

Abstract
Protein production is driven by protein translation and relies on ribosomal biogenesis, globally essential for cell growth, proliferation, and animal development. Deregulation of these sophisticated cellular processes leads to abnormal homeostasis and carcinogenesis. Thus, their tight regulation is vitally important for a cell to warrant normal growth and proliferation. One newly-identified key regulator for ribosomal biogenesis and translation is the oncoprotein c-Myc, whose aberrantly excessive level and activity are highly associated with human cancers, too. Recently, we have shown that ribosomal protein L11 functions as a feedback regulator of c-Myc. Hence, in this review, we will provide some prospects on the interplay between c-Myc and ribosomal proteins during ribosomal biogenesis and discuss its implications in cancer.
Keywords: c-Myc, ribosomal biogenesis, ribosomal proteins, L11, transcription, cell cycle
Introduction: Ribosomal biogenesis and translation
Ribosomal biogenesis is a highly-ordered cellular process for producing the ribosome, the mRNA-to-protein translational machinery of the cell. This event consumes a vast portion of cellular energy and metabolites, and is essential for cell growth and proliferation [Warner, 1999]. In principle, ribosomal biogenesis includes the synthesis and import of ribosomal proteins, synthesis and processing of rRNA, the concomitant assembly of ribosomal proteins into the pre-ribosomal subunits and their subsequent transport [Fatica and Tollervey, 2002]. Most of these events occur in a coordinated fashion with the help of a number of auxiliary factors for rRNA processing and ribosome assembly in the nucleolus [Hannan et al., 1998], a subnuclear compartment without a membrane, except for 5S rRNA synthesis in the nucleoplasm and synthesis of ribosomal proteins in the cytoplasm.
All three RNA polymerases (I, II and III) are utilized for highly efficient and accurate ribosome production. RNA polymerase I (Pol I) catalyzes the synthesis of a single 47S rRNA precursor (pre-rRNA) from multiple copies of the genes (rDNA), and the pre-rRNA is in turn processed through sequential endonucleolytic and exonucleolytic cleavages into 18S, 5.8S, and 28S rRNA species [Boisvert et al., 2007; Hannan et al., 1998]. Pol II transcribes the mRNAs for ribosomal proteins and auxiliary factors. Pol III synthesizes 5S rRNA, which is used for the assembly of a 60S pre-ribosomal subunit. In mammalian cells, the mature 40S ribosomal subunit contains 18S rRNA and approximately 32 small ribosomal proteins (RPS), whereas the 60S subunit is composed of 5S, 5.8S, and 28S rRNAs and approximately 47 large ribosomal proteins (RPL). These ribosome subunits are then transported to the cytoplasm for protein translation.
Protein translation is a high energy-consuming intracellular biosynthesis with mRNAs as templates, and also essential for cell growth, proliferation and differentiation. The basic translation machinery is composed of ribosomes, mRNA, tRNAs, as well as translational initiation and elongation factors. The translation takes place in the rough endoplasmic reticulum and is predominantly regulated at its initiation. The translational initiation begins with several critical steps: incorporation of the initiation factors eIF-2, eIF-3, tRNAiMet, and GTP into a 40S ribosomal subunit to form a 43 S complex; Then the eIF-4E factor is recruited into the 43 S complex with target mRNA to form a 48 S complex; finally a 60 S ribosomal subunit joins the 48 S complex to form a complete 80 S complex. Among them, the recruitment of the eIF-4E factor to the 43 S complex is a rate-limiting step (reviewed by [De Benedetti and Graff, 2004; Scheper et al., 2007]).
Ribosomal biogenesis and protein translation are finely coordinated with cell proliferation. It is believed that the increase of global translation could facilitate the cell cycle. Also, the increase of cell mass (cell growth), which is largely composed of proteins, must be aligned with the cell cycle (cell proliferation). Thus, interfering with the ribosomal biogenesis can severely retard cell growth, proliferation and consequently animal development [Dai et al., 2007b]. However, increasing evidence has suggested that both deregulated over-production and hapoloinsuffciency of ribosomal biogenesis could lead to tumorigenesis (see more discussion below). Hence, both ribosomal biogenesis and protein translation need to be tightly regulated in order for a cell to sustain normal cell homeostasis and proliferation. There are a number of tumor suppressors or oncoproteins that play regulatory roles in ribosomal biogenesis [Dai et al., 2007b] and protein translation [Holland, 2004; Ruggero and Pandolfi, 2003]. One of these frequently studied oncoproteins for regulation of ribosomal biogenesis is c-Myc.
In general, c-Myc promotes cell growth and proliferation by enhancing ribosomal biogenesis and protein translation largely due to its key function in stimulating transcription of a number of genes encoding proteins essential for ribosomal biogenesis and protein translation including ribosomal proteins [Oskarsson and Trumpp, 2005]. Recently, we have demonstrated that one of the ribosomal L proteins, RPL11, acts as a feedback regulator of c-Myc, providing the first example for the regulation of c-Myc by a ribosomal protein during ribosomal biogenesis [Dai et al., 2007a]. Therefore, in this review, we will focus on the crosstalk between c-Myc and ribosome by highlighting the current views on the contribution of ribosomal biogenesis and translation to cancer, and offering some prospects about the RPL11-c-Myc feedback regulation and the implications of this regulation in cancers.
Do deregulated ribosomal biogenesis and translation contribute to cancer development?
Cancer cells undergo uncontrolled and infinite proliferation, which requires more production of ribosomes for the need of protein translation. It is clear that ribosomal biogenesis and translation rates are generally elevated in cancer cells. Although it has been the question of whether these increases are merely the consequences or side effects of cancer cell transformation or they might have an active or causal role in cell transformation and tumorigenesis, accumulating evidence shows that reinforced global translation results in transformation and tumorigenesis in cells and in animals (Fig. 1A), suggesting that excessively active translation could have a direct role in tumorigenesis.
Figure 1.
