

Abstract
As we age, the majority of our cells gradually lose the capacity to divide because of replicative senescence that results from the inability to replicate the ends of chromosomes. The timing of senescence is dependent on the length of telomeric DNA, which elicits a checkpoint signal when critically short. Critically short telomeres also become vulnerable to deleterious rearrangements, end degradation and telomere-telomere fusions. Here we report a novel role of non-homologous end joining (NHEJ), a pathway of double-strand break (DSB) repair in influencing both the kinetics of replicative senescence and the rate of chromosome loss in telomerase-deficient Saccharomyces cerevisiae. In telomerase-deficient cells, the absence of NHEJ delays replicative senescence, decreases loss of viability during senescence, and suppresses senescence-associated chromosome loss and telomere-telomere fusion. Differences in mating-type gene expression in haploid and diploid cells affect NHEJ function, resulting in distinct kinetics of replicative senescence. These results suggest that the differences in the kinetics of replicative senescence in haploid and diploid telomerase-deficient yeast is determined by changes in NHEJ-dependent telomere fusion, perhaps through the initiation of the breakage-fusion-bridge (BFB) cycle.
Introduction
Telomeres are the structures located at the ends of eukaryotic chromosomes that protect them from end-to end fusions and translocations (Muller, 1938, McClintock, 1941). In budding yeast, loss of even a single telomere can cause cell-cycle arrest followed by either chromosome loss or acquisition of a new telomere, which is usually associated with a loss of heterozygosity (LOH) (Zakian, 1996). Telomeres are established and maintained by a variety of cellular factors including telomerase, a ribonucleoprotein complex required for the replication and protection of telomeric DNA (Greider and Blackburn, 1985; Blackburn, 2005). Consequently, the absence of telomerase leads to a progressive shortening of chromosomes and to a loss of DNA end protection, or capping (Chen and Kolodner, 1999; DuBois et al., 2002). Ultimately, telomerase-deficient cells undergo replicative senescence, a process in which the majority of cells in the culture cease to grow, and a variety of deleterious genome destabilizing events increase in frequency (Chen and Kolodner, 1999; Hackett et al., 2001; DuBois et al., 2002; Hackett and Greider, 2003; Mieczkowski et al., 2003; Meyer and Bailis, 2007). However, a small fraction of the cells escape senescence by using homologous recombination (HR) to reestablish and maintain telomeres (Lundblad and Blackburn, 1993).
In S. cerevisiae, HR is primarily controlled by genes in the RAD52 epistasis group (McKee and Lawrence, 1980). However, genes that specify factors involved in DNA replication, cell cycle control and a variety of DNA repair pathways other than HR are also known to affect HR (Paques and Haber, 1999; Symington, 2002). Among these is the mating type locus, MAT (Friis and Roman, 1968; Heude and Fabre, 1993; Fasullo and Dave, 1994; Lowell et al., 2003). Haploid and diploid strains expressing both MATa and MATα display greater resistance to a variety of DNA damaging agents than haploids or diploids expressing only MATa or MATα, and this resistance is mediated by the RAD52 epistasis group members RAD51, RAD52 and RAD54 (Saeki et al., 1980). Recent work has shown that the MAT locus exerts this control through a complex network of genes that have distinct interactions with the various components of HR (Valencia-Burton et al., 2006).
MAT has also been shown to influence the response to uncapped chromosome ends in telomerase-deficient cells. Simultaneous expression of MATa and MATα in telomerase-defective est1Δ or tlc1Δ mutant haploids was found to suppress the loss of viability normally associated with replicative senescence in these cells (Lowell et al., 2003). Further, work done with MATa/MATα heterozygous diploids that lack telomerase showed that they undergo normal replicative senescence, but that telomerase-deficient MATa/MATa or MATα/MATα homozygous diploids display accelerated senescence. Although this work strongly suggests that mating type influences replicative senescence, the precise nature of this effect remains unclear.
The MAT locus is known to directly control the expression of an array of genes important for specifying cell type and cell-cell interaction (Strathern et al., 1981). Haploid MATa or MATα cells express haploid-specific genes, while MATa/MATα heterozygous diploids express the Mata1-Mata2 corepressor that represses expression of haploid-specific genes and derepresses the expression of diploid-specific genes. A number of both haploid- and diploid-specific genes have been discovered through microarray analysis (Galitski et al., 1999; Valencia et al., 2001), including the haploid-specific gene, NEJ1, which is required for the function of DNA ligase IV, and limits the action of nonhomologous end-joining (NHEJ) to haploid cells (Kegel et al., 2001; Valencia et al., 2001).
NHEJ has been shown to cause telomere-telomere fusions in mammals, plants and yeast cells that have lost telomere capping (Espejel et al., 2002; Chan & Blackburn, 2003; Mieczkowski et al., 2003; Carter et al., 2007; Heacock et al., 2007). Telomere fusions create dicentric chromosomes that feed into the BFB cycle (McClintock, 1941), which results in serial genome instability that can, in turn lead to tumorigenesis and loss of cell viability (Maser et al., 2002; Feldser et al., 2003; DeLange, 2005; Johnson and Broccoli, 2007). The different characteristics of replicative senescence in telomerase-defective haploids and diploids may, therefore, be related to the differential capacity of these cells to generate telomere fusions by NHEJ when the chromosome ends are uncapped.
In this manuscript we demonstrate that the loss of viability observed in telomerase-deficient haploids and diploids is less severe when both MATa and MATα are expressed simultaneously than when only a single MAT allele is expressed. The increased inviability associated with the expression of only a single MAT allele was suppressed by a mutation in DNL4, a gene encoding a subunit of DNA ligase IV, which suggests that the loss of viability is a result of NHEJ. Importantly, the dnl4 mutation also suppressed elevated levels of chromosome loss and telomere fusion associated with mono-allelic expression of MAT, suggesting that the loss of viability associated with telomerase deficiency may, in part, be due to the loss, or rearrangement of chromosomes that follow NHEJ-dependent telomere fusion.
