, Hirata Y, Nagano T. Fluorescent indicators for imaging nitric oxide production. Angew Chem Int Ed Engl. 1999;38:3209–3212. doi: 10.1002/(sici)1521-3773(19991102)38:21<3209::aid-anie3209>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 24.Kimura K, Tsuda H, Kwangseok Y, Tamura N, Kanai Y, Kobayashi S. Study of plasma levels of soluble CD40 ligand in systemic lupus erythematosus patients who have undergone plasmapheresis. Ther Apher Dial. 2005;9:64–68. doi: 10.1111/j.1774-9987.2005.00221.x. [DOI] [PubMed] [Google Scholar]
- 25.Genova ML, Pich MM, Bernacchia A, et al. The mitochondrial production of reactive oxygen species in relation to aging and pathology. Ann N Y Acad Sci. 2004;1011:86–100. doi: 10.1007/978-3-662-41088-2_10. [DOI] [PubMed] [Google Scholar]
- 26.Madamanchi NR, Hakim ZS, Runge MS. Oxidative stress in atherogenesis and arterial thrombosis: the disconnect between cellular studies and clinical outcomes. J Thromb Haemost. 2005;3:254–267. doi: 10.1111/j.1538-7836.2004.01085.x. [DOI] [PubMed] [Google Scholar]
- 27.Matés JM, Sanchez-Jimenez F. Antioxidant enzymes and their implications in pathophysiologic processes. Front Biosci. 1999;4:D339–D345. doi: 10.2741/mates. [DOI] [PubMed] [Google Scholar]
- 28.Chai H, Yang H, Yan S, et al. Effects of 5 HIV protease inhibitors on vasomotor function and superoxide anion production in porcine coronary arteries. J Acquir Immune Defic Syndr. 2005;40:12–19. doi: 10.1097/01.qai.0000172368.05327.7b. [DOI] [PubMed] [Google Scholar]
- 29.Monaco C, Paleolog E. Nuclear factor κB: a potential therapeutic target in atherosclerosis and thrombosis. Cardiovasc Res. 2004;61:671–682. doi: 10.1016/j.cardiores.2003.11.038. [DOI] [PubMed] [Google Scholar]
- 30.National Center for Biotechnology Information. [Accessed June 10, 2008];GenBank. http://www.ncbi.nlm.nih.gov/Genbank/
- 31.Wienhold D, Berger N, Armengol E, Büttner M, Saalmüller A, Pfaff E. Cloning, sequencing and expression of porcine CD40 ligand in Escherichia coli and human and porcine cells. Cytokine. 2002;20:274–282. doi: 10.1006/cyto.2003.2000. [DOI] [PubMed] [Google Scholar]
- 32.Choi I, Kim SD, Cho B, et al. Xenogeneic interaction between human CD40L and porcine CD40 activates porcine endothelial cells through NF-kappaB signaling. Mol Immunol. 2008;45:575–580. doi: 10.1016/j.molimm.2007.06.161. [DOI] [PubMed] [Google Scholar]
- 33.Fleming I, Bauersachs J, Fisslthaler B, Busse R. Ca2+-independent activation of the endothelial nitric oxide synthase in response to tyrosine phosphatase inhibitors and fluid shear stress. Circ Res. 1998;82:686–695. doi: 10.1161/01.res.82.6.686. [DOI] [PubMed] [Google Scholar]
- 34.Kawashima S, Yokoyama M. Dysfunction of endothelial nitric oxide synthase and atherosclerosis. Arterioscler Thromb Vasc Biol. 2004;24:998–1005. doi: 10.1161/01.ATV.0000125114.88079.96. [DOI] [PubMed] [Google Scholar]
- 35.Sessa WC. eNOS at a glance. J Cell Sci. 2004;117:2427–2429. doi: 10.1242/jcs.01165. [DOI] [PubMed] [Google Scholar]
- 36.Chai H, Yan S, Wang H, et al. CD40 ligand increases expression of its receptor CD40 in human coronary artery endothelial cells. Surgery. 2006;140:236–242. doi: 10.1016/j.surg.2006.03.016. [DOI] [PubMed] [Google Scholar]
- 37.Schäfer A, Alp NJ, Cai S, et al. Reduced vascular NO bioavailability in diabetes increases platelet activation in vivo. Arterioscler Thromb Vasc Biol. 2004;24:1720–1726. doi: 10.1161/01.ATV.0000138072.76902.dd. [DOI] [PubMed] [Google Scholar]
- 38.Wassmann S, Wassmann K, Nickenig G. Modulation of oxidant and antioxidant enzyme expression and function in vascular cells. Hypertension. 2004;44:381–386. doi: 10.1161/01.HYP.0000142232.29764.a7. [DOI] [PubMed] [Google Scholar]