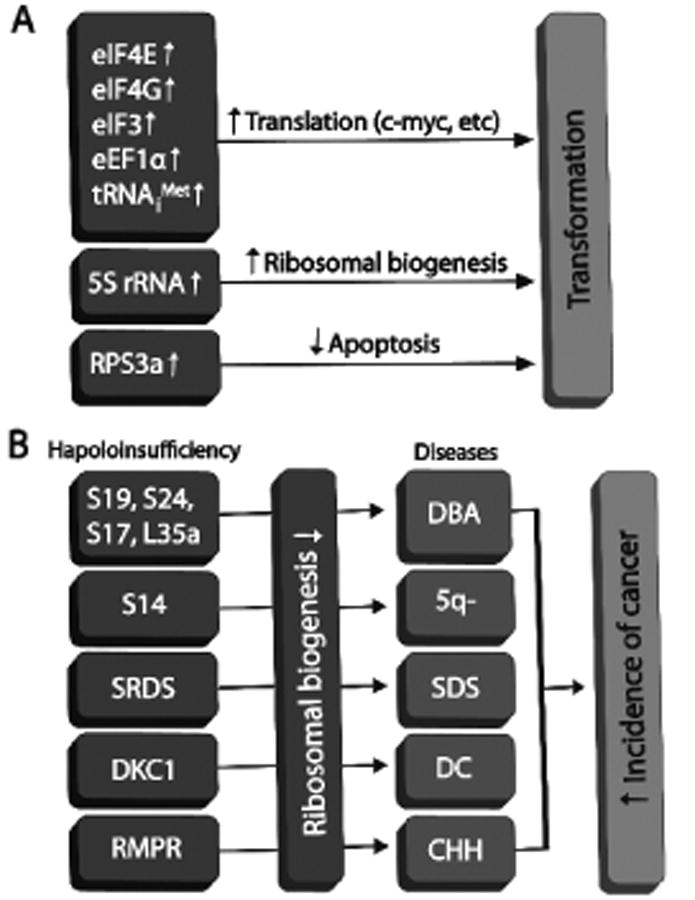
Possible contributions of deregulated ribosomal biogenesis and translation to tumorigenesis. (A). Over-production of translational factors and ribosomal biogenesis components leads to cellular transformation. (B). Haploinsufficiency of ribosomal proteins and other factors involved in ribosomal biogenesis leads to clinical syndromes with increased susceptibility to cancer. Abbreviations: 5q-: 5q deletion syndrome; CHH: Cartilage-Hair hypoplasia; DBA: Diamond-Blackfan anemia; DC: dyskeratosis congenital; SDS: Shwachman-Diamond syndrome.
One of the more intensively-studied examples in this aspect is the key translational initiation factor eIF4E. Overexpression of eIF4E in NIH3T3 fibroblasts enhanced colony formation in soft agar and transformation and tumorigenesis in nude mice [Lazaris-Karatzas et al., 1990]. Conversely, inhibition of eIF4E expression by antisense RNA [De Benedetti et al., 1991] or inhibition of eIF4E activity by overexpression of the eIF4E binding proteins (4E-BPs), a negative regulator of eIF4E, reduced these oncogenic outcomes [Rousseau et al., 1996]. In line with these findings, overexpression of eIF4E was found in various primary human cancers, such as breast, head and neck, colon, prostate, bladder, cervix, and lung cancers [De Benedetti and Graff, 2004]. Also, eIF-4E transgenic mice show marked increase of tumorigenesis, including lymphomas, angiosarcomas, lung adenocarcinomas, and hepatocellular adenomas [Ruggero et al., 2004], which match several types of human cancers as mentioned above. In addition, overexpression of eIF4E accelerated lymphomagenesis in mice transplanted with hematopoietic stem cells (HSCs) derived from Eμ-Myc mice [Wendel et al., 2004]. These studies have firmly established eIF4E as an oncogene. Its oncogenic activity appears to attribute to its translational function specific for a certain group of growth-promoting proteins, such as c-Myc, cyclin D1, vascular endothelial growth factor, or etc, which under normal conditions are translated less efficiently due to high-structured 5′-untranslated region (UTR) of their mRNAs (so-called weak mRNAs), though eIF4E also moderately enhances the global translation [De Benedetti and Graff, 2004; Mamane et al., 2007; Mamane et al., 2004].
More translation factors have been found to possess similar potential oncogenic activity. For instance, overexpression of eIF4G [Fukuchi-Shimogori et al., 1997] or eEF1α2 [Anand et al., 2002] induced transformation of immortalized NIH3T3 cells and tumorigenesis in nude mice. Interestingly, the eEF1α2 gene is amplified in 25% of primary ovarian tumors and established cancer cell lines. Recently, overexpression of individual human translation initiation factors of eIF3, such as eIF3a, -3b, -3c, -3h, or -3i was also reported to induce cellular transformation in NIH3T3 cells [Zhang et al., 2007]. These studies support the notion that the overly increased translation perhaps due to forced expression of individual translation factors would favor mammalian cell transformation and eventually tumorigenesis.
Another possible mechanism for abnormally increased translation-driven cell transformation and tumorigenicity is the induced overexpression of RNA polymerase III- specific transcription factor, Brf1 [Marshall et al., 2008]. Over production of Brf1 resulted in high levels of tRNAs and 5S rRNA. This effect was dependent on the enhanced expression of Pol III targets tRNA and 5S rRNA, as depletion of RPC39, a specific subunit of Pol III that interacts with Brf1 in order to recruit the polymerase to its genetic templates, abolished Brf1-induced cell proliferation. Also, elevated expression of tRNAiMet alone, a Pol III-catalyzed target gene that is required for polypeptide chain initiation, is sufficient to induce cell proliferation, cell transformation and tumorigenicity in mice [Marshall et al., 2008].
Not only the regulatory factors for translation, but also the ribosomal proteins themselves play a critical role in cell transformation and tumorigenicity. One early-reported example was the ribosomal protein RPS3a (also called the v-fos transformation effector Fte-1). Overexpression of RPS3a induced cell transformation in NIH3T3 cells and tumor formation in nude mice [Naora et al., 1998]. It still remains unclear how overexpression of a single ribosomal protein could contribute to cell transformation. It is likely that high levels of RPS3a may enhance the production of anti-apoptotic proteins, as its overexpression inhibits apoptosis [Naora et al., 1998]. Whether it enhances global translation or specifically the translation of “weak mRNAs” still remains unanswered.
In addition to the contribution of overly active ribosomal biogenesis and translation to tumorigenesis, reduction of ribosomal biogenesis and translation also plays a role in tumorigenesis (Fig. 1B). The first example involved the rps19 gene. It has been shown that its heterozygous null mutations occur in 25% of patients with Diamond-Blackfan anemia (DBA), a syndrome characterized by a chronic constitutional regenerative anemia, various degree of congenital abnormalities, and an increased susceptibility to hematopoietic malignancies [Draptchinskaia et al., 1999]. Thereafter, hapaloinsufficiency of other ribosomal proteins including RPS24, RPS17, and RPL35A via mutations and deletions was also reported in patients with DBA [Cmejla et al., 2007; Farrar et al., 2008; Gazda et al., 2006]. It is predicted that hepaloinsufficiency of other ribosomal proteins may exist in DBA as well and that DBA may be caused by global reduction of ribosomal biogenesis. Another example is 5q- syndrome. This is also an anemia syndrome with increased incidence of hematopoietic tumors, and highly associated with deletion of one allele of the rps14 gene [Ebert et al., 2008]. One dominant phenotype of all these syndromes is severe anemia; this can be explained by the fact that erythropoiesis, the production of red blood cells, is considerably rapid and the cell proliferation rate of erythroid progenitor cells is significantly high [Dai et al., 2000], both of which demand more efficient and productive ribosomal biogenesis and translation. Then, how to explain the increased incidence of tumors in these syndromes with the haploinsufficiency of ribosomal proteins? One possibility would be that these ribosomal proteins might be required for p53 response to diverse cellular stresses. We and others have recently shown that several ribosomal proteins including RPL5, RPL11, RPL23, and RPS7 induce the activity of the tumor suppressor p53 by binding to MDM2 and inhibiting its ubiquitin E3 ligase activity toward p53 [Chen et al., 2007; Dai and Lu, 2004; Dai et al., 2006b; Dai et al., 2004; Lohrum et al., 2003; Zhang et al., 2003]. Interestingly, RPL5, RPL11, and RPL23 are essential for p53 activation in response to ribosomal or nucleolar stresses, such as those caused by treatment of actinomycin D, 5-Fluorouracil, and mycophenolic acid [Dai and Lu, 2004; Dai et al., 2004; Sun et al., 2007; Sun et al., 2008; Zhang et al., 2003]. Nucleolar stress could be one general outcome of different cellular stresses [Rubbi and Milner, 2003]. Thus, it is possible that the above ribosomal proteins associated with DBA and 5q- syndrome may also play a role in p53 activation in response to nucleolar stress. As such, haploinsufficiency of these ribosomal proteins might impair the nucleolar stress-p53 pathway, consequently leading to higher incidences of tumorigenesis. Another testable idea is that insufficient ribosomal biogenesis may lower the production of some important tumor suppressor proteins, including p53. On the other hand, possible alternations in the c-Myc pathway caused by reduction of the disease-associated ribosomal proteins may partially account for the cancer mechanism in these syndromes, too, as will be discussed in the following sections.