Results
The influence of MAT on the kinetics of replicative senescence is dependent on DNL4
We initially examined the influence of mating type on replicative senescence by determining the effects of expressing either MATa alone, or both MATa and MATα in est2 mutant haploid strains. In est2 haploids that expressed MATa alone viability was reduced to 0.9% of wild type viability at 40 generations, while strains expressing both MATa and MATα displayed a minimum viability of 33% at 60 generations (Figure 1A). These results indicate that mating type influences both the severity and the rate of onset of replicative senescence, similar to previously published reports (Lowell et al., 2003). Experiments with est2/est2 homozygous diploid strains revealed substantially similar results, as est2 haploid strains expressing MATa alone displayed a minimum viability of 4.5% at 40 generations, while strains expressing both MATa and MATα displayed a minimum viability of 17% at 60 generations (Figure 1B). These results suggest that mating type exerts relatively equivalent effects on both the rate and severity of replicative senescence in haploids and diploids, while ploidy has relatively minor effects.
Figure 1.
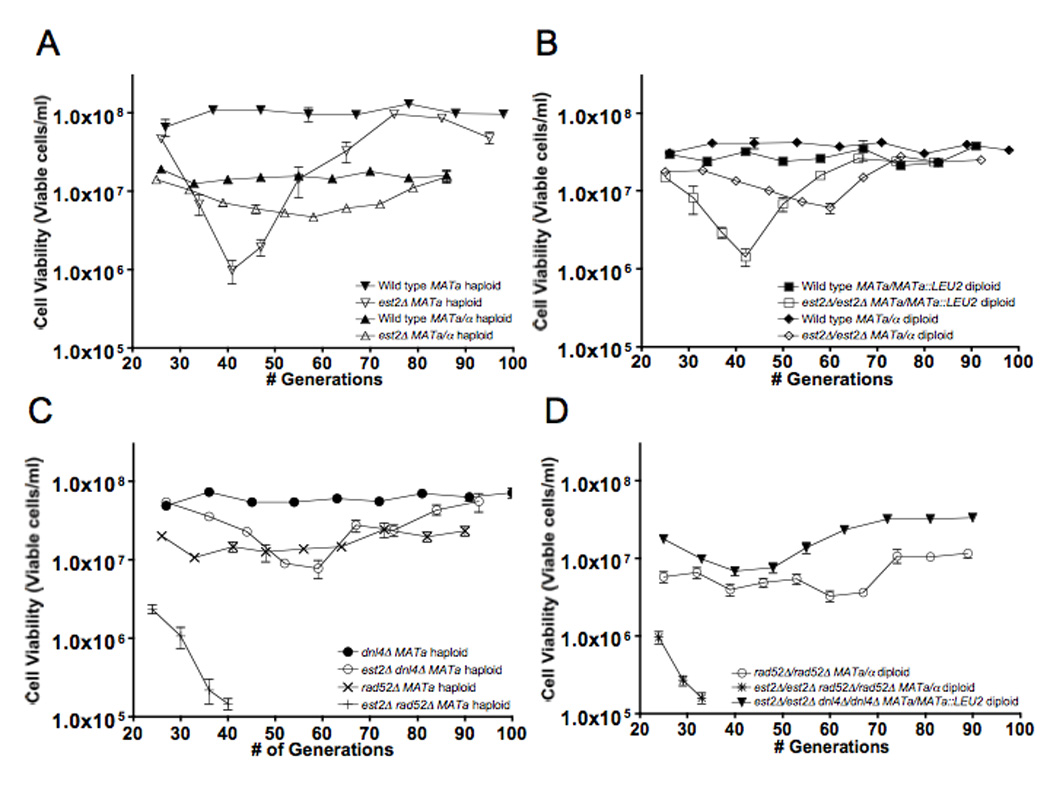
Viability of telomerase deficient cells in serial liquid culture is influenced by NHEJ. Serial liquid growth was performed as described previously (Meyer and Bailis 2007). After each day of serial liquid growth, hemocytometer counts were performed to determine the number of cell bodies, after which 500 cells were plated to YPD medium and incubated at 30° for 3 days. The resulting colonies were counted and the number divided by 500 to determine plating efficiency. Viability was determined by multiplying the number of cell bodies plated by the plating efficiency. Each data point represents the mean viability ±2 SE determined from at least eight independent cultures. The trough of cell viability appearing at 40 or 60 generations corresponds to the point of maximum cell death prior to recovery by HR-dependent telomere recovery. (A) (▲) Wild type MATa/α haploid, (△) est2Δ MATa/α haploid, (▼) Wild type MATa haploid, (▽) est2Δ MATa haploid. (B) (■) Wild type MATa/MATa::LEU2 diploid, (□) est2Δ/est2Δ MATa/MATa::LEU2 diploid, (◆) est2Δ/est2Δ MATa/α diploid, (◇)Wild type MATa/α diploid. (C) (●) dnl4Δ MATa haploid, (○) est2Δ dnl4Δ MATa haploid, (×) rad52Δ MATa haploid, (+) est2Δ rad52Δ MATa haploid. (D) (✱) est2Δ /est2Δ rad52Δ /rad52Δ MATa/α diploid, (○) rad52Δ /rad52Δ MATa/α diploid, (▼) est2Δ/est2Δ dnl4Δ/dnl4Δ MATa/MATa::LEU2 diploid.
