

Abstract
Signaling by the peptide ligand apelin and its cognate G-protein coupled receptor APJ has a potent inotropic effect on cardiac contractility and modulates systemic vascular resistance through nitric oxide dependent signaling. In addition, there is evidence for counter-regulation of the angiotensin and vasopressin pathways. Regulatory stimuli of the apelin-APJ pathway are of obvious importance, but remain to be elucidated. To better understand the physiological response of apelin-APJ to disease states such as heart failure and elucidate the mechanism by which such a response might occur, we have utilized the murine model of LAD-ligation induced ischemic cardiac failure. To identify the key cells responsible for modulation and production of apelin in vivo, we have created a novel apelin-lacZ reporter mouse. Data from these studies demonstrate apelin and APJ are upregulated in the heart and skeletal muscle following myocardial injury, and suggest that apelin expression remains restricted to the endothelium. In cardiac failure, endothelial apelin expression correlates with other hypoxia-responsive genes, and in healthy animals both apelin and AJP are markedly upregulated in various tissues following systemic hypoxic exposure. Experiments with cultured endothelial cells in vitro show apelin mRNA and protein levels to be increased by hypoxia, through a HIF-mediated pathway. These studies suggest that apelin-expressing endothelial cells respond to conditions associated with heart failure, possibly including local tissue hypoxia, and modulate apelin-APJ expression to regulate cardiovascular homeostasis. The apelin-APJ pathway may thus provide a mechanism for systemic endothelial monitoring of tissue perfusion and adaptive regulation of cardiovascular function.
Keywords: congestive heart failure, endothelium, gene expression
Introduction
APJ is a seven transmemberane domain G-coupled protein receptor for which apelin remains the only known ligand (24). Apelin is a highly conserved 77 amino-acid pre-pro peptide, cleaved to shorter peptides in various tissues (33). Given the cell and developmental-specific pattern of expression of apelin and APJ in vascular and cardiac structures, and initial studies in developmental model organisms, it is likely that this pathway has a fundamental role in embryogenesis of the cardiovascular system (9, 12, 15, 19, 31, 38). In the adult cardiovascular system, both APJ and apelin are expressed in the endothelium of heart, kidney, and lung, and APJ is expressed by myocardial cells and some vascular smooth muscle cells (6, 20, 21).
A growing body of literature suggests that the apelin-APJ pathway has direct effects on both cardiac and vascular functions. Data from experimental models indicate that signaling through the apelin-APJ pathway increases cardiac contractility. In the isolated rat heart, Szokodi et al. described a positive and potent effect of apelin on externally developed tension and pre-load recruitable maximum rate of developed pressure (32). Others have found that echo-derived load dependent measures of contractile function are significantly increased with chronic administration of apelin in normal mice, and that load independent measures of left ventricular performance show increased contractility in normal mice and rats that were administered exogenous apelin (2, 5). Interestingly, chronic infusion of apelin was found to improve cardiac function without concomitant hypertrophy of the heart, a seemingly unique characteristic of this peptide compared to other known inotropic agents, either endogenous or synthetic (2). Experiments employing myocardial injury models have suggested that apelin has a salutary positive inotropic effect on failing myocardium (3, 18). Beyond modulating cardiac inotropy, the apelin pathway also regulates vasomotor tone. While early in vitro studies demonstrated that apelin acts as a venous vasoconstrictor, recent studies support a vasodepressor role in both the arterial and venous circulation (7, 23, 34). Apelin-induced hypotension was abrogated by co-administration of nitric oxide synthase blockers, implicating the nitric oxide-system as one downstream effector of the apelin-APJ signaling pathway in the vasculature (16, 34). Recent studies of a mouse model lacking endogenous apelin have suggested that the apelin-APJ pathway is important for maintaining cardiac function with ageing and pressure overload (22).
Aside from its direct effects on the heart and circulation, the apelin-APJ pathway has additional actions that impact cardiovascular physiology. Apelin and APJ are expressed in central regulatory areas of the brain, and central administration of apelin peptide has suggested this pathway is involved in fluid homeostasis and central autonomic regulation of cardiovascular function (10, 11, 26, 27). In addition, there are data suggesting that the apelin-APJ pathway functionally counter-regulates the actions of vasopressin and angiotensin (1, 11, 17).
Although studies in animal models and humans suggest cardiac failure results in an altered balance of apelin and APJ expression, the precise disease-related regulatory mechanisms for apelin and APJ expression have not been investigated. The primary goals of the present study were to (1) use a well characterized murine model of ischemic heart failure to establish the longitudinal changes in apelin-APJ expression by various tissues in the setting of chronic heart failure, (2) to identify the cells responsible for apelin production and their response to cardiac failure in vivo, and (3) explore hypoxia as one of the key in vivo stimuli responsible for failure-induced perturbation of apelin-APJ by utilizing both in vivo and cell culture models of hypoxic stress.