Besides the above-mentioned ribosomal proteins, other regulatory proteins for ribosomal biogenesis have also been identified to be associated with tumor formation. For example, mutation of the DKC1 gene was found in patients with dyskeratosis congenital (DC), a disease characterized by premature aging, including bone marrow failure and hyperkeratosis of the skin, and an increased susceptibility to cancers [Ruggero et al., 2003]. The DKC1 gene product dyskerin is a putative pseudouridine synthase that mediates posttranslational modification of ribosomal RNA (rRNA) through site-specific conversion of uridine to pseudouridine. Thus, loss of function mutation of the DKC1 gene would impair ribosomal biogenesis. Supporting this idea is that hypomorphic DKC1 mutant mice not only recapitulate the clinical features of DC, but also show defects of rRNA modification and processing [Ruggero et al., 2003].
Another case is Shwachman-Diamond syndrome (SDS), which is an autosomal recessive disorder characterized by hematological dysfunction, pancreatic exocrine insufficiency, skeleton abnormalities and short stature. Up to one-third of SDS patients develop leukemia, mostly acute myeloid leukemia. The disease is caused by mutations in the SBDS gene [Zhang et al., 2006]. The yeast SBDS ortholog sdo1 has been shown to be critical for the release and recycling of the nucleolar shuttling factor Tif6 from pre-60S ribosomes, a key step in 60S ribosome maturation and translational activation of the ribosome. TIF6 gain-of-function alleles suppressed the pre-60S nuclear export defects and sdo1-deletion phenotype [Menne et al., 2007]. These data suggest that defects in 60S ribosomal maturation may contribute to this inherited bone marrow failure syndrome associated with leukemia predisposition.
Lastly, mutations of another gene that encodes an RNase called RMRP involved in pre-rRNA cleavage cause a pleiotropic human disease, Cartilage-Hair hypoplasia (CHH) [Ridanpaa et al., 2001]. CHH is a recessive and highly pleiotropic disorder characterized by short statue, defective cellular immunity, and predisposition to several cancers. RMRP is a component of the endoribonuclease RNse MRP complex containing one RNA molecule bound to several proteins and essential for rRNA processing [Ridanpaa et al., 2001]. Therefore, it is also possible that defects in rRNA processing caused by mutations in the RMRP gene may be responsible for predisposition of CHH patients to multiple cancers.
In summary, all of these studies suggest that the deregulated ribosomal biogenesis and translation may play a crucial role in the molecular pathogenesis of tumors, some of which are highly associated with specific genetic defects or syndromes. However, the exact molecular mechanisms or pathways underlying these tumor-prone genetic defects are still far from being well understood. Numerous studies over the past half a decade have demonstrated an essential role for one of the major oncoproteins, c-Myc, in regulating ribosomal biogenesis and protein translation. Thus, it is logical to imagine the link of c-Myc with the ribosomal biogenesis-associated cancers (see more details below), although it is still immature to conclude that c-Myc is responsible for the development of the above-discussed cancers.
The role of c-Myc in ribosomal biogenesis
c-Myc is a transcriptional factor essential for normal cell and stem cell growth, proliferation, self-renewal, and animal development [Adhikary and Eilers, 2005; Pelengaris et al., 2002a]. c-Myc heterodimerizes with its partner protein Max. The c-Myc/Max heterodimer binds to cognate E-box (CACGTG) DNA elements at target gene promoters through the C-terminal bHLH/LZ domain of c-Myc and activates transcription of these genes [Adhikary and Eilers, 2005]. The N-terminal transcriptional activation domain (TAD) of c-Myc contains two conserved segments termed Myc box (MB) I and II, which are crucial for all biological activity [Sakamuro and Prendergast, 1999]. The essential role of c-Myc in cell growth and animal development is demonstrated by the fact that homozygous deletion of the c-myc gene is lethal to mice at E9.5-10.5 days [Davis et al., 1993].
However, its deregulated overproduction contributes to many types of human cancers (reviewed by [Adhikary and Eilers, 2005; Dai et al., 2006a; Pelengaris et al., 2002a]. Constitutive, inducible or conditional expression of a c-myc transgene leads to neoplastic, pre-malignant and malignant phenotypes in mice [Adams et al., 1985; Felsher and Bishop, 1999; Pelengaris et al., 2002b]. Interestingly, when c-myc expression is turned off in these mice, these tumorigenic phenotypes spontaneously remit [Felsher and Bishop, 1999; Pelengaris et al., 2002b; Pelengaris et al., 1999]. These studies demonstrate that the excess level and activity of c-Myc endorse cell transformation and tumorigenesis.
The proliferation-promoting and tumor-promoting activity of the c-Myc is well tied with its role in enhancing ribosomal biogenesis (Fig. 2). Consistent with this statement, genetically, c-Myc transgenic mice display an increase in cell size corresponding to elevated ribosome biogenesis [Iritani and Eisenman, 1999; Kim et al., 2000]. Biochemically, c-Myc regulates transcription by all three RNA polymerases [Adhikary and Eilers, 2005; Oskarsson and Trumpp, 2005] with an ultimate goal of boosting ribosomal biogenesis. However, mechanistically, c-Myc interacts with different regulatory factors in regulation of these RNA polymerases. For instance, c-Myc enhances Pol I-catalyzed synthesis of rRNA precursor (pre-rRNA) by binding to TBP and TBP-associated factors (TAFs), thereby facilitating the recruitment of Pol I to the rDNA promoter [Arabi et al., 2005; Grandori et al., 2005; Grewal et al., 2005]. c-Myc also augments Pol III-mediated 5S and tRNA transcription by directly interacting with and activating TFIIIB [Gomez-Roman et al., 2003]. c-Myc plays a role in rRNA processing as well [Schlosser et al., 2003]. In addition, c-Myc activates Pol II-catalyzed transcription of a large number of genes that encode proteins involved in ribosomal biogenesis and translation, such as ribosomal proteins, ribosome assembly proteins, and translation initiation and elongation factors [Boon et al., 2001; Coller et al., 2000; Guo et al., 2000; Menssen and Hermeking, 2002]. According to genome wide and microarray studies, c-Myc may be critical for the expression of almost 15 % of all human genes, and many of them are involved in ribosomal biogenesis and protein translation [Patel et al., 2004]. Thus, it is not surprising that c-Myc plays a pivotal role in controlling ribosomal biogenesis, cell growth and proliferation.