The MAT locus has been shown to control the expression of a large network of genes (Strathern et al., 1981; Galitski et al., 1999; Valencia et al., 2001) making it likely that mating type-dependent expression of one or more genes may influence the kinetics of replicative senescence. One of these is the haploid-specific gene, NEJ1, which is necessary for the function of DNA ligase IV (Kegel et al., 2001; Valencia et al., 2001). MATa/MATα heterozygous diploids suppress the expression of NEJ1, severely reducing the efficiency of DNA ligase IV and blocking NHEJ. This suggests that MAT could be influencing the kinetics of replicative senescence in telomerase deficient cells through its effect on NHEJ. We addressed this possibility by examining the effect of a mutation in the DNL4 gene, which encodes a subunit of DNA ligase IV (Wilson et al., 1997), on the serial growth characteristics of est2 mutant haploid strains. Our results show that the dnl4 allele both delayed and reduced the severity of replicative senescence in est2 dnl4 double mutant haploids, as they reached a minimum viability of 14% at 60 generations (Figure 1C), similar to the effects of expressing both MATa and MATα in est2 haploid strains (Figure 1A). Consistent with these results, we also found that the dnl4 allele reduced the severity of replicative senescence in est2/est2 dn4/dnl4 double homozygotes that expressed MATa alone, a minimum viability of 24% at 40 generations (Figure 1D), which is similar to the minimum viability observed in the est2/est2 homozgotes that expressed both MATa and MATα (Figure 1B). These results are consistent with MAT influencing replicative senescence through its control of NHEJ.
In contrast to disabling NHEJ, mutating the central yeast HR gene, RAD52 (Resnick and Martin, 1976), significantly accelerated replicative senescence in both est2 rad52 double mutant haploid (Figure1C) and est2/est2 rad52/rad52 doubly homozygous diploid (Figure 1D) strains. As expected, neither set of strains survived replicative senescence, presumably due to their inability to activate the HR-dependent telomere recovery pathway (Le et al., 1999). Taken together these results suggest that NHEJ both accelerates and increases the severity of replicative senescence in telomerase-deficient cells, while HR delays senescence and facilitates recovery from it.
NHEJ influences chromosome loss in the absence of telomerase
The absence of telomerase function has been shown to lead to a senescence-associated increase in genome instability in both haploid and diploid strains (Chen and Kolodner, 1999; Hackett et al., 2001; DuBois et al., 2002; Hackett and Greider, 2003; Mieczkowski et al., 2003; Meyer and Bailis, 2007). We have shown previously that senescent est2 mutant haploids display increased rates of mutation and gross chromosomal rearrangement (GCR) at the CAN1 locus which lies near the left end of chromosome V (Meyer and Bailis, 2007). Since both chromosome loss (CL) and interhomolog recombination (IHR) have previously been shown to be stimulated during senescence in est2/est2 homozygous diploids (Hacket and Greider, 2003; Ijpma and Greider, 2003), we sought to determine the rates of these events in our strains using the CAN1 locus and chromosome V. We found that the rate of CL increased almost 7.5-fold and the rate of IHR increased 13-fold during replicative senescence in est2/est2 homozygotes, but that the rates of these events were not significantly different from wild type before and after replicative senescence (Table 1). In contrast, the rate of IHR at the CYH2 locus that lies near the centromere on the left arm of chromosome VII was not significantly different from wild type before, during or after senescence in the est2/est2 homozygotes. These results confirm that replicative senescence in telomerase deficient diploids results in levels of instability that are similar to those observed in haploids, and that this instability is limited to telomere proximal loci.
TABLE 1.
Analysis of rates of interhomolog recombination (IHR) and chromsome loss (CL) in wild type and mutant diploid strains in serial liquid culture a
| Pre-senescence b | Senescence c | Post-senescence d | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Genotype | IHR (C1) e | IHR (C2) f | CLg | IHR (C1) | IHR (C2) | CL | IHR (C1) | IHR (C2) | CL |
| Wild type | 4.8×10−5(1) h | 9.7×10−5(1) | 3.0×10−5(1) | 9.2×10−5(1) | 2.0×10−4(1) | 1.6×10−5 (1) | 2.6×10−4(1) | 2.4×10−4(1) | 4.2×10−5(1) |
| [3.7–5.8] i | [6.7–13.0] | [1.7–3.6] | [8.9–10.0] | [1.0–3.2] | [1.1–3.0] | [1.7–4.7] | [1.7–2.8] | [2.7–4.7] | |
| est2/est2 | 5.3×10−5(1.1) | 1.2×10−4(1.2) | 3.0×10−5(1) | 1.2×10−3(13) | 2.4×10−4(1.2) | 1.2×10−4(7.5) | 4.0×10−4(1.5) | 2.4×10−4(1) | 2.6×10−5(0.6) |
| [3.5–1.2] | [0.8–1.6] | [1.7–6.4] | [0.6–3.2] | [1.6–4.5] | [0.9–1.8] | [2.3–5.1] | [1.7–3.2] | [1.8–4.5] | |
| rad52/rad52 | 4.0×10−5(0.8) | - | 6.0×10−4 (20) | 5.0×10−5(0.5) | - | 7.2×10−4 (45) | 2.7×10−5(0.1) | - | 9.0×10−4(21) |
| [2.0–4.3] | [3.0–11.0] | [2.8–9.4] | [4.6–7.8] | [1.6–8.1] | [4.2–10.0] | ||||
| est2/est2, rad52/rad52j | 3.6×10−6(0.07) | - | 5.3×10−5(1.8) | 3.0×10−6(0.03) | - | 7.7×10−5(5) | NRk | - | NR |
| MATa/- | 7.4×10−5(1.5) | - | 4.3×10−5(1.4) | 7.8×10−5(0.8) | - | 4.7×10−5(3.0) | 8.8×10−5(0.3) | - | 5.3×10−5(1.3) |
| [3.7–9.6] | [3.1–5.0] | [5.0–10.0] | [2.9–6.9] | [6.6–19.0] | [4.2–9.9] | ||||
| est2/est2 MATa/- | 1.2×10−4(2.5) | - | 9.6×10−5(3.2) | 8.3×10−4(9) | - | 5.2×10−4(33) | 1.9×10−4 (0.7) | - | 5.6×10−5(1.3) |
| [3.7–9.6] | [6.0–21.0] | [6.6–23.0] | [2.8–9.3] | [1.5–4.4] | [4.0–6.8] | ||||
| est2/est2, MATa/-, dnl4/dn14 | 1.9×10−4(3.9) | - | 2.5×10−5(0.8) | 1.7×10−3 (18) | - | 1.3×10−4(7.8) | 1.6×10−4(0.6) | - | 2.1×10−5 (0.5) |
| [0.7–3.0] | [1.6–3.0] | [0.5–2.9] | [0.8–1.6] | [1.3–2.5] | [1.2–2.4] | ||||
Wildtype and mutant diploids transformed with a wild-type EST2 gene on a URA3 selectable plasmid were grown to single colonies on YPD medium and then replica plated to medium lacking uracil to select for those that had evicted the plasmid. Freshly isolated Ura− colonies were dispersed in water, cell number assessed by hemacytometer, viability determined by plating on to YPD medium, and interhomolog recombination and chromosome loss determined by plating to either synthetic medium lacking arginine and containing canavanine or complete synthetic medium containing cycloheximide. Canr colonies were replica plated to synthetic medium lacking threonine. Serial liquid cultures were started by inoculating five ml YPD cultures with 5×105 cells from the freshly suspended Ura− colonies, and grown for 20 hours at 30°C. Each successive five ml culture was inoculated with 5×105 cells and processed as above. Diploids designated as MATa/- possess one intact MATa allele and a MATa allele that has been disrupted with a LEU2 marker. All other strains possess intact MATa and MATα alleles.
