

Summary
Since lipopolysaccharide endotoxins of Gram-negative bacteria were last reviewed in this series in 1990, much has been learned about the assembly and signaling functions of these remarkable glycoconjugates. Lipopolysaccharides typically consist of a hydrophobic domain known as lipid A (or endotoxin), a non-repeating “core” oligosaccharide, and a distal polysaccharide (or O-antigen). The flood of recent genomic data has made it possible to study lipopolysaccharide assembly in diverse Gram-negative bacteria, many of which are human or plant pathogens, and to create mutants or hybrid constructs with novel properties. Unexpectedly, key genes for lipid A biosynthesis have also been found in higher plants, indicating that eucaryotic lipid A-like molecules may exist. The carbohydrate diversity of lipopolysaccharides is better appreciated now than ten years ago, but much remains to be learned about function. Sequence comparisons suggest that extensive lateral transfer of genes for the assembly of O-antigens has occurred among bacteria. The most significant finding in the field of endotoxin biology since 1990 has been the identification of the plasma membrane protein TLR4 as the lipid A signaling receptor of animal cells. The latter belongs to a family of innate immunity receptors, all of which possess a large extracellular domain of leucine-rich repeats, a single trans-membrane segment and a smaller cytoplasmic signaling region that engages the adaptor protein MyD88. The expanding knowledge of TLR4 specificity and its downstream signaling pathways should provide new opportunities for blocking the inflammatory side effects of sepsis. Future progress will require insights into lipopolysaccharide-protein recognition at the atomic level, greater understanding of intra- and inter-cellular lipopolysaccharide trafficking, and incisive biological approaches that combine the tools of bacterial and animal genetics.
Endotoxins as Activators of Innate Immunity
Lipid A (endotoxin), the hydrophobic anchor of lipopolysaccharide (LPS), is a glucosamine-based phospholipid that makes up the outer monolayer of the outer membranes of most Gram-negative bacteria (1-5). There are ∼106 lipid A residues and ∼107 glycerophospholipids in an Escherichia coli cell (6). The minimal LPS required for the growth of E. coli consists of the lipid A and Kdo (3-deoxy-D-manno-oct-2-ulosonic acid) domains (Figs. 1 and 2) (1, 7, 8). In wild type strains, additional core and O-antigen sugars are present (Fig. 1) (5, 7, 9-11). These are generally not required for growth in the laboratory, but help bacteria resist antibiotics, the complement system, and other environmental stresses.
Figure 1. Model of the inner and outer membranes of E. coli K-12.

Only the Kdo and lipid A regions of LPS are required for the growth of E. coli and most other Gram-negative bacteria (2). Exceptions to this general rule include certain spirochetes in which all lipid A biosynthesis genes are absent (141), Thermotoga maritima (137), and Neisseria meningitidis type B in which lipid A-deficient lpxA knockouts can be constructed (133), provided the polysialic acid capsule is present.
Figure 2. Structure and biosynthesis of Kdo2-lipid A in E. coli K-12.

The symbols indicate the relevant structural genes encoding each of the enzymes (2, 48). A single enzyme catalyzes each reaction. The lipid A system may have evolved only once, as judged by the available genomes. In almost all cases, as illustrated by E. coli (367), the genes encoding the enzymes of lipid A biosynthesis are present in single copy. At the protein level, orthologs of LpxA and LpxC are the most highly conserved among bacteria. The acyl chain incorporated by LpxA is highlighted in red. The LpxC inhibitor L-161,240 displays antibiotic activity against E. coli that is comparable in potency to ampicillin (64, 65).
Many Gram-negative bacteria, including pathogens, synthesize lipid A species resembling the one found in E. coli (Fig. 2) (1, 3, 4). Early ambiguities concerning the structure of lipid A have generally been resolved, and are discussed elsewhere (1, 3, 4). Given their conserved architecture, most types of lipid A molecules are detected at picomolar levels by an ancient receptor of the innate immune system present on macrophages and endothelial cells of animals (12, 13). The receptor, recently identified as TLR4 (toll-like receptor 4) (14, 15), is a membrane spanning protein that is distantly related to the IL1 receptor (12, 13).
In macrophages, lipid A activation of TLR4 triggers the biosynthesis of diverse mediators of inflammation, such as TNF-α and IL1-β (16, 17), and activates the production of co-stimulatory molecules required for the adaptive immune response (13). In mononuclear and endothelial cells, lipid A also stimulates tissue factor production (18, 19). These events are desirable for clearing local infections, and they act in synergy. When overproduced systemically in the setting of severe sepsis, however, the various mediators and clotting factors can damage small blood vessels and precipitate Gram-negative septic shock, accompanied by disseminated intra-vascular coagulation and multiple organ failure (20-23). Synthetic E. coli lipid A by itself causes a similar spectrum of effects when injected into animals (3, 24), supporting the proposed role of lipid A in Gram-negative sepsis. The characteristic structural features of E. coli lipid A (Fig. 2), especially its two phosphate groups and its two acyloxyacyl moieties, are needed to trigger the endotoxin response in human cells (3, 25-27).
Many of the initial events in the interaction of lipid A with animal cells have been elucidated in the past ten years (14, 15, 28, 29). A special lipid transfer protein in plasma delivers lipid A from bacteria (or bacterial membrane fragments) to CD14 on the surfaces of animal cells (Fig. 3) (30, 31). The subsequent recognition of lipid A (or perhaps of the lipid A/CD14 complex) by the receptor protein TLR4 represents the earliest known step in signal transduction (Fig. 3) (14, 15, 28, 29, 32). The interaction of lipid A with TLR4 likely involves other proteins, including not only the phosphatidylinositol glycan-linked surface protein CD14 (33, 34) but also the soluble accessory protein MD2 (Fig. 3) (35). Despite very persuasive genetic evidence for the function of TLR4 (14, 15), direct biochemical evidence for binding of lipid A and related molecules to TLR4 is still lacking.
Figure 3. Detection of lipid A by the TLR4 innate immunity receptor of animal cells.

The paradigm is based mainly on studies of the human, mouse and hamster systems (12-15, 27, 31, 368). We speculate that the TLR4 receptor may be oligomerized upon binding of lipid A. The TRL4 mediated inflammatory response is beneficial in combating localized infections, but may be detrimental in overwhelming systemic sepsis. Novel therapies of sepsis directed against the disseminated intravascular coagulation component using recombinant activated protein C have recently shown efficacy in human trials (23). Combination therapy with TLR4 antagonists (369) remains to be explored.
The first protein of the toll receptor family was discovered in the context of insect development, but was later shown to play an additional role in protecting insects against fungal infections (36-38). Subsequently, more than ten Toll-like receptor protein homologues were discovered in the mouse and human genomes (12, 13, 39). Although much remains to be learned about the functions of the TLR proteins, TLR2 appears to be less specific than TLR4 in that it is activated by diverse ligands, including bacterial lipoproteins and peptidoglycan fragments (12, 40). TLR6 may function together with TLR2 to recognize a subset of bacterial membrane proteins and lipopeptides (41). TLR3 is activated by double-stranded RNA (42), and TLR5 is activated by bacterial flagellin (43). TLR9 responds to bacterial DNA sequences containing the CpG motif (44). As suggested for TLR4 in Fig. 3, it may be that all Toll-like receptors function as dimers or perhaps as heterodimers, in analogy to the IL-1 receptor (45).
Although the three dimensional structures of the leucine-rich extracellular domains of the TLR receptors have not yet been determined, x-ray structures of the intracellular signaling domains of the TLR1 and TLR2 receptors have recently been reported (46). These receptors show significant sequence homology to the intracellular domain of the IL-1 receptor, and closely resemble TLR4 in their ability to engage the MyD88 adaptor protein (Fig. 3).
Lipid A biosynthesis in Escherichia coli and Salmonella typhimurium
The constitutive lipid A (endotoxin) pathway
The enzymology and molecular genetics of the conserved steps of lipid A biosynthesis are best characterized in E. coli (1, 2, 47, 48). The first reaction of lipid A biosynthesis is the acylation of the sugar nucleotide UDP-GlcNAc (Fig. 2) (49). The E. coli UDP-GlcNAc acyltransferase (LpxA) is selective for β-hydroxymyristate, consistent with the composition of E. coli lipid A (49). UDP-GlcNAc acyltransferase has an absolute requirement for an acyl carrier protein (ACP) thioester as its donor substrate (49, 50).
E. coli LpxA (a homotrimer) contains multiple contiguous hexad repeats (51), which are also found in certain other acyltransferases and acetyltransferases (52-54). The x-ray structure of LpxA shows that these hexads specify a novel protein fold, consisting of a left-handed helix of short parallel β-sheets (51). Three hexads form one coil of the β-helix, and each LpxA monomer contains ten such coils stacked on top of each other. The active site of E. coli LpxA, which is located between the coils of adjacent subunits, functions as an accurate hydrocarbon ruler that incorporates 14-carbon acyl chains two orders of magnitude faster than 12- or 16-carbon acyl chains (55, 56). In Pseudomonas aeruginosa, the ruler is reset to measure ten carbons (57). Single amino acid substitutions can convert P. aeruginosa LpxA to a fourteen carbon specific acyltransferase or E. coli LpxA to a ten carbons specific enzyme (55). Chlaymidia trachomatis LpxA is unique in that it utilizes myristoyl-ACP instead of hydroxymyristoyl-ACP, generating novel hybrid lipid A species when substituted for E. coli LpxA in living cells (58). The structural basis for this selectivity is unknown.
The equilibrium constant (∼0.01) for the acylation of UDP-GlcNAc is unfavorable (50). The biological implication is that the deacetylation of the product, UDP-3-O-(acyl)-GlcNAc (Fig. 2), by the zinc metalloenzyme LpxC is the committed reaction of the pathway (59-63). LpxC is the product of a conserved, single-copy gene present in diverse Gram-negative bacteria, and it displays no sequence similarity to other deacetylases or amidases. LpxC is an excellent target for the design of novel, Gram-negative specific antibiotics (64, 65).
Following deacetylation, a second β-hydroxymyristate moiety is incorporated by LpxD to generate UDP-2,3-diacylglucosamine (Fig. 2) (66). Again, only ACP thioesters are substrates (66). The sequences of LpxA and LpxD are distantly related because of the multiple, contiguous hexad repeats that are present in both proteins (66).
UDP-2,3-diacylglucosamine (Fig. 2) is cleaved at its pyrophosphate bond by the highly selective pyrophosphatase LpxH to form 2,3-diacylglucosamine-1-phosphate (lipid X) (67). LpxH is missing in a few Gram-negative genomes, suggesting additional enzymes of this kind may exist. A β, 1′-6 linked disaccharide is then generated by the condensation of another molecule of UDP-2,3-diacylglucosamine with lipid X (Fig. 2) (68, 69). The disaccharide synthase is encoded by lpxB, which is co-transcribed with lpxA in E. coli (70-72), and many other organisms.
A specific kinase next phosphorylates the 4′ position of the disaccharide to form lipid IVA (Fig. 2) (73). The kinase structural gene (lpxK) (74, 75) is located downstream in an operon with the msbA gene of E. coli. MsbA (see below) is an essential, inner membrane ABC transporter (76) that functions in the initial stages of lipid A (77, 78) and phospholipid (78, 79) export.
The kinase product, lipid IVA (Fig. 2), is of great interest in its own right because it possesses some of the properties of endotoxins (80). In mouse cells it is a potent endotoxin-like agonist, but in human cells it is an endotoxin antagonist (26). This unusual pharmacology of lipid IVA is determined by whether the target cells express mouse or human TLR4 (Fig. 3) (81, 82).
E. coli LPS contains two Kdo residues that are transferred to lipid IVA by a bifunctional enzyme (Fig. 2), encoded by waaA (also known as kdtA) (83). The related waaA gene of H. influenzae specifies an enzyme that adds one Kdo moiety (84). It is not possible as yet to determine whether a Kdo transferase is mono-, bi-, or even tri-functional (as in Chlamydia trachomatis) (85) by inspection of its sequence. The labile sugar nucleotide, CMP-Kdo (1, 2), synthesized by KdsA and KdsB (not shown in Fig. 2), serves as the Kdo donor.
The last steps of E. coli lipid A biosynthesis involve the addition of lauroyl and myristoyl residues to the distal glucosamine unit (Fig. 2), generating acyloxyacyl moieties (86). The E. coli “late” acyltransferases require the presence of the Kdo disaccharide in their substrate for activity (86-88). This sequence of events is not strictly conserved in P. aeruginosa (89), however, in which the ACP dependent late acyltransferases add one secondary laurate chain to each glucosamine unit (Fig. 4) in a symmetrical pattern. Like LpxA and LpxD, the late acyltransferases utilize acyl-ACPs exclusively as donors (86-88). The E. coli lauroyl and the myristoyl transferases are encoded by lpxL and lpxM respectively (Fig. 2), known as htrB and msbB prior to elucidation of their functions (87, 88, 90, 91), and display significant sequence similarity to each other, but not to LpxA or LpxD. The lpxM(msbB) gene is not required for growth (91) in E. coli, but interestingly, lpxM mutants are greatly attenuated in their ability to activate human macrophages (92). In Salmonella, lpxM mutants are much less lethal than wild-type bacteria in animal shock models (93, 94), supporting a central role for lipid A in sepsis physiology. Mutants with a reduced number secondary acyl chains on their lipid A moieties may be useful for the development of new vaccines (95).
Figure 4. Regulated modifications of lipid A in E. coli and S. typhimurium and unusual lipid A structures in other bacteria.

Partial covalent modifications are indicated with the dashed bonds. Panel A. Modifications of E. coli and S. typhimurium lipid A under the control of PmrA are blue, whereas modifications primarily under the control of PhoP are red. For a recent discussion of the structures of the modified lipid A species that can be isolated from various mutants or under different growth conditions see Zhou et al. (112). When present, the L-Ara4N (dark blue) moiety is located mainly at the 4′ position, whereas the phosphoethanolamine (light blue) is mostly at position 1. Certain lipid A species exist in which the same substituents are attached at both sites, or in which their locations are reversed (112, 370). In cells grown with 1-10 mM Mg++ above pH 7.4, the modifications are suppressed, and a pyrophosphate group (the origin of which is unknown) is present at position 1 in about one third of the lipid A molecules (112). Panel B. Lipid A modifications in Pseudomonas under the control of PmrA are shown in blue, whereas modifications primarily under the control of PhoP are red (109). The portion of the lipid A molecule generated by the constitutive pathway is shown in black. Panel C. R. etli lipid A, which lacks phosphate groups, includes a major species in which aminogluconate (magenta) replaces the proximal glucosamine. Aminogluconate is formed in the outer membrane from a lipid A species containing glucosamine (149, 150, 156). The 4′ galacturonic acid moiety is green. Panel D. Like R. etli, Aquifex aeolicus lipid A lacks phosphate moieties (126), but contains two galacturonic acid residues (green). The hydroxyacyl chains at positions 3 and 3′ differ from E. coli in that they are amide-linked (magenta NH atoms).
Regulated pathways for the covalent modification of lipid A
E. coli contains one additional gene that is similar to lpxL, termed lpxP, which is activated under conditions of cold shock (12 °C) (96). LpxP inserts palmitoleate in place of laurate (not shown in Fig. 2), perhaps reflecting the need to adjust outer membrane fluidity at low temperatures (96). By deleting the lpxL, lpxM and lpxP genes simultaneously, it is possible to obtain strains of E. coli with lipid A species that lack all secondary acyl chains (M. Vorachek-Warren and C. R. H. Raetz, in preparation). These triple mutants grow slowly on minimal medium, but are not viable in nutrient broth. However, second site suppressors (M. Vorachek-Warren and C. R. H. Raetz, in preparation) or multi-copy suppressors (see below) can be introduced that allow these E. coli mutants to grow almost normally under all conditions.
E. coli and S. typhimurium contain additional enzymes for modifying lipid A with phosphoethanolamine, 4-amino-4-deoxy-L-arabinose (L-Ara4N) and/or palmitate groups (Fig. 4A). These enzymes are normally latent in E. coli K-12, but are expressed in the presence of metavanadate (97). Attachment of the phosphoethanolamine and L-Ara4N moieties is also induced by activation of the PmrA transcription factor following exposure to mildly acidic conditions, or by mutation (98-102). The presence of the L-Ara4N group protects bacteria against killing by polymyxin and certain cationic anti-bacterial peptides (98, 99, 103) (Fig. 4). Polymyxin-resistance has provided a means to select mutants in the biosynthesis and regulatory pathways.
The modification of lipid A with palmitate is under control of the PhoP/PhoQ system (103, 104), which is activated by low concentrations of Mg++, as would be encountered inside phagolysomes (105, 106). S. typhimurium mutants unable to add palmitate to their lipid A are sensitive to the NP-2 defensin (103). Interestingly, palmitate transfer to lipid A occurs in the outer membrane of S. typhimurium and E. coli (104), and is catalyzed by the unusual palmitoyl transferase (PagP) (104) that uses glycerophospholipids as its palmitoyl donor (107). The occurrence of PagP in eubacteria is limited to E. coli, Salmonella, Bordetella, Yersinia pestis and Legionella pneumophila. Its absence in mutants of Legionella may interfere with pathogenesis (108).
Pseudomonas aeruginosa lipid A can also be modified with palmitate during conditions of PhoP activation (Fig. 4B) (109), but the palmitoyl group is added to a different site on lipid A than in E. coli (Fig. 4A). P. aeruginosa does not contain a PagP ortholog.
S. typhimurium contains two additional lipid A modifying enzymes, not found in E. coli. The first of these hydroxylates the 3′ secondary acyl chain in an O2-dependent manner (110) to generate S-2-hydroxymyristate (Fig. 4A). The other selectively removes the β-hydroxymyristoyl residue at position 3 (111) (Fig. 4A). The functions of these modifications are unknown. Removal of the β-hydroxymyristoyl residue at position 3 might reduce the cytokine inducing potential of bacterial cells.
The enzymes that add phosphoethanolamine to lipid A in polymyxin resistant mutants (112) have not been characterized. However, E. coli cells grown with 5 mM CaCl2 are induced to make a distinct phosphoethanolamine transferase (113) that modifies the outer Kdo residue (not shown). Phosphatidylethanolamine is the likely phosphoethanolamine donor in all cases (102, 113), including the modification of the heptose region (114), which occurs in a constitutive manner (Fig. 1) (see below). The relevant genes have not been identified.
Origin of L-Ara4N modified lipid A in polymyxin resistant mutants
The proposed biosynthesis and mode of attachment of the L-Ara4N moiety to lipid A is shown in Fig. 5. Our hypothesis for the existence of this scheme (97) was based on the discovery by Gunn et al. (99) of gene clusters in S. typhimurium and E. coli required for the maintenance of polymyxin resistance. In accordance with the system of Reeves et al. (9), the genes of the polymyxin resistance operon have been renamed arn (Fig. 5), given their recently demonstrated functions in the biosynthesis and transfer of the L-Ara4N moiety (102, 115, 116). The pathway starts with the conversion of UDP-glucose to UDP-glucuronic acid by the well-characterized dehydrogenase, Ugd (Fig. 5). Next, ArnA (previously Orf3 or PmrI) catalyzes the oxidative decarboxylation of UDP-glucuronic acid to generate a novel UDP-4-keto-pyranose intermediate, which can be isolated in milligram quantities (116). ArnB (previously Orf1 or PmrH) then catalyzes a transamination using glutamic acid as the amine donor to form UDP-L-Ara4N (116). Based upon its homology to dolichyl phosphate-mannose synthase of yeast, it is believed that ArnC (PmrF) (99) transfers the L-Ara4N moiety to undecaprenyl phosphate (97), forming the novel compound undecaprenyl phosphate-α-L-Ara4N. The existence of this important intermediate, which accumulates in polymyxin resistant mutants, is firmly established (102). After its presumed translocation to the outer surface of the inner membrane by unknown mechanisms (Fig. 5), ArnT (previously Orf5, PmrK or YfbI) transfers the L-Ara4N unit to lipid A (115). Robust in vitro assays for ArnT have recently been developed (115). Other genes of the polymyxin resistance operon (pmrJ, pmrL, and pmrM), as well as the adjacent pmrG gene, cannot yet be assigned specific functions (99, 117).
Figure 5. Pathway for L-Ara4N biosynthesis and mechanism of polymyxin resistance in E. coli and S. typhimurium.

In accordance with the proposal of Reeves et al. (9), we have renamed the genes of the polymyxin resistance operon “arn”, given their function in the biosynthesisof the L-Ara4N moiety and its transfer to lipid A (97, 102, 115). The proposed pathway starts with the conversion of UDP-glucose to UDP-glucuronic acid by the well-characterized dehydrogenase, Ugd. Next, ArnA (previously Orf3 or PmrI) (97, 102, 115) catalyzes the oxidative decarboxylation of UDP-glucuronic acid to generate a novel UDP-4-keto-pyranose intermediate, which can be isolated in mg quantities using ArnA (116). In contrast to the proposal of Baker et al. (371), a separate enzyme to catalyze the decarboxylation step is not necessary in our scheme (97, 116). ArnB (previously Orf1 or PmrH) (97, 99, 117) then catalyzes a further transamination to form UDP-L-Ara4N (116). Based upon its homology to dolichyl phosphate-mannose synthase of yeast, we propose that ArnC (PmrF) (97, 99, 117) transfers the L-Ara4N moiety to undecaprenyl phosphate, forming the novel compound undecaprenyl phosphate-α-L-Ara4N (102). After translocation to the outer surface of the inner membrane by unknown mechanisms, ArnT (previously Orf5, PmrK or YfbI) (115) transfers the L-Ara4N unit to lipid A. Other genes of the polymyxin operon (pmrJ, pmrL, and pmrM), as well as the adjacent pmrG gene (99, 117), cannot yet be assigned specific enzymatic or transport functions in our scheme. The ArnA protein has a second catalytic domain (reaction not shown) that can transfer a formyl group from N10-tetrahydrofolate to UDP-L-Ara4N (116), but the significance of this modification is unclear. Addition of the L-Ara4N moiety to lipid A reduces the affinity of lipid A for polymyxin and other cationic anti-microbial peptides.
Given that palmitate addition to lipid A occurs within the outer membrane (104) and L-Ara4N incorporation may take place within the periplasm (102, 115), it may be possible to utilize the presence or absence of these covalent modifications as markers for the transport of nascent lipid A from its site of biosynthesis on the inner surface of the inner membrane to the outer membrane (Fig. 1).
Role of the ABC transporter MsbA in lipid A and phospholipid export
The outer membrane is an asymmetric bilayer with phospholipids on its inner surface, and lipid A, the hydrophobic anchor of lipopolysaccharide on the outside (Fig. 1). The constitutive enzymes that make phospholipids and lipid A (Fig. 2) in E. coli are located in the cytoplasm or on the inner surface of the inner membrane (1, 118). How E. coli lipids cross the inner membrane and are transported to the outer membrane is not understood at the molecular level.
A clue to lipopolysaccharide transport has recently emerged from studies of E. coli lpxL(htrB) mutants and their suppression by multiple copies of the msbA gene (77-79, 87, 119). LpxL is the lauroyl transferase that functions late in E. coli lipid A biosynthesis (Fig. 2) (87, 119). Lipopolysaccharides bearing tetra-acylated lipid A species accumulate in the inner membranes of lpxL mutants at 42 °C, and unless second site suppressors are introduced, cell growth is inhibited (78). MsbA is an essential ABC transporter (Figs. 6 and 7), closely related to eucaryotic MDR proteins (76). MsbA over-expression represents one way in which to restore growth of lpxL mutants at 42 °C without restoring laurate incorporation (78), resulting in the export of lipopolysaccharides with tetra-acylated lipid A anchors to the outer membrane (78). E. coli msbA knockouts are lethal. Their biochemical analysis is complicated by the long times (4-8 h) needed to dilute out pre-existing MsbA supplied in trans from a temperature-sensitive plasmid (78), and by the fact that the lpxK gene (Fig. 2), which is immediately downstream in an operon with MsbA in E. coli, is also essential for cell growth (75).