Figure 2.
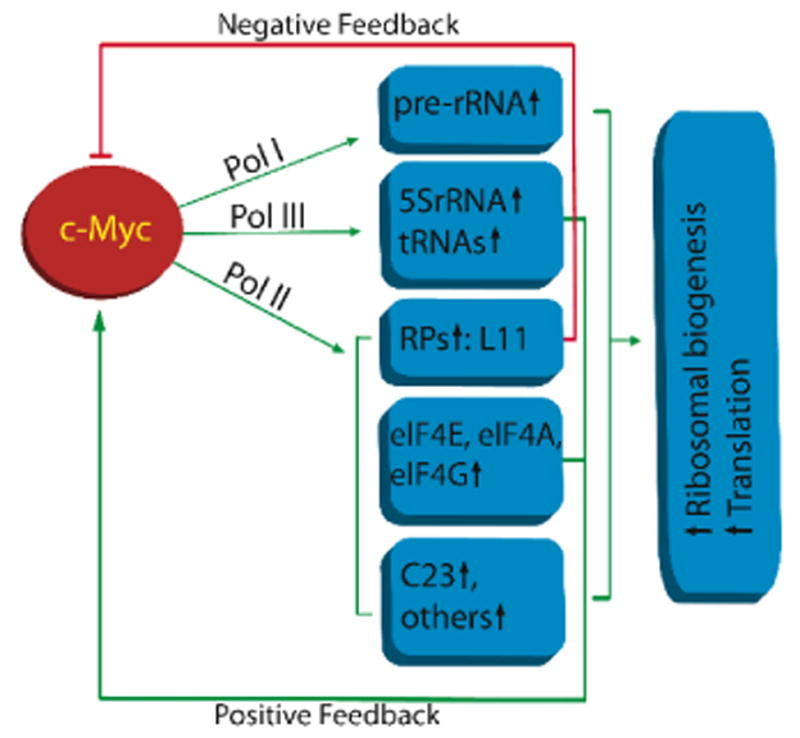
A schematic diagram illustrating the interplay between c-Myc and ribosomal biogenesis and translation. Arrows indicate activation whereas a bar denotes inhibition.
Regulation of c-Myc by ribosomal protein L11
For the dual reason that c-Myc is vital for normal cell proliferation but could be tumorigenic once over-produced or active, cells have developed multiple mechanisms to control c-Myc level and activity in order to avoid undergoing hyperplasia and consequent neoplasia. These mechanisms include transcriptional, posttranscriptional (mRNA stability and translation), translational, and posttranslational (protein stability) regulations (For details, see these review articles:[Dai et al., 2006a; Sears, 2004; Spencer and Groudine, 1991]). In our initial attempt to search potential regulators of c-Myc by screening some of its ribosomal target genes involved in ribosomal biogenesis, we fortunately identified RPL11 as a feedback regulator of c-Myc [Dai et al., 2007a]. First, we have verified the rpl11 gene as a bona fide transcriptional target of c-Myc. Also, we have demonstrated that RPL11 suppresses the transcriptional activity of c-Myc in cells (Fig. 2). These findings provide the first paradigm for a ribosomal protein to play a regulatory role in monitoring the activity of c-Myc as a negative feedback regulator perhaps during ribosomal biogenesis [Dai et al., 2007a]. Hence, this finding is physiologically significant.
To our surprise, RPL11 regulates c-Myc activity via multiple mechanisms. First, RPL11 physically interacts with c-Myc at c-Myc target gene promoters and inhibits the recruitment of one essential c-Myc coactivator called TRRAP to the promoters, as both RPL11 and TRRAP binds to the same MB II motif of c-Myc, and thus they compete with each other for binding to c-Myc [Dai et al., 2007a]. By preventing the binding of TRRAP to c-Myc and the promoters, RPL11 can reduce histone acetylation of the target gene promoters and consequently inhibits the transcription of the target genes. The competition of RPL11 with TRRAP for binding to c-Myc and its target promoters is also recapitulated in chromatin immunoprecipitation (ChIP) analyses of cellular c-Myc response to growth signals as their ChIP profiles on c-Myc target promoters are exactly inverse to each other. Our studies suggest a physiologically role of RPL11 in downregulating c-Myc activity.
Also, we have found that when overexpressed, RPL11 can re-localize ectopic c-Myc into the nucleolus [Dai et al., 2007b], consistent with the observation that more c-Myc molecules are retained in non-NP40-extracted (insoluble) fractions in the presence of ectopic RPL11. Thus, it is possible that RPL11 may inactivate c-Myc by associating with it in the nucleolus [Dai et al., 2007b], and may inhibit the c-Myc-boosted RNA Pol I activity in this subnuclear compartment as well.
It has been shown that the dynamic binding of c-Myc to target gene promoters as mediated by another c-Myc coactivator SCFSkp2 E3 ligase is also critical for regulation of c-Myc activity and turnover [Kim et al., 2003; von der Lehr et al., 2003]. Because Skp2 also binds to the MB II of c-Myc [Kim et al., 2003; von der Lehr et al., 2003], we speculate that RPL11 may inhibit the binding of Skp2 to c-Myc, resulting in the reduced turnover of c-Myc at target gene promoters. This would be an interesting question to address in the near future.
Lastly and more intriguingly, RPL11 appears to affect c-myc mRNA level as knockdown of RPL11 drastically increased the level of c-myc mRNA in cells [Dai et al., 2007b]. Although it is still unclear how RPL11 does that, this effect should be independent of the RPL11-c-Myc binding, but may involve the binding of RPL11 to the c-myc mRNA or c-myc gene promoter. One plausible idea would be that RPL11 might bind to c-myc gene promoters and act as a repressor of c-myc gene transcription. By doing so, RPL11 might directly, or indirectly through other unknown co-repressors, interfere with the transcriptional machinery or with the remodeling of chromatin structure in the promoter region of the c-myc gene. Supporting this hypothesis is that several ribosomal proteins including RPL11 have been shown to bind to linker histone H1 and suppress transcription of a set of genes in drosophila [Panic et al., 2006]. Also, we have recently purified an RPL11-associated complex that contains the linker histone H1 (data not shown).