The pre-senescence timepoint corresponds to approximately 25 generations.
The number of generations at senescence was between 40 and 60 generations for est2/est2 mutants with the exception of the est2/est2 rad52/rad52 double mutant that reached senescence at approximately 32 generations (see Figure 1).
The post-senescence timepoint corresponds to approximately 100 generations.
Rates of interhomolog recombination (IHR) at the CAN1 locus were determined by the method of the median from a minimum of nine independent cultures for each strain and at each timepoint.
Rates of interhomolog recombination at the CYH2 locus were determined by the method of the median from a minimum of nine independent cultures for each strain and at each timepoint.
Rates of the loss of chromosome V were determined by the method of the median from a minimum of nine independent cultures for each strain and at each timepoint.
Numbers in parentheses are the fold differences of the rates of IHR in each strain and at each stage in the growth of the culture from the rate in the wild type strain at the equivalent stage.
Numbers in brackets are the 95% confidence intervals and were determined using a table: http://www.math.unb.ca/~knight/utility/MedInt95.htm.
Rates determined using fluctuation analysis.
NR = Not recovered.
Our observation that MAT influences replicative senescence through its effect on NHEJ led us to investigate how it might affect the increased genome instability that occurs concomitantly with replicative senescence. We found that est2/est2 homozygotes expressing only MATa, which displayed more rapid and severe replicative senescence than est2/est2 homozygotes expressing both MATa and MATα (Figure 1B), also displayed a 4.4-fold increased rate of senescence-associated CL, but a rate of IHR that was not statistically different (Table 1). The stimulation of CL was found to be completely dependent on NHEJ as the CL rate in senescent est2/est2 dnl4/dnl4 double homozygotes expressing only MATa was essentially identical to that in the senescent est2/est2 homozygotes expressing both MATa and MATα. In contrast, loss of HR had only a minimal effect on senescence-associated CL but a substantial effect on IHR as the rate of CL in senescent est2/est2 rad52/rad52 double homozygotes was very similar to that in senescent est2/est2 homozygotes but the rate of IHR was reduced 400-fold. These results suggest that senescence-associated CL is NHEJ-dependent but HR-independent, while senescence-associated IHR is NHEJ-independent but HR-dependent.
Replicative senescence stimulates NHEJ-dependent telomere fusion
Telomere fusion has been associated with the uncapping of telomeres in a broad spectrum of eukaryotic cells, including yeast (Espejel et al., 2002; Chan & Blackburn, 2003; Mieczkowski et al., 2003; Carter et al., 2007; Heacock et al., 2007). This process has previously been shown to be dependent upon the NHEJ apparatus, which is thought to recognize uncapped telomeres as substrates for DSB repair (Riha et al., 2006). Given that telomere fusion could affect cell viability by initiating the BFB cycle (McClintock, 1941) we investigated whether such events were occurring during replicative senescence in our strains, and whether they were under the control of NHEJ. Genomic DNA extracted from serially grown cultures of wild type, est2 single, and est2 dnl4 double mutant haploid strains was subjected to PCR using a single primer complementary to the YRF1 sequence that is present in the same orientation at seven telomeres in the yeast genome (SGD). In the event of a telomere fusion involving any two of these telomeres, the single primer would amplify a sequence that spans the distance between the complementary sequences that would then be oriented toward each other. We found that PCR signals appeared when using genomic DNA extracted from cells from the third day of serial growth of the est2 single mutants (Figure 2), concomitant with replicative senescence (Figure 1A), but not before or after. We also failed to obtain PCR signals using genomic DNA obtained from wild type, or est2 dnl4 double mutant cells at any point during serial liquid growth. Cloning and sequencing of the PCR bands verified that they represent genuine telomere fusions (Figure S1). The DNA sequences at the junctions suggested that little if any homology between the parent molecules was utilized to make the fusions, consistent with the involvement of NHEJ. In addition, the majority of the telomere fusions that were isolated and characterized were between YRF1 and unidentified sequences that may be heterogeneous subtelomeric DNA (Mefford and Trask, 2002). These data confirm that telomere fusion accompanies replicative senescence in telomerase-deficient cells, and suggest that these events are dependent upon NHEJ.
Figure 2.