Figure 6. Structure of the E. coli MsbA dimer at 4.5 Å resolution.

This backbone tracing was made from protein data bank file 1 JSQ (120). Transmembrane helices 1-6 are colored purple, blue, yellow, green, red and orange, respectively. The intracellular domain (ICD) is brown, and the nucleotide-binding domain is cyan (120). A schematic model of lipid A is shown in panel A for size comparison. The location of the A270 residue, which is changed to threonine in the temperature sensitive lipid transport mutant WD2 (79), is shown as a red sphere. The putative chamber for binding lipids on the inner surface of the inner membrane is lined with basic residues (120) (not shown).
Figure 7. Model for MsbA mediated lipid export in E. coli.

Following the MsbA mediated transport at the inner membrane, additional proteins are likely to be involved in steps 2 and 3, but these have not yet been identified (79).
To gain a clearer understanding of the function of MsbA, a new temperature-sensitive mutant of E. coli (WD2) was recently isolated in which there is a single A270T substitution in the fifth trans-membrane region of MsbA, and the impact of this mutation has been studied in detail (79) (Fig. 6). This MsbA variant is rapidly inactivated at 44 °C. As shown by 32Pi and 14C-acetate labeling, export of all major lipids (i.e. both LPS and phospholipids) to the outer membrane is inhibited by ∼90 % in WD2 after a 30 minute shift to 44 °C. Transport of newly synthesized proteins is not impaired under the same conditions. Electron microscopy reveals inner membrane reduplications and invaginations in WD2 at 44 °C, consistent with an increased surface area secondary to the complete block of lipid export (79).
His-tagged versions of MsbA have been constructed. The protein was over-expressed, solubilized with non-ionic detergents, purified, and reconstituted. Purified MsbA catalyzes lipid stimulated ATP hydrolysis in vitro. Kdo2-lipid A is an especially potent activator (W. Doerrler and C. R. H. Raetz, unpublished), suggesting that this molecule is transported. Cell-free assays to evaluate MsbA catalyzed lipid flip-flop and/or inter-vesicular lipid transport remain to be established.
Chang and Roth recently purified and crystallized E. coli MsbA using a similar preparation method, and they were able to solve its structure at a resolution of 4.5 Å (Fig. 6) (120). This remarkable achievement should provide insights into the functions of lipid pumps, including the mammalian MDR proteins, which are about twice as long as MsbA but are internally duplicated (120, 121). Based upon the crystal structure (Fig. 6), MsbA is a homodimer (120). It contains six trans-membrane helices (consistent with hydropathy analysis) (79). There is a large cavity between the subunits that could accommodate lipid A and/or phospholipid molecules (Fig. 6A). This space is lined with basic residues (120). Exactly how MsbA would mediate lipid flip-flop by binding and hydrolyzing ATP in the nucleotide binding domains (Fig. 6) is unclear. It is speculated that the nucleotide binding domains may come together following lipid entry into the putative transport chamber present in the trans-membrane region (Fig. 6), forcing the lipid to move to the periplasmic side of the inner membrane (as suggested in Fig. 7).
MsbA may indeed function as a flippase within the inner membrane, but in principle, it could also be a pump that ejects lipid A and phospholipids from the outer surface of the inner membrane during the transit of these substances to the outer membrane (Fig. 7). The location of the A270T mutation at the critical MsbA dimer interface on the periplasmic surface of the inner membrane (Fig. 6) is compatible with the lipid export defect observed in mutant WD2 (79). We anticipate that additional, as yet unidentified, protein components are needed to shuttle lipids across the periplasm, and to assemble them properly in the outer membrane (Fig. 7, steps 2 and 3). While the details will surely differ, the TolC hemolysin secretion system represents an interesting paradigm with respect to the involvement of multiple membrane proteins in the export process, including an ABC transporter with similarity to MsbA (76, 122).
Although many proteins in the MDR family appear to catalyze lipid flip-flop in vitro, mouse knockout mutants lacking the three major MDR proteins show no generalized defects in lipid trafficking, and the mice are viable (121). MDR2 knockouts do display a specific phospholipid transport deficiency in that they cannot pump phosphatidylcholine into their bile (123). Several distinct ABC transporters have recently been shown to be involved in reverse cholesterol flow and bile acid secretion (124). We believe that the many additional MDR-like proteins, present in all eucaryotes, may function in catalyzing lipid trafficking when the three known MDR proteins are inactivated. It may be necessary to isolate mutants defective in more than just the three major MDR proteins to obtain generalized lipid transport defects in animal cells, resembling those seen in WD2.
Other bacterial ABC transporters play key roles in exporting some types of O-antigens and bacterial capsules (see below). As with MsbA, robust in vitro systems to study the biochemistry of these transporters need to be developed.
Genomic insights into lipid A biosynthesis and diversity
The constitutive lipid A pathway as a target for new antibiotics
The genomic sequences of about 100 Gram-negative bacteria are either complete or nearing completion. In almost every case, each of the constitutive enzymes of lipid A biosynthesis (Fig. 2) is encoded by a single-copy gene. LpxH (67) is present in only ∼70 % of the available genomes, suggesting that additional isoenzymes catalyzing UDP-2,3,-diacylglucosamine hydrolysis (Fig. 2) may exist. A few of the genomes also lack homologues of lpxL and/or lpxM (125), though at least one acyloxyacyl moiety is invariably present in all lipid As when analyzed chemically (126). LpxA and LpxC are the most highly conserved of the constitutive enzymes (Fig. 2). The enzymes that catalyze regulated or other special modifications of lipid A (Figs. 4 and 5) are more variable, and are restricted in their distribution.
Lipid A is required for growth of E. coli and most other Gram-negative bacteria (6, 64) studied so far, and also, it is essential for the maintenance of an effective outer membrane barrier (127). The outer membrane protein FhuA interacts in a highly specific manner with LPS (128, 129), and lipid A may be required for the proper folding of some porins (130). Inhibitors of lipid A biosynthesis are themselves good antibiotics against E. coli, and sensitize bacteria to other antibiotics that normally do not penetrate the outer membrane (64). Onishi et al. have described L-161,240 (64), a specific LpxC inhibitor with a Ki of about 50 nM (Fig. 2, inset) that is bactericidal against E. coli at concentrations comparable to ampicillin (64, 65). This compound also kills strains of Enterobacter and Kebsiella, but not Pseudomonas (64, 65). Resistance in the latter case is due to the fact that L-161,240 and related compounds are poor inhibitors of P. aeruginosa LpxC. Novel inhibitors that are active against diverse LpxCs have recently been designed, but have not yet been optimized (65). An x-ray structure of LpxC would be very helpful in this regard. LpxC is a soluble zinc containing metallo-enzyme (62, 63), implying that far more potent inhibitors might yet be found, as demonstrated in earlier studies with angiotensin converting enzyme (131) and carboxypeptidase A (132).
In contrast to E. coli and most other Gram-negative human pathogens, strains of Neisseria meningitidis that possess a polysialic acid capsule can grow slowly without lipid A when their lpxA gene is inactivated (133). Such cells no longer activate TLR4, but do activate TLR2 at ∼100 fold higher doses (134). In such mutants the polysialic capsule becomes essential for growth (P. van der Ley, personal communication) and appears to substitute for lipid A. The hydrophobic anchor of the polysialic acid capsule of N. menigitidis is not fully characterized (135). These remarkable observations are important in that they may provide new insights into the functions of lipid A in Gram-negative bacteria.
A few types of diverse bacteria that possess outer membranes inherently lack the lipid A system, as defined by the absence of the lpx genes (Fig. 2). These include Sphingomonas paucimobilis, which appears to contain glycosphingolipids in place of lipid A (136), Thermotoga maritima, the genome of which contains about 25 % archaebacterial sequences (137), Deinococcus radiodurans (138, 139), and the spirochetes Treponema pallidum and Borrelia burgdorferi (140, 141). The genomic information for B. burgdorferi is consistent with the failure to detect lipid A in this organism by chemical methods (142). Whether or not other lipids substitute for lipid A in these organisms (as in Sphinogmonas) is unknown. Some bacteria that lack the lpx genes, such as Deinococcus radiodurans, do contain “alien gene” clusters encoding putative glycosyl transferases, resembling O-polysaccharide biosynthesis modules (143, 144). Such O-polysaccharide-like polymers, which could function as receptors for bacteriophages (see below), might be grafted onto novel lipid anchor(s) in these systems. The underlying biochemistry has not yet been explored. Unlike O-polysaccharide modules, the lpx genes themselves (Fig. 2) do not qualify as “alien” (143), suggesting that they have not undergone extensive lateral transfer.
Treponema pallidum and Borrelia burgdorferi have relatively small genomes (140, 141), and are restricted in their ability to grow outside of their hosts. On the other hand, Leptospira interrogans is a spirochete with a much larger genome that can grow on a defined medium (145, 146). It contains the genes encoding the constitutive lipid A pathway (Fig. 2). The final product may be modified in a unique manner, however, given that Leptospira interrogans lipid A appears to activate TLR2 instead of TLR4 (146).
The constitutive pathway in bacteria with unusual lipid A structures
As shown in Fig. 4, significant variations in lipid A structure occur in some Gram-negative bacteria. These changes (relative to E. coli and S. typhimurium) include differences in acyl chain length and distribution, as in Pseudomonas aeruginosa (Fig. 4B) (109, 147), the absence of phosphate moieties in organisms like Rhizobium etli (148-150) and Aquifex aeolicus (126) (Fig. 4C and 4D), the presence of galacturonic acid residues (Fig. 4C and D), and subtle modifications of one or both glucosamine units (Fig. 4C and D) (126, 148-150). Despite these differences, the key enzymes of the constitutive lipid A pathway (Fig. 2) remain operative (65, 125, 151). The observed structural variations are generally explained by the existence of additional enzymes (152-157) that are not present in E. coli.
In Pseudomonas aeruginosa, the altered substrate specificities of LpxA and LpxD account for the shorter R-3-hydroxyacyl chains (Fig. 4B) (158). The LpxL ortholog that incorporates the secondary laurate on the proximal glucosamine unit is Kdo independent (89). The secondary laurate chain on the distal unit is incorporated by a more typical Kdo-dependent LpxL ortholog (N. Que and C. Raetz, in preparation). Two LpxO orthologs (110) are present in P. aeruginosa, possibly accounting for the two alternative sites at which 2-hydroxylaurate may be generated (159) (Fig. 4B, red X) in an O2 dependent manner (160). In contrast to the secondary laurate residues, the secondary palmitoyl chain of P. aeruginosa lipid A (Fig. 4B, red) is not incorporated by an ACP-dependent mechanism (S. Trent and C. R. H. Raetz, unpublished), and it is absent unless the PhoP system is activated (109). However, a PagP ortholog is not present in P. aeruginosa. Similarly, although there is no apparent PagL ortholog in Pseudomonas, much of the 3-position is deacylated in mature LPS species isolated from cells (109, 147, 161) (Fig. 4B). A 3-O-deacylase activity is present in cell extracts (S. Trent and C. R. H. Raetz, unpublished). In contrast to the situation with PagP and PagL, the orthologs responsible for L-Ara4N modifications (Fig. 4, dark blue) of P. aeruginosa appear to be very similar to those of E. coli and S. typhimurium (Fig. 5).
Rhizobium etli lipid A consists of several molecular species (149, 150), the most unusual of which are shown in Fig. 4C. The absence of phosphate residues is explained by late-functioning phosphatases (152, 153), perhaps located on the outer surface of the inner membrane (157). The oxidation of the dephosphorylated proximal residue to generate the aminogluconate moiety (Fig. 4C, magenta) occurs in the outer membrane (156). The long secondary (C28) acyl chain at position 2′ is incorporated by a distant ortholog of LpxL (S. Basu and C. R. H. Raetz, in preparation), which requires a specialized acyl carrier protein (ACP-XL) (155). The origins of the pendant 3-hydroxybutryate moiety and the 4′ galacturonic acid residue (Fig. 4C, green) are unknown.
Like R. etli, Aquifex aeolicus lipid A lacks phosphate residues and contains galacturonic acid moieties (Fig. 4D, green) (126). In addition, the glucosamine disaccharide is replaced with two 2,3-diamino-2,3-dideoxy-D-glucose residues (Fig 4D, magenta NH atoms) (126). The latter are also found in many other Gram-negative bacteria (162), like Thiobacillus ferrooxidans. In the latter organism, the 2,3-diamino-2,3-dideoxy-D-glucose residue arises by NAD dependent oxidation of UDP-GlcNAc at the GlcN 3-position, followed by a transamination with glutamic acid as the amine donor (163). The resulting “UDP-3-amino-3-deoxy-GlcNAc” is then utilized preferentially by Thiobacillus LpxA, in analogy to what occurs with UDP-GlcNAc in the E. coli pathway (Fig. 2) (C. Sweet and C. R. H. Raetz, unpublished).
The biological significance of these and other lipid A structural variations will have to be evaluated by genetic methods. The unusual R. etli lipid A might be advantageous during nodulation and symbiosis with plants, whereas A. aeolicus lipid A might confer thermo-tolerance. Elucidation of the relevant enzymology is necessary as a prelude to genetic studies.
Presence of lipid A biosynthesis genes in plants
Recent versions of protein, DNA and EST databases indicate that Arabidopsis thaliana and other plants contain significant homologues of E. coli genes encoding enzymes of lipid A biosynthesis. These genomic observations imply that plants synthesize lipid A-like substance(s). Lipid A or closely related molecules may be minor constituents of plant membranes that were overlooked in earlier biochemical studies. The lipid A pathway may have appeared in plants following symbiosis with cyanobacteria. Nuclear genes encoding apparent orthologs of LpxA, LpxC, LpxB, LpxD, LpxK, KdtA, KdsA and KdsB are present in the genomic DNA database of A. thaliana. These genes have introns and display plant codon usage. ESTs encoding key portions of LpxA, LpxC, LpxD, LpxB, LpxK and KdtA have also been recovered from many other plants, including cotton, pine, rice, soy bean, pepper and corn. Known active site residues of LpxA (56) and LpxC (63) are conserved.
The plant homologues of the E. coli lpx gene products display 30-45 % sequence identity over the predicted lengths of each of the proteins. Given the enzymatic studies of diverse bacterial Lpx proteins (58, 65, 85), this degree of sequence identity between plants and E. coli is indicative of a closely related enzymatic function. The A. thaliana lpxA gene can in fact complement a temperature-sensitive mutant of E. coli with a point mutation in its own lpxA gene (D. Liu and C. R. H. Raetz, unpublished) (6). A mutant of A. thaliana containing a T-DNA insert in the middle of the lpxA gene has recently been identified (D. Liu and C. R. H. Raetz, unpublished).
The sub-cellular localization of the plant lpx gene products is uncertain. LpxA contains an N-terminal leader sequence in A. thaliana suggestive of targeting to chloroplasts or mitochondria. Interestingly, full-length lpxA and lpxC genes are actually found next to each other in the chloroplast genome of the red alga Cyanidium caldarium (164). Perhaps, the first few steps of lipid A biosynthesis occur in chloroplasts, where ACP and fatty acids are abundantly available (165).
Given these considerations, it is reasonable to hypothesize that plants have the capacity to make a lipid A-like molecule, minimally consisting of Kdo-lipid IVA (Fig. 2). However, extensive additional modifications to this scaffold might occur, as is observed in some other Gram-negative bacteria (Fig. 4C and 4D) (126, 149, 150). Orthologs of the lipid A genes are not present in the human, mouse, worm, fly, or yeast databases.
What functions might plant lipid A serve? One scenario is that it is a structural component of chloroplast outer membranes. Alternatively, lipid A could function as a signaling molecule in plants, given that homologues of lipopolysaccharide binding protein and of the extra-cellular domains of the Toll-like receptors are found in A. thaliana. One TLR homologue of Arabidopsis has been identified as a brassinosteroid receptor (166), and is required for normal growth. The brassinosteroid receptor is homologous to other plant gene products needed for disease resistance (166).
Characterization of the lpxA gene knockouts in A. thaliana (D. Liu and C. R. H. Raetz, unpublished), in conjunction with a systematic search for lipid A-like substances, should provide incisive information concerning the function of the Lpx orthologs in plants. Given that lipid A-like molecules are potent adjuvants in humans, the possible existence of such molecules in plants may prove to be of considerable medical/toxicological interest with regard to the development of allergies or respiratory diseases induced by plant dust (167).
Structure and biosynthesis of core oligosaccharides
Structure of core oligosaccharides
As with lipid A, much of our understanding of the structure and biosynthesis of the core oligosaccharide is founded on work in E. coli and Salmonella but detailed structures are now available from a variety of organisms with different lifestyles, and the scope of biosynthesis data is rapidly expanding. The structures of core oligosaccharides are described in detail elsewhere (reviewed in 168) and only a general overview is provided here. In bacteria that produce smooth LPS (S-LPS), the core oligosaccharides are conceptually divided into two regions: inner core (lipid A proximal) and outer core. The outer core region provides an attachment site for O polysaccharide (O antigen). Mucosal pathogens often lack O polysaccharides. Instead they produce lipooligosaccharides (LOSs) that contain a recognizable inner core from which extend one or more mono- or oligosaccharide branches (equivalent to the outer core). These extensions determine serological specificity. Several excellent reviews are available for detailed coverage of the structure and function of LOS (169-171).
Within a genus or family, the structure of the inner core tends to be well conserved, and the fact that the core oligosaccharides from distantly related bacteria share structural features in the inner core is a reflection of the importance of the core in outer membrane integrity. The inner core typically contains residues of Kdo and L-glycero-D-mannoheptose (L,D-Hep) (Figure 8 inset). The Kdo residue is the only component found in all known cores, although in some cases a derivative, D-glycero-D-talo-oct-ulosonic acid (Ko), is also present (5). Some bacteria contain D-glycero-D-mannoheptose (D,D-Hep) alone or in combination with the more prevalent L,D-Hep (see Figure 9) while others, like Rhizobium, lack heptose entirely (154, 172, 173). The base structure of inner core is often decorated with non-stoichiometric additions of other sugars and with phosphate (P), pyrophosphorylethanolamine (2-aminoethanol; PPEtN), or phosphorylcholine (PCho) residues. The varying extent of these modifications contributes to the heterogeneity of LPS molecules extracted from a culture. In many cases, only the structure of the predominant carbohydrate backbone is known for a given core oligosaccharide, and the extent of phosphorylation and non-stoichiometric additions are unknown. This is a reflection of the difficulties encountered in structural analyses.
Figure 8. Structures of the known lipopolysaccharide core oligosaccharides from E. coli and Salmonella.
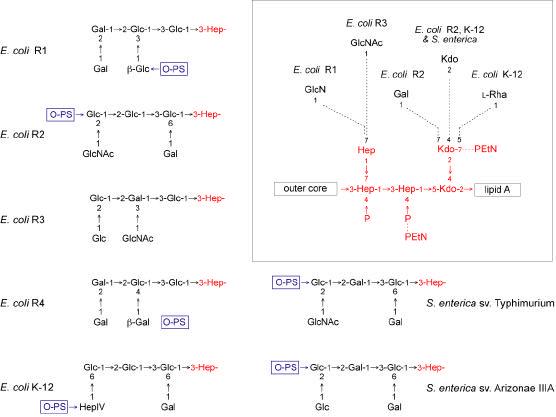
The outer cores are shown, together with one heptose residue of the inner core. The ligation sites for O polysaccharides (O-PS) are indicated where known (reviewed in 201). The inset shows the conserved base structure of the inner core and type-specific non-stoichiometric additions to the inner core are identified by dotted lines. Residues and linkages that are conserved in each of the core oligosaccharides are shown in red. Details of the structures are described elsewhere (168, 236, 372). Unless otherwise noted, all linkages are in the α-anomeric configuration.
Figure 9. Structures of the core oligosaccharides from Klebsiella pneumoniae O1 and P. aeruginosa O5.
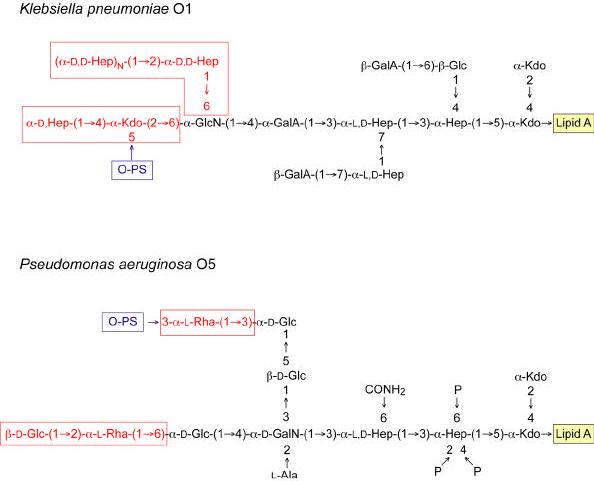
The β-galacturonic acid residues on the K. pneumoniae core are non-stoichiometric and details of the structures are described elsewhere (178, 179, 232, 373) (and references therein). The boxed regions of the core structures are those that differ between S-LPS and R-LPS, and the details are discussed in the text.
The outer core shows more structural diversity as might be expected for a region with more exposure to the selective pressures of host responses, bacteriophages, and environmental stresses. However, the extent of structural variation in core oligosaccharides within a given species, or even a genus, is still limited. For example, in E. coli there are 5 known core types (R1, R2, R3, R4 and K-12) (Figure 8). All are found among commensal isolates (174) while the R1 type predominates among strains that cause extraintestinal infections (175, 176) and the R3 type is found in most verotoxigenic isolates such as O157:H7 (174, 177). These cores differ in the non-stoichiometric inner core substituents found in a minor fraction of isolated LPS molecules, but the most substantial changes are evident in the outer core (Figure 8). The two known cores from Salmonella are quite similar to those of E. coli (Fig. 8), and genetic data indicate that the structure found in serovar Typhimurium is common to isolates that are isolated from routine human infections (S. enterica subspecies I) whereas the structure found in Arizonae IIIA predominates in other subspecies (N. Kaniuk and C. Whitfield, unpublished). In K. pneumoniae only one major core structure has been discovered (Figure 9), and this is distributed among different serotypes (178, 179) (and references therein). However, there is some variation in the extent of non-stoichiometric substitutions among isolates, and there may be some additional core diversity based on reactivity with a core-specific monoclonal antibody (180). The K. pneumoniae core with its outer core α-Kdo provides an example of a structure with lability to both mild acid and alkaline deacylation, two treatments commonly used to generate carbohydrate backbones for structural analysis. As a result, some features of the outer core structure were not identified in initial analyses (178). The limited structural variation in the core oligosaccharide within a genus is in striking contrast to the hypervariable O polysaccharides, and has stimulated interest in the possibility of targeting the core OSs for the generation of immunotherapeutic antibodies (181-185). Similarly, the enzymes responsible for the conserved structural features in inner cores provide potential targets for novel therapeutics.
In the LOS from mucosal pathogens, the inner core is also conserved within a species. However, differential expression of the attached oligosaccharides leads to phase variation and multiple glycoforms and immunotypes in the LOS (170, 171). The LOS of Neisseria meningitidis provides a good example, where the varying composition of the α-chain and differential substitution of the side branch heptose (β and γ chains), and sialylation all generate heterogeneity (Figure 10). In several cases LOS contain structures that mimic host-cell antigens.
Figure 10. Structure of lipooligosaccharides from Neisseria meningitidis.