Alternatively, RPL11 might influence c-myc mRNA stability. As discussed in the previous section, two cis-acting sequence elements have been shown to regulate c-myc mRNA turnover: an AU-rich element (ARE) in 3′-untranslated region (3′-UTR) [Bonnieu et al., 1988; Yeilding et al., 1996] and an ∼250 nt coding region instability determinant (CRD) [Doyle et al., 1998]. CRD binding protein (CRD-BP), a member of a family of KH domain containing RNA-binding proteins, binds to the CRD of c-myc mRNA, leading to protection of c-myc mRNA from endoribonuclease cleavage within CRD [Doyle et al., 1998; Lee et al., 1998]. Although this regulation has been implicated in the stabilization of c-myc mRNA in response to β-catenin signaling [Noubissi et al., 2006], our preliminary data shows that RPL11 does not apparently bind to CRD-BP (data not shown). Thus, it is less likely that RPL11 regulates c-myc mRNA stability via CRD-BP, but more likely that RPL11 might interplay with the c-myc 3-UTR. Several ARE binding proteins, including AUF1 [Zhang et al., 1993] and HuR [Ma et al., 1996], have been found to bind to the c-myc ARE and act as c-myc mRNA destabilizing factors. An immediate thought would be if RPL11 might regulate c-myc mRNA stability through interaction with these components. It is also possible that RPL11 may regulate c-myc mRNA stability through microRNA-mediated gene silencing pathways. Supporting this idea are two lines of indirect evidence: RPL11, together with RPL5 and RISC components such as Dicer, Ago2 and P68, have been shown to associate with drosophila FMR1, an ortholog of human FMRP protein that is associated with Fragile X syndrome, and this complex contains microRNAs [Ishizuka et al., 2002]; Also, microRNA has been implicated in regulating mRNA stability as well as translation in other system [Jing et al., 2005]. Hence it will be tempting and quite informative to determine whether RPL11 facilitates the targeting of certain microRNAs to c-myc 3′-UTR. Although it still remains entirely perplexing how RPL11 regulates c-myc mRNA levels, it is clear that RPL11 does play a feedback regulatory role in controlling c-Myc level and activity during ribosomal biogenesis.
More questions
Identification of RPL11 as a c-Myc feedback regulator not only establishes a new bridge between the ribosome and c-Myc and opens a new research avenue for more explorations as partially discussed above, but also raises more questions. One of the obvious questions is whether other ribosomal proteins also regulate c-Myc activity and level. It is clear that not all of the ribosomal proteins bind to and regulate c-Myc, as we have shown that several ribosomal proteins including RPL29, RPL30, and RPS12 do not bind to c-Myc [Dai et al., 2007a]. Neither overexpression nor knockdown of RPL29 affects the level and activity of c-Myc [Dai et al., 2007a]. However, this does not exclude the possibility of that other ribosomal proteins may act like RPL11 in regulating c-Myc. Indeed, our trial experiments show that several other tested ribosomal proteins, such as RPL5, RPL23, and RPS7, bind to c-Myc as well. Although too preliminary, this result suggests that RPL11 might not be the only ribosomal component that works on c-Myc during ribosomal biogenesis, and more ribosomal proteins are involved in this regulation, too. This result also brings up several more testable questions: Do they cooperate with RPL11 in negating c-Myc activity during ribosomal biogenesis; Do they regulate c-Myc in response to physiological signals and/or pathological stresses; Are there more ribosomal or translational proteins that regulate c-Myc and if so, how do they execute their regulatory functions? Addressing these questions is certainly important for our better understanding of the molecular details for the regulation by c-Myc of ribosomal biogenesis.
Another question is whether RPL11 has a tumor suppression function, given its dual ability to activate p53 and inactivate c-Myc as shown in substantial biochemical and cellular studies [Dai et al., 2007a; Dai et al., 2006b; Lohrum et al., 2003; Zhang et al., 2003]. It will be enormously challenging to employ genetically manipulated animal models for demonstrating the role of RPL11 in tumorigenesis, as this protein is particularly essential for ribosomal biogenesis and thus cell proliferation and animal development. However, this difficulty would not prevent us from exploring the possibility of identifying possible mutants or single nucleotide polymorphisms (SNP) of RPL11 in human cancers, which might be defective in MDM2- or c-Myc-binding, but still active as a subunit of the ribosome for translation. Discovering these mutants or SNPs would be especially crucial for our better understanding the biological role of RPL11 in tumor development.
Finally, it is rational to relate c-Myc with cancer-prone and ribosomal biogenesis-defective syndromes or genetic defects as described in the second section, since knocking down RPL11 results in increased level and activity of c-Myc. It is interesting to ask whether haploinsufficiency of above-mentioned RPs implicated in DBA would also enhance c-Myc activity and level. If this is true, c-Myc may contribute, at least partly, to the increased incidence of cancer in these patients. Altogether, more systematic and painstaking dissections of the ribosome-c-Myc pathway will certainly yield important information for our better understanding of the feedback interplay between c-Myc and ribosome during ribosomal biogenesis and tumorigenesis. This feedback regulation would have a translational impact on development of strategies that target c-Myc for cancer therapy.
Acknowledgments
We thank Timothy Lu for the figure art work and Arif Khan for proofreading. This work was supported in part by NCI grants CA93614, CA095441, CA 129828 to H. L. and a NCI grant K99-CA127134 and an Indiana University Biomedical Research Grant (BRG) to M.-S. D.