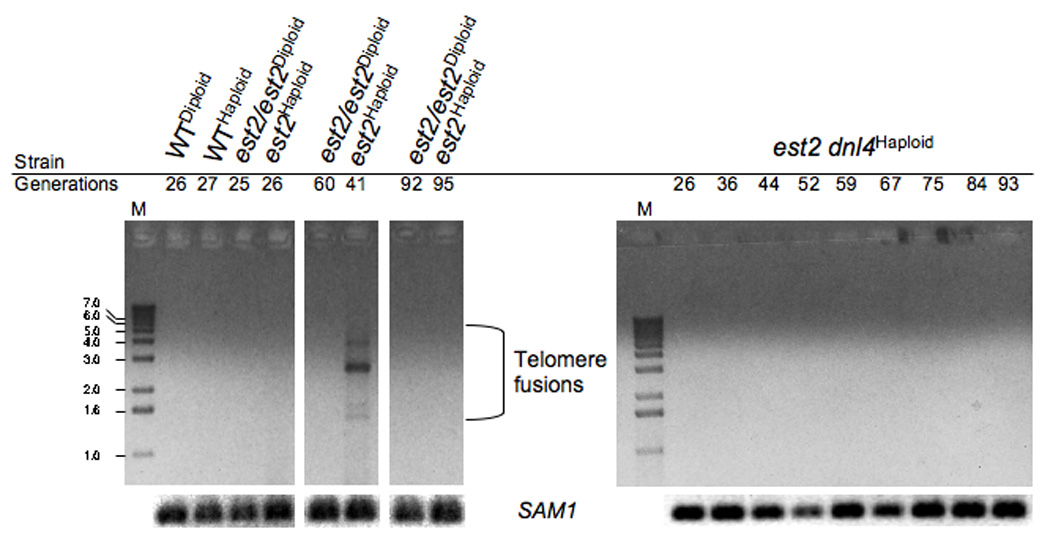
Telomere fusion in telomerase deficient cells is dependent on Dnl4. Cells from cultures with the indicated genotypes and collected after the indicated number of generations in serial liquid culture, were used to prepare genomic DNA. PCR was performed with 30 ng of the DNA samples and the products run on 0.8% agarose gels along with 1 kb markers (M) before visualization by staining with ethidium bromide. Potential telomere fusions were detected using a single primer complementary to a sequence located in the YRF1 open reading frame that lies adjacent to several telomeres. PCR signals generated using primers complementary to sequences in the SAM1 gene were used as a positive control.
Discussion
The results described here confirm that mating type influences the rate of onset and the severity of replicative senescence in telomerase-deficient yeast cells (Lowell et al., 2003), but further establishes that this control is through the effect of MAT on NHEJ. Additionally, this analysis suggests that the increased loss of viability during senescence that is associated with mono-allelic expression of MAT is the result of NHEJ-dependent telomere fusion that may also fuel senescence-associated CL. However, we cannot rule out the possibility that other MAT regulated genes also influence replicative senescence in the absence of telomerase. The absence of telomerase has been shown to lead to telomere dysfunction and genome instability in a variety of eukaryotic systems (Espejel et al., 2002; Smogorzewska et al., 2002; Chan and Blackburn, 2003; Hackett and Greider, 2003; Mieczkowski et al., 2003; Carter et al., 2007; Heacock et al., 2007). Interestingly, NHEJ seems to play a prominent role in promoting genome instability through fusions of dysfunctional telomeres in higher eukaryotes (Riha et al., 2006), perhaps by initiating the BFB cycle first proposed by Barbara McClintock (McClintock, 1941). The consequences of the BFB cycle are LOH, gene duplication, chromosome loss and non-reciprocal translocation. Emerging evidence is pointing increasingly toward telomere dysfunction as being one of the driving forces behind tumorigenesis in mammals (Maser et al., 2002; Feldser et al., 2003; DeLange, 2005; Johnson and Broccoli, 2007).
Although our results suggesting the importance of NHEJ in yeast replicative senescence are consistent with observations in other systems (Riha et al., 2006), a previous study in yeast suggested that NHEJ has no impact on replicative sencescence (Hackett et al., 2001). In their study, Hackett and colleagues observed no difference in the kinetics of replicative senescence in est1 single mutant and est1 lig4 double mutant haploid strains. One potential source of the discrepancy may be that the previous study monitored cell growth potential by determining cell density instead of cell viability, a more sensitive measure of proliferative capacity. Another potential issue is that the previous study used a switch of carbon source to initiate replicative senescence by inhibiting the expression of a catabolite-repressible EST1 gene. Since the authors found that these conditions allowed rad52 mutants to recover from replicative senescence it seems likely that breakthrough expression of EST1 reduced the stringency of the telomerase defect, potentially obscuring the effect of losing NHEJ. Finally, differences in the genetic backgrounds of the strains used in these studies may have contributed to the dissimilar results. Despite these differences, it is important to note that overall, the two studies report similar findings regarding the link between replicative senescence and genome instability, including that replicative senescence results in the appearance of telomere fusions.
Another important observation from the current analysis is that replicative senescence stimulates CL and IHR at telomere proximal loci. This confirms the results of other studies that have tied telomere dysfunction to increased LOH in diploid yeast (Hackett et al., 2001; Hackett et al., 2003). Interestingly, while senescence-associated CL is at least in part, Dnl4-dependent, sensescence-associated IHR is dependent on Rad52 (Table 1). This suggests that senescence-associated LOH is the product of both NHEJ and HR. It also suggests that CL and IHR may be mechanistically separate. We speculate that CL may be the result of the NHEJ-dependent fusion of uncapped telomeres and subsequent chromosomal non-disjunction, while IHR may be the result of the recombinational repair of replication lesions (Meyer and Bailis, 2007), or exonucleolytically digested chromosome ends (Hackett et al. 2001).
The observation that increases in the rates of both mutation and LOH at telomere proximal loci accompany replicative senescence in yeast suggests a role for these processes in the progression toward cancer in aging mammalian cells. Mutagenesis of tumor suppressor genes may increase as cells approach senescence if they lay near a telomere. But, since most of these mutations would be expected to confer a recessive loss of function, the mutated cells would remain phenotypically normal. However, if these mutations are subsequently either homozygosed by IHR, or rendered hemizygous by CL, a mutant phenotype would emerge. Senescence-associated loss of tumor suppression via sequential genetic change may help explain the precipitous increase in cancer observed in elderly humans (DePinho, 2000).