Materials and Methods
Animal models
Animal care was provided in accordance with the Laboratory Animal Welfare Act, the Guide for the Care and Use of Laboratory Animals (National Institutes of Health, publication 78-23, revised 1978) and the Stanford University School of Medicine guidelines and policies for the use of laboratory animals for research and teaching.
Eighty-six adult, female SVJ129 mice (Jackson Laboratories, Bar Harbor, ME) and 12 female apelin-LacZ gene targeted (apelin+/lacZ) mice were used. Targeted apelin+/lacZ animals were created on the SVJ129 background by insertion of the bacterial lacZ gene with a nuclear localizing signal in the first apelin exon immediately upstream of the translation start site (Charo et al., submitted). Specific gene targeting replaced the murine apelin ATG and leader sequence with the lacZ gene containing a nuclear localization signal, with no deletion of upstream or intronic sequence that might serve to regulate transcription.
For the surgical heart failure model, wildtype (WT) control animals were randomized to sham operation (n=13), or underwent left anterior descending coronary artery (LAD) ligation (n=39) as previously described (4). To assess cellular localization of post-LAD ligation apelin expression, hearts were harvested 8 weeks post-operatively from additional apelin+/lacZ reporter mice that underwent sham procedure (n=3) or LAD ligation (n=3).
A separate cohort of 26 female mice was used to study the effects of systemic hypoxia upon apelin and APJ expression in vivo. Mice were randomized to hypoxia (n=10 WT, 3 apelin+/lacZ) or control group (n=10 WT, 3 apelin+/lacZ). Hypoxic exposure was achieved by housing animals in a tightly sealed normobaric hypoxic chamber with an oxygen fraction of 10% for 7 days. Control animals were kept at room air (FiO2 of 21%).
Echocardiography was performed by two independent, blinded operators using the Siemens-Acuson Sequioa C512 system equipped with a multi-frequency (8–14 MHz) 15L8 transducer. Mice were assessed pre-operatively, and 4, 8, and 12 weeks post LAD-ligation. Animals were induced with isoflourane and received continuous inhaled anesthetic (1.5–2%) for the duration of the imaging session (10–15 minutes). Analysis of the M-Mode images was performed in a blinded fashion using Siemens built-in analysis software. Left ventricular end diastolic diameter (EDD) and end-systolic diameter (ESD) were measured and used to calculate fractional shortening (FS) by the following formula: FS= [EDD-ESD]/EDD.
Peripheral oxyhemoglobin saturation was measured using the veterinary Nonin 8600V pulse oximeter equipped with the 2000SL small animal sensor (Nonin Medical, Plymouth, MN). Measurements were carried out by two blinded technicians on a cohort of 14 wildtype (WT) animals pre-operatively and then at 4 and 8 weeks following LAD ligation (n=7) or sham procedure (n=7). Animals were induced as above and maintained on 3–4% inhaled isoflorane in 100% oxygen via nose cone. The right lower extremity was prepared by application of topical depilatory agent followed by placement of the sensor over the inguinal region. Detection of a valid signal was confirmed by comparing sensor measured heart rate to concomitant electrocardiographic assesement of heart rate. Ten to fifteen readings were acquired per animal.
Tissue processing and quantitation of mRNA
Quantitation of RNA was performed on heart, lung, and quadriceps muscle tissue collected at weeks 4, 8, and 12 following LAD ligation or 4 weeks following the sham procedure. For animals in the systemic hypoxia study, tissues were harvested after one week of continual hypoxia or normoxia. The apelin+/lacZ animals underwent perfusion fixation for Xgal staining and histological analysis.
Tissue samples (heart, lung, quadriceps) were thawed and homogenized in RLT lysis buffer (Qiagen, Valencia, CA), followed by RNA isolation using the RNeasy Midi Kit (Qiagen). For RNA isolation from in vitro experiments, cells were lysed using Trizol (Invitrogen, Carlsbad, Calif.), followed by chloroform extraction and purification using the RNeasy Mini Kit (Qiagen). Purified RNA was reverse transcribed with Superscript II (Invitrogen). Real-time polymerase chain reaction (RT-PCR) was performed on a 7900HT Sequence Detection System with TaqMan Assays on Demand gene expression probes (systems and probes from Applied Biosystems, Foster City, Calif).
Immunohistochemical analysis
Animals were intubated and perfusion fixation was carried out for ~2 minutes at 120mm Hg with 4% paraformaldehyde (Sigma, St. Louis, Mo) in phosphate buffered saline (PBS) at pH 7.4. Tissues were harvested for immersion fixation for 1–2 hours and then processed for histology by embedding in paraffin. Blocks were sectioned and stained with Hematoxylin and Eosin, or Masson-Trichrome.