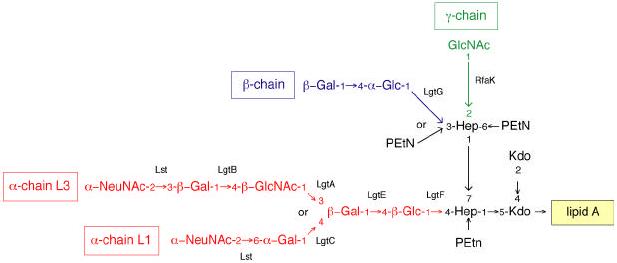
The structures and immunotypes have been reviewed elsewhere (168, 246). The structure of the single α-chain in each LOS varies and shown here are the sialylated α-chains from immunotypes L1 (374) and L3. Some of the additions to the inner core are missing in some LOS species. Relevant glycosyltransferases involved in the synthesis are identified, and the basis for the structural variations is described in the text. The RfaK enzyme should not be confused with the WaaK (formerly RfaK) glycosyltransferases that adds the terminal GlcNAc residue to the outer core of S. enterica serovar Typhimurium LPS.
Role of core in outer membrane stability: the “deep-rough” phenotype
The deep rough phenotype is displayed by E. coli and Salmonella mutants that lack the heptose region of the inner core. The phenotype is actually a series of characteristics that collectively reflect changes in both structure and composition of the outer membrane leading to its instability (reviewed in 7, 186). These characteristics include changes in surface hydrophilicity, resulting in hypersensitivity to hydrophobic dyes, detergents, hydrophobic antibiotics, fatty acids, phenols, and polycyclic hydrocarbons. Deep-rough mutants also typically release significant amounts of periplasmic enzymes into the culture media unless high concentrations of Mg2+ are included in the medium. The outer membrane of some deep-rough mutants has also been reported to have a decreased protein content with a concomitant increase in phospholipid. Finally, “deep rough” mutants of E. coli activate expression of colanic acid exopolysaccharide, lose expression of pili and flagella (187) and secrete a form of hemolysin with reduced hemolytic activity (188, 189). These factors can also influence biofilm formation (190).
Early studies on the deep-rough phenotype used Salmonella mutants with multiple LPS defects leading to truncated molecules with reduced phosphorylation. Phosphoryl groups were implicated in the formation of a stable membrane through the provision of sites that would enable adjacent LPS molecules to be cross-linked by divalent cations, and it is well established that treatment of cells with EDTA has a detrimental effect on outer membrane integrity and viability (186). In fact, for some time it was thought that mutants lacking heptose-P residues were not viable unless the core was truncated. Discovery and biochemical characterization of the LPS kinase WaaP (see below) allowed construction of defined mutants lacking core phosphorylation but retaining a complete carbohydrate backbone and, where relevant, the O polysaccharide (191, 192). In the laboratory, waaP mutants show the same growth rate as the parent. Interestingly, waaP mutants in E. coli show some characteristics of the “deep-rough” phenotype (e.g. increased sensitivity to hydrophobic compounds) but do not show altered outer membrane protein profiles (193). Mutations that eliminate synthesis of the outer core result in increased susceptibility to some hydrophobic compounds due to an indirect effect on core phosphorylation (193). The absence of core phosphorylation attenuates the virulence of S. enterica serovar Typhimurium and this is thought to be due in part to increased sensitivity to polycationic peptides. In this respect, pmrAB-regulated modifications of the lipid A-core that accompany polymyxin resistance cannot overcome the defects resulting from lost phosphorylation (191). Collectively these results have confirmed the critical roles played by phosphoryl groups in the LPS of E. coli and Salmonella and have helped clarify their association with the deep-rough phenotype.
While E. coli and Salmonella can accommodate LPS lacking core phosphorylation, P. aeruginosa, mutants with defects in waaP or inner core heptose assembly are not viable (194). The P. aeruginosa core has a multiply phosphorylated inner-core heptose (195) (Figure 9), and this organism is highly susceptible to lysis in EDTA (196). Thus phosphorylation is more critical in some bacteria than in others, and more examples are needed before we can determine whether the ability of E. coli and Salmonella to tolerate phosphorylation defects is broadly representative. Other bacteria apparently accommodate the requirement for a negatively charged inner core in different ways. In K. pneumoniae, there is no phosphorylation of the heptose region, and equivalent negative charges may be provided by uronic acids (Figure 9). The same is true of Rhizobium etli (173). Structural heterogeneity in the LPS extracted from K. pneumoniae results from structural variants lacking one or both of the terminal β-galacturonic acid residues (178, 179). Notably K. pneumoniae typically sheds significant amounts of an extracellular toxic complex comprising capsular polysaccharide, LPS, and proteins, and this complex is implicated in the damage to lung tissues that characterizes the pneumonia caused by this organism (197, 198). It will be interesting to determine whether these β-galacturonic acid substitutions are essential for viability or if their elimination results in a deep-rough phenotype. The B. pertussis core also lacks heptose-P but does have Kdo-P and uronic acids (199). “Deep rough” mutants have also been constructed in Bordetella pertussis, parapertussis and bronchiseptica by mutating the gene (waaF) whose product adds the second heptose residue in the core backbone (200). These mutants lack the remainder of the core that, in this organism, is attached to the remaining heptose residue. The mutants are viable but properties reflecting the stability of the outer membrane have not been reported.
Genetics and biosynthesis of core oligosaccharide
The completed lipid A-Kdo2 serves as the acceptor on which the core oligosaccharide and LOS chains are assembled via sequential glycosyl transfer from nucleotide sugar precursors. Endogenous in vitro glycosyltransferase activity can be detected in membrane fractions and many of the enzymes are predicted to be peripheral proteins. It is assumed that rapid and efficient core oligosaccharide synthesis reflects a coordinated complex of membrane-associated glycosyltransferases, although the existence of such a complex needs to be tested experimentally. The complex would act at the cytoplasmic face of the inner membrane, where acceptor and nucleotide sugars are available. Given the diversity of known structures, it is perhaps not surprising that biosynthetic functions have not been assigned in all cases.
The chromosomal waa region (formerly rfa) contains the major core-oligosaccharide assembly operons, and E. coli K-12 provided the first waa region sequenced in its entirety. Although some waa-encoded functions have been defined biochemically in E. coli K-12, assignment of others relies heavily on LPS structure and LPS-specific phage-receptor data for Salmonella mutants and on heterologous complementation of those mutations. Comparative sequence analysis of the waa regions from type strains representing the known core oligosaccharide types of E. coli and S. enterica serovar Typhimurium has been reported and the insertions, deletions, and rearrangements that result in the different core structures have been discussed (reviewed in 201). Homologs of core biosynthetic genes are now being identified in a variety of non-enteric bacteria and advances in whole-genome sequencing will result in a rapid expansion of data in this area. Since many gene assignments currently rely heavily on E. coli and Salmonella prototypes, a consideration of the limitations of the functional assignments in these prototypes is critical. Arguably the best characterized system involves the E. coli R1 core where all of the genes in major core biosynthesis operon have been mutated and the structure of the resulting LPS has been determined (Figure 11). Some of the R1 enzymes have also been studied at a biochemical level. From this prototype, and using data for conserved enzymes in other E. coli and Salmonella core systems, a comprehensive picture is emerging.
Figure 11. Structure and biosynthesis of the E. coli R1 core.
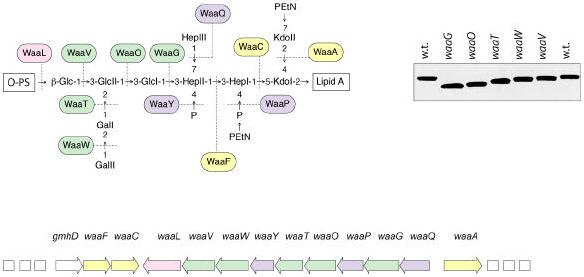
Organization of the waa locus is shown. Each gene has been mutated with a non-polar insertion, and the structures of LPSs from the resulting mutants were determined to generate the enzyme assignments shown with the structure (192, 228). Glycosyltransferases that form the inner core backbone are denoted by the yellow boxes and enzymes that modify the structure are in blue. Green boxes identify outer core glycosyltransferases and the ligase enzyme is in pink. The mobility of the mutant LPSs on a silver stained tricine-PAGE gel is also shown.
In E. coli, Salmonella, and K. pneumoniae (201, 202) the identification of core biosynthesis genes is aided by the fact that they are clustered on the chromosome. These loci generally encode all of the activities required for outer core assembly as well as the transferases needed for inner core synthesis. In E. coli and Salmonella, the waa locus consists of three operons (Figure 11) mapping between cysE and pyrE on the chromosome. The operons are defined by the first gene in each transcriptional unit: gmhD-, waaQ-, and waaA. The gmhD-waaFC genes are required for biosynthesis and transfer of L,D-heptose (see below). Transcription of the gmhD operon in E. coli K-12 is regulated by a heat-shock promoter, perhaps indicating a requirement for the heptose domain of LPS, at least in E. coli K-12, for growth at elevated temperatures (2, 7). The long central waaQ operon contains genes necessary for the biosynthesis of the outer core and for modification and/or decoration of core. In E. coli isolates with the R1 and R4 cores this operon also contains the “ligase” structural gene (waaL), whose product is required to link O polysaccharide to the completed core. The waaQ operon is preceded by a JUMPStart (Just Upstream of Many Polysaccharide-associated gene Starts) sequence that includes the conserved 8-bp region known as ops (operon polarity suppressor) that, together with RfaH (a NusG homolog), is required for operon polarity suppression (reviewed in 203, 204). As expected, Salmonella and E. coli K-12 mutants deficient in RfaH produce truncated LPS molecules (reviewed in 7) due to premature termination within the operon and reduced amounts of some outer core synthesis enzymes. The waaA transcript contains the structural gene (waaA formerly kdtA) for the bifunctional Kdo transferase (83), and a “non-LPS” gene encoding phosphopantetheine adenylyltransferase (coaD formerly kdtB) (205).
Examination of annotated genomes from more distantly related organisms reveals some have clusters of subsets of core genes; examples include Campylobacter jejuni, Vibrio cholerae, and P. aeruginosa. Determination of the range of functions encoded by these loci, and identification of “missing” genes, are limited by the number of ORFs with no currently known function. In mucosal pathogens including Haemophilus influenzae and Neisseria meningitidis there is no significant clustering of inner-core biosynthesis genes but many of the genes encoding glycosyltransferases that synthesize serologically important oligosaccharide extensions (e.g. LgtA-E, Figure 10) tend to be found in the same locus.
Assembly of the inner core
The Kdo transferase, WaaA (formerly KdtA) has been discussed above because, while Kdo is a core component, its assembly in the nascent LPS molecule precedes completion of lipid A in E. coli.
In the early 1980s, ADP-D,D-heptose and ADP-L,D-heptose were isolated from Salmonella enterica serovar Minnesota and Shigella sonnei (206, 207), identifying the precursor for heptose residues. Using synthetic substrates and in vitro reactions, it has been established that the configuration of L,D-Hep residues in the core is established by the specificity of the heptosyltransferases, WaaC and WaaF. These enzymes exhibit preference for ADP-L,D-heptose as their substrate, with ADP-D,D-heptose being used at low efficiency (208, 209). Interestingly, ADP-D-mannose can also serve as a surrogate substrate for WaaC (210). WaaC and WaaF homologs have been identified in a variety of bacteria, and in most cases, convincing assignments have been made by complementation of the corresponding mutations in Salmonella and E. coli.
Despite identification of the transferases and the L,D-heptose precursor, the pathway for ADP-L,D-heptose synthesis has only recently been resolved (Figure 12). The first step was predicted in a pathway for NDP-heptose synthesis first proposed in 1971 (211), and involves conversion of sedheptulose-7-phosphate to D,D-heptose-7-phosphate by the isomerase, GmhA. This activity has been confirmed in both E. coli and H. influenzae, (212, 213). The first clue that the originally proposed pathway was not strictly correct came from work on RfaE, an enzyme acting downstream of GmhA that is essential for formation of heptose-containing LPS (214). The sequence of this enzyme predicted 2 domains, one with similarity to ribokinases and the other resembling nucleotidyltransferases. The bifunctional nature of this enzyme, now renamed HldE, was subsequently confirmed (215). These activities correlated with data for the parallel pathway for formation of GDP-D,D-heptose, a precursor for the glycosylation of S-layer proteins in Aneurinibacillus thermoaerophilus (216). The authors have proposed a modified pathway (Figure 12) involving a kinase reaction to generate a D,D-heptose-1,7- bisphosphate intermediate, and invoking an additional phosphatase step to generate D,D-heptose-1-phosphate. The previously undetected E. coli phosphatase (GmhB) was found by database searches using the corresponding A. thermoaerophilus sequence, and its activity was confirmed biochemically and by analysis of a gmhB chromosomal mutation (215). Sequential activities of GmhA-HldE-GmhB-HldE lead to the formation of ADP-D-glycero-D-manno-heptose. The terminal epimerase enzyme (GmhD formerly RfaD) converts this compound to the preferred heptosyltransferase substrate ADP-L-glycero-D-manno-heptose. GmhD been characterized at the biochemical (217) and structural (218) levels.
Figure 12. Biosynthesis pathway for ADP-L-glycero-D-manno-heptose in E. coli.
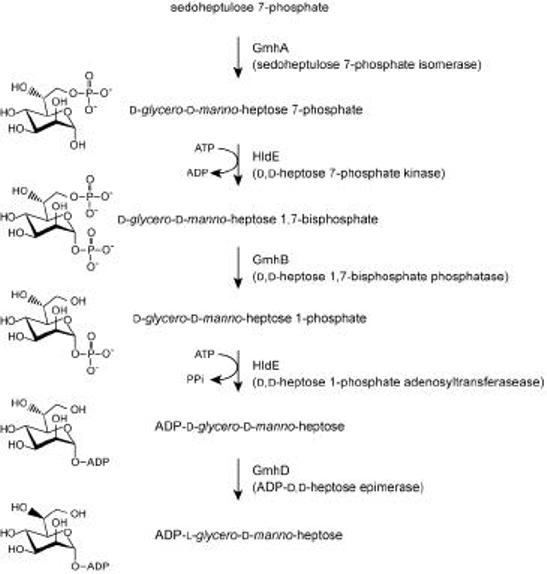
The figure is adapted from recent studies that necessitate revision of the previously proposed pathway (2, 215).
Elucidation of the A. thermoaerophilus pathway was aided by the fact that the genes encoding the enzymes for GDP-D,D-heptose synthesis are clustered on the chromosome. The same seems to be the case for the ADP-heptose genes in Campylobacter jejuni and Helicobacter pylori, whereas the homologs from E. coli, P. aeruginosa and H. influenzae are spread throughout the chromosome. Interestingly, while most of the homologs are well conserved in Gram-negative bacteria, N. meningitidis has the two domains of HldE in separate polypeptides (214). Vibrio cholerae contains the isomerase and phosphatase but lacks obvious candidates for the kinase and nucleotidytransferase activities (216), despite the presence of L,D-heptose residues in its core oligosaccharide (reviewed in 168) and homologs of known heptosyltransferases in the genome sequence. Nevertheless, the widespread distribution of these enzymes in important pathogens, the requirement for heptose-containing LPS in the formation of a stable outer membrane, and the now established synthetic pathway, offers new avenues for the development of therapeutic compounds.
The modification of the core region of E. coli and Salmonella LPS requires the action of three enzymes, WaaP (an LPS kinase), WaaY (an enzyme required for a second phosphorylation) and WaaQ (a transferase that adds the side-branch heptose) (Figure 11). The WaaP enzyme is the most important of these activities since the modifications must proceed in the strict order WaaPQY (192) and, as already indicated, mutants lacking WaaP activity exhibit the “deep rough” phenotype and are avirulent. The WaaP protein has been purified and its kinase action established in vitro using radiolabeled ATP and an acceptor consisting of isolated waaP mutant LPS (219). Although WaaP shares little primary sequence homology with eukaryotic kinases, PSI-BLAST searches did identify some conservation in catalytic residues inferred from functional and structural studies of eukaryotic kinases. Site-directed mutagenesis confirmed that these residues are also essential for catalysis in WaaP. The WaaP enzyme from P. aeruginosa shares extensive similarity with the homologs from E. coli and Salmonella and they are functionally interchangeable (194). Additional putative kinases have been identified in P. aeruginosa and these are presumably responsible for the remaining phosphates in the core oligosaccharide of that organism. What remains to be determined is the timing of these modifications during synthesis and the nature of the preferred acceptor. The ordered events in WaaPQY activity suggest complex acceptor requirements for the modifying enzymes. Mutant LPSs lacking the outer core (waaG in Figure 11) are phosphorylated inefficiently by WaaP (approximately 40% of wild type levels) (193). Conversely, the loss of phosphorylation results in a decreased efficiency of core extension, leading to more truncated cores than seen in the wild type, so there may be complex protein:protein interactions required for optimal core assembly. The WaaP kinase is not homologous to the Kdo kinase (KdkA) that phosphorylates the 4′ position of the single Kdo residue of Haemophilus influenzae (220, 221).
The modifications of inner core involving addition of glycose substituents to Kdo residues (Figure 8) have received less attention. The WaaZ enzyme is found in S. enterica and in E. coli isolates with the R2 and K-12 core types and is responsible for the addition of the α-2,4 Kdo transferase activity that adds the third Kdo residue found only in the inner cores of these bacteria (E. Frirdich, B. Lindner, O. Holst, and C. Whitfield, unpublished results). Putative glycosyltransferases have been identified for the other inner core modification reactions, but functional data are currently limited (reviewed in 201). In laboratory-grown cultures, the LPS contains only minor amounts of molecules with these modifications, making their identification particularly difficult.
Assembly of the outer core
All of the E. coli and Salmonella cores have a glucose residue as the first sugar in the outer core. Biochemical data, LPS structure, and heterologous gene complementation experiments identify WaaG as the UDP-glucose:(heptosyl) lipopolysaccharide α1,3-glucosyltransferase in E. coli K-12 (222) and Salmonella (reviewed in 201). As expected, the homologs of WaaG from each of the core types are highly conserved. The subsequent two transferases form α1,3 and α1,2-linkages, respectively and, depending on the resulting structure (Figure 8), the enzymes transfer glucose or galactose to an acceptor of either glucose or galactose. The corresponding transferases are all members of family 8 in the glycosyltransferases classification system (223) (http://afmb.cnrs-mrs.fr/~pedro/CAZY/) and share several highly conserved motifs (201), making assignments of their functions is[difficult in the absence of either an LPS structure from a defined mutant, or biochemical evidence with a purified protein. These enzymes are retaining glycosyltransferases, catalyzing formation of an α-linkage in the product from an α-linked donor. The mechanism of action of these transferases is not fully understood despite the interesting insight provided by the recent structural elucidation of a family 8 representative, LgtC (224). LgtC is an α1,4-galactosyltransferase involved in LOS synthesis in Neisseria meningitidis, and the protein was crystallized in the presence of a both sugar nucleotide donor and acceptor analogues, allowing mechanistic predictions to be tested. The prevailing model for the activity of retaining transferases was inferred from detailed studies concerning glycosylhydrolases and was proposed to involve a double displacement reaction involving a transient covalent intermediate between the donor sugar and the enzyme. However, an appropriately placed catalytic nucleophile could not be identified on the face of LgtC in proximity to the donor. The authors proposed an alternative model in which departing and attacking groups exist on the same side of the sugar ring. Their simultaneous action would result in a SNi-like internal return transition state.
In general, the enzymes catalyzing conserved linkages in E. coli and S. enterica cores are closely related, and the genes occupy similar positions in the loci. One exception is the glycosyltransferases responsible for the terminal α-1,2-linked glucose in E. coli R3 and S. enterica serovar Arizonae IIIA (Figure 8). The enzymes responsible in these strains share no significant similarity and are located at different positions within the corresponding waaQ operons (N. Kaniuk and C. Whitfield, unpublished results).
Within E. coli and Salmonella, the R1 and R4 core types are the only ones involving a β-glycosyltransferase. These inverting enzymes use the α-linked donor to generate a β-linked product. They are also identified by sequence similarity and two-dimensional features evident in Hydrophobic Cluster Analysis (HCA) (225) and the structures for several examples have been reported (reviewed in 226, 227). These enzymes require (at least) two catalytic carboxylates, where one serves to coordinate a divalent cation associated with the donor sugar nucleotide and another serves as a base to activate the donor. The R1 and R4 systems add a different β-linked sugar at the same site in an otherwise identical outer core (Figures 8 and 11), and the enzymes (WaaV and WaaX, respectively) were identified by interchanging them and determining the resulting LPS structure (228). These enzymes belong to different glycosyltransferase families (WaaV, family 2; WaaX, family 25).
Ligation of O polysaccharide to lipid A-core acceptor
Completion of the S-LPS molecule involves the addition of O polysaccharide to the nascent lipid A-core, and the ligation reaction occurs at the periplasmic face of the cytoplasmic membrane (reviewed in 11, 201). Lipid A-core is delivered to the periplasmic face by MsbA (see above), and the three pathways for O-polysaccharide synthesis take different approaches to deliver completed O polysaccharide to this location; these will be described below. The ligation reaction mechanism has not been determined, and the waaL gene product is currently the only enzyme presumed to be required for the process. However, in vitro assays with well-defined donor and acceptor substrates have not been reported.
WaaL primary sequences offer no obvious clues as to function. Collectively they share only low levels of similarity in their primary sequences, but they are all predicted to be integral membrane proteins with 8 or more membrane-spanning domains. The resulting hydropathy profiles are virtually identical with hydrophobic domains of similar length and distribution. Although ligase enzymes form glycosidic linkages, the donor is an undecaprenol pyrophosphate-linked oligo- or polysaccharide and, consequently, ligases share no similarity with any of the families of glycosyltransferases that utilize sugar nucleotide donors. The ligase enzyme presumably functions as part of a complex that involves highly specific interactions with the lipid-linked O-polysaccharide intermediates and lipid A-core acceptor. The ligase enzyme from E. coli K-12 can link to its lipid A-core O polysaccharides with diverse structures, arising from any of the known biosynthesis pathways. The lack of discrimination for donor structures suggests that the ligase enzyme may recognize the undecaprenol pyrophosphate carrier rather than the polymer attached to it. The variations in ligase sequences may then reflect specificity for the lipid A-core acceptor structure, or required protein:protein interactions in the biosynthesis complex, or both. Acceptor specificity has been demonstrated and involves not just the core residue providing the attachment site, but also other proximal residues in the core (229-231). The ligation sites in the E. coli cores are variable (Figure 8). There are no significant correlations between the structure of the linkage site and WaaL sequence. And heterologous complementation of waaL defects is only possible if the complementing gene product acts on a closely related core structure.
Contributions of core biosynthesis to LPS heterogeneity
LPS molecules extracted from a given isolate show variation in the extent of core completion. The extent of capping of lipid A-cores with O polysaccharide (i.e. the amount of rough versus smooth LPS) can also vary in a strain-dependent manner. It has been suggested that the heterogeneity of LPS is affected by modifications in the inner core Kdo region (Figure 8) (reviewed in 7). Analysis of SDS-PAGE profiles of LPS from mutants of E. coli K-12 led to speculation that WaaS (a putative 1,5 rhamnosyltransferase), WaaZ (the putative distal α-2,4 Kdo transferase), and WaaQ play an integral role in the production of two forms of LPS. In this scenario, the classical LPS form provides an acceptor for O polysaccharide, whereas the “LOS form” (distinct from R-LPS) is destined to terminate without O-polysaccharide addition. There are no current biochemical data that directly support this interesting hypothesis. The WaaZ enzyme is conserved in E. coli isolates with K-12 and R2 cores and in S. enterica, and all three are reported to contain some LPS molecules bearing a Kdo3 moiety (Figure 8). Conversely, the R1 core type lacks the waaZ gene and has no third Kdo in its natural structure. When plasmid-encoded WaaZ activity is introduced into a background with R1 core LPS, the Kdo3 structure becomes evident and most core oligosaccharides truncate prematurely in the outer core (E. Frirdich et al., unpublished results). While these results suggest a role for the inner core modification in control of core extension, the data could also be explained by WaaZ overexpression disrupting important protein:protein interactions and stoichiometry in a core biosynthesis enzyme complex. Further research is required to resolve these alternatives.