Materials and Methods
Yeast strains and plasmids
All strains used in this study are isogenic with W303-1A (Thomas and Rothstein 1989) but are wild type at the RAD5 locus. All strains were constructed using conventional methods (Burke et al., 2000). The est2::URA3 allele used in this study was the generous gift of Victoria Lundblad (Rizki and Lundblad, 2001). The est2::ura3::LEU2 allele was created by single-step gene disruption of the est2::URA3 allele (Rothstein, 1991) using a ura3::LEU2 construct from pLAY315. The MATa::LEU2 allele used in this analysis was introduced by single-step gene disruption using a DNA fragment derived from the plasmid pJH124 which was the kind gift of Jim Haber. The dnl4::LEU2 allele was constructed as previously described (Pannunzio et al., 2008). The hom3-10 and CAN1 alleles were derived from strain RKY2672 (Tishkoff et al., 1997), which was the generous gift of Richard Kolodner. The plasmid pLAY263 was constructed by cloning a 4.0 kb BamHI/ClaI fragment containing the wild type MATα sequence into the polylinker of the single-copy plasmid pRS413, which carries a HIS3 selectable marker (Sikorski and Hieter, 1989). The plasmid pSD196, a generous gift from Dan Gottschling, bears the wild type EST2 gene on a 4.5 kb SalI fragment cloned into the single-copy vector pRS316, which carries a URA3 selectable marker (Sikorski and Hieter, 1989).
Serial liquid growth
Serial liquid growth was performed as described previously (Meyer and Bailis, 2007). Mean cell densities were reported and were calculated from at least eight independent cultures of each indicated genotype. Error bars represent two standard errors. Selection for the plasmid containing MATα (pLAY263) and the control plasmid, pRS413 were maintained throughout the serial liquid growth analysis by growth in liquid synthetic medium lacking histidine. During each day of serial liquid growth, hemocytometer counts were performed to determine the number of cell bodies, after which approximately 500 cells were plated to YPD medium and incubated at 30°C for three days. Colonies were then counted and divided by 500 to determine plating efficiency. Finally, viability was determined following each day of growth in liquid culture by taking the number of cell bodies, and multiplying by the plating efficiency.
Determination of the rates of chromosome loss (CL) and interhomolog recombination (IHR)
The rates of interhomolog recombination (IHR) and chromosome loss were determined as described previously (Hartwell and Smith, 1985). Freshly dissected segregants containing copies of chromosome V with either, CAN1 and HOM3 or can1-100 and hom3-10 alleles were crossed to generate CAN1/can1-100, HOM3/hom3-10 doubly heterozygous diploid strains. Segregants containing CYH2 and cyh2r were also crossed to generate diploids heterozygous at the CYH2 locus on chromosome VII in order to facilitate a second, independent assay of IHR. At least five independent diploids of each genotype were prepared. Cultures were initiated from homozygous wild type and mutant diploid strains that contained pSD196, a single-copy plasmid containing a wild type allele of EST2 and a URA3 selectable marker. Seven fresh Ura+ diploid colonies of each genotype were dispersed in sterile water, plated onto YPD medium, and the resulting colonies lightly replica plated to medium lacking uracil to identify those that had lost pSD196. Colonies that had lost the plasmid were used to inoculate cultures containing five ml of YPD medium. These cultures were grown for 24 hours at 30°C and cell density determined by hemocytometer counts. Serial cultures were initiated by inoculating fresh five ml cultures with appropriate dilutions.
With the initial liquid cultures and with each successive culture appropriate dilutions were also plated to YPD medium to determine the total number of viable cells. Dilutions were also plated onto either synthetic medium lacking arginine and containing 60 µg/ml of canavanine to determine the number of canavanine resistant (Canr) colonies, or onto synthetic medium containing 1µg/ml of cycloheximide to determine the number of cycloheximide resistant (Cyhr) colonies. The total numbers of Canr or Cyhr colonies were counted after growth at 30°C for four days. Cyhr colonies were scored directly as IHR events. The Canr colonies were replica plated onto synthetic medium lacking threonine and incubated for two days at 30°C to score for the presence of the HOM3 marker and distinguish between CL and IHR events. Colonies that were Canr and Thr− were scored as CL events while the Canr and Thr+ colonies were scored as IHR events. While Canr Thr+ colonies may also represent mutation events it was previously found that these colonies result predominantly from mitotic recombination (Golin and Esposito, 1977). Rates and 95% confidence intervals for both IHR and CL were determined each day in liquid culture over 9 days from a minimum of 12 trials using the method of the median (Lea and Coulson, 1948) or fluctuation analysis (Luria and Delbruck, 1943).
Detecting telomere fusion by PCR
Genomic DNA was prepared from aliquots of cells from independent serial cultures by glass bead extraction. Thirty ng of genomic DNA were used to detect telomere fusions by PCR using a single primer, YRF (5’ –AAC GTT GAA GTT CTC GCT GC- 3’) that is complementary to a sequence located within the YRF1 open reading frame located adjacent to the end of the telomeres on the right arms of chromosomes IV, V, VII and XII, and the left arms of chromosomes XIV and XVI (SGD http://www.yeastgenome.org/). Positive controls were generated by amplifying sequences from the SAM1 locus using the primers SAM1-264F (5’–GCC CTT GCC TAC TAG TGC ATT T- 3’) and SAM1-88R (5’-CGA AGC TAA CCG AAA AAC AAC G-3’). PCR products were run on 0.8% agarose gels and visualized with ethidium bromide staining.
Characterization of telomere fusions by DNA sequencing
The PCR amplified telomere fusion products were treated with T4 DNA polymerase to polish the ends before inserting them into the polylinker of EcoRV digested pBluescript (Stratagene, Inc.), and transformation into E. coli. Blue/white screening was utilized to identify insert-bearing clones. Plasmids containing inserts were purified and their DNA sequences determined by automated DNA sequencing using T3 and T7 primers complementary to sequences flanking the polylinker in pBluescript.
Acknowledgements
We would like to thank D.E. Gottschling, J. E. Haber and V. Lundblad for supplying strains and plasmids. This work was supported by grants from the National Institutes of Health (GM057484 to A. B.), the Department of Defense Breast Cancer Research Program (W81XWH0410407 to D. M.), and funds from the Beckman Institute of the City of Hope. We would also like to thank R. J. Lin, T. O’Connor, J. Stark, J. Termini, and members of the Bailis lab for constructive comments and suggestions.