- 39.Azumi H, Inoue N, Ohashi Y, et al. Superoxide generation in directional coronary atherectomy specimens of patients with angina pectoris: important role of NAD(P)H oxidase. Arterioscler Thromb Vasc Biol. 2002;22:1838–1844. doi: 10.1161/01.atv.0000037101.40667.62. [DOI] [PubMed] [Google Scholar]
- 40.Harrison DG. Cellular and molecular mechanisms of endothelial cell dysfunction. J Clin Invest. 1997;100:2153–2157. doi: 10.1172/JCI119751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barry-Lane PA, Patterson C, van der Merwe M, et al. p47phox is required for atherosclerotic lesion progression in ApoE(-/-) mice. J Clin Invest. 2001;108:1513–1522. doi: 10.1172/JCI11927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cyrus T, Witztum JL, Rader DJ, et al. Disruption of the 12/15-lipoxygenase gene diminishes atherosclerosis in apo E-deficient mice. J Clin Invest. 1999;103:1597–1604. doi: 10.1172/JCI5897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brandes RP, Kreuzer J. Vascular NADPH oxidases: molecular mechanisms of activation. Cardiovasc Res. 2005;65:16–27. doi: 10.1016/j.cardiores.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 44.Nordberg J, Arner ES. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free Radic Biol Med. 2001;31:1287–1312. doi: 10.1016/s0891-5849(01)00724-9. [DOI] [PubMed] [Google Scholar]
- 45.Allan CB, Lacourciere GM, Stadtman TC. Responsiveness of selenoproteins to dietary selenium. Annu Rev Nutr. 1999;19:1–16. doi: 10.1146/annurev.nutr.19.1.1. [DOI] [PubMed] [Google Scholar]
- 46.Safaya R, Chai H, Kougias P, et al. Effect of lysophosphatidylcholine on vasomotor functions of porcine coronary arteries. J Surg Res. 2005;126:182–188. doi: 10.1016/j.jss.2005.01.015. [DOI] [PubMed] [Google Scholar]
- 47.Hu Y, Dietrich H, Metzler B, Wick G, Xu Q. Hyperexpression and activation of extracellular signal-regulated kinases (ERK1/2) in atherosclerotic lesions of cholesterol-fed rabbits. Arterioscler Thromb Vasc Biol. 2000;20:18–26. doi: 10.1161/01.atv.20.1.18. [DOI] [PubMed] [Google Scholar]
- 48.Varo N, de Lemos JA, Libby P, et al. Soluble CD40L: risk prediction after acute coronary syndromes. Circulation. 2003;108:1049–1052. doi: 10.1161/01.CIR.0000088521.04017.13. [DOI] [PubMed] [Google Scholar]
- 49.Garlichs CD, John S, Schmeiber A, et al. Upregulation of CD40 and CD40 ligand (CD154) in patients with moderate hypercholesterolemia. Circulation. 2001;104:2395–2400. doi: 10.1161/hc4501.099312. [DOI] [PubMed] [Google Scholar]
- 50.Semb AG, van Wissen S, Ueland T, et al. Raised serum levels of soluble CD40 ligand in patients with familial hypercholesterolemia: downregulatory effect of statin therapy. J Am Coll Cardiol. 2003;41:275–279. doi: 10.1016/s0735-1097(02)02718-3. [DOI] [PubMed] [Google Scholar]
- 51.Damås JK, Otterdal K, Yndestad A, et al. Soluble CD40 ligand in pulmonary arterial hypertension: possible pathogenic role of the interaction between platelets and endothelial cells. Circulation. 2004;110:999–1005. doi: 10.1161/01.CIR.0000139859.68513.FC. [DOI] [PubMed] [Google Scholar]
- 52.Häkkinen T, Karkola K, Ylä-Herttuala S. Macrophages, smooth muscle cells, endothelial cells, and T-cells express CD40 and CD40L in fatty streaks and more advanced human atherosclerotic lesions: colocalization with epitopes of oxidized low-density lipoprotein, scavenger receptor, and CD16 (Fc gammaRIII). Virchows Arch. 2000;437:396–405. doi: 10.1007/s004280000239. [DOI] [PubMed] [Google Scholar]
- 53.Mulhaupt F, Matter CM, Kwak BR, et al. Statins (HMG-CoA reductase inhibitors) reduce CD40 expression in human vascular cells. Cardiovasc Res. 2003;59:755–766. doi: 10.1016/s0008-6363(03)00515-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}