Differences in outer core can also dictate the efficiency of ligation of O antigen to the core, in turn influencing LPS heterogeneity. In P. aeruginosa there are significant differences in the structures of core with, and without, attached O polysaccharides ((232); Figure 9). R-LPS cores contain a β-D-Glc-(1→2)-α-L-Rha- moiety that is absent in the smooth LPS. Conversely, only the S-LPS contains an essential α-(1→3)-linked L-Rha residue that provides the ligation site (233). Recent studies have also identified core modifications involving quorum sensing-dependent expression of migA, encoding a putative glycosyltransferase (234), but the resulting structures have not been determined. Overexpression of MigA results in loss of molecules containing single repeat units of O polysaccharide, with no apparent effect on longer chain molecules, and the gene is highly expressed in the cystic fibrosis lung, suggesting a biologically significant role. In K. pneumoniae, R-LPS molecules contain a heptan structure and lack the α-Kdo residue in the outer core that serves as the ligation site (178, 179, 235). In Salmonella enterica serovar Arizonae IIIA, smooth LPS molecules have been reported to lack the terminal α-(1→2)-linked Glc residue (Figure 8), and the possibility that this residue may be trimmed away during the ligation step in some other core OS-type acceptor molecules has been suggested for S. enterica serovar Arizonae IIIa (discussed in 236). However, in the related core structure of E. coli R2, the terminal α-(1→2)-linked GlcNAc is present in S-LPS (237) and the corresponding transferase is essential for ligation in S. enterica (229). Further experimentation is required to determine whether this reflects a structural requirement in the ligation-competent acceptor or the need for protein:protein interactions in an assembly complex.
It is clear that the heterogeneity in LPS molecules isolated from a strain is complex and many of the modifications described here may be profoundly affected by growth conditions. It would be particularly interesting to know the extent of heterogeneity in LPS of bacteria growing within the host.
Phase variation and the biosynthesis of lipo-oligosaccharide
The biosynthesis of lipo-oligosaccharide (LOS) in mucosal pathogens represents a special case in terms of structural heterogeneity because many are subject to high-frequency phase variation (reviewed in 169, 170). The addition of the oligosaccharide extensions in LOS follows essentially the same mechanism used for the outer core. Identification of the glycosyltransferases has involved a comparison of LOS structures arising from different mutations (for examples see 238, 239-242), as well as overexpression of the appropriate enzymes and analysis of their activities using defined acceptors (243-245). In Neisseria (Figure 10) structural studies with an rfaK mutant have provided some insight into the early order of events in synthesis (242). Addition of PEtN residues to the O-3 and O-6 positions of the side-branch heptose residue is thought to precede addition of the α-GlcNAc (γ-chain) at O-2, the extension of the α-chain on the first heptose, and substitution of the glycosyl residue at position O-3 of the side-branch heptose (i.e. β-chain) (240). The addition of the β-chain cannot occur if PEtN already occupies the O-3 position, so an unidentified PEtN hydrolase has been invoked. The extension of the α-chain requires the prior addition of the α-GlcNAc (γ-chain), but because some LOS structures possess the α-chain and lack the γ-chain, it is conceivable that another processing activity removes the α-GlcNAc residue later in the pathway. These putative processing pathways may provide a new dimension in LPS biosynthesis and merit further study.
The precise structure of α-chain produced in the different immunotypes is determined by the combination of glycosyltransferases activities expressed from the lgt locus (246) (Figure 10). Differential synthesis of LOS structures in both N. meningitidis and N. gonorrhoeae is complicated by phase-variation mediated by poly(G) tracts in lgtA, C and D (247-249). The expression of lgtG (β-chain) is regulated in a similar fashion by a poly(C) tract (240). Variations in the numbers of the C’s or G’s occur during replication, altering the reading frame and leading to premature termination of translation. This seems to be a common theme in mucosal pathogens and poly(C) tracts have been implicated in the phase variation in the Lewis antigens presented in the LOS of H. pylori (250, 251). The ganglioside GM1-like antigen in Campylobacter jejuni LOS is also phase variable and contains a poly(G) tract (252). High-frequency phase variation occurs by a slightly different method in H. influenzae and affects both the carbohydrate structure of the LOS and its modification with PCho substituents, leading to altered LOS immunoreactivity and diminished colonization and virulence (for examples see 253, 254-257). In this organism, phase variation is a result of tandem repeats of the sequence 5′-CAAT that lead to slipped-strand mismatches during replication and a translational switch. Collectively, these variations affect the recognition of the LOS structure by the host immune system and can have a significant impact on the biological properties of the organism.
The addition of sialic acid residues to the α-chain in N. meningitidis LOS involves an enzyme (Lst) that can exhibit relaxed specificity (258). Lst from an immunotype L1 isolate can form either α-2,3 or α-2,6 linkages with a terminal galactose acceptor; in vivo this enzyme generates an α-2,6 linkage. The Lst from an L3 isolate is monofunctional with α-2,3 specificity but a single amino acid replacement can convert it into a bifunctional form. This provides an extreme example of the difficulty and potential pitfalls in assigning activities of transferases from sequence data alone.
Structure and biosynthesis of O polysaccharides
Structure of O polysaccharides
Smooth (S)-LPSs are typically produced by isolates of the families Enterobacteriaceae, Pseudomonadaceae, Pasteurellaceae and Vibrionaceae, as well as in many other Gram-negative bacteria. The structural diversity of O antigens is remarkable with more than 60 monosaccharides and 30 different non-carbohydrate components being recognized (reviewed in 259, 260). The O-polysaccharide repeat unit structures can differ in the monomer glycoses, the position and stereochemistry of the O-glycosidic linkages, and the presence or absence of noncarbohydrate substituents. O-repeat units from different structures may comprise varying numbers of monosaccharides, they may be linear or branched, and they can form homopolymers (i.e. a single monosaccharide component) or, more frequently, heteropolymers. In some cases, nonstoichiometric modifications (e.g. O-acetylation or glycosylation) complicate the identification of a precise O-repeat unit. When the LPS molecules extracted from any S-LPS-containing strain are separated by SDS-PAGE, it is apparent that there is extensive heterogeneity in the sizes of the molecules due to variations in the chain length of the O polysaccharides. This is evident in the classical “ladder” patter in SDS-PAGE, where each “rung” up the ladder represents a lipid A-core molecule substituted with an increment of one additional O unit. The spacing between the “rungs” is determined by the size of the O unit. The S-LPS from most isolates exhibits a preferred and strain-specific size distribution pattern (i.e. modal distribution).
The structure of the O polysaccharide defines the O-antigen serological specificity in an organism, but the numbers of unique O antigens within a species vary considerably. E. coli, for example, produces approximately 170 O serotypes. In S. enterica there are 46 serogroups but modifications to these basal O-repeat unit structures creates many additional “O factors”. O-antigen specificity is only relevant when discussed within a species, because there are several examples where all, or part, of an O-repeat unit structure is found in taxonomically distant species. A given isolate generally expresses one O antigen, but there are several exceptions where bacteria contain multiple lipid A-core-linked polymers, complicating both structural analysis and O-antigen terminology. E. coli provides good examples, since its lipid A-core can act as an anchor for O antigens, for one form of the Enterobacterial common antigen (ECA) polymer (261), and for a subset of group 1 and group 4 capsular K-antigens (reviewed in 262). In some isolates of Salmonella, lipid A-core serves as an acceptor for the classical O antigen as well as the unstable minor antigens T1 and T2 (263). Other isolates of S. enterica produce a chromosomally encoded O antigen, together with one resulting from a mobile plasmid carrying a biosynthesis locus (264, 265). Pseudomonas aeruginosa also expresses two lipid A-core-linked polymers: a conserved polyrhamnose homopolymer known as A-band LPS that is analogous to lipid A-core-linked ECA and a serotype-specific O antigen known as B-band LPS (reviewed in 266). In summary, lipid A-core is a versatile anchor for the presentation of cell-surface polysaccharides.
The location of O polysaccharide at the cell surface places it at the interface between the bacterium and its environment. Early interest in the chemistry, biosynthesis and genetics of O polysaccharides was stimulated by their roles as essential virulence determinants, and their potential application in vaccine development, but information is now available for a wide range of species with different lifestyles. The primary role(s) of the O polysaccharides appear to be protective. In animal pathogens, O polysaccharides may contribute to bacterial evasion of host immune responses, particularly the alternative complement cascade. Assembly of the membrane attack complex is affected by the chemistry of O polysaccharide, its chain length (267), and the relative amounts of long chain S-LPS (reviewed in 268). Chain length also affects the sensitivity of E. coli to neutrophil bactericidal/permeability-increasing protein (BPI) (269). In V. cholerae, R-LPS mutants are impaired in colonization of intestinal epithelia, reflecting their increased sensitivity to complement and cationic peptides (270). The presence and chain length of O polysaccharide in Shigella flexneri is also essential for invasiveness and subsequent inter- and intracellular spread (271-273), because S-LPS influences the polar localization of the invasion protein (IcsA). The exact role played by O polysaccharide in different bacteria is therefore variable, and this is potentially magnified in bacteria that can produce more than one lipid A-core-linked polymer.
Biosynthesis of O polysaccharides
With a few exceptions, the enzymes involved in O polysaccharide assembly are genes at the locus historically known as rfb. While the majority of systems are chromosomally encoded, there are notable exceptions such as the plasmid encoded O:54 polysaccharide of S. enterica (264, 265). These loci encode enzymes for the synthesis of novel sugar nucleotide precursors, glycosyltransferases that synthesize the O polysaccharide, and enzymes required for export processes. As expected, the diversity of O polysaccharide structures is reflected in highly polymorphic rfb loci. There is a significant literature devoted to the impact of lateral gene transfer, and insertion and deletion events, on the evolution and diversification of O-antigen types, much resulting from the efforts of P. Reeves and colleagues. For a detailed survey of the known loci, the reader is referred to the Bacterial Polysaccharide Gene Database (BPGD; http:www.microbio.usyd.edu.au/BPGD/default.htm). Most of the rfb operons appear to be constitutively expressed, and they are often preceded by a JUMPStart/ops sequence indicating that an antitermination process is active, as has been described above for the major core biosynthesis operon. In one exception, the O-polysaccharide biosynthesis locus of Y. enterocolitica O:3 is transcriptionally regulated by temperature (274, 275), and regulation is mediated by a repressor encoded outside of the cluster. The core biosynthesis genes are not thermoregulated in this organism.
Unlike biosynthesis the core oligosaccharide, the repeating unit structures of O polysaccharides are assembled on the membrane-bound carrier, undecaprenyl phosphate (und-P). This is the same C55-isoprenoid alcohol derivative used for synthesis of peptidoglycan, L-Ara4N modification of lipid A (Fig. 5), and capsular polysaccharides, and it participates in a trans-cytoplasmic membrane assembly process. O polysaccharides are synthesized from sugar nucleotides by glycosyltransferase enzymes that are often soluble or peripheral membrane proteins, indicating that assembly of lipid-linked O-repeat units occurs at the inner face of the cytoplasmic membrane. However, O polysaccharides are transferred from the carrier lipid and ligated to lipid A-core at the periplasmic face of the cytoplasmic membrane (276). As a result, the assembly processes must include a mechanism whereby either lipid-linked O polysaccharides or lipid-linked O units are delivered to the periplasm. The three currently known pathways for O-polysaccharide biosynthesis are distinguished by their respective export mechanisms, and the corresponding characteristic enzyme classes provide preliminary identification of the pathway used. The pathways are called Wzy-dependent, ABC-transporter dependent, and synthase-dependent, and each will be discussed below. The first two processes are widespread, but the synthase pathway has very limited distribution in O-polysaccharide biosynthesis. Despite the export differences, the pathways have similar initiation reactions and are completed by the same ligation process. This can lead to problems when heterologous genes are expressed in E. coli K-12, because functions from the host’s cryptic Wzy-dependent pathway (277) have unexpected interactions with cloned gene products, complicating interpretation of the results (278, 279). For this reason, cloning and functional analyses are best done in a K-12 strain such as E. coli SØ874, where the entire O-polysaccharide biosynthesis locus has been deleted (280).
Despite the growing body of genetic information for many O-polysaccharide biosynthesis gene clusters, detailed biochemical investigations are relatively limited and many of the individual enzymatic mechanisms are often speculative and based on sequence similarities. The pathways themselves are also represented among systems for the biosynthesis of capsular or exopolysaccharides in Gram-negative and Gram-positive bacteria. The O antigens are distinguished from other polysaccharides by the ligation step that links them to lipid A-core, and the translocation machinery required to translocate the completed S-LPS molecule to the surface of the outer membrane. In the following discussion, we focus on prototype enzymes for which some biochemical data is available.
Initiation reactions
The first step in O-polysaccharide biosynthesis involves the formation of an und-PP-linked glycose by transfer of a sugar-1-P residue to und-P. The energy of the sugar-phosphate linkage in the donor molecule is conserved in the resulting und-PP-linked intermediate and is available to drive the post-polymerization lipid A-core ligation reaction. The initiating enzymes recognize a hydrophobic lipid carrier rather than the sugar acceptor used by most other glycosyltransferases. Although there are many different O-repeat structures, the initiation reactions are relatively well conserved. The best-characterized reactions are catalyzed by the enzymes WbaP (formerly RfbP) from S. enterica (281, 282) and WecA (formerly Rfe) from E. coli (283). WbaP catalyzes the reversible transfer of galactose-1-P (Gal-1-P) from UDP-Gal to und-P and was first characterized in the Wzy-dependent synthesis of O antigens from S. enterica serovars Typhimurium and Anatum (Figure 13). Homologs of WbaP showing glucosyl or galactosyltransferase activity are widespread in capsule and exopolysaccharide synthesis systems. WecA (Rfe) is a GlcNAc-1-phosphate transferase that was first characterized in the Wzy-dependent pathway for ECA synthesis, but it also initiates a variety of Wzy-dependent O polysaccharides from E. coli (284) and Shigella species (279, 285). The enzyme appears to have relaxed specificity because it can also initiate polymers by the formation of und-PP-GalNAc in some E. coli isolates (286) and perhaps also in Yersinia enterocolitica O:8 antigen (287). This type of relaxed specificity is not confined to WecA; it also occurs in the WbpL enzyme from P. aeruginosa. WbpL shares similarity with WecA and is required for initiation of both A-band and B-band polysaccharides (288). In the formation of B-band, WbpL appears to act as a Fuc2NAc transferase, but available information points to the enzyme catalyzing a different (and as yet unidentified) transfer in the initiation of A-band synthesis.
Figure 13. Biosynthesis and assembly of O polysaccharides in a Wzy-dependent pathway.
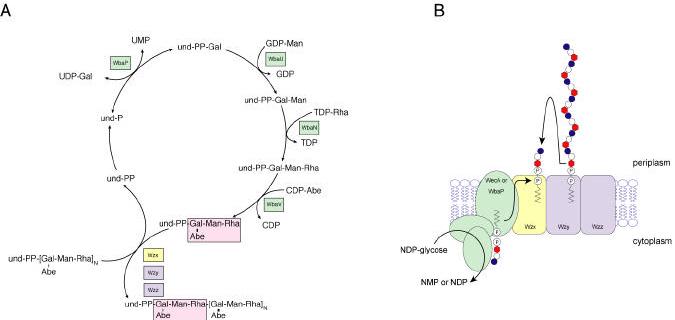
Panel A shows the sequence of reactions involved in the formation of the undecaprenyl-linked repeat units and their polymerization in S. enterica serovar Typhimurium. The individual glycosyltransferase enzymes are identified in green enzymes are identified in the boxes and structural details are presented elsewhere (2, 9). Panel B shows a model for the events in polymerization. Individual undecaprenyl-linked O-repeat units are transferred across the membrane by a process involving Wzx (yellow). These intermediates provide the substrates for the putative polymerase (Wzy- identified in blue) acting in the periplasm. Chain extension occurs at the reducing terminus with the nascent chain being transferred from its undecaprenyl carrier to the non-reducing terminus of the “new” undecaprenyl-linked subunit. The chain-length modality (i.e. the extent of polymerization) is determined by Wzz (also in blue). The nascent polymer is then ligated to lipid A-core and translocated to the outer membrane.
The Wzy-dependent pathway
The classical prototype systems for the Wzy-dependent pathway are the O polysaccharides from S. enterica serogroups B (Typhimurium) and E1 (Anatum). These O-repeat units have identical Man-Rha-Gal trisaccharide backbones, but they differ in the presence of a side-branch dideoxyhexose (abequose) substitution in serogroup B and in the linkage between the O-repeat units (2, 5, 9). Early studies by M.J. Osborn, H. Nikaido, A. Wright, and P. Robbins and others established the sequence of glycosyl transfer, the required sugar nucleotide precursors, and the direction of chain growth (reviewed in 7, 10, 263, 289), and the corresponding genes have now been identified (290) (Figure 13). Following initiation by WbaP, subsequent steps are performed by enzymes that are usually predicted to be peripheral membrane proteins. However, there is still no information concerning the manner in which the various enzymes interact with the membrane and with one another, to facilitate their organization into functional complexes.
Following their assembly, the individual und-PP-linked O units are exported to the site of polymerization at the periplasmic face of the plasma membrane (Figure 13). This process minimally requires a Wzx protein homolog and the corresponding structural genes are found in gene clusters for all Wzy-dependent O polysaccharides. In vivo labeling and detection/localization of intermediates have been used to investigate the effects of a Wzx defect (291). A hybrid strain lacking wzx accumulated und-PP-linked O units that appeared to be located at the cytoplasmic face of the plasma membrane, consistent with the proposal that Wzx is the O unit transporter (“flippase”). However, the small amount of material involved makes definitive localization difficult. Subsequent experiments have shown that several Wzx homologs from O-polysaccharide biosynthesis systems can function interchangeably, indicating that Wzx operates independently of repeat-unit structure and of initiating sugar (292). Feldman et al. speculated that a parallel process may be involved in the transmembrane transfer of dolichol-linked oligosaccharides in microsomes for glycoprotein biosynthesis, but the mechanism that flips molecules as large as und-PP-linked O units are across the lipid bilayer remains an intriguing unresolved question. It is interesting to note that predicted Wzx proteins are all highly hydrophobic proteins with 12 potential transmembrane domains and, while they share little primary sequence similarity, they do share structural features with bacterial permeases (293). Is Wzx the only protein required for “flippase” activity? Earlier studies had predicted that the N-terminal domain of the initiating WbaP enzyme might play a role in the process (294), but this was not supported in later studies (291), and Wzx is currently the only known participant. Mechanistic analyses of Wzx are currently hampered by difficulties in overexpressing the protein (291, 293).
At the periplasmic face of the cytoplasmic membrane und-PP-linked O units are polymerized (Figure 13). The reaction involves transfer of nascent polymer from its und-PP carrier to the nonreducing end of the new und-PP-linked O repeat (295, 296). The net effect is chain-length increase by one new repeat unit added to the reducing end of the nascent polymer. Polymerization is catalyzed by the O-polysaccharide polymerase, Wzy, and mutants affected in the wzy gene produce LPS consisting of lipid A-core capped with a single O unit (SR-LPS) (297). This is a defining feature of the pathway. The released und-PP must be recycled to the active monophosphoryl form. Due to the intensive use of und-P carriers in the Wzy-dependent biosynthetic pathway, it is characteristically inhibited by the antibiotic bacitracin, a compound that inhibits the dephosphorylation recycling reaction (298). The O polysaccharides synthesized using the Wzy-dependent pathway appear to be exclusively heteropolysaccharides, often with side-branch residues. This mechanism of assembly provides a relatively efficient process for maintaining the fidelity of synthesis of such complex structures so its involvement in the synthesis of other cell-surface heteropolysaccharides is perhaps not surprising (reviewed in 262, 289).
Wzy proteins are all predicted to be integral membrane proteins with 11-13 transmembrane domains and, like the Wzx proteins, they exhibit little primary sequence similarity (299, 300). However, Wzy proteins are specific for the cognate O unit, or for structures containing the same linkage between O-repeat units (reviewed in 7, 10, 289). The absence of conserved features has complicated the identification of catalytic and binding residues in Wzy and, consequently, the mechanism of its activity has not been resolved. For example, it is unclear whether Wzy is the only component of the biosynthesis machinery required for polymerization. Since its function has only been studied in vivo, it is not possible to determine whether contact with other proteins (e.g. Wzx) is also required. Ultimately, resolution of the mechanism of action will require in vitro studies in a reconstituted system but, as with Wzx, such approaches are currently limited by the difficulty in expressing Wzy in practical amounts (300).
The final component of the Wzy-dependent pathway is the Wzz protein (formerly Cld or Rol). The wzz gene was first reported in E. coli and S. enterica serovar Typhimurium (301), but homologs are involved all Wzy-dependent O-polysaccharide biosynthesis systems. Wzz generates the strain-specific modal distribution of O-polysaccharide chain lengths that is reflected in characteristic clusters of bands in SDS-PAGE (reviewed in 11, 302). Models have been proposed for the underlying process that implicate Wzz, Wzy and WaaL. A quantitative analysis of the distribution of chain lengths in the presence and absence of a functional Wzz determinant led Bastin et al. to conclude that Wzz confers a preference for chain length on either the polymerase, or the ligase, or both (303). In the absence of Wzz, the distribution of chain lengths was shown to be independent of chain length, correlating instead with a constant probability of transfer to lipid A-core. Wzz was hypothesized to act as a timing “clock”, interacting with the Wzy polymerase and modulating its activity between two states that favor either elongation or transfer to the ligase (i.e. chain termination). Morona et al. have suggested that Wzz acts as a molecular chaperone to assemble a complex consisting of Wzy, the WaaL ligase and und-PP-linked O polysaccharide (304). The specific modality is determined by different kinetics resulting from a Wzz-dependent ratio of Wzy to WaaL. A role for WaaL is also inferred from the observation that Wzz can only regulate chain length of polymers that are linked to lipid A-core (reviewed in 11). In some cases, bacteria have more than one wzz gene. P. aeruginosa has two chromosomal wzz genes (266), and in S. flexneri, the chromosomal wzz gene product is accompanied by a plasmid-encoded homolog (305). The biological impact of possessing two Wzz activities remains unknown.
Localized regions of Wzz primary sequences are relatively well conserved, as are the predicted secondary structures (reviewed in 11, 302). Wzz homologs are located in the cytoplasmic membrane, and all have two transmembrane helices flanking a periplasmic loop with a predicted propensity for coiled-coil structure (302). The ability of Wzz to somehow modulate the kinetics of the polymerization and/or ligation reactions suggests that the protein must recognize and form a complex with either Wzy, or WaaL, or both. The coiled-coil domains may be important for such interactions, and oligomers of Wzz have been identified by chemical cross-linking experiments (306). However, there is currently no definitive evidence of cross-linking of Wzz to either Wzy or WaaL. The function of Wzz homologs is not restricted by a specific O-repeat unit structure. Attempts to define specific regions required for Wzz modality have involved expression of heterologous wzz genes, comparative sequence analyses, construction of hybrid proteins, and site-directed mutagenesis of specific residues (306-308). To date no clear picture has emerged concerning the regions of the protein that confer specificity.
The ABC-transporter dependent pathway
This pathway is currently confined to linear (unbranched) O-polysaccharide structures and involves chain extension by processive addition of glycosyl residues to the non-reducing terminus of the und-PP-linked growing chain. There is no requirement for a specific polymerase enzyme, and the polymer is completed at the inner face of the cytoplasmic membrane. Export of the polymer to the outer face for ligation requires an ABC (ATP-binding cassette) transporter, precluding the involvement of Wzx.