References
- Blackburn EH. Telomeres and telomerase: their mechanisms of action and the effects of altering their functions. FEBS Lett. 2005;579:859–862. doi: 10.1016/j.febslet.2004.11.036. [DOI] [PubMed] [Google Scholar]
- Burke D, Dawson DS, Stearns T. Methods in Yeast Genetics: A Cold Spring Harbor Laboratory Course. CSHL Press; 2000. [Google Scholar]
- Carter SD, Iyer S, Xu J, McEachern MJ, Astrom SU. The role of nonhomologous end-joining components in telomere metabolism in Kluyveromyces lactis. Genetics. 2007;175:1035–1045. doi: 10.1534/genetics.106.067447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SW, Blackburn EH. Telomerase and ATM/Tel1p protect telomeres from nonhomologous end joining. Mol Cell. 2003;11:1379–1387. doi: 10.1016/s1097-2765(03)00174-6. [DOI] [PubMed] [Google Scholar]
- Chen C, Kolodner RD. Gross chromosomal rearrangements in Saccharomyces cerevisiae replication and recombination defective mutants. Nat Genet. 1999;23:81–85. doi: 10.1038/12687. [DOI] [PubMed] [Google Scholar]
- De Lange T. Telomere-related genome instability in cancer. Cold Spring Harb Symp Quant Biol. 2005;70:197–204. doi: 10.1101/sqb.2005.70.032. [DOI] [PubMed] [Google Scholar]
- DePinho RA. The age of cancer. Nature. 2000;408:248–254. doi: 10.1038/35041694. [DOI] [PubMed] [Google Scholar]
- DuBois ML, Haimberger ZW, McIntosh MW, Gottschling DE. A quantitative assay for telomere protection in Saccharomyces cerevisiae. Genetics. 2002;161:995–1013. doi: 10.1093/genetics/161.3.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espejel S, Franco S, Rodriguez-Perales S, Bouffler SD, Cigudosa JC, Blasco MA. Mammalian Ku86 mediates chromosomal fusions and apoptosis caused by critically short telomeres. Embo J. 2002;21:2207–2219. doi: 10.1093/emboj/21.9.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasullo M, Dave P. Mating type regulates the radiation-associated stimulation of reciprocal translocation events in Saccharomyces cerevisiae. Mol Gen Genet. 1994;243:63–70. doi: 10.1007/BF00283877. [DOI] [PubMed] [Google Scholar]
- Feldser DM, Hackett JA, Greider CW. Telomere dysfunction and the initiation of genome instability. Nat Rev Cancer. 2003;3:623–627. doi: 10.1038/nrc1142. [DOI] [PubMed] [Google Scholar]
- Friis J, Roman H. The effect of the mating-type alleles on intragenic recombination in yeast. Genetics. 1968;59:33–36. doi: 10.1093/genetics/59.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galitski T, Saldanha AJ, Styles CA, Lander ES, Fink GR. Ploidy regulation of gene expression. Science. 1999;285:251–254. doi: 10.1126/science.285.5425.251. [DOI] [PubMed] [Google Scholar]
- Gallego ME, White CI. DNA repair and recombination functions in Arabidopsis telomere maintenance. Chromosome Res. 2005;13:481–491. doi: 10.1007/s10577-005-0995-4. [DOI] [PubMed] [Google Scholar]
- Golin JE, Esposito MS. Evidence for joint genic control of spontaneous mutation and genetic recombination during mitosis in Saccharomyces. Mol Gen Genet. 1977;150:127–135. doi: 10.1007/BF00695392. [DOI] [PubMed] [Google Scholar]
- Greider CW, Blackburn EH. Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell. 1985;43:405–413. doi: 10.1016/0092-8674(85)90170-9. [DOI] [PubMed] [Google Scholar]
- Hackett JA, Feldser DM, Greider CW. Telomere dysfunction increases mutation rate and genomic instability. Cell. 2001;106:275–286. doi: 10.1016/s0092-8674(01)00457-3. [DOI] [PubMed] [Google Scholar]
- Hackett JA, Greider CW. End resection initiates genomic instability in the absence of telomerase. Mol Cell Biol. 2003;23:8450–8461. doi: 10.1128/MCB.23.23.8450-8461.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwell LH, Smith D. Altered fidelity of mitotic chromosome transmission in cell cycle mutants of S. cerevisiae. Genetics. 1985;110:381–395. doi: 10.1093/genetics/110.3.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heacock ML, Idol RA, Friesner JD, Britt AB, Shippen DE. Telomere dynamics and fusion of critically shortened telomeres in plants lacking DNA ligase IV. Nucleic Acids Res. 2007;35:6490–6500. doi: 10.1093/nar/gkm472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heude M, Fabre F. a/alpha-control of DNA repair in the yeast Saccharomyces cerevisiae: genetic and physiological aspects. Genetics. 1993;133:489–498. doi: 10.1093/genetics/133.3.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IJpma AS, Greider CW. Short telomeres induce a DNA damage response in Saccharomyces cerevisiae. Mol Biol Cell. 2003;14:987–1001. doi: 10.1091/mbc.02-04-0057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JE, Broccoli D. Telomere maintenance in sarcomas. Curr Opin Oncol. 2007;19:377–382. doi: 10.1097/CCO.0b013e3281214423. [DOI] [PubMed] [Google Scholar]
- Kegel A, Sjostrand JO, Astrom SU. Nej1p, a cell type-specific regulator of nonhomologous end joining in yeast. Curr Biol. 2001;11:1611–1617. doi: 10.1016/s0960-9822(01)00488-2. [DOI] [PubMed] [Google Scholar]
- Le S, Moore JK, Haber JE, Greider CW. RAD50 and RAD51 define two pathways that collaborate to maintain telomeres in the absence of telomerase. Genetics. 1999;152:143–152. doi: 10.1093/genetics/152.1.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lea DE, Coulson CA. The distribution of the numbers of mutants in bacterial populations. Journal of Genetics. 1949;49:264–285. doi: 10.1007/BF02986080. [DOI] [PubMed] [Google Scholar]