Apelin+/lacZ reporter mice were perfusion fixed with either 0.25% glutaraldehyde (for whole mount staining) or 4% paraformaldehyde (PFA, for immunohistochemistry) followed by immersion fixation of tissues for 1–2 hours. For whole mount staining, tissues were immersed in Xgal substrate solution for 4–12 hours at 29° C. Tissues were then post- fixed in 0.25% glutaraldehyde overnight, followed by embedding in paraffin, sectioning and counterstaining with nuclearfast red (Biomedia, Foster City, CA). LacZ expressing cells were quantified in randomly selected, high power (20x) views of tissue sections by two individual technicians blinded to the study.
For co-staining of tissues with CD31 and Xgal, whole mount stained tissues were embedded in OCT and 10µm thick sections created. Sections were then stained with anti-CD31 (clone MEC13.3, Pharmingen) at 5 ng/ml using Biocare Medical Rat Detection Kit (Biocare Medical, Concord, CA) per manufacturer’s instructions.
For immunohistochemical analysis of pulmonary nuclear lacZ expression, lungs were excised en-bloc and inflated with 4% PFA at 25mm H20 of pressure. Fixed lungs were immersed in 30% sucrose overnight, embedded into OCT, frozen, and prepared into 10-micron thick frozen sections. Anti-β-galactosidase (rabbit polyclonal, Chemicon, Foster City, CA) or chicken polyclonal, (Immunology Consultants Lab, Newburg, OR) staining was carried out in combination with either anti-CD31 (clone MEC13.3, Pharmingen), anti-surfactant protein C (rabbit polyclonal, Seven Hills Bioreagents, Cincinatti, Ohio), or anti-RAGE (mouse type I pneumocyte specific; rat monoclonal 175410, R&D Systems, Minneapolis, MN). Primary antibodies were all used at 1 µg/mL with secondary antibodies conjugated with either FITC or Cy3 (Jackson Immunoresearch, West Grove, PA). Confocal microscopy was performed on a Leica SP5 confocal system (Leica, Wetzlar, Germany).
Cell culture studies
Human coronary artery endothelial cells (HCAEC), human dermal microvascular endothelial cells (HMVEC-D), human pulmonary artery endothelial cells (HPAEC), and supplemented EGM-2 MV media were purchased from Cambrex Bio Science (Wakersville, MD). Cells were maintained per manufacturer’s instructions and used at P5-9. Human embryonic kidney 293 (HEK-293) and ECV-304 cell lines were cultured in supplemented D-MEM media.
In vitro hypoxia experiments were performed with cells grown to 80–90% confluency and serum starved for 16 hours prior to hypoxic exposure. Plates were placed in a humidified Billups-Rothenberg modular incubation chamber (model MIC-101, Billups-Rothenberg, Del Mar, CA), charged with a gas mixture of 1% O2/5%CO2/94%N2 and sealed prior to placement in a tissue culture incubator. Hypoxic exposure was carried out for 4 (ECV-304), 24 (HCAEC, HMVEC-D), or 48 hours (HPAEC), followed by isolation of RNA. Control cells were kept at ambient oxygen concentrations.
HEK-293 cells were transfected with DNA expression vectors encoding either a constitutively active form of Hif-1α (HIF-ODD) or HIF-2α as previously described (25). RNA was isolated from cells 48 hours following transfection and apelin mRNA quantitated as above.
For measurement of soluble apelin in cell culture, media collected from cultured HCAEC following hypoxic exposure was assayed for soluble apelin concentration with Pheonix Pharmaceutical’s Apelin-12 ELISA (Pheonix Pharmaceuticals, Burlingame, CA) per manufacturer’s instructions (6).
Statistical analysis
Experimental results were expressed in graphs and text as mean ± 95% CI (for data from the animal cohort) or mean ± SD (for in vitro data). Normal distribution was tested for all the experimental variables by Kolmogorov-Smirnov test and non-parametrically distributed variables normalized by rescaling to 10-based logarithm or square root, as appropriate.
Presence of significant differences in fractional shortening change and gene expression among the four experimental groups was assessed by ANOVA. Presence of linear or quadratic trends in parameters distribution was checked by error bar graph and confirmed by the appropriate polynomial contrast model. Post-hoc comparisons were performed using LSD multiple comparisons approach after verification of the homogeneity of variance assumption.
Pearson’s linear regression test was used to assess the presence of bivariate correlations between BNP expression and apelin-APJ expression, as well as fractional shortening.
Correlation heat map was generated by using the Heatmapbuilder software developed in this laboratory (http://quertermous.stanford.edu/heatmap.htm).
In vitro data were compared using ANOVA or two-tailed, non-paired Student’s t-test where appropriate. The level of significance was set at p < 0.05 and software package SPSS 12.0 for Windows (SPSS Inc., Chicago) was used for computations.