To date, the O polysaccharides formed by this pathway are all initiated by a homolog of WecA (Figure 14). In the characterized pathways, the resulting und-PP-GlcNAc acts as a primer for chain extension, and while the GlcNAc residue will be transferred to lipid A-core during ligation, this residue occurs only once per chain. This role contrasts to the involvement of WecA in the Wzy-dependent pathway where the enzyme is required for each lipid-linked O repeat, and GlcNAc is therefore found within the repeat unit structure itself. The involvement of WecA is well established for the polymannose O polymers of E. coli O8, O9 and O9a (and their structural counterparts in K. pneumoniae O5 and O3) and for the galactose-containing O polysaccharides (D-galactan I) from K. pneumoniae. Transfer of GlcNAc-1-P has been unequivocally identified as the initial reaction in vivo for E. coli O8 (309), and this correlates with the demonstrated activity of the enzyme in ECA synthesis as well as the preferred reaction in vitro (310). Furthermore, definitive structural analysis of the core-O polysaccharide linkage region in K. pneumoniae O3 and O5 identifies the GlcNAc residue resulting from WecA (235). In the next step in biosynthesis, an O-polysaccharide-specific glycosyltransferase adds the adaptor between the und-PP-GlcNAc primer and the repeat unit domain and commits the lipid intermediate to the chain extension pathway. In E. coli O9 adaptor formation involves the addition of a single mannose residue by WbdC (formerly MtfC) (310) (Figure 14). As with the WecA-mediated transfer, this reaction occurs only once per chain, and the residue is evident in the linkage regions for K. pneumoniae O3 and O5 LPSs (235). In the biosynthesis of D-galactan I, the corresponding activity is mediated by the bifunctional galactosyltransferase, WbbO. Both activities of WbbO are involved in formation of the adaptor, but one also participates in subsequent chain extension reactions (311, 312) (Figure 14). In the chain extension phase, the repeating-unit domain is assembled by processive transfer of residues to the non-reducing terminus of the und-PP-linked acceptor (310, 312, 313). The transferases involved in the elongation reaction can be monofunctional or, as in the case of the E. coli O9 and O9 apolymannans, can add two or more residues of a given linkage type (310, 314, 315). This may be useful for the maintenance of fidelity of O-repeat unit structure that may be defined by the linkage distribution rather than by different monomers. Overall the pathway requires the participation of only one und-P molecule per polymer chain, and so und-PP recycling is not a limiting factor and the process is resistant to bacitracin (298).
Figure 14. Biosynthesis and assembly of O polysaccharides in an ABC-transporter-dependent pathway.
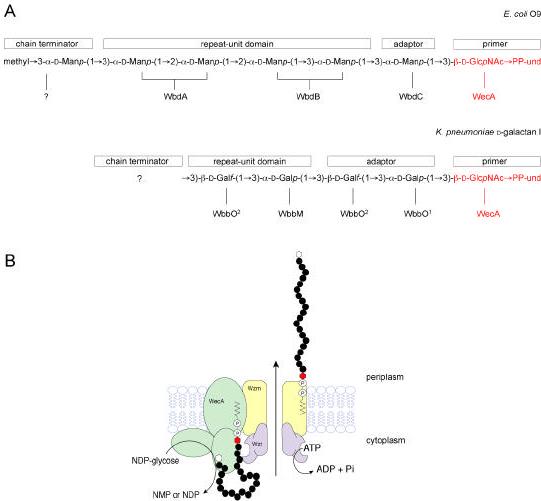
Panel A shows the predicted structures of undecaprenyl-linked intermediates in the biosynthesis of the O9a antigen of E. coli and the D-galactan I polymer found in several serotypes of K. pneumoniae. The glycosyltransferase enzymes involved in each step are indicated below the structures. Biosynthesis is broken down into the formation of a common primer (shown in red), addition of an adaptor region, chain extension of the repeat unit, and addition of a chain terminator (for details see the text). In the O9a structure, the chain is terminated by addition of 3-O-methylmannose but the enzyme responsible has not been identified. In D-galactan I, the structure of the chain terminator has not been established. Chain elongation occurs by processive glycosyl transfer to the non-reducing terminus. Several enzymes in these pathways are bifunctional. In the case of the galactosyltransferase WbbO, both of its activities are required for adaptor synthesis, but only one (WbbO2) participates in chain extension. Panel B provides a model for the trans-membrane assembly system. The glycosyltransferases are shown in green. The ABC-transporter formed by Wzm and Wzt is required for transfer of the undecaprenyl-linked polymer to the periplasmic face of the membrane, where it is ligated to lipid A-core and translocated to the outer membrane. It is presumed that the polymer remains attached to the undecaprenyl carrier throughout the export process. While chain extension and export are separable by mutations in the ABC-transporter, it is conceivable that the two processes are temporally coupled in vivo. Within the nascent polymer, the primer/adaptor is identified by the black-filled hexagon, the residues of the repeating-unit domain by gray-filled circles, and the chain terminator by the open hexagon.
Following polymerization, the nascent O polysaccharide must be exported across the cytoplasmic membrane for ligation. Studies with Yersinia enterocolitica serotype O:3 first identified pathway mutants that accumulated intracellular O polysaccharide (316), but the same phenomenon has been described in K. pneumoniae O1 (317) and P. aeruginosa A-band LPS synthesis (318). In these systems, surface assembly requires an ABC transporter encoded by the O-polysaccharide biosynthesis cluster. The genes for the transporters were initially identified by protein sequence similarities shared with the ABC-2 subfamily of ATP-binding cassette transporters (319). ABC-2 transporters consist of an integral membrane protein with an average of six membrane-spanning domains, and a hydrophilic protein containing an ATP-binding motif or Walker box. The genes for these two components have now been identified in a number of O-polysaccharide biosynthesis clusters. As with other ABC-transporters involved in transmembrane export, the membrane-spanning (Wzm) homologs for O-polysaccharide biosynthesis generally exhibit little primary sequence identity but have nearly identical hydropathy plots. In contrast, primary sequences of the ATP-binding (Wzt) homologues are much more highly conserved, particularly (and predictably) in the nucleotide-binding region.
Little is known about the organization and exact mode of action of ABC-transporters for O polysaccharides and, unlike MsbA (see above) no structural data is available. Much of the information is inferred from studies and hypothetical models for with comparable systems involved in group 2 capsule synthesis in E. coli and other bacteria (reviewed in 320). Other ABC-transporters, such as the Sec system for protein export (reviewed in 321), have been particularly influential. In the group 2 capsule systems, the ABC-transporter forms part of an assembly complex that may allow polymerization and export processes to proceed in a coordinated manner (reviewed in 262). The export process is driven by ATP-hydrolysis and the Wzt protein may undergo an ATP-binding-induced conformational change, resulting in its insertion into the membrane via an interaction with the Wzm, introducing polymer into the channel (Figure 14). A series of insertion-deinsertion events would then drive the polymer through the membrane. Experiments that directly address this hypothesis are difficult to formulate. Since E. coli K-12 can ligate to its lipid A-core polymers from any of the three known assembly pathways, we assume that the material is presented to the K-12 ligase in the same form (i.e. attached to und-PP). If this is indeed the case, the export machinery must handle und-PP-linked polymer.
The ABC-transport dependent pathway does not involve a Wzz chain length determinant, yet S-LPSs in bacteria with the pathway do exhibit strain-specific modal distributions (11). In one extreme example, the Aeromonas salmonicida O polysaccharides have a uniform chain length (322). The observation that modality is conserved when O polysaccharides are expressed from cloned genes in heterologous hosts (i.e. E. coli K-12) demonstrates that modal regulation is a function of one or more of the components encoded by the biosynthesis cluster itself. The fact that chain-length distribution is altered when Wzm/Wzt are expressed at higher levels than the glycosyltransferases (317) favors a competition between the termination-export and chain extension activities, but what terminates the processive chain extension process? An intriguing feature of several O polysaccharides from ABC-transporter-dependent pathways is the presence of novel non-reducing terminal residues on the glycan chains. In the polymannan O8 and O9 glycans of E. coli (and their K. pneumoniae O3 and O5 counterparts), the chains end in a 3-O-methylmannose residue (235, 323). 2-O-methyl groups are found at the termini of V. cholerae O1 chains and these are associated with the seroconversion to Inaba from Ogawa type (324, 325). Recent structural studies have identified α-Kdo and β-Kdo at the termini of the K. pneumoniae O4 and O12 antigens (polymers with no Kdo residues in their repeat units), respectively (235). In the ABC transporter-dependent O polysaccharide of Bordetella bronchiseptica (326), a unique 2,3,4-triamino-2,3,4-trideoxy-α-galacturonamide derivative is found at the chain terminus (327). It therefore appears that terminal modifications are a widespread phenomenon in this pathway, and it is tempting to speculate that they provide an essential component of the ABC-transporter-dependent systems. The frequency by which these residues are incorporated could alter the assembly complex from a chain elongation to a chain termination mode, initiating the export process. Detailed analysis of mutants lacking the terminal modifying enzymes should provide insight into the mechanism(s), and candidates have been identified (B.R. Clarke and C. Whitfield, unpublished results).
The synthase-dependent pathway
Synthases are processive glycosyltransferases that have the capacity to synthesize polymers within a single polypeptide. Included among their members are enzymes involved in biosynthesis of cellulose and chitin. Other well-documented examples occur in the biosynthesis of capsular polysaccharides including hyaluronic acid by Streptococcus pyogenes and other bacteria (reviewed in 328), and the type 3 capsules of Streptococcus pneumoniae (329, 330). These enzymes have 2 conserved domains. Domain A is a region found in both processive and monofunctional β-glycosyltransferase enzymes, whereas domain B is confined to the processive examples. Chain extension occurs at the non-reducing terminus (330, 331), but the identity of the carrier on which the nascent chains are extended, and the process of initiation, have not been resolved for the bacterial capsule synthases.
The plasmid-encoded O:54 antigen of S. enterica serovar Borreze is currently the only known example of a synthase-dependent O polysaccharide (265, 332, 333). Synthesis of the poly-N-acetylmannosamine homopolymer that comprises the O:54 antigen is initiated by WecA (265) (Figure 15). The first ManNAc residue commits the und-PP-GlcNAc primer to O:54 biosynthesis and is transferred by the non-processive ManNAc transferase WbbE (formerly RfbA) (334). The first two steps in this pathway are therefore reminiscent of those in the ABC-transporter-dependent process. The second transferase encoded by the O:54 biosynthesis cluster, WbbF (formerly RfbB) is a member of the HasA (hyaluronan synthase) family of enzymes, and it is proposed that this enzyme performs the chain extension steps. Synthases are integral membrane proteins with similar topology (332, 335) and are believed to catalyze a vectorial polymerization reaction, simultaneously extending the polysaccharide chain and extruding the nascent polymer across the plasma membrane (328, 332). As with the ABC-transporter-dependent pathway, the product of the synthase pathway is ligated to lipid A-core by the E. coli K-12 ligase (265). While this is consistent with the possibility that the exported form of the polymer is und-PP-linked, there is no information that casts light on the exact mechanism of export. Also unknown is the process involved in chain termination. Studies on biosynthesis of the streptococcal type 3 capsule have led to a proposal that chain termination in that synthase results from an abortive translocation step in which the normal passage of nascent polymer between sites that add new residues is disrupted (330). When one of the sugar nucleotide donors for the type 3 polymer (which has a disaccharide repeat unit) is limiting, chain termination is favored, followed by release of the nascent polymer from the enzyme. While a very interesting proposal, firm conclusions are complicated by the uncertainty surrounding the exact mechanism of glycosyltransfer by the synthases. Early speculation about a two-glycosyltransfer site model has been challenged by more recent structural data for β-glycosyltransferases (reviewed in 226, 227, 336).
Figure 15. Biosynthesis and assembly of O polysaccharides in a synthase-dependent pathway.
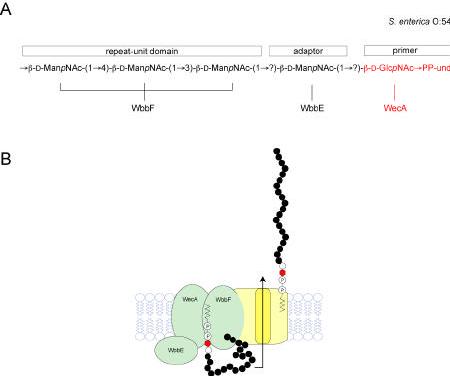
The only current example is the plasmid-encoded O:54 antigen of S. enterica serovar Borreze. Panel A shows the predicted structure of the undecaprenyl-linked intermediate. The primer is made by WecA. The WbbE enzyme then adds a single ManNAc residue as an adaptor, but the precise linkage has not been determined. The synthase (WbbF) is then required for chain extension, generating a repeat-unit domain with alternating β(1,3) and β(1,4) linkages. Growth is at the non-reducing terminus. Panel B shows a model for the transmembrane assembly process, with glycosyltransferases identified in green and putative transport functions in yellow. Available data for this system, and information for other bacterial synthases, is consistent with the conclusion that WbbF serves as both a processive glycosyltransferase with dual linkage specificity, as well as an exporter that moves undecaprenyl-linked intermediates to the periplasm. While chain extension and export are shown as separate processes, it is conceivable that they are temporally coupled in vivo. The nascent polymer is then ligated to lipid A-core and translocated to the outer membrane.
Seroconversion reactions
The impact of non-stoichiometric substitutions on O antigenicity is widely recognized. The phenomenon is well studied in Salmonella and Shigella, where additions of acetyl groups or glucose residues create novel O factors. In many cases, the modifying enzymes are carried by lysogenic or cryptic bacteriophages.
The oafA gene product encodes the O-acetyltransferase conferring O:5 specificity in Salmonella (337). Other well characterized O-acetyltransferases include Oac, encoded by the Shigella seroconverting bacteriophage SF6 (338), and the O-acetyltransferase carried by P. aeruginosa bacteriophage D3 (339). Acetyl-CoA provides the donor for O-acetylation, and early experiments suggested that the acceptor for the O-acetyltransferase reaction is the und-PP-linked O unit (discussed in 263). The location of the reaction and the precise extent of acetylation of individual O-repeat units have not been definitively established. The use of acetyl-CoA as the initial donor suggested a cytoplasmic location, but recent analysis suggests the possibility of a periplasmic reaction (reviewed in 340). In general, O-acetyltransferases fall within 4 families. The larger members such as OafA are integral membrane proteins and contain an N-terminal domain that shares some similarity with human acetyl-CoA transporter. It is proposed that these enzymes export acetate and acetylate the acceptor. Soluble (and putatively extracellular) O-acetyltransferases are accompanied by an integral membrane protein (a member of the AlgI family) that is proposed to export acetate for the modification reaction. Further experimentation is now required to address these interesting hypotheses. An extracellular location would allow the use of acceptors comprising either und-PP-O repeat or und-PP-O-polysaccharide.
The donor for non-stoichiometric glucosyl substituents in S. enterica O polysaccharides is an unusual lipid intermediate, α-glucosylmonophosphorylundecaprenol (und-P-Glc) (341-343), resembling the donor for the L-Ara4N residues that modify lipid A (Fig. 5). In serogroups B and D, glucosylation is a post-polymerization reaction and is assumed to occur at the level of the und-PP-linked O polysaccharide (341, 343, 344). In contrast, in serogroup C1 and C2, glucosylation is reported to occur before polymerization, at the level of individual und-PP-linked O units (345), and these contradictory data need to be revisited with additional experiments. Glucosylation is particularly prevalent among the O antigens of Shigella where the genetic determinants have been described (reviewed in 346). Each Shigella glucosylation locus contains three genes and is contained within DNA from a lysogenic or cryptic phage. Homologues are recognized in the sequence of the Salmonella bacteriophage P22 and in a cryptic prophage from E. coli. Preliminary functional assignments of the genetic determinants have been made (347, 348). The GtrB enzyme appears to be a glucosyltransferase that synthesizes und-P-Glc by transferring glucose from UDP-glucose to und-P. A serotype-specific transferase (Gtr) is responsible for the transfer of the glucose residue from the und-P-Glc donor to the acceptor. The gtrA gene encodes an integral membrane protein that is proposed to serve as a flippase to transfer und-P-Glc to the periplasmic face of the membrane, where glucosylation may occur. Whether this protein operates alone or in conjunction with other O polysaccharide biosynthesis machinery (e.g. Wzx) is unknown. As expected, the gtrA and B genes are interchangeable among systems, but the gtr genes are specific for a given modification.
Another well-documented bacteriophage-encoded modification involves a change in the specificity of the Wzy polymerase, resulting in an alteration in the anomeric linkage between O-repeat units in the final polymer. The classical example of this modification is the ε15-mediated seroconversion in Salmonella (349), but recent detailed insight into the process has been provided by studies with the D3 lysogenic seroconverting phage of P. aeruginosa. The D3 lysogen acetylates the host O polysaccharide as well as directing an α to β switch in the linkage between O repeats (339). The end result is a seroconversion from serotype O5 to O16. The D3 phage carries a β-specific Wzy but also encodes a highly hydrophobic 31-residue α-polymerase inhibitor (Iap) that inactivates the “normal” chromosomal Wzy protein. The ε15-seroconversion also involves a putative small polypeptide inhibitor (350). The relationships (if any) between the inhibitors have not been established, and more work is required to unravel the fascinating mechanism by which they act.
The examples above all involve modifications to a Wzy-dependent pathway but there is evidence for similar and often non-stoichiometric glycosylation and O-acetylation modifications in ABC-transporter pathways too. For example, a number of Klebsiella O serotypes express galactose-containing O polysaccharides based on a common D-galactan I backbone but differ in modifications to this structure (351-357). The O-polysaccharide biosynthesis loci only contain genes for D-galactan I synthesis (317, 358, 359) and the location(s) of the genes responsible for the modifications have not been identified yet so mechanistic information is not available.
Export of lipopolysaccharide to the cell surface
The method by which completed LPS molecules are translocated to the cell surface represents one of the most interesting open issues in LPS biosynthesis. The LPS molecules must pass across the periplasm and through the outer membrane. The translocation processes do not discriminate between LPS with different O-polysaccharide structures, and S-LPS and R-LPS molecules are apparently translocated with equal efficiency (360). The targeting of LPS to the outer membrane is predicted to require a pathway, rather than being mediated by structural features of the LPS molecule itself.
It has been known for some time from work in Salmonella that new LPS molecules appear on the cell surface at a limited number of sites (361, 362). Membrane-adhesion sites, regions where the inner and outer membranes come into apposition, were implicated. These structures are only visualized by specific electron microscopy and have been quite controversial (for discussion see 289, 363, 364). However, current models for the assembly and translocation of capsular polysaccharides do invoke a molecular “scaffold” across the periplasm comprising proteins involved in the translocation process, and there is experimental evidence that lends support to the hypotheses (reviewed in 262, 320). The type II and III protein secretion systems in bacteria offer similar examples of multi-protein translocation machineries (365). Membrane-adhesion sites may reflect points where the translocation machinery effectively creates continuity between a biosynthesis complex and the outer membrane, providing an organized domain for the translocation of macromolecules.
The capsule biosynthesis loci from Gram-negative bacteria encode proteins involved in translocation across the periplasm and outer membrane, but no corresponding proteins are encoded by the LPS core or O-polysaccharide biosynthesis loci. Translocation systems for LPS potentially involve generalized outer membrane assembly functions in the cell. Unlike the capsules, LPS are essential for growth in the laboratory, and so mutations affecting dedicated translocation components would be lethal, limiting the isolation of informative mutants from random screens. While there are enough “unknowns” in the bacterial genomes, to potentially accommodate translocation functions, it is also possible that other well known systems participate in the process. For example, the TolA protein has been implicated in the processing or translocation of S-LPS in E. coli (366). The four-protein Tol system is required for import of bacteriocins and bacteriophages across the outer membrane. E. coli K-12 tolA mutants are viable but in a wild-type strain making S-LPS, a tolA mutation is lethal. One intriguing interpretation of this data is that the translocation systems contain essential components needed for all LPSs, and additional components specifically required for S-LPS. This finding may provide a useful starting point for resolution of the translocation pathways.
Future directions for lipopolysaccharide research
As is apparent from the discussion of the biosynthesis of LPS, detailed mechanistic understanding of the biosynthesis of the core and O polysaccharide regions lags behind the lipid A area. One reason for this is undoubtedly the structural variation of these regions of the LPS molecule and the corresponding diversity of the underlying enzymes. However, genomic and other sequence-based studies have shown that despite the variations in details, the overall pathways from different bacteria are quite conserved. This will allow the development of broadly representative prototype systems and a focused approach to the elucidation of critical enzymes. The mechanistic details can now be addressed. Clearly the area where there remains the most intriguing open questions in lipid A and LPS assembly involves the transport of the various LPS components across the cytoplasmic membrane and translocation of the completed molecules to the cell surface. Examination of these potentially multienzyme processes is experimentally challenging. However, the progress that has been made in recent years with similarly complex systems in protein export and secretion points to possible experimental avenues and provides confidence that these processes will be resolved in time. The development of well-defined in vitro enzymatic systems for O-polysaccharide polymerization and ligation to core-lipid A would serve as a useful prelude to biochemical studies of LPS export and assembly into the outer membrane.
To ensure substantial progress, an interdisciplinary experimental approach will be needed in future studies of lipopolysaccharide endotoxins. The application of structural biology, bacterial genomics, and animal knockout models to lipopolysaccharide biology is still in its infancy. These newer strategies need to be utilized in conjunction with older approaches, like enzymology, carbohydrate chemistry, and membrane biochemistry. The remarkable images provided by the x-ray structures of the LpxA trimer (51), the outer membrane protein FhuA bound to a single LPS molecule (128) and the MsbA lipid A/phospholipid transporter (120) hint at what the future has in store. Novel antibiotics, vaccines and anti-inflammatory agents are likely to emerge from an in depth understanding of lipopolysaccharide-protein interaction at the atomic level.
Acknowledgments
Research in the laboratory of C. R. H. Raetz is supported by NIH Grants R01-GM-51310 and R37-GM-51796. C. Whitfield is recipient of a Canada Research Chair, and his research in this area is funded by the Natural Sciences and Engineering Research Council and the Canadian Bacterial Diseases Network (NCE program). We thank all the members of our laboratories for their critical comments and stimulating discussions. This review is dedicated to Professor Laurens Anderson of the University of Wisconsin-Madison.