- Lowell JE, Roughton AI, Lundblad V, Pillus L. Telomerase-independent proliferation is influenced by cell type in Saccharomyces cerevisiae. Genetics. 2003;164:909–921. doi: 10.1093/genetics/164.3.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundblad V, Blackburn EH. An alternative pathway for yeast telomere maintenance rescues est1- senescence. Cell. 1993;73:347–360. doi: 10.1016/0092-8674(93)90234-h. [DOI] [PubMed] [Google Scholar]
- Luria SE, Delbruck M. Mutations of Bacteria from Virus Sensitivity to Virus Resistance. Genetics. 1943;28:491–511. doi: 10.1093/genetics/28.6.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClintock B. The Stability of Broken Ends of Chromosomes in Zea Mays. Genetics. 1941;26:234–282. doi: 10.1093/genetics/26.2.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee RH, Lawrence CW. Genetic analysis of gamma-ray mutagenesis in yeast. III. Double-mutant strains. Mutat Res. 1980;70:37–48. doi: 10.1016/0027-5107(80)90056-1. [DOI] [PubMed] [Google Scholar]
- Mefford HC, Trask BJ. The complex structure and dynamic evolution of human subtelomeres. Nat Rev Genet. 2002;3:91–102. doi: 10.1038/nrg727. [DOI] [PubMed] [Google Scholar]
- Meyer DH, Bailis AM. Telomere dysfunction drives increased mutation by error-prone polymerases Rev1 and zeta in Saccharomyces cerevisiae. Genetics. 2007;175:1533–1537. doi: 10.1534/genetics.106.068130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mieczkowski PA, Mieczkowska JO, Dominska M, Petes TD. Genetic regulation of telomere-telomere fusions in the yeast Saccharomyces cerevisae. Proc Natl Acad Sci U S A. 2003;100:10854–10859. doi: 10.1073/pnas.1934561100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller HJ. The Remaking of Chromosomes. The Collecting Net. 1938;13:181–198. [Google Scholar]
- Pannunzio NR, Manthey GM, Bailis AM. RAD59 is required for efficient repair of simultaneous double-strand breaks resulting in translocations in Saccharomyces cerevisiae. DNA Repair (Amst) 2008 doi: 10.1016/j.dnarep.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardo B, Marcand S. Rap1 prevents telomere fusions by nonhomologous end joining. Embo J. 2005;24:3117–3127. doi: 10.1038/sj.emboj.7600778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resnick MA, Martin P. The repair of double-strand breaks in the nuclear DNA of Saccharomyces cerevisiae and its genetic control. Mol Gen Genet. 1976;143:119–129. doi: 10.1007/BF00266917. [DOI] [PubMed] [Google Scholar]
- Riha K, Heacock ML, Shippen DE. The role of the nonhomologous end-joining DNA double-strand break repair pathway in telomere biology. Annu Rev Genet. 2006;40:237–277. doi: 10.1146/annurev.genet.39.110304.095755. [DOI] [PubMed] [Google Scholar]
- Rizki A, Lundblad V. Defects in mismatch repair promote telomerase-independent proliferation. Nature. 2001;411:713–716. doi: 10.1038/35079641. [DOI] [PubMed] [Google Scholar]
- Saeki T, Machida I, Nakai S. Genetic control of diploid recovery after gamma-irradiation in the yeast Saccharomyces cerevisiae. Mutat Res. 1980;73:251–265. doi: 10.1016/0027-5107(80)90192-x. [DOI] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smogorzewska A, de Lange T. Different telomere damage signaling pathways in human and mouse cells. Embo J. 2002;21:4338–4348. doi: 10.1093/emboj/cdf433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strathern J, Hicks J, Herskowitz I. Control of cell type in yeast by the mating type locus. The alpha 1-alpha 2 hypothesis. J Mol Biol. 1981;147:357–372. doi: 10.1016/0022-2836(81)90488-5. [DOI] [PubMed] [Google Scholar]
- Thomas BJ, Rothstein R. The genetic control of direct-repeat recombination in Saccharomyces: the effect of rad52 and rad1 on mitotic recombination at GAL10, a transcriptionally regulated gene. Genetics. 1989;123:725–738. doi: 10.1093/genetics/123.4.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tishkoff DX, Filosi N, Gaida GM, Kolodner RD. A novel mutation avoidance mechanism dependent on S. cerevisiae RAD27 is distinct from DNA mismatch repair. Cell. 1997;88:253–263. doi: 10.1016/s0092-8674(00)81846-2. [DOI] [PubMed] [Google Scholar]
- Valencia M, Bentele M, Vaze MB, Herrmann G, Kraus E, Lee SE, Schar P, Haber JE. NEJ1 controls non-homologous end joining in Saccharomyces cerevisiae. Nature. 2001;414:666–669. doi: 10.1038/414666a. [DOI] [PubMed] [Google Scholar]
- Valencia-Burton M, Oki M, Johnson J, Seier TA, Kamakaka R, Haber JE. Different mating-type-regulated genes affect the DNA repair defects of Saccharomyces RAD51, RAD52 and RAD55 mutants. Genetics. 2006;174:41–55. doi: 10.1534/genetics.106.058685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson JD. Origin of concatemeric T7 DNA. Nat New Biol. 1972;239:197–201. doi: 10.1038/newbio239197a0. [DOI] [PubMed] [Google Scholar]
- Wilson TE, Grawunder U, Lieber MR. Yeast DNA ligase IV mediates non-homologous DNA end joining. Nature. 1997;388:495–498. doi: 10.1038/41365. [DOI] [PubMed] [Google Scholar]
- Zakian VA. Telomere functions: lessons from yeast. Trends Cell Biol. 1996;6:29–33. doi: 10.1016/0962-8924(96)81035-x. [DOI] [PubMed] [Google Scholar]