References
- Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, Palmiter RD, Brinster RL. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature. 1985;318:533–8. doi: 10.1038/318533a0. [DOI] [PubMed] [Google Scholar]
- Adhikary S, Eilers M. Transcriptional regulation and transformation by Myc proteins. Nat Rev Mol Cell Biol. 2005;6:635–45. doi: 10.1038/nrm1703. [DOI] [PubMed] [Google Scholar]
- Anand N, Murthy S, Amann G, Wernick M, Porter LA, Cukier IH, Collins C, Gray JW, Diebold J, Demetrick DJ, Lee JM. Protein elongation factor EEF1A2 is a putative oncogene in ovarian cancer. Nat Genet. 2002;31:301–5. doi: 10.1038/ng904. [DOI] [PubMed] [Google Scholar]
- Arabi A, Wu S, Ridderstrale K, Bierhoff H, Shiue C, Fatyol K, Fahlen S, Hydbring P, Soderberg O, Grummt I, Larsson LG, Wright AP. c-Myc associates with ribosomal DNA and activates RNA polymerase I transcription. Nat Cell Biol. 2005;7:303–10. doi: 10.1038/ncb1225. [DOI] [PubMed] [Google Scholar]
- Boisvert FM, van Koningsbruggen S, Navascues J, Lamond AI. The multifunctional nucleolus. Nat Rev Mol Cell Biol. 2007;8:574–85. doi: 10.1038/nrm2184. [DOI] [PubMed] [Google Scholar]
- Bonnieu A, Piechaczyk M, Marty L, Cuny M, Blanchard JM, Fort P, Jeanteur P. Sequence determinants of c-myc mRNA turn-over: influence of 3′ and 5′ non-coding regions. Oncogene Res. 1988;3:155–66. [PubMed] [Google Scholar]
- Boon K, Caron HN, van Asperen R, Valentijn L, Hermus MC, van Sluis P, Roobeek I, Weis I, Voute PA, Schwab M, Versteeg R. N-myc enhances the expression of a large set of genes functioning in ribosome biogenesis and protein synthesis. Embo J. 2001;20:1383–93. doi: 10.1093/emboj/20.6.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Zhang Z, Li M, Wang W, Li Y, Rayburn ER, Hill DL, Wang H, Zhang R. Ribosomal protein S7 as a novel modulator of p53-MDM2 interaction: binding to MDM2, stabilization of p53 protein, and activation of p53 function. Oncogene. 2007;26:5029–37. doi: 10.1038/sj.onc.1210327. [DOI] [PubMed] [Google Scholar]
- Cmejla R, Cmejlova J, Handrkova H, Petrak J, Pospisilova D. Ribosomal protein S17 gene (RPS17) is mutated in Diamond-Blackfan anemia. Hum Mutat. 2007;28:1178–82. doi: 10.1002/humu.20608. [DOI] [PubMed] [Google Scholar]
- Coller HA, Grandori C, Tamayo P, Colbert T, Lander ES, Eisenman RN, Golub TR. Expression analysis with oligonucleotide microarrays reveals that MYC regulates genes involved in growth, cell cycle, signaling, and adhesion. Proc Natl Acad Sci U S A. 2000;97:3260–5. doi: 10.1073/pnas.97.7.3260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai MS, Arnold H, Sun XX, Sears R, Lu H. Inhibition of c-Myc activity by ribosomal protein L11. Embo J. 2007a;26:3332–45. doi: 10.1038/sj.emboj.7601776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai MS, Jin Y, Gallegos JR, Lu H. Balance of Yin and Yang: ubiquitylation-mediated regulation of p53 and c-Myc. Neoplasia. 2006a;8:630–44. doi: 10.1593/neo.06334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai MS, Lu H. Inhibition of MDM2-mediated p53 ubiquitination and degradation by ribosomal protein L5. J Biol Chem. 2004;279:44475–82. doi: 10.1074/jbc.M403722200. [DOI] [PubMed] [Google Scholar]
- Dai MS, Mantel CR, Xia ZB, Broxmeyer HE, Lu L. An expansion phase precedes terminal erythroid differentiation of hematopoietic progenitor cells from cord blood in vitro and is associated with up-regulation of cyclin E and cyclin-dependent kinase 2. Blood. 2000;96:3985–7. [PubMed] [Google Scholar]
- Dai MS, Sears R, Lu H. Feedback regulation of c-Myc by ribosomal protein L11. Cell Cycle. 2007b;6:2735–41. doi: 10.4161/cc.6.22.4895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai MS, Shi D, Jin Y, Sun XX, Zhang Y, Grossman SR, Lu H. Regulation of the MDM2-p53 pathway by ribosomal protein L11 involves a post-ubiquitination mechanism. J Biol Chem. 2006b;281:24304–13. doi: 10.1074/jbc.M602596200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai MS, Zeng SX, Jin Y, Sun XX, David L, Lu H. Ribosomal protein L23 activates p53 by inhibiting MDM2 function in response to ribosomal perturbation but not to translation inhibition. Mol Cell Biol. 2004;24:7654–68. doi: 10.1128/MCB.24.17.7654-7668.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis AC, Wims M, Spotts GD, Hann SR, Bradley A. A null c-myc mutation causes lethality before 10.5 days of gestation in homozygotes and reduced fertility in heterozygous female mice. Genes Dev. 1993;7:671–82. doi: 10.1101/gad.7.4.671. [DOI] [PubMed] [Google Scholar]
- De Benedetti A, Graff JR. eIF-4E expression and its role in malignancies and metastases. Oncogene. 2004;23:3189–99. doi: 10.1038/sj.onc.1207545. [DOI] [PubMed] [Google Scholar]
- De Benedetti A, Joshi-Barve S, Rinker-Schaeffer C, Rhoads RE. Expression of antisense RNA against initiation factor eIF-4E mRNA in HeLa cells results in lengthened cell division times, diminished translation rates, and reduced levels of both eIF-4E and the p220 component of eIF-4F. Mol Cell Biol. 1991;11:5435–45. doi: 10.1128/mcb.11.11.5435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle GA, Betz NA, Leeds PF, Fleisig AJ, Prokipcak RD, Ross J. The c-myc coding region determinant-binding protein: a member of a family of KH domain RNA-binding proteins. Nucleic Acids Res. 1998;26:5036–44. doi: 10.1093/nar/26.22.5036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draptchinskaia N, Gustavsson P, Andersson B, Pettersson M, Willig TN, Dianzani I, Ball S, Tchernia G, Klar J, Matsson H, Tentler D, Mohandas N, Carlsson B, Dahl N. The gene encoding ribosomal protein S19 is mutated in Diamond-Blackfan anaemia. Nat Genet. 1999;21:169–75. doi: 10.1038/5951. [DOI] [PubMed] [Google Scholar]
- Ebert BL, Pretz J, Bosco J, Chang CY, Tamayo P, Galili N, Raza A, Root DE, Attar E, Ellis SR, Golub TR. Identification of RPS14 as a 5q- syndrome gene by RNA interference screen. Nature. 2008;451:335–9. doi: 10.1038/nature06494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrar JE, Nater M, Caywood E, McDevitt MA, Kowalski J, Takemoto CM, Talbot CC, Jr, Meltzer P, Esposito D, Beggs AH, Schneider HE, Grabowska A, Ball SE, Niewiadomska E, Sieff CA, Vlachos A, Atsidaftos E, Ellis SR, Lipton JM, Gazda HT, Arceci RJ. Abnormalities of the large ribosomal subunit protein, Rpl35A, in diamond-blackfan anemia. Blood. 2008 doi: 10.1182/blood-2008-02-140012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatica A, Tollervey D. Making ribosomes. Curr Opin Cell Biol. 2002;14:313–8. doi: 10.1016/s0955-0674(02)00336-8. [DOI] [PubMed] [Google Scholar]