References
- 1.Raetz CRH. Annual Reviews in Biochemistry. 1990;59:129–70. doi: 10.1146/annurev.bi.59.070190.001021. [DOI] [PubMed] [Google Scholar]
- 2.Raetz CRH. Escherichia coli and Salmonella. In: Niedhardt FC, editor. Cellular and Molecular Biology. American Society for Microbiology; Washington, D.C.: 1996. pp. 1035–63. [Google Scholar]
- 3.Rietschel ET, Kirikae T, Schade FU, Mamat U, Schmidt G, et al. FASEB Journal. 1994;8:217–25. doi: 10.1096/fasebj.8.2.8119492. [DOI] [PubMed] [Google Scholar]
- 4.Zähringer U, Lindner B, Rietschel ET. In: Endotoxin in Health and Disease. Brade H, Opal SM, Vogel SN, Morrison DC, editors. Marcel Dekker, Inc.; New York: 1999. pp. 93–114. [Google Scholar]
- 5.Brade H, Opal SM, Vogel SN, Morrison DC, editors. Endotoxin in Health and Disease. Marcel Dekker, Inc.; New York: 1999. p. 950. [Google Scholar]
- 6.Galloway SM, Raetz CRH. J. Biol. Chem. 1990;265:6394–402. [PubMed] [Google Scholar]
- 7.Schnaitman CA, Klena JD. Microbiological Reviews. 1993;57:655–82. doi: 10.1128/mr.57.3.655-682.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brabetz W, Muller-Loennies S, Holst O, Brade H. Eur. J. Biochem. 1997;247:716–24. doi: 10.1111/j.1432-1033.1997.00716.x. [DOI] [PubMed] [Google Scholar]
- 9.Reeves PR, Hobbs M, Valvano M, Skurnik M, Whitfield C, et al. Trends in Microbiology. 1996;4:495–503. doi: 10.1016/s0966-842x(97)82912-5. [DOI] [PubMed] [Google Scholar]
- 10.Whitfield C. Trends in Microbiology. 1995;3:178–85. doi: 10.1016/s0966-842x(00)88917-9. [DOI] [PubMed] [Google Scholar]
- 11.Whitfield C, Amor PA, Köplin R. Molecular Microbiology. 1997;23:629–38. doi: 10.1046/j.1365-2958.1997.2571614.x. [DOI] [PubMed] [Google Scholar]
- 12.Aderem A, Ulevitch RJ. Nature. 2000;406:782–7. doi: 10.1038/35021228. [DOI] [PubMed] [Google Scholar]
- 13.Medzhitov R, Janeway C., Jr. N. Engl. J. Med. 2000;343:338–44. doi: 10.1056/NEJM200008033430506. [DOI] [PubMed] [Google Scholar]
- 14.Poltorak A, He X, Smirnova I, Liu MY, Huffel CV, et al. Science. 1998;282:2085–8. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 15.Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, et al. J. Immunol. 1999;162:3749–52. [PubMed] [Google Scholar]
- 16.Beutler B, Cerami A. Annu. Rev. Biochem. 1988;57:505–18. doi: 10.1146/annurev.bi.57.070188.002445. [DOI] [PubMed] [Google Scholar]
- 17.Dinarello CA. Blood. 1991;77:1627–52. [PubMed] [Google Scholar]
- 18.Drake TA, Cheng J, Chang A, Taylor FB., Jr. Am. J. Pathol. 1993;142:1458–70. [PMC free article] [PubMed] [Google Scholar]
- 19.Li A, Chang AC, Peer GT, Hinshaw LB, Taylor FB., Jr. Shock. 1996;5:274–9. doi: 10.1097/00024382-199604000-00007. [DOI] [PubMed] [Google Scholar]
- 20.Parillo JE. N. Engl. J. Med. 1993;328:1471–7. doi: 10.1056/NEJM199305203282008. [DOI] [PubMed] [Google Scholar]
- 21.Esmon CT. Biochim. Biophys. Acta. 2000;1477:349–60. doi: 10.1016/s0167-4838(99)00266-6. [DOI] [PubMed] [Google Scholar]
- 22.van Deuren M, Brandtzaeg P, van der Meer JW. Clin. Microbiol. Rev. 2000;13:144–66. doi: 10.1128/cmr.13.1.144-166.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bernard GR, Vincent JL, Laterre PF, LaRosa SP, Dhainaut JF, et al. N. Engl. J. Med. 2001;344:699–709. doi: 10.1056/NEJM200103083441001. [DOI] [PubMed] [Google Scholar]
- 24.Galanos C, Lüderitz O, Rietschel ET, Westphal O, Brade H, et al. Eur. J. Biochem. 1985;148:1–5. doi: 10.1111/j.1432-1033.1985.tb08798.x. [DOI] [PubMed] [Google Scholar]
- 25.Loppnow H, Brade H, Dürrbaum I, Dinarello CA, Kusumoto S, et al. J. Immunol. 1989;142:3229–38. [PubMed] [Google Scholar]
- 26.Golenbock DT, Hampton RY, Qureshi N, Takayama K, Raetz CRH. J. Biol. Chem. 1991;266:19490–8. [PubMed] [Google Scholar]
- 27.Alexander C, Rietschel ET. J. Endotoxin Res. 2001;7:167–202. [PubMed] [Google Scholar]
- 28.Chow JC, Young DW, Golenbock DT, Christ WJ, Gusovsky F. J. Biol. Chem. 1999;274:10689–92. doi: 10.1074/jbc.274.16.10689. [DOI] [PubMed] [Google Scholar]
- 29.Qureshi ST, Lariviere L, Leveque G, Clermont S, Moore KJ, et al. J. Exp. Med. 1999;189:615–25. doi: 10.1084/jem.189.4.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tobias PS, Soldau K, Ulevitch RJ. Journal of Experimental Medicine. 1986;164:777–93. doi: 10.1084/jem.164.3.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ulevitch RJ, Tobias PS. Curr. Opin. Immunol. 1999;11:19–22. doi: 10.1016/s0952-7915(99)80004-1. [DOI] [PubMed] [Google Scholar]
- 32.Medzhitov R, Preston-Hurlburt P, Janeway CA., Jr. Nature. 1997;388:394–7. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- 33.Shumann RR, Leong SR, Flaggs GW, Gray PW, Wright SD, et al. Science. 1990;249:1429–31. doi: 10.1126/science.2402637. [DOI] [PubMed] [Google Scholar]
- 34.Wright SD, Ramso RA, Tobias PS, Ulevitch RJ, Mathison JC. Science. 1990;249:1431–3. doi: 10.1126/science.1698311. [DOI] [PubMed] [Google Scholar]
- 35.Shimazu R, Akashi S, Ogata H, Nagai Y, Fukudome K, et al. J. Exp. Med. 1999;189:1777–82. doi: 10.1084/jem.189.11.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Anderson KV, Jurgens G, Nusslein-Volhard C. Cell. 1985;42:779–89. doi: 10.1016/0092-8674(85)90274-0. [DOI] [PubMed] [Google Scholar]
- 37.Hashimoto C, Hudson KL, Anderson KV. Cell. 1988;52:269–79. doi: 10.1016/0092-8674(88)90516-8. [DOI] [PubMed] [Google Scholar]
- 38.Anderson KV. Curr. Opin. Immunol. 2000;12:13–9. doi: 10.1016/s0952-7915(99)00045-x. [DOI] [PubMed] [Google Scholar]
- 39.Rock FL, Hardiman G, Timans JC, Kastelein RA, Bazan JF. Proc. Natl. Acad. Sci. USA. 1998;95:588–93. doi: 10.1073/pnas.95.2.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lien E, Sellati TJ, Yoshimura A, Flo TH, Rawadi G, et al. J. Biol. Chem. 1999;274:33419–25. doi: 10.1074/jbc.274.47.33419. [DOI] [PubMed] [Google Scholar]
- 41.Hajjar AM, O’Mahony DS, Ozinsky A, Underhill DM, Aderem A, et al. J. Immunol. 2001;166:15–9. doi: 10.4049/jimmunol.166.1.15. [DOI] [PubMed] [Google Scholar]
- 42.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Nature. 2001;413:732–8. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 43.Hayashi F, Smith KD, Ozinsky A, Hawn TR, Yi EC, et al. Nature. 2001;410:1099–103. doi: 10.1038/35074106. [DOI] [PubMed] [Google Scholar]
- 44.Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, et al. Nature. 2000;408:740–5. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 45.Greenfeder SA, Nunes P, Kwee L, Labow M, Chizzonite RA, Ju G. J. Biol. Chem. 1995;270:13757–65. doi: 10.1074/jbc.270.23.13757. [DOI] [PubMed] [Google Scholar]
- 46.Xu Y, Tao X, Shen B, Horng T, Medzhitov R, et al. Nature. 2000;408:111–5. doi: 10.1038/35040600. [DOI] [PubMed] [Google Scholar]
- 47.Raetz CRH. Journal of Bacteriology. 1993;175:5745–53. doi: 10.1128/jb.175.18.5745-5753.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wyckoff TJO, Raetz CRH, Jackman JE. Trends in Microbiology. 1998;6:154–9. doi: 10.1016/s0966-842x(98)01230-x. [DOI] [PubMed] [Google Scholar]
- 49.Anderson MS, Raetz CRH. J. Biol. Chem. 1987;262:5159–69. [PubMed] [Google Scholar]
- 50.Anderson MS, Bull HS, Galloway SM, Kelly TM, Mohan S, et al. J. Biol. Chem. 1993;268:19858–65. [PubMed] [Google Scholar]
- 51.Raetz CRH, Roderick SL. Science. 1995;270:997–1000. doi: 10.1126/science.270.5238.997. [DOI] [PubMed] [Google Scholar]
- 52.Vaara M. FEMS Microbiol. Letts. 1992;97:249–54. doi: 10.1016/0378-1097(92)90344-n. [DOI] [PubMed] [Google Scholar]
- 53.Beaman TW, Binder DA, Blanchard JS, Roderick SL. Biochemistry. 1997;36:489–94. doi: 10.1021/bi962522q. [DOI] [PubMed] [Google Scholar]
- 54.Olsen LR, Roderick SL. Biochemistry. 2001;40:1913–21. doi: 10.1021/bi002503n. [DOI] [PubMed] [Google Scholar]
- 55.Wyckoff TJ, Lin S, Cotter RJ, Dotson GD, Raetz CR. Journal of Biological Chemistry. 1998;273:32369–72. doi: 10.1074/jbc.273.49.32369. [DOI] [PubMed] [Google Scholar]
- 56.Wyckoff TJ, Raetz CRH. J. Biol. Chem. 1999;274:27047–55. doi: 10.1074/jbc.274.38.27047. [DOI] [PubMed] [Google Scholar]
- 57.Dotson GD, Kaltashov IA, Cotter RJ, Raetz CR. J. Bacteriol. 1998;180:330–7. doi: 10.1128/jb.180.2.330-337.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sweet CR, Lin S, Cotter RJ, Raetz CRH. J. Biol. Chem. 2001;276:19565–74. doi: 10.1074/jbc.M101868200. [DOI] [PubMed] [Google Scholar]
- 59.Anderson MS, Robertson AD, Macher I, Raetz CRH. Biochemistry. 1988;27:1908–17. doi: 10.1021/bi00406a017. [DOI] [PubMed] [Google Scholar]
- 60.Young K, Silver LL, Bramhill D, Cameron P, Eveland SS, et al. J. Biol. Chem. 1995;270:30384–91. doi: 10.1074/jbc.270.51.30384. [DOI] [PubMed] [Google Scholar]
- 61.Sorensen PG, Lutkenhaus J, Young K, Eveland SS, Anderson MS, Raetz CRH. J. Biol. Chem. 1996;271:25898–905. doi: 10.1074/jbc.271.42.25898. [DOI] [PubMed] [Google Scholar]
- 62.Jackman JE, Raetz CRH, Fierke CA. Biochemistry. 1999;38:1902–11. doi: 10.1021/bi982339s. [DOI] [PubMed] [Google Scholar]
- 63.Jackman JE, Raetz CRH, Fierke CA. Biochemistry. 2001 doi: 10.1021/bi001872g. submitted. [DOI] [PubMed] [Google Scholar]
- 64.Onishi HR, Pelak BA, Gerckens LS, Silver LL, Kahan FM, et al. Science. 1996;274:980–2. doi: 10.1126/science.274.5289.980. [DOI] [PubMed] [Google Scholar]
- 65.Jackman JE, Fierke CA, Tumey LN, Pirrung M, Uchiyama T, et al. J. Biol. Chem. 2000;275:11002–9. doi: 10.1074/jbc.275.15.11002. [DOI] [PubMed] [Google Scholar]
- 66.Kelly TM, Stachula SA, Raetz CRH, Anderson MS. J. Biol. Chem. 1993;268:19866–74. [PubMed] [Google Scholar]
- 67.Babinski KJ, Raetz CRH. FASEB J. 1998;12:A1288. [Google Scholar]
- 68.Bulawa CE, Raetz CRH. J. Biol. Chem. 1984;259:4846–51. [PubMed] [Google Scholar]
- 69.Ray BL, Painter G, Raetz CRH. J. Biol. Chem. 1984;259:4852–9. [PubMed] [Google Scholar]
- 70.Crowell DN, Anderson MS, Raetz CRH. J. Bacteriol. 1986;168:152–9. doi: 10.1128/jb.168.1.152-159.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Crowell DN, Reznikoff WS, Raetz CRH. J. Bacteriol. 1987;169:5727–34. doi: 10.1128/jb.169.12.5727-5734.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Radika K, Raetz CRH. J. Biol. Chem. 1988;263:14859–67. [PubMed] [Google Scholar]
- 73.Ray BL, Raetz CRH. J. Biol. Chem. 1987;262:1122–8. [PubMed] [Google Scholar]
- 74.Garrett TA, Kadrmas JL, Raetz CRH. J. Biol. Chem. 1997;272:21855–64. doi: 10.1074/jbc.272.35.21855. [DOI] [PubMed] [Google Scholar]
- 75.Garrett TA, Que NL, Raetz CRH. J. Biol. Chem. 1998;273:12457–65. doi: 10.1074/jbc.273.20.12457. [DOI] [PubMed] [Google Scholar]
- 76.Karow M, Georgopoulos C. Mol. Microbiol. 1993;7:69–79. doi: 10.1111/j.1365-2958.1993.tb01098.x. [DOI] [PubMed] [Google Scholar]
- 77.Polissi A, Georgopoulos C. Molecular Microbiology. 1996;20:1221–33. doi: 10.1111/j.1365-2958.1996.tb02642.x. [DOI] [PubMed] [Google Scholar]
- 78.Zhou Z, White KA, Polissi A, Georgopoulos C, Raetz CRH. J. Biol. Chem. 1998;273:12466–75. doi: 10.1074/jbc.273.20.12466. [DOI] [PubMed] [Google Scholar]
- 79.Doerrler WT, Reedy MC, Raetz CRH. J. Biol. Chem. 2001;276:11461–4. doi: 10.1074/jbc.C100091200. [DOI] [PubMed] [Google Scholar]
- 80.Raetz CRH, Purcell S, Meyer MV, Qureshi N, Takayama K. J. Biol. Chem. 1985;260:16080–8. [PubMed] [Google Scholar]
- 81.Lien E, Means TK, Heine H, Yoshimura A, Kusumoto S, et al. J. Clin. Invest. 2000;105:497–504. doi: 10.1172/JCI8541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Poltorak A, Ricciardi-Castagnoli P, Citterio S, Beutler B. Proc. Natl. Acad. Sci. U S A. 2000;97:2163–7. doi: 10.1073/pnas.040565397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Clementz T, Raetz CRH. Journal of Biological Chemistry. 1991;266:9687–96. [PubMed] [Google Scholar]
- 84.White KA, Kaltashov IA, Cotter RJ, Raetz CRH. J. Biol. Chem. 1997;272:16555–63. doi: 10.1074/jbc.272.26.16555. [DOI] [PubMed] [Google Scholar]
- 85.Belunis CJ, Mdluli KE, Raetz CRH, Nano FE. Journal of Biological Chemistry. 1992;267:18702–7. [PubMed] [Google Scholar]
- 86.Brozek KA, Raetz CRH. J. Biol. Chem. 1990;265:15410–7. [PubMed] [Google Scholar]
- 87.Clementz T, Bednarski JJ, Raetz CRH. J. Biol. Chem. 1996;271:12095–102. doi: 10.1074/jbc.271.20.12095. [DOI] [PubMed] [Google Scholar]
- 88.Clementz T, Zhou Z, Raetz CRH. J. Biol. Chem. 1997;272:10353–60. doi: 10.1074/jbc.272.16.10353. [DOI] [PubMed] [Google Scholar]
- 89.Mohan S, Raetz CRH. J. Bacteriol. 1994;176:6944–51. doi: 10.1128/jb.176.22.6944-6951.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Karow M, Georgopoulos C. Mol. Microbiol. 1991;5:2285–92. doi: 10.1111/j.1365-2958.1991.tb02159.x. [DOI] [PubMed] [Google Scholar]
- 91.Karow M, Georgopoulos C. J. Bacteriol. 1992;174:702–10. doi: 10.1128/jb.174.3.702-710.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Somerville JE, Jr., Cassiano L, Bainbridge B, Cunningham MD, Darveau RP. J. Clin. Invest. 1996;97:359–65. doi: 10.1172/JCI118423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Khan SA, Everest P, Servos S, Foxwell N, Zahringer U, et al. Mol. Microbiol. 1998;29:571–9. doi: 10.1046/j.1365-2958.1998.00952.x. [DOI] [PubMed] [Google Scholar]
- 94.Low KB, Ittensohn M, Le T, Platt J, Sodi S, et al. Nat. Biotechnol. 1999;17:37–41. doi: 10.1038/5205. [DOI] [PubMed] [Google Scholar]
- 95.van der Ley P, Steeghs L, Hamstra HJ, ten Hove J, Zomer B, van Alphen L. Infect. Immun. 2001;69:5981–90. doi: 10.1128/IAI.69.10.5981-5990.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Carty SM, Sreekumar KR, Raetz CRH. J. Biol. Chem. 1999;274:9677–85. doi: 10.1074/jbc.274.14.9677. [DOI] [PubMed] [Google Scholar]
- 97.Zhou Z, Lin S, Cotter RJ, Raetz CRH. J. Biol. Chem. 1999;274:18503–14. doi: 10.1074/jbc.274.26.18503. [DOI] [PubMed] [Google Scholar]
- 98.Guo L, Lim KB, Gunn JS, Bainbridge B, Darveau RP, et al. Science. 1997;276:250–3. doi: 10.1126/science.276.5310.250. [DOI] [PubMed] [Google Scholar]
- 99.Gunn JS, Lim KB, Krueger J, Kim K, Guo L, et al. Molecular Microbiology. 1998;27:1171–82. doi: 10.1046/j.1365-2958.1998.00757.x. [DOI] [PubMed] [Google Scholar]
- 100.Helander IM, Kilpeläinen I, Vaara M. Mol. Microbiol. 1994;11:481–7. doi: 10.1111/j.1365-2958.1994.tb00329.x. [DOI] [PubMed] [Google Scholar]
- 101.Nummila K, Kilpeläinen I, Zähringer U, Vaara M, Helander IM. Mol. Microbiol. 1995;16:271–8. doi: 10.1111/j.1365-2958.1995.tb02299.x. [DOI] [PubMed] [Google Scholar]
- 102.Trent MS, Ribeiro AA, Doerrler WT, Lin S, Cotter RJ, Raetz CRH. J. Biol. Chem. 2001;276:43132–44. doi: 10.1074/jbc.M106962200. [DOI] [PubMed] [Google Scholar]
- 103.Guo L, Lim KB, Poduje CM, Daniel M, Gunn JS, et al. Cell. 1998;95:189–98. doi: 10.1016/s0092-8674(00)81750-x. [DOI] [PubMed] [Google Scholar]
- 104.Bishop RE, Gibbons HS, Guina T, Trent MS, Miller SI, Raetz CRH. Embo J. 2000;19:5071–80. doi: 10.1093/emboj/cdd507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ohl ME, Miller SI. Annu. Rev. Med. 2001;52:259–74. doi: 10.1146/annurev.med.52.1.259. [DOI] [PubMed] [Google Scholar]
- 106.Groisman EA. J. Bacteriol. 2001;183:1835–42. doi: 10.1128/JB.183.6.1835-1842.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Brozek KA, Bulawa CE, Raetz CRH. J. Biol. Chem. 1987;262:5170–9. [PubMed] [Google Scholar]
- 108.Robey M, O’Connell W, Cianciotto NP. Infect. Immun. 2001;69:4276–86. doi: 10.1128/IAI.69.7.4276-4286.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ernst RK, Yi EC, Guo L, Lim KB, Burns JL, et al. Science. 1999;286:1561–5. doi: 10.1126/science.286.5444.1561. [DOI] [PubMed] [Google Scholar]
- 110.Gibbons HS, Lin S, Cotter RJ, Raetz CRH. J. Biol. Chem. 2000;275:32940–9. doi: 10.1074/jbc.M005779200. [DOI] [PubMed] [Google Scholar]
- 111.Trent MS, Pabich W, Raetz CRH, Miller SI. J. Biol. Chem. 2001;276:9083–92. doi: 10.1074/jbc.M010730200. [DOI] [PubMed] [Google Scholar]
- 112.Zhou Z, Ribeiro AA, Lin S, Cotter RJ, Miller SI, Raetz CRH. J. Biol. Chem. 2001;276:43111–21. doi: 10.1074/jbc.M106960200. [DOI] [PubMed] [Google Scholar]
- 113.Kanipes MI, Lin S, Cotter RJ, Raetz CRH. J. Biol. Chem. 2001;276:1156–63. doi: 10.1074/jbc.M009019200. [DOI] [PubMed] [Google Scholar]
- 114.Hasin M, Kennedy EP. Journal of Biological Chemistry. 1982;257:12475–7. [PubMed] [Google Scholar]
- 115.Trent MS, Ribeiro AA, Lin S, Cotter RJ, Raetz CRH. J. Biol. Chem. 2001;276:43122–31. doi: 10.1074/jbc.M106961200. [DOI] [PubMed] [Google Scholar]
- 116.Breazeale S, Ribeiro AA, Raetz CRH. J. Biol. Chem. 2002;277 doi: 10.1074/jbc.M109377200. in press. [DOI] [PubMed] [Google Scholar]
- 117.Gunn JS, Ryan SS, Van Velkinburgh JC, Ernst RK, Miller SI. Infect. Immun. 2000;68:6139–46. doi: 10.1128/iai.68.11.6139-6146.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Raetz CRH, Dowhan W. J. Biol. Chem. 1990;265:1235–8. [PubMed] [Google Scholar]
- 119.Clementz T, Bednarski J, Raetz CRH. FASEB J. 1995;9:A1311. [Google Scholar]
- 120.Chang G, Roth CB. Science. 2001;293:1793–800. doi: 10.1126/science.293.5536.1793. [DOI] [PubMed] [Google Scholar]
- 121.Borst P, Zelcer N, van Helvoort A. Biochim. Biophys. Acta. 2000;1486:128–44. doi: 10.1016/s1388-1981(00)00053-6. [DOI] [PubMed] [Google Scholar]
- 122.Andersen C, Hughes C, Koronakis V. EMBO Reports. 2000;1:313–8. doi: 10.1093/embo-reports/kvd075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Smit JJM, Schinkel AH, Oude Elferink RPJ, Groen AK, Wagenaar E, et al. Cell. 1993;75:451–62. doi: 10.1016/0092-8674(93)90380-9. [DOI] [PubMed] [Google Scholar]
- 124.Lu TT, Repa JJ, Mangelsdorf DJ. J. Biol. Chem. 2001;276:37735–8. doi: 10.1074/jbc.R100035200. [DOI] [PubMed] [Google Scholar]
- 125.Deckert G, Warren PV, Gaasterland T, Young WG, Lenox AL, et al. Nature. 1998;392:353–8. doi: 10.1038/32831. [DOI] [PubMed] [Google Scholar]
- 126.Plotz BM, Lindner B, Stetter KO, Holst O. J. Biol. Chem. 2000;275:11222–8. doi: 10.1074/jbc.275.15.11222. [DOI] [PubMed] [Google Scholar]
- 127.Vaara M. Antimicrob. Agents Chemother. 1993;37:2255–60. doi: 10.1128/aac.37.11.2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ferguson AD, Hofmann E, Coulton JW, Diederichs K, Welte W. Science. 1998;282:2215–20. doi: 10.1126/science.282.5397.2215. [DOI] [PubMed] [Google Scholar]
- 129.Ferguson AD, Welte W, Hofmann E, Lindner B, Holst O, et al. Structure Fold. Des. 2000;8:585–92. doi: 10.1016/s0969-2126(00)00143-x. [DOI] [PubMed] [Google Scholar]
- 130.de Cock H, Brandenburg K, Wiese A, Holst O, Seydel U. J. Biol. Chem. 1999;274:5114–9. doi: 10.1074/jbc.274.8.5114. [DOI] [PubMed] [Google Scholar]
- 131.Patchett AA, Cordes EH. Adv. Enzymol. Relat. Areas of Mol. Biol. 1985;57:1–83. doi: 10.1002/9780470123034.ch1. [DOI] [PubMed] [Google Scholar]
- 132.Hanson JE, Kaplan AP, Bartlett PA. Biochemistry. 1989;28:6294–305. doi: 10.1021/bi00441a022. [DOI] [PubMed] [Google Scholar]
- 133.Steeghs L, den Hartog R, den Boer A, Zomer B, Roholl P, van der Ley P. Nature (London) 1998;392:449–50. doi: 10.1038/33046. [DOI] [PubMed] [Google Scholar]
- 134.Pridmore AC, Wyllie DH, Abdillahi F, Steeghs L, van der Ley P, et al. J. Infect. Dis. 2001;183:89–96. doi: 10.1086/317647. [DOI] [PubMed] [Google Scholar]
- 135.Gotschlich EC, Fraser BA, Nishimura O, Robbins JB, Liu T-Y. Journal of Biological Chemistry. 1981;256:8915–21. [PubMed] [Google Scholar]
- 136.Kawasaki S, Moriguchi R, Sekiya K, Nakai T, Ono E, et al. Journal of Bacteriology. 1994;176:284–90. doi: 10.1128/jb.176.2.284-290.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Nelson KE, Clayton RA, Gill SR, Gwinn ML, Dodson RJ, et al. Nature. 1999;399:323–9. doi: 10.1038/20601. [DOI] [PubMed] [Google Scholar]
- 138.Thompson BG, Murray RG. Can. J. Microbiol. 1981;27:729–34. doi: 10.1139/m81-111. [DOI] [PubMed] [Google Scholar]
- 139.White O, Eisen JA, Heidelberg JF, Hickey EK, Peterson JD, et al. Science. 1999;286:1571–7. doi: 10.1126/science.286.5444.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Fraser CM, Casjens S, Huang WM, Sutton GG, Clayton R, et al. Nature. 1997;390:580–6. doi: 10.1038/37551. [DOI] [PubMed] [Google Scholar]
- 141.Fraser CM, Norris SJ, Weinstock GM, White O, Sutton GG, et al. Science. 1998;281:375–88. doi: 10.1126/science.281.5375.375. [DOI] [PubMed] [Google Scholar]
- 142.Takayama K, Rothenberg RJ, Barbour AG. Infect. Immun. 1987;55:2311–3. doi: 10.1128/iai.55.9.2311-2313.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Karlin S. Trends Microbiol. 2001;9:335–43. doi: 10.1016/s0966-842x(01)02079-0. [DOI] [PubMed] [Google Scholar]
- 144.Karlin S, Mrazek J. Proc. Natl. Acad. Sci. U S A. 2001;98:5240–5. doi: 10.1073/pnas.081077598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Vinh T, Adler B, Faine S. J. Gen. Microbiol. 1986;132:103–9. doi: 10.1099/00221287-132-1-103. [DOI] [PubMed] [Google Scholar]
- 146.Werts C, Tapping RI, Mathison JC, Chuang TH, Kravchenko V, et al. Nat. Immunol. 2001;2:346–52. doi: 10.1038/86354. [DOI] [PubMed] [Google Scholar]
- 147.Kulshin VA, Zahringer U, Lindner B, Jager KE, Dmitriev BA, Rietschel ET. Eur. J. Biochem. 1991;198:697–704. doi: 10.1111/j.1432-1033.1991.tb16069.x. [DOI] [PubMed] [Google Scholar]
- 148.Bhat UR, Forsberg LS, Carlson RW. J. Biol. Chem. 1994;269:14402–10. [PubMed] [Google Scholar]
- 149.Que NLS, Lin S, Cotter RJ, Raetz CRH. J. Biol. Chem. 2000;275:28006–16. doi: 10.1074/jbc.M004008200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Que NLS, Ribeiro AA, Raetz CRH. J. Biol. Chem. 2000;275:28017–27. doi: 10.1074/jbc.M004009200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Price NPJ, Kelly TM, Raetz CRH, Carlson RW. J. Bacteriol. 1994;176:4646–55. doi: 10.1128/jb.176.15.4646-4655.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Price NJP, Jeyaretnam B, Carlson RW, Kadrmas JL, Raetz CRH, Brozek KA. Proc. Natl. Acad. Sci. U. S. A. 1995;92:7352–6. doi: 10.1073/pnas.92.16.7352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Brozek KA, Kadrmas JL, Raetz CRH. J. Biol. Chem. 1996;271:32112–8. [PubMed] [Google Scholar]
- 154.Kadrmas JL, Brozek KA, Raetz CRH. J. Biol. Chem. 1996;271:32119–25. [PubMed] [Google Scholar]
- 155.Brozek KA, Carlson RW, Raetz CRH. J. Biol. Chem. 1996;271:32126–36. doi: 10.1074/jbc.271.50.32126. [DOI] [PubMed] [Google Scholar]
- 156.Que NLS, Karbarz MJ, Lin SH, Cotter RJ, Raetz CRH. Glycobiology. 2000;10:177. [Google Scholar]
- 157.Karbarz MJ, Raetz CRH. Glycobiology. 2000;10:179. [Google Scholar]
- 158.Williamson JM, Anderson MS, Raetz CRH. J. Bacteriol. 1991;173:3591–6. doi: 10.1128/jb.173.11.3591-3596.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Goldman RC, Doran CC, Kadam SK, Capobianco JO. J. Biol. Chem. 1988;263:5217–23. [PubMed] [Google Scholar]
- 160.Kawahara K, Uchida K, Aida K. Biochim. Biophys. Acta. 1979;572:1–8. [PubMed] [Google Scholar]
- 161.Karunaratne DN, Richards JC, Hancock RE. Arch. Biochem. Biophys. 1992;299:368–76. doi: 10.1016/0003-9861(92)90289-9. [DOI] [PubMed] [Google Scholar]
- 162.Weckesser J, Mayer H. FEMS Microbiol Rev. 1988;4:143–53. doi: 10.1111/j.1574-6968.1988.tb02740.x. [DOI] [PubMed] [Google Scholar]
- 163.Sweet CR, Raetz CRH. FASEB J. 2001;15:A194. [Google Scholar]
- 164.Glockner G, Rosenthal A, Valentin K. J. Mol. Evol. 2000;51:382–90. doi: 10.1007/s002390010101. [DOI] [PubMed] [Google Scholar]
- 165.Ohlrogge JB, Browse J, Somerville CR. Biochim. Biophys. Acta. 1991;1082:1–26. doi: 10.1016/0005-2760(91)90294-r. [DOI] [PubMed] [Google Scholar]
- 166.Li J, Chory J. Cell. 1997;90:929–38. doi: 10.1016/s0092-8674(00)80357-8. [DOI] [PubMed] [Google Scholar]
- 167.Jagielo PJ, Thorne PS, Watt JL, Frees KL, Quinn TJ, Schwartz DA. Chest. 1996;110:263–70. doi: 10.1378/chest.110.1.263. [DOI] [PubMed] [Google Scholar]
- 168.Holst O. In: Endotoxin in Health and Disease. Brade H, Opal SM, Vogel SN, Morrison DC, editors. Marcel Dekker Inc.; New York: 1999. pp. 305–30. [Google Scholar]
- 169.Hood DW, Moxon ER. In: Endotoxin in Health and Disease. Brade H, Opal SM, Vogel SN, Morrison DC, editors. Marcel Dekker, Inc.; New York, Basel: 1999. pp. 179–94. [Google Scholar]
- 170.Giardina PC, Apicella MA, B. G, Preston A. In: Endotoxin in Health and Disease. Brade H, Opal SM, Vogel SN, Morrison DC, editors. Marcel Dekker, Inc.; New York, Basel: 1999. pp. 55–65. [Google Scholar]
- 171.Griffiss JM, Schneider H. In: Endotoxin in Health and Disease. Brade H, Opal SM, Vogel SN, Morrison DC, editors. Marcel Dekker, Inc.; New York, Basel: 1999. pp. 179–94. [Google Scholar]
- 172.Kadrmas JL, Allaway D, Studholme RE, Sullivan JT, Ronson CW, et al. J. Biol. Chem. 1998;273:26432–40. doi: 10.1074/jbc.273.41.26432. [DOI] [PubMed] [Google Scholar]
- 173.Forsberg LS, Carlson RW. J. Biol. Chem. 1998;273:2747–57. doi: 10.1074/jbc.273.5.2747. [DOI] [PubMed] [Google Scholar]
- 174.Amor K, Heinrichs DE, Frirdich E, Ziebell K, Johnson RP, Whitfield C. Infection and Immunity. 2000;68:1116–24. doi: 10.1128/iai.68.3.1116-1124.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Appelmelk BJ, An Y-Q, Hekker TAM, Thijs LG, MacLaren DM, deGraaf J. Microbiology. 1994;140:1119–24. doi: 10.1099/13500872-140-5-1119. [DOI] [PubMed] [Google Scholar]
- 176.Gibb AR, Barclay GR, Poxton IR, di Padova F. Journal of Infectious Diseases. 1992;166:1051–7. doi: 10.1093/infdis/166.5.1051. [DOI] [PubMed] [Google Scholar]
- 177.Currie CG, Poxton IR. FEMS Immunology and Medical Microbiology. 1999;24:57–62. doi: 10.1111/j.1574-695X.1999.tb01265.x. [DOI] [PubMed] [Google Scholar]
- 178.Vinogradov E, Cedzynski M, Ziolkowski A, Swierzko A. European Journal of Biochemistry. 2001;268:1722–9. doi: 10.1046/j.1432-1033.2001.02047.x. [DOI] [PubMed] [Google Scholar]
- 179.Vinogradov E, Perry MB. European Journal of Biochemistry. 2001 doi: 10.1046/j.1432-1327.2001.02264.x. in press. [DOI] [PubMed] [Google Scholar]
- 180.Trautmann M, Ruhnke M, Rukavina T, Held TK, Cross AS, et al. Clinical and Diagnostic Laboratory Immunology. 1997;4:550–5. doi: 10.1128/cdli.4.5.550-555.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Di Padova FE, Brade H, Barclay GR, Poxton IR, Liehl E, et al. Infection and Immunity. 1993;61:3863–72. doi: 10.1128/iai.61.9.3863-3872.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Di Padova FE, Mikol V, Barclay GR, Poxton IR, Brade H, Rietschel ET. Progress in Clinical and Biological Research. 1994;388:85–94. [PubMed] [Google Scholar]
- 183.Bennett-Guerrero E, McIntosh TJ, Barclay GR, Snyder DS, Gibbs RJ, et al. Infection and Immunity. 2000;68:6202–8. doi: 10.1128/iai.68.11.6202-6208.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Stanislavsky ES, Lam JS. FEMS Microbiol. Rev. 1997;21:243–77. doi: 10.1111/j.1574-6976.1997.tb00353.x. [DOI] [PubMed] [Google Scholar]
- 185.Currie CG, McCallum K, Poxton IR. Journal of Medical Microbiology. 2001;50:345–54. doi: 10.1099/0022-1317-50-4-345. [DOI] [PubMed] [Google Scholar]
- 186.Nikaido H, Vaara M. Microbiological Reviews. 1985;49:1–32. doi: 10.1128/mr.49.1.1-32.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Parker CT, Kloser AW, Schnaitman CA, Stein MA, Gottesman S, Gibson BW. Journal of Bacteriology. 1992;174:2525–38. doi: 10.1128/jb.174.8.2525-2538.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Bauer ME, Welch RA. Infection and Immunity. 1997;65:2218–24. doi: 10.1128/iai.65.6.2218-2224.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Stanley PL, Diaz P, Bailey MJ, Gygi D, Juarez A, Hughes C. Molecular Microbiology. 1993;10:781–7. doi: 10.1111/j.1365-2958.1993.tb00948.x. [DOI] [PubMed] [Google Scholar]
- 190.O’Toole G, Kaplan HB, Kolter R. Annual Reviews in Microbiology. 2000;54:49–79. doi: 10.1146/annurev.micro.54.1.49. [DOI] [PubMed] [Google Scholar]
- 191.Yethon JA, Gunn JS, Malo D, Laroche L, Ernst RK, et al. Infection and Immunity. 2000;68:4485–91. doi: 10.1128/iai.68.8.4485-4491.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Yethon JA, Heinrichs DE, Monteiro MA, Perry MB, Whitfield C. Journal of Biological Chemistry. 1998;273:26310–6. doi: 10.1074/jbc.273.41.26310. [DOI] [PubMed] [Google Scholar]
- 193.Yethon JA, Vinogradov E, Perry MB, Whitfield C. Journal of Bacteriology. 2000;182:5620–3. doi: 10.1128/jb.182.19.5620-5623.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Walsh AG, Matewish MJ, Burrows LL, Monteiro MA, Perry MB, Lam JS. Molecular Microbiology. 2000;35:718–27. doi: 10.1046/j.1365-2958.2000.01741.x. [DOI] [PubMed] [Google Scholar]
- 195.Sanchez Carballo PM, Rietschel ET, Kosma P, Zahringer U. European Journal of Biochemistry. 1999;261:500–8. doi: 10.1046/j.1432-1327.1999.00299.x. [DOI] [PubMed] [Google Scholar]
- 196.Watt SR, Clarke AJ. FEMS Microbiology Letters. 1994;124:113–9. doi: 10.1111/j.1574-6968.1994.tb07270.x. [DOI] [PubMed] [Google Scholar]
- 197.Straus DC. Infection and Immunity. 1987;55:44–8. doi: 10.1128/iai.55.1.44-48.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Straus DC, Atkisson DL, Garner CW. Infection and Immunity. 1985;50:787–95. doi: 10.1128/iai.50.3.787-795.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Caroff M, Brisson J, Martin A, Karibian D. FEBS Letters. 2000;477:8–14. doi: 10.1016/s0014-5793(00)01720-8. [DOI] [PubMed] [Google Scholar]
- 200.Allen AG, Isobe T, Maskell DJ. Journal of Bacteriology. 1998;180:35–40. doi: 10.1128/jb.180.1.35-40.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Heinrichs DE, Yethon JA, Whitfield C. Molecular Microbiology. 1998;30:221–32. doi: 10.1046/j.1365-2958.1998.01063.x. [DOI] [PubMed] [Google Scholar]
- 202.Regué M, Climent N, Abitiu N, Coderch N, Merino S, et al. Journal of Bacteriology. 2001;183:3564–73. doi: 10.1128/JB.183.12.3564-3573.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Bailey MJ, Hughes C, Koronakis V. Molecular Microbiology. 1997;26:845–51. doi: 10.1046/j.1365-2958.1997.6432014.x. [DOI] [PubMed] [Google Scholar]
- 204.Marolda CL, Valvano MA. Journal of Bacteriology. 1998;180:3070–9. doi: 10.1128/jb.180.12.3070-3079.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Geerlof A, Lewendon A, Shaw WV. Journal of Biological Chemistry. 1999;274:27105–11. doi: 10.1074/jbc.274.38.27105. [DOI] [PubMed] [Google Scholar]
- 206.Kontrohr T, Kocsis B. Journal of Biological Chemistry. 1981;256:7715–8. [PubMed] [Google Scholar]
- 207.Kocsis B, Kontrohr T. J Biol Chem. 1984;259:11858–60. [PubMed] [Google Scholar]
- 208.Zamyatina A, Gronow S, Oertelt C, Puchberger M, Brade H, Kosma P. Agnew. Chem. Int. Ed. 2000;39:4150–3. doi: 10.1002/1521-3773(20001117)39:22<4150::aid-anie4150>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 209.Gronow S, Brabetz W, Brade H. European Journal of Biochemistry. 2000;267:6602–11. doi: 10.1046/j.1432-1327.2000.01754.x. [DOI] [PubMed] [Google Scholar]
- 210.Kadrmas JL, Raetz CR. Journal of Biological Chemistry. 1998;273:2799–807. doi: 10.1074/jbc.273.5.2799. [DOI] [PubMed] [Google Scholar]
- 211.Eidels L, Osborn MJ. Proceedings of the National Academy of Sciences, USA. 1971;68:1673–7. doi: 10.1073/pnas.68.8.1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Brooke JS, Valvano MA. Journal of Biological Chemistry. 1996;271:3608–14. doi: 10.1074/jbc.271.7.3608. [DOI] [PubMed] [Google Scholar]
- 213.Brooke JS, Valvano MA. Journal of Bacteriology. 1996;178:3339–41. doi: 10.1128/jb.178.11.3339-3341.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Valvano MA, Marolda CL, Bittner M, Glaskin-Clay M, Simon TL, Klena JD. Journal of Bacteriology. 2000;182:488–97. doi: 10.1128/jb.182.2.488-497.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Kneidinger B, Marolda CL, Graninger M, Zamyatina A, McArthur F, et al. Molecular Microbiology. 2001 submitted. [Google Scholar]
- 216.Kneidinger B, Graninger M, Puchberger M, Kosma P, Messner P. Journal of Biological Chemistry. 2001;276:20935–44. doi: 10.1074/jbc.M100378200. [DOI] [PubMed] [Google Scholar]
- 217.Coleman WG., Jr. J Biol Chem. 1983;258:1985–90. [PubMed] [Google Scholar]
- 218.Deacon AM, Ni YS, Coleman WG, Jr., Ealick SE. Structure and Fold Design. 2000;8:453–62. doi: 10.1016/s0969-2126(00)00128-3. [DOI] [PubMed] [Google Scholar]
- 219.Yethon JA, Whitfield C. Journal of Biological Chemistry. 2001;276:5498–504. doi: 10.1074/jbc.M008255200. [DOI] [PubMed] [Google Scholar]
- 220.White KA, Lin S, Cotter RJ, Raetz CR. J. Biol. Chem. 1999;274:31391–400. doi: 10.1074/jbc.274.44.31391. [DOI] [PubMed] [Google Scholar]
- 221.Brabetz W, Muller-Loennies S, Brade H. J. Biol. Chem. 2000;275:34954–62. doi: 10.1074/jbc.M005204200. [DOI] [PubMed] [Google Scholar]
- 222.Creeger ES, Rothfield LI. Journal of Biological Chemistry. 1979;254:804–10. [PubMed] [Google Scholar]
- 223.Campbell JA, Davies GJ, Bulone V, Henrissat B. Biochemical Journal. 1997;326:929–39. doi: 10.1042/bj3260929u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Persson K, Ly HD, Dieckelmann M, Wakarchuk WW, Withers SG, Strynadka NC. Nature Structural Biology. 2001;8:166–75. doi: 10.1038/84168. [DOI] [PubMed] [Google Scholar]
- 225.Saxena IM, Brown RM, Fevre M, Geremia RA, Henrissat B. Journal of Bacteriology. 1995;177:1419–24. doi: 10.1128/jb.177.6.1419-1424.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Charnock SJ, Henrissat B, Davies GJ. Plant Physiology. 2001;125:527–31. doi: 10.1104/pp.125.2.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Unligil UM, Rini JM. Current Opinions in Structural Biology. 2000;10:510–7. doi: 10.1016/s0959-440x(00)00124-x. [DOI] [PubMed] [Google Scholar]
- 228.Heinrichs DE, Yethon JA, Amor PA, Whitfield C. Journal of Biological Chemistry. 1998;273:29497–505. doi: 10.1074/jbc.273.45.29497. [DOI] [PubMed] [Google Scholar]
- 229.Heinrichs DE, Monteiro MA, Perry MB, Whitfield C. Journal of Biological Chemistry. 1998;273:8849–59. doi: 10.1074/jbc.273.15.8849. [DOI] [PubMed] [Google Scholar]
- 230.Klena JD, Ashford RSI, Schnaitman CA. Journal of Bacteriology. 1992;174:7297–307. doi: 10.1128/jb.174.22.7297-7307.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.MacLachlan PR, Kadam SK, Sanderson KE. Journal of Bacteriology. 1991;173:7151–63. doi: 10.1128/jb.173.22.7151-7163.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Sadovskaya I, Brisson JR, Thibault P, Richards JC, Lam JS, Altman E. European Journal of Biochemistry. 2000;267:1640–50. doi: 10.1046/j.1432-1327.2000.01156.x. [DOI] [PubMed] [Google Scholar]
- 233.Rahim R, Burrows LL, Monteiro MA, Perry MB, Lam JS. Microbiology. 2000;146:2803–14. doi: 10.1099/00221287-146-11-2803. [DOI] [PubMed] [Google Scholar]
- 234.Yang H, Matewish M, Loubens I, Storey DG, Lam JS, Jin S. Microbiology. 2000;146:2509–19. doi: 10.1099/00221287-146-10-2509. [DOI] [PubMed] [Google Scholar]
- 235.Vinogradov E, MacLean LL, Perry MB, Petersen O, Frirdich E, Whitfield C. manuscript in preparation. [Google Scholar]
- 236.Olsthoorn MM, Petersen BO, Schlecht S, Haverkamp J, Bock K, et al. Journal of Biological Chemistry. 1998;273:3817–29. doi: 10.1074/jbc.273.7.3817. [DOI] [PubMed] [Google Scholar]
- 237.Gamian A, Romanowska E. European Journal of Biochemistry. 1982;129:105–9. doi: 10.1111/j.1432-1033.1982.tb07027.x. [DOI] [PubMed] [Google Scholar]
- 238.Kahler CM, Carlson RW, Rahman MM, Martin LE, Stephens DS. Journal of Bacteriology. 1996;178:6677–84. doi: 10.1128/jb.178.23.6677-6684.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Kahler CM, Carlson RW, Rahman MM, Martin LE, Stephens DS. Journal of Bacteriology. 1996;178:1265–73. doi: 10.1128/jb.178.5.1265-1273.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Banerjee A, Wang R, Uljon SN, Rice PA, Gotschlich EC, Stein DC. Proceedings of the National Academy of Sciences, USA. 1998;95:10872–7. doi: 10.1073/pnas.95.18.10872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Rahman MM, Stephens DS, Kahler CM, Glushka J, Carlson RW. Carbohydrate Research. 1998;307:311–24. doi: 10.1016/s0008-6215(98)00012-3. [DOI] [PubMed] [Google Scholar]
- 242.Rahman MM, Kahler CM, Stephens DS, Carlson RW. Glycobiology. 2001;11:703–9. doi: 10.1093/glycob/11.8.703. [DOI] [PubMed] [Google Scholar]
- 243.Gilbert M, Watson DC, Cunningham AM, Jennings MP, Young NM, Wakarchuk WW. Journal of Biological Chemistry. 1996;271:28271–6. doi: 10.1074/jbc.271.45.28271. [DOI] [PubMed] [Google Scholar]
- 244.Gilbert M, Cunningham AM, Watson DC, Martin A, Richards JC, Wakarchuk WW. European Journal of Biochemistry. 1997;249:187–94. doi: 10.1111/j.1432-1033.1997.t01-1-00187.x. [DOI] [PubMed] [Google Scholar]
- 245.Wakarchuk W, Martin A, Jennings MP, Moxon ER, Richards JC. Journal of Biological Chemistry. 1996;271:19166–73. doi: 10.1074/jbc.271.32.19166. [DOI] [PubMed] [Google Scholar]
- 246.Kahler CM, Stephens DS. Critical Reviews in Microbiology. 1998;24:281–334. doi: 10.1080/10408419891294216. [DOI] [PubMed] [Google Scholar]
- 247.Yang QL, Gotschlich EC. Journal of Experimental Medicine. 1996;183:323–7. doi: 10.1084/jem.183.1.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Burch CL, Danaher RJ, Stein DC. Journal of Bacteriology. 1997;179:982–6. doi: 10.1128/jb.179.3.982-986.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Danaher RJ, Levin JC, Arking D, Burch CL, Sandlin R, Stein DC. Journal of Bacteriology. 1995;177:7275–9. doi: 10.1128/jb.177.24.7275-7279.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Appelmelk BJ, Martino MC, Veenhof E, Monteiro MA, Maaskant JJ, et al. Infection and Immunity. 2000;68:5928–32. doi: 10.1128/iai.68.10.5928-5932.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Wang G, Rasko DA, Sherburne R, Taylor DE. Molecular Microbiology. 1999;31:1265–74. doi: 10.1046/j.1365-2958.1999.01268.x. [DOI] [PubMed] [Google Scholar]
- 252.Linton D, Gilbert M, Hitchen PG, Dell A, Morris HR, et al. Molecular Microbiology. 2000;37:501–14. doi: 10.1046/j.1365-2958.2000.02020.x. [DOI] [PubMed] [Google Scholar]
- 253.High NJ, Jennings MP, Moxon ER. Molecular Microbiology. 1996;20:165–74. doi: 10.1111/j.1365-2958.1996.tb02498.x. [DOI] [PubMed] [Google Scholar]