- Felsher DW, Bishop JM. Reversible tumorigenesis by MYC in hematopoietic lineages. Mol Cell. 1999;4:199–207. doi: 10.1016/s1097-2765(00)80367-6. [DOI] [PubMed] [Google Scholar]
- Fukuchi-Shimogori T, Ishii I, Kashiwagi K, Mashiba H, Ekimoto H, Igarashi K. Malignant transformation by overproduction of translation initiation factor eIF4G. Cancer Res. 1997;57:5041–4. [PubMed] [Google Scholar]
- Gazda HT, Grabowska A, Merida-Long LB, Latawiec E, Schneider HE, Lipton JM, Vlachos A, Atsidaftos E, Ball SE, Orfali KA, Niewiadomska E, Da Costa L, Tchernia G, Niemeyer C, Meerpohl JJ, Stahl J, Schratt G, Glader B, Backer K, Wong C, Nathan DG, Beggs AH, Sieff CA. Ribosomal protein S24 gene is mutated in Diamond-Blackfan anemia. Am J Hum Genet. 2006;79:1110–8. doi: 10.1086/510020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Roman N, Grandori C, Eisenman RN, White RJ. Direct activation of RNA polymerase III transcription by c-Myc. Nature. 2003;421:290–4. doi: 10.1038/nature01327. [DOI] [PubMed] [Google Scholar]
- Grandori C, Gomez-Roman N, Felton-Edkins ZA, Ngouenet C, Galloway DA, Eisenman RN, White RJ. c-Myc binds to human ribosomal DNA and stimulates transcription of rRNA genes by RNA polymerase I. Nat Cell Biol. 2005;7:311–8. doi: 10.1038/ncb1224. [DOI] [PubMed] [Google Scholar]
- Grewal SS, Li L, Orian A, Eisenman RN, Edgar BA. Myc-dependent regulation of ribosomal RNA synthesis during Drosophila development. Nat Cell Biol. 2005;7:295–302. doi: 10.1038/ncb1223. [DOI] [PubMed] [Google Scholar]
- Guo QM, Malek RL, Kim S, Chiao C, He M, Ruffy M, Sanka K, Lee NH, Dang CV, Liu ET. Identification of c-myc responsive genes using rat cDNA microarray. Cancer Res. 2000;60:5922–8. [PubMed] [Google Scholar]
- Hannan KM, Hannan RD, Rothblum LI. Transcription by RNA polymerase I. Front Biosci. 1998;3:d376–98. doi: 10.2741/a282. [DOI] [PubMed] [Google Scholar]
- Holland EC. Regulation of translation and cancer. Cell Cycle. 2004;3:452–5. [PubMed] [Google Scholar]
- Iritani BM, Eisenman RN. c-Myc enhances protein synthesis and cell size during B lymphocyte development. Proc Natl Acad Sci U S A. 1999;96:13180–5. doi: 10.1073/pnas.96.23.13180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizuka A, Siomi MC, Siomi H. A Drosophila fragile X protein interacts with components of RNAi and ribosomal proteins. Genes Dev. 2002;16:2497–508. doi: 10.1101/gad.1022002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing Q, Huang S, Guth S, Zarubin T, Motoyama A, Chen J, Di Padova F, Lin SC, Gram H, Han J. Involvement of microRNA in AU-rich element-mediated mRNA instability. Cell. 2005;120:623–34. doi: 10.1016/j.cell.2004.12.038. [DOI] [PubMed] [Google Scholar]
- Kim S, Li Q, Dang CV, Lee LA. Induction of ribosomal genes and hepatocyte hypertrophy by adenovirus-mediated expression of c-Myc in vivo. Proc Natl Acad Sci U S A. 2000;97:11198–202. doi: 10.1073/pnas.200372597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SY, Herbst A, Tworkowski KA, Salghetti SE, Tansey WP. Skp2 regulates Myc protein stability and activity. Mol Cell. 2003;11:1177–88. doi: 10.1016/s1097-2765(03)00173-4. [DOI] [PubMed] [Google Scholar]
- Lazaris-Karatzas A, Montine KS, Sonenberg N. Malignant transformation by a eukaryotic initiation factor subunit that binds to mRNA 5′ cap. Nature. 1990;345:544–7. doi: 10.1038/345544a0. [DOI] [PubMed] [Google Scholar]
- Lee CH, Leeds P, Ross J. Purification and characterization of a polysome-associated endoribonuclease that degrades c-myc mRNA in vitro. J Biol Chem. 1998;273:25261–71. doi: 10.1074/jbc.273.39.25261. [DOI] [PubMed] [Google Scholar]
- Lohrum MA, Ludwig RL, Kubbutat MH, Hanlon M, Vousden KH. Regulation of HDM2 activity by the ribosomal protein L11. Cancer Cell. 2003;3:577–87. doi: 10.1016/s1535-6108(03)00134-x. [DOI] [PubMed] [Google Scholar]
- Ma WJ, Cheng S, Campbell C, Wright A, Furneaux H. Cloning and characterization of HuR, a ubiquitously expressed Elav-like protein. J Biol Chem. 1996;271:8144–51. doi: 10.1074/jbc.271.14.8144. [DOI] [PubMed] [Google Scholar]
- Mamane Y, Petroulakis E, Martineau Y, Sato TA, Larsson O, Rajasekhar VK, Sonenberg N. Epigenetic activation of a subset of mRNAs by eIF4E explains its effects on cell proliferation. PLoS ONE. 2007;2:e242. doi: 10.1371/journal.pone.0000242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamane Y, Petroulakis E, Rong L, Yoshida K, Ler LW, Sonenberg N. eIF4E--from translation to transformation. Oncogene. 2004;23:3172–9. doi: 10.1038/sj.onc.1207549. [DOI] [PubMed] [Google Scholar]
- Marshall L, Kenneth NS, White RJ. Elevated tRNA(iMet) synthesis can drive cell proliferation and oncogenic transformation. Cell. 2008;133:78–89. doi: 10.1016/j.cell.2008.02.035. [DOI] [PubMed] [Google Scholar]
- Menne TF, Goyenechea B, Sanchez-Puig N, Wong CC, Tonkin LM, Ancliff PJ, Brost RL, Costanzo M, Boone C, Warren AJ. The Shwachman-Bodian-Diamond syndrome protein mediates translational activation of ribosomes in yeast. Nat Genet. 2007;39:486–95. doi: 10.1038/ng1994. [DOI] [PubMed] [Google Scholar]
- Menssen A, Hermeking H. Characterization of the c-MYC-regulated transcriptome by SAGE: identification and analysis of c-MYC target genes. Proc Natl Acad Sci U S A. 2002;99:6274–9. doi: 10.1073/pnas.082005599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naora H, Takai I, Adachi M, Naora H. Altered cellular responses by varying expression of a ribosomal protein gene: sequential coordination of enhancement and suppression of ribosomal protein S3a gene expression induces apoptosis. J Cell Biol. 1998;141:741–53. doi: 10.1083/jcb.141.3.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noubissi FK, Elcheva I, Bhatia N, Shakoori A, Ougolkov A, Liu J, Minamoto T, Ross J, Fuchs SY, Spiegelman VS. CRD-BP mediates stabilization of betaTrCP1 and c-myc mRNA in response to beta-catenin signalling. Nature. 2006;441:898–901. doi: 10.1038/nature04839. [DOI] [PubMed] [Google Scholar]
- Oskarsson T, Trumpp A. The Myc trilogy: lord of RNA polymerases. Nat Cell Biol. 2005;7:215–7. doi: 10.1038/ncb0305-215. [DOI] [PubMed] [Google Scholar]