- 254.Weiser JN, Love JM, Moxon ER. Cell. 1989;59:657–65. doi: 10.1016/0092-8674(89)90011-1. [DOI] [PubMed] [Google Scholar]
- 255.Weiser JN, Shchepetov M, Chong ST. Infection and Immunity. 1997;65:943–50. doi: 10.1128/iai.65.3.943-950.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Maskell DJ, Szabo MJ, Butler PD, Williams AE, Moxon ER. Molecular Microbiology. 1991;5:1013–22. doi: 10.1111/j.1365-2958.1991.tb01874.x. [DOI] [PubMed] [Google Scholar]
- 257.Maskell DJ, Szabo MJ, Butler PD, Williams AE, Moxon ER. Journal of Infectious Diseases. 1992;165(Suppl 1):S90–2. doi: 10.1093/infdis/165-supplement_1-s90. [DOI] [PubMed] [Google Scholar]
- 258.Wakarchuk WW, Watson D, St Michael F, Li J, Wu Y, et al. Journal of Biological Chemistry. 2001;276:12785–90. doi: 10.1074/jbc.M011293200. [DOI] [PubMed] [Google Scholar]
- 259.Knirel YA, Kochetkov NK. Biochemistry (Moscow) 1994;59:1325–83. [Google Scholar]
- 260.Jansson P-E. In: Endotoxin in Health and Disease. Brade H, Opal SM, Vogel SN, Morrison DC, editors. Marcel Dekker, Inc.; New York, Basel: 1999. pp. 155–78. [Google Scholar]
- 261.Kuhn H-M, Meier-Dieter U, Mayer H. FEMS Microbiology Reviews. 1988;54:195–222. doi: 10.1111/j.1574-6968.1988.tb02743.x. [DOI] [PubMed] [Google Scholar]
- 262.Whitfield C, Roberts IS. Molecular Microbiology. 1999;31:1307–19. doi: 10.1046/j.1365-2958.1999.01276.x. [DOI] [PubMed] [Google Scholar]
- 263.Mäkelä PH, Stocker BAD. In: Handbook of Endotoxin. Rietschel ET, editor. Elsevier Science Publishers, B.V.; Amsterdam: 1984. pp. 59–137. [Google Scholar]
- 264.Popoff MY, Le Minor L. Annales of the Institut Pasteur/Microbiology. 1985;136B:169–79. doi: 10.1016/s0769-2609(85)80042-9. [DOI] [PubMed] [Google Scholar]
- 265.Keenleyside WJ, Perry MB, MacLean LL, Poppe C, Whitfield C. Molecular Microbiology. 1994;11:437–48. doi: 10.1111/j.1365-2958.1994.tb00325.x. [DOI] [PubMed] [Google Scholar]
- 266.Rocchetta HL, Burrows LL, Lam JS. Microbiological and Molecular Biological Reviews. 1999;63:523–53. doi: 10.1128/mmbr.63.3.523-553.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Burns SM, Hull SI. Infection and Immunity. 1998;66:4244–53. doi: 10.1128/iai.66.9.4244-4253.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Joiner KA. Annual Reviews in Microbiology. 1988;42:201–30. doi: 10.1146/annurev.mi.42.100188.001221. [DOI] [PubMed] [Google Scholar]
- 269.Weiss J, Hutzler M, Kao L. Infection and Immunity. 1986;51:594–9. doi: 10.1128/iai.51.2.594-599.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Nesper J, Lauriano CM, Klose KE, Kapfhammer D, Kraiss A, Reidl J. Infection and Immunity. 2001;69:435–45. doi: 10.1128/IAI.69.1.435-445.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Van den Bosch L, Manning PA, Morona R. Molecular Microbiology. 1997;23:765–75. doi: 10.1046/j.1365-2958.1997.2541625.x. [DOI] [PubMed] [Google Scholar]
- 272.Sandlin RC, Lampel KA, Keasler SP, Goldberg MB, Stolzer AL, Maurelli AT. Infection and Immunity. 1995;63:229–37. doi: 10.1128/iai.63.1.229-237.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Sandlin RC, Goldberg MB, Maurelli AT. Molecular Microbiology. 1996;22:63–73. doi: 10.1111/j.1365-2958.1996.tb02656.x. [DOI] [PubMed] [Google Scholar]
- 274.al-Hendy A, Toivanen P, Skurnik M. Microbial Pathogenesis. 1991;10:81–6. doi: 10.1016/0882-4010(91)90068-l. [DOI] [PubMed] [Google Scholar]
- 275.Skurnik M, Toivanen P. Trends in Microbiology. 1993;1:148–52. doi: 10.1016/0966-842x(93)90130-j. [DOI] [PubMed] [Google Scholar]
- 276.McGrath BC, Osborn MJ. Journal of Bacteriology. 1991;173:649–54. doi: 10.1128/jb.173.2.649-654.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Liu D, Reeves PR. Microbiology. 1994;140:49–57. doi: 10.1099/13500872-140-1-49. [DOI] [PubMed] [Google Scholar]
- 278.Kogan G, Haraguchi G, Hull SI, Hull RA, Shashkov A, et al. European Journal of Biochemistry. 1993;214:259–65. doi: 10.1111/j.1432-1033.1993.tb17919.x. [DOI] [PubMed] [Google Scholar]
- 279.Yao Z, Valvano MA. Journal of Bacteriology. 1994;176:4133–43. doi: 10.1128/jb.176.13.4133-4143.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Neuhard J, Thomassen E. Journal of Bacteriology. 1976;126:999–1001. doi: 10.1128/jb.126.2.999-1001.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Osborn MJ, Cynkin MA, Gilbert JM, Muller L, Singh M. Methods in Enzymology. 1972;28:583–601. [Google Scholar]
- 282.Wang L, Liu D, Reeves PR. Journal of Bacteriology. 1996;178:2598–604. doi: 10.1128/jb.178.9.2598-2604.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Meier-Dieter U, Barr K, Starman R, Hatch L, Rick PD. Journal of Biological Chemistry. 1992;267:746–53. [PubMed] [Google Scholar]
- 284.Alexander DC, Valvano MA. Journal of Bacteriology. 1994;176:7079–84. doi: 10.1128/jb.176.22.7079-7084.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Klena JD, Schnaitman CA. Molecular Microbiology. 1993;9:393–402. doi: 10.1111/j.1365-2958.1993.tb01700.x. [DOI] [PubMed] [Google Scholar]
- 286.Amor PA, Whitfield C. Molecular Microbiology. 1997;26:145–61. doi: 10.1046/j.1365-2958.1997.5631930.x. [DOI] [PubMed] [Google Scholar]
- 287.Zhang L, Radziejewska-Lebrecht J, Krajewska-Pietrasik D, Toivanen P, Skurnik M. Molecular Microbiology. 1997;23:63–76. doi: 10.1046/j.1365-2958.1997.1871558.x. [DOI] [PubMed] [Google Scholar]
- 288.Rocchetta HL, Burrows LL, Pacan JC, Lam JS. Molecular Microbiology. 1998;28:1103–19. doi: 10.1046/j.1365-2958.1998.00871.x. [DOI] [PubMed] [Google Scholar]
- 289.Whitfield C, Valvano MA. Advances in Microbial Physiology. 1993;35:135–246. doi: 10.1016/s0065-2911(08)60099-5. [DOI] [PubMed] [Google Scholar]
- 290.Liu D, Haase AM, Lindqvist L, Lindberg AA, Reeves PR. Journal of Bacteriology. 1993;175:3408–13. doi: 10.1128/jb.175.11.3408-3413.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Liu D, Cole R, Reeves PR. Journal of Bacteriology. 1996;178:2102–7. doi: 10.1128/jb.178.7.2102-2107.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Feldman MF, Marolda CL, Monteiro MA, Perry MB, Parodi AJ, Valvano MA. Journal of Biological Chemistry. 1999;274:35129–38. doi: 10.1074/jbc.274.49.35129. [DOI] [PubMed] [Google Scholar]
- 293.MacPherson DF, Manning PA, Morona R. Gene. 1995;155:9–17. doi: 10.1016/0378-1119(94)00918-i. [DOI] [PubMed] [Google Scholar]
- 294.Wang L, Reeves PR. Journal of Bacteriology. 1994;176:4348–56. doi: 10.1128/jb.176.14.4348-4356.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Bray D, Robbins PW. Biochemical and Biophysical Research Communications. 1967;28:334–9. doi: 10.1016/0006-291x(67)90314-2. [DOI] [PubMed] [Google Scholar]
- 296.Robbins PW, Bray D, Dankert M, Wright A. Science. 1967;158:1536–42. doi: 10.1126/science.158.3808.1536. [DOI] [PubMed] [Google Scholar]
- 297.Collins LV, Hackett J. Journal of Bacteriology. 1991;173:2521–9. doi: 10.1128/jb.173.8.2521-2529.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Jann K, Jann B. In: Handbook of Endotoxin. Rietschel ET, editor. Elsevier Science Publishers, B.V.; Amsterdam: 1984. pp. 138–86. [Google Scholar]
- 299.Morona R, Mavris M, Fallarino A, Manning PA. Journal of Bacteriology. 1994;176:733–47. doi: 10.1128/jb.176.3.733-747.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Daniels C, Vindurampulle C, Morona R. Molecular Microbiology. 1998;28:1211–22. doi: 10.1046/j.1365-2958.1998.00884.x. [DOI] [PubMed] [Google Scholar]
- 301.Batchelor RA, Haraguchi GE, Hull RA, Hull SI. Journal of Bacteriology. 1991;173:5699–704. doi: 10.1128/jb.173.18.5699-5704.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Morona R, Van Den Bosch L, Daniels C. Microbiology. 2000;146:1–4. doi: 10.1099/00221287-146-1-1. [DOI] [PubMed] [Google Scholar]
- 303.Bastin DA, Stevenson G, Brown PK, Haase A, Reeves PR. Molecular Microbiology. 1993;7:725–34. doi: 10.1111/j.1365-2958.1993.tb01163.x. [DOI] [PubMed] [Google Scholar]
- 304.Morona R, Van Den Bosch L, Manning PA. Journal of Bacteriology. 1995;177:1059–68. doi: 10.1128/jb.177.4.1059-1068.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Stevenson G, Kessler A, Reeves PR. FEMS Microbiology Letters. 1995;125:23–30. doi: 10.1111/j.1574-6968.1995.tb07330.x. [DOI] [PubMed] [Google Scholar]
- 306.Daniels C, Morona R. Molecular Microbiology. 1999;34:181–94. doi: 10.1046/j.1365-2958.1999.01591.x. [DOI] [PubMed] [Google Scholar]
- 307.Klee SR, Tzschaschel BD, Timmis KN, Guzman CA. Journal of Bacteriology. 1997;179:2421–5. doi: 10.1128/jb.179.7.2421-2425.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Franco AV, Liu D, Reeves PR. Journal of Bacteriology. 1998;180:2670–5. doi: 10.1128/jb.180.10.2670-2675.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Rick PD, Hubbard GL, Barr K. Journal of Bacteriology. 1994;176:2877–84. doi: 10.1128/jb.176.10.2877-2884.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Kido N, Torgov VI, Sugiyama T, Uchiya K, Sugihara H, et al. Journal of Bacteriology. 1995;177:2178–87. doi: 10.1128/jb.177.8.2178-2187.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Clarke BR, Bronner D, Keenleyside WJ, Severn WB, Richards JC, Whitfield C. Journal of Bacteriology. 1995;177:5411–8. doi: 10.1128/jb.177.19.5411-5418.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Guan S, Clarke AJ, Whitfield C. Journal of Bacteriology. 2001;183 in press. [Google Scholar]
- 313.Weisgerber C, Jann K. European Journal of Biochemistry. 1982;127:165–8. doi: 10.1111/j.1432-1033.1982.tb06851.x. [DOI] [PubMed] [Google Scholar]
- 314.Kido N, Kobayashi H. Journal of Bacteriology. 2000;182:2567–73. doi: 10.1128/jb.182.9.2567-2573.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Kido N, Sugiyama T, Yokochi T, Kobayashi H, Okawa Y. Molecular Microbiology. 1998;27:1213–21. doi: 10.1046/j.1365-2958.1998.00765.x. [DOI] [PubMed] [Google Scholar]
- 316.Zhang L, Al-Hendy A, Toivanen P, Skurnik M. Molecular Microbiology. 1993;9:309–21. doi: 10.1111/j.1365-2958.1993.tb01692.x. [DOI] [PubMed] [Google Scholar]
- 317.Bronner D, Clarke BR, Whitfield C. Molecular Microbiology. 1994;14:505–19. doi: 10.1111/j.1365-2958.1994.tb02185.x. [DOI] [PubMed] [Google Scholar]
- 318.Rocchetta HL, Lam JS. Journal of Bacteriology. 1997;179:4713–24. doi: 10.1128/jb.179.15.4713-4724.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Reizer J, Reizer A, Saier MHJ. Protein Science. 1992;1:1326–32. doi: 10.1002/pro.5560011012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Bliss JM, Silver RP. Molecular Microbiology. 1996;21:221–31. doi: 10.1046/j.1365-2958.1996.6461357.x. [DOI] [PubMed] [Google Scholar]
- 321.Manting EH, Driessen AJ. Molecular Microbiology. 2000;37:226–38. doi: 10.1046/j.1365-2958.2000.01980.x. [DOI] [PubMed] [Google Scholar]
- 322.Chu S, Noonan B, Cavaignac S, Trust TJ. Proceedings of the National Academy of Sciences, USA. 1995;92:5754–8. doi: 10.1073/pnas.92.12.5754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Jansson P-E, Lönngren J, Widmalm G. Carbohydrate Research. 1985;145:59–66. doi: 10.1016/s0008-6215(00)90412-9. [DOI] [PubMed] [Google Scholar]
- 324.Ito T, Higuchi T, Hirobe M, Hiramatsu K, Yokota T. Carbohydrate Research. 1994;256:113–28. doi: 10.1016/0008-6215(94)84231-0. [DOI] [PubMed] [Google Scholar]
- 325.Stroeher UH, Karageorgos LE, Morona R, Manning PA. Proceedings of the National Academy of Sciences, USA. 1992;89:2566–70. doi: 10.1073/pnas.89.7.2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Preston A, Allen AG, Cadisch J, Thomas R, Stevens K, et al. Infection and Immunity. 1999;67:3763–7. doi: 10.1128/iai.67.8.3763-3767.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Vinogradov E, Peppler MS, Perry MB. European Journal of Biochemistry. 2000;267:7230–7. doi: 10.1046/j.1432-1327.2000.01835.x. [DOI] [PubMed] [Google Scholar]
- 328.Deangelis PL. Cellular and Molecular Life Sciences. 1999;56:670–82. doi: 10.1007/s000180050461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Forsee WT, Cartee RT, Yother J. Journal of Biological Chemistry. 2000;275:25972–8. doi: 10.1074/jbc.M002613200. [DOI] [PubMed] [Google Scholar]
- 330.Cartee RT, Forsee WT, Schutzbach JS, Yother J. Journal of Biological Chemistry. 2000;275:3907–14. doi: 10.1074/jbc.275.6.3907. [DOI] [PubMed] [Google Scholar]
- 331.DeAngelis PL. Journal of Biological Chemistry. 1999;274:26557–62. doi: 10.1074/jbc.274.37.26557. [DOI] [PubMed] [Google Scholar]
- 332.Keenleyside WJ, Whitfield C. Journal of Biological Chemistry. 1996;271:28581–92. doi: 10.1074/jbc.271.45.28581. [DOI] [PubMed] [Google Scholar]
- 333.Keenleyside WJ, Whitfield C. Journal of Bacteriology. 1995;177:5247–53. doi: 10.1128/jb.177.18.5247-5253.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Keenleyside WJ, Clarke AJ, Whitfield C. Journal of Bacteriology. 2000;182 doi: 10.1128/JB.183.1.77-85.2001. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Heldermon C, DeAngelis PL, Weigel PH. Journal of Biological Chemistry. 2001;276:2037–46. doi: 10.1074/jbc.M002276200. [DOI] [PubMed] [Google Scholar]
- 336.Breton C, Imberty A. Current Opinions in Structural Biology. 1999;9:563–71. doi: 10.1016/s0959-440x(99)00006-8. [DOI] [PubMed] [Google Scholar]
- 337.Slauch JM, Lee AA, Mahan MJ, Mekalanos JJ. Journal of Bacteriology. 1996;178:5904–9. doi: 10.1128/jb.178.20.5904-5909.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Verma NK, Brandt JM, Verma DJ, Lindberg AA. Molecular Microbiology. 1991;5:71–5. doi: 10.1111/j.1365-2958.1991.tb01827.x. [DOI] [PubMed] [Google Scholar]
- 339.Newton GJ, Daniels C, Burrows LL, Kropinski AM, Clarke AJ, Lam JS. Molecular Microbiology. 2001;39:1237–47. doi: 10.1111/j.1365-2958.2001.02311.x. [DOI] [PubMed] [Google Scholar]
- 340.Clarke AJ, Strating H, Blackburn NT. In: Glycomicrobiology. Doyle RJ, editor. Kluwer Academic/ Plenum Publishers; New York, NY: 2000. p. 187224. [Google Scholar]
- 341.Takeshita M, Makela PH. Journal of Biological Chemistry. 1971;246:3920–7. [PubMed] [Google Scholar]
- 342.Nikaido K, Nikaido H. Journal of Biological Chemistry. 1971;246:3912–9. [PubMed] [Google Scholar]
- 343.Nikaido H, Nikaido K, Nakae T, Makela PH. Journal of Biological Chemistry. 1971;246:3902–11. [PubMed] [Google Scholar]
- 344.Wright A. Journal of Bacteriology. 1971;105:927–36. doi: 10.1128/jb.105.3.927-936.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Shibaev VN, Druzhinina TN, Popova AN, Rozhnova SS, Kilesso VA. European Journal of Biochemistry. 1979;101:309–16. doi: 10.1111/j.1432-1033.1979.tb04244.x. [DOI] [PubMed] [Google Scholar]
- 346.Allison GE, Verma NK. Trends in Microbiology. 2000;8:17–23. doi: 10.1016/s0966-842x(99)01646-7. [DOI] [PubMed] [Google Scholar]
- 347.Guan S, Bastin DA, Verma NK. Microbiology. 1999;145:1263–73. doi: 10.1099/13500872-145-5-1263. [DOI] [PubMed] [Google Scholar]
- 348.Mavris M, Manning PA, Morona R. Molecular Microbiology. 1997;26:939–50. doi: 10.1046/j.1365-2958.1997.6301997.x. [DOI] [PubMed] [Google Scholar]
- 349.Losick R, Robbins PW. Journal of Molecular Biology. 1967;30:445–55. doi: 10.1016/0022-2836(67)90361-0. [DOI] [PubMed] [Google Scholar]
- 350.Losick R. Journal of Molecular Biology. 1969;42:237–46. doi: 10.1016/0022-2836(69)90040-0. [DOI] [PubMed] [Google Scholar]
- 351.Whitfield C, Richards JC, Perry MB, Clarke BR, MacLean LL. Journal of Bacteriology. 1991;173:1420–31. doi: 10.1128/jb.173.4.1420-1431.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Whitfield C, Perry MB, MacLean LL, Yu S-H. Journal of Bacteriology. 1992;174:4913–9. doi: 10.1128/jb.174.15.4913-4919.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Kelly J, Masoud H, Perry MB, Richards JC, Thibault P. Analytical Biochemistry. 1996;233:15–30. doi: 10.1006/abio.1996.0002. [DOI] [PubMed] [Google Scholar]
- 354.Kelly RF, MacLean LL, Perry MB, Whitfield C. Journal of Endotoxin Research. 1995;2:131–40. [Google Scholar]
- 355.Kelly RF, Severn WB, Richards JC, Perry MB, MacLean LL, et al. Molecular Microbiology. 1993;10:615–25. doi: 10.1111/j.1365-2958.1993.tb00933.x. [DOI] [PubMed] [Google Scholar]
- 356.MacLean LL, Whitfield C, Perry MB. Carbohydrate Research. 1993;239:325–8. doi: 10.1016/0008-6215(93)84231-t. [DOI] [PubMed] [Google Scholar]
- 357.Kol O, Wieruszeski J-M, Strecker G, Fournet B, Zalisz R, Smets P. Carbohydrate Research. 1992;236:339–44. doi: 10.1016/0008-6215(92)85028-x. [DOI] [PubMed] [Google Scholar]
- 358.Clarke BR, Whitfield C. Journal of Bacteriology. 1992;174:4614–21. doi: 10.1128/jb.174.14.4614-4621.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Kelly RF, Whitfield C. Journal of Bacteriology. 1996;178:5205–14. doi: 10.1128/jb.178.17.5205-5214.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Osborn MJ, Rick PD, Rasmussen NS. Journal of Biological Chemistry. 1980;255:4246–51. [PubMed] [Google Scholar]
- 361.Mühlradt PF, Menzel J, Golecki JR, Speth V. European Journal of Biochemistry. 1973;35:471–81. doi: 10.1111/j.1432-1033.1973.tb02861.x. [DOI] [PubMed] [Google Scholar]
- 362.Kulpa CF, Jr., Leive L. Journal of Bacteriology. 1976;126:467–77. doi: 10.1128/jb.126.1.467-477.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Kellenberger E. Molecular Microbiology. 1990;4:697–705. doi: 10.1111/j.1365-2958.1990.tb00640.x. [DOI] [PubMed] [Google Scholar]
- 364.Bayer ME. Journal of Structural Biology. 1991;107:268–80. doi: 10.1016/1047-8477(91)90052-x. [DOI] [PubMed] [Google Scholar]
- 365.Sandkvist M. Mol. Microbiol. 2001;40:271–83. doi: 10.1046/j.1365-2958.2001.02403.x. [DOI] [PubMed] [Google Scholar]
- 366.Gaspar JA, Thomas JA, Marolda CL, Valvano MA. Molecular Microbiology. 2000;38:262–75. doi: 10.1046/j.1365-2958.2000.02094.x. [DOI] [PubMed] [Google Scholar]
- 367.Blattner FR, Plunkett G, Bloch CA, Perna NT, Burland V, et al. Science. 1997;277:1453–74. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 368.Beutler B. Curr. Opin. Microbiol. 2000;3:23–8. doi: 10.1016/s1369-5274(99)00046-6. [DOI] [PubMed] [Google Scholar]
- 369.Christ WJ, Asano O, Robidoux AL, Perez M, Wang Y, et al. Science. 1995;268:80–3. doi: 10.1126/science.7701344. [DOI] [PubMed] [Google Scholar]
- 370.Zhou Z, Ribeiro AA, Raetz CRH. J. Biol. Chem. 2000;275:13542–51. doi: 10.1074/jbc.275.18.13542. [DOI] [PubMed] [Google Scholar]
- 371.Baker SJ, Gunn JS, Morona R. Microbiology. 1999;145:367–78. doi: 10.1099/13500872-145-2-367. [DOI] [PubMed] [Google Scholar]
- 372.Vinogradov EV, van der Drift K, Thomas-Oates JE, Meshkov S, Brade H, Holst O. European Journal of Biochemistry. 1999;261 doi: 10.1046/j.1432-1327.1999.00280.x. [DOI] [PubMed] [Google Scholar]
- 373.Susskind M, Brade L, Brade H, Holst O. Journal of Biological Chemistry. 1998;273:7006–17. doi: 10.1074/jbc.273.12.7006. [DOI] [PubMed] [Google Scholar]
- 374.Wakarchuk WW, Gilbert M, Martin A, Wu Y, Brisson JR, et al. European Journal of Biochemistry. 1998;254:626–33. doi: 10.1046/j.1432-1327.1998.2540626.x. [DOI] [PubMed] [Google Scholar]