- Panic L, Tamarut S, Sticker-Jantscheff M, Barkic M, Solter D, Uzelac M, Grabusic K, Volarevic S. Ribosomal protein S6 gene haploinsufficiency is associated with activation of a p53-dependent checkpoint during gastrulation. Mol Cell Biol. 2006;26:8880–91. doi: 10.1128/MCB.00751-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel JH, Loboda AP, Showe MK, Showe LC, McMahon SB. Analysis of genomic targets reveals complex functions of MYC. Nat Rev Cancer. 2004;4:562–8. doi: 10.1038/nrc1393. [DOI] [PubMed] [Google Scholar]
- Pelengaris S, Khan M, Evan G. c-MYC: more than just a matter of life and death. Nat Rev Cancer. 2002a;2:764–76. doi: 10.1038/nrc904. [DOI] [PubMed] [Google Scholar]
- Pelengaris S, Khan M, Evan GI. Suppression of Myc-induced apoptosis in beta cells exposes multiple oncogenic properties of Myc and triggers carcinogenic progression. Cell. 2002b;109:321–34. doi: 10.1016/s0092-8674(02)00738-9. [DOI] [PubMed] [Google Scholar]
- Pelengaris S, Littlewood T, Khan M, Elia G, Evan G. Reversible activation of c-Myc in skin: induction of a complex neoplastic phenotype by a single oncogenic lesion. Mol Cell. 1999;3:565–77. doi: 10.1016/s1097-2765(00)80350-0. [DOI] [PubMed] [Google Scholar]
- Ridanpaa M, van Eenennaam H, Pelin K, Chadwick R, Johnson C, Yuan B, van Venrooij W, Pruijn G, Salmela R, Rockas S, Makitie O, Kaitila I, de la Chapelle A. Mutations in the RNA component of RNase MRP cause a pleiotropic human disease, cartilage-hair hypoplasia. Cell. 2001;104:195–203. doi: 10.1016/s0092-8674(01)00205-7. [DOI] [PubMed] [Google Scholar]
- Rousseau D, Kaspar R, Rosenwald I, Gehrke L, Sonenberg N. Translation initiation of ornithine decarboxylase and nucleocytoplasmic transport of cyclin D1 mRNA are increased in cells overexpressing eukaryotic initiation factor 4E. Proc Natl Acad Sci U S A. 1996;93:1065–70. doi: 10.1073/pnas.93.3.1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubbi CP, Milner J. Disruption of the nucleolus mediates stabilization of p53 in response to DNA damage and other stresses. Embo J. 2003;22:6068–77. doi: 10.1093/emboj/cdg579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggero D, Grisendi S, Piazza F, Rego E, Mari F, Rao PH, Cordon-Cardo C, Pandolfi PP. Dyskeratosis congenita and cancer in mice deficient in ribosomal RNA modification. Science. 2003;299:259–62. doi: 10.1126/science.1079447. [DOI] [PubMed] [Google Scholar]
- Ruggero D, Montanaro L, Ma L, Xu W, Londei P, Cordon-Cardo C, Pandolfi PP. The translation factor eIF-4E promotes tumor formation and cooperates with c-Myc in lymphomagenesis. Nat Med. 2004;10:484–6. doi: 10.1038/nm1042. [DOI] [PubMed] [Google Scholar]
- Ruggero D, Pandolfi PP. Does the ribosome translate cancer? Nat Rev Cancer. 2003;3:179–92. doi: 10.1038/nrc1015. [DOI] [PubMed] [Google Scholar]
- Sakamuro D, Prendergast GC. New Myc-interacting proteins: a second Myc network emerges. Oncogene. 1999;18:2942–54. doi: 10.1038/sj.onc.1202725. [DOI] [PubMed] [Google Scholar]
- Scheper GC, van der Knaap MS, Proud CG. Translation matters: protein synthesis defects in inherited disease. Nat Rev Genet. 2007;8:711–23. doi: 10.1038/nrg2142. [DOI] [PubMed] [Google Scholar]
- Schlosser I, Holzel M, Murnseer M, Burtscher H, Weidle UH, Eick D. A role for c-Myc in the regulation of ribosomal RNA processing. Nucleic Acids Res. 2003;31:6148–56. doi: 10.1093/nar/gkg794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sears RC. The life cycle of C-myc: from synthesis to degradation. Cell Cycle. 2004;3:1133–7. [PubMed] [Google Scholar]
- Spencer CA, Groudine M. Control of c-myc regulation in normal and neoplastic cells. Adv Cancer Res. 1991;56:1–48. doi: 10.1016/s0065-230x(08)60476-5. [DOI] [PubMed] [Google Scholar]
- Sun XX, Dai MS, Lu H. 5-fluorouracil activation of p53 involves an MDM2-ribosomal protein interaction. J Biol Chem. 2007;282:8052–9. doi: 10.1074/jbc.M610621200. [DOI] [PubMed] [Google Scholar]
- Sun XX, Dai MS, Lu H. Mycophenolic acid activation of p53 requires ribosomal proteins L5 and L11. J Biol Chem. 2008;283:12387–92. doi: 10.1074/jbc.M801387200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von der Lehr N, Johansson S, Wu S, Bahram F, Castell A, Cetinkaya C, Hydbring P, Weidung I, Nakayama K, Nakayama KI, Soderberg O, Kerppola TK, Larsson LG. The F-box protein Skp2 participates in c-Myc proteosomal degradation and acts as a cofactor for c-Myc-regulated transcription. Mol Cell. 2003;11:1189–200. doi: 10.1016/s1097-2765(03)00193-x. [DOI] [PubMed] [Google Scholar]
- Warner JR. The economics of ribosome biosynthesis in yeast. Trends Biochem Sci. 1999;24:437–40. doi: 10.1016/s0968-0004(99)01460-7. [DOI] [PubMed] [Google Scholar]
- Wendel HG, De Stanchina E, Fridman JS, Malina A, Ray S, Kogan S, Cordon-Cardo C, Pelletier J, Lowe SW. Survival signalling by Akt and eIF4E in oncogenesis and cancer therapy. Nature. 2004;428:332–7. doi: 10.1038/nature02369. [DOI] [PubMed] [Google Scholar]
- Yeilding NM, Rehman MT, Lee WM. Identification of sequences in c-myc mRNA that regulate its steady-state levels. Mol Cell Biol. 1996;16:3511–22. doi: 10.1128/mcb.16.7.3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Pan X, Hershey JW. Individual overexpression of five subunits of human translation initiation factor eIF3 promotes malignant transformation of immortal fibroblast cells. J Biol Chem. 2007;282:5790–800. doi: 10.1074/jbc.M606284200. [DOI] [PubMed] [Google Scholar]
- Zhang S, Shi M, Hui CC, Rommens JM. Loss of the mouse ortholog of the shwachman-diamond syndrome gene (Sbds) results in early embryonic lethality. Mol Cell Biol. 2006;26:6656–63. doi: 10.1128/MCB.00091-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Wagner BJ, Ehrenman K, Schaefer AW, DeMaria CT, Crater D, DeHaven K, Long L, Brewer G. Purification, characterization, and cDNA cloning of an AU-rich element RNA-binding protein, AUF1. Mol Cell Biol. 1993;13:7652–65. doi: 10.1128/mcb.13.12.7652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Wolf GW, Bhat K, Jin A, Allio T, Burkhart WA, Xiong Y. Ribosomal protein L11 negatively regulates oncoprotein MDM2 and mediates a p53-dependent ribosomal-stress checkpoint pathway. Mol Cell Biol. 2003;23:8902–12. doi: 10.1128/MCB.23.23.8902-8912.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]