Results
Apelin and APJ are upregulated in the heart and peripheral muscle with ischemic heart failure
To profile apelin and APJ gene expression changes in a clinically relevant and reliable model of progressive heart failure, we utilized the murine LAD-ligation heart failure model. Cardiac failure following LAD-ligation was confirmed by serial echocardiography, RT-PCR analysis of the myocardium for β-natriuretic peptide (BNP, a clinically used marker of heart failure), and histological evaluation of the hearts at different time points. All three assays revealed progressive heart failure similar to that observed in patients, with a 41.7% reduction in mean left ventricular shortening and significant increase in BNP by post-LAD ligation week 8 (Supplemental Figure 1).
We chose to focus our expression analysis on three specific tissues: heart, lung, and skeletal muscle (quadriceps). The heart and lungs were chosen as they are integral in the pathophysiology of congestive heart failure. The quadriceps muscle was included as a representative model tissue affected by decreased systemic perfusion and an ideal site where vasomotor tone may be significantly modulated by changes in apelin-APJ expression.
Evaluation of apelin expression in the myocardium revealed a modest but significant increase in cardiac apelin expression as cardiac failure progressed into 8 weeks (Figure 1, top row). The upregulation persisted through week 12, although a trend towards diminished apelin expression was present at that time. Cardiac APJ upregulation paralleled that of apelin, with a significant near 2-fold upregulation at weeks 8 and 12. For both apelin and APJ, we observed a non-significant diminution of expression at 4 weeks compared to sham, similar to that observed for BNP mRNA levels, likely resulting from the loss of myocardial (APJ expressing) cells and endothelial (apelin and APJ expressing) cells in the sizable infarct zones. Although both apelin and APJ expression in the lung demonstrated trends similar to those observed in the myocardium (Figure 1, bottom row), these expression changes did not reach statistical significance. Expression of another protein known to regulate vasomotor tone, pulmonary angiotensin converting enzyme (ACE), also did not demonstrate significant changes over time (data not shown).
Figure 1.

Ischemic cardiac failure induces upregulation of apelin and APJ mRNA in the heart and skeletal muscle. Apelin (left column) and APJ (right column) mRNA levels (as measured by RT-PCR) in the heart (top row) increase as heart failure progresses, with significantly elevated levels by week 8 following ischemic injury (n=11 per group; bars represent means ± 95% CI; * P<0.001; **P<0.05). This pattern is consistent with known markers of heart failure in this model, such as β-natreutic peptide (Supplemental Figure 1). Apelin expression in the quadriceps muscle also increases, as early as week 4, reaching a peak by 8 weeks (* P<0.0001, **P<0.03). Quadriceps APJ is upregulated in a similar fashion following LAD ligation, but continues to increase through 12 weeks (*P<0.016, **P<0.0001, ***P<0.049). Pulmonary apelin and APJ levels (bottom row) are not significantly altered in ischemic heart failure.
Within the quadriceps, apelin was significantly upregulated as early as 4 weeks post-LAD ligation (as opposed to the 8 week response in the heart). Specifically, quadriceps apelin mRNA increased by 2.4 fold by week 4 and remained elevated through week 8 compared to sham (Figure 1, middle row). Expression tapered somewhat to 2.1 fold by week 12, although still significantly elevated compared to sham. APJ expression was similarly elevated at 4 weeks, and continued to show a linear increase up to 12 weeks, with a 3-fold increase over sham at that time.
Localization of apelin expression using a lacZ reporter mouse
To identify the cells responsible for apelin expression and evaluate their response to disease states such as congestive heart failure, we created a transgenic apelin-lacZ (apelin+/lacZ) reporter mouse in which the bacterial lacZ gene was integrated into the apelin locus.
We first localized reporter gene expression in the three tissues analyzed in the LAD ligation model (heart, lung, and skeletal muscle) utilizing the well-characterized Xgal assay. Study of all three tissue types from healthy transgenic animals with Xgal staining and CD31 immunostaining revealed lacZ to be primarily expressed by CD31 labeled endothelium (Figure 2, lower row panels). Xgal staining was specifically restricted to capillaries and veins (Figure 2, upper and middle row panels). Notably, in both the heart and skeletal muscle, post-arterial capillaries and venous endothelium universally expressed the reporter protein, while large arteries and arterioles did not demonstrate reporter expression. Within the lung, alveolar septal capillaries and pulmonary veins all expressed the reporter protein as well, with no arterial expression observed. These patterns of expression for the lacZ reporter are consistent with published in situ hybridization and immunohistochemistry studies of apelin expression (20, 30). Tissues from WT animals processed for Xgal staining demonstrated no blue staining (data not shown).
Figure 2.

Tissues from the transgenic apelin+/lacZ reporter mouse demonstrate the apelin reporter gene is primarily expressed by the endothelium. Top two rows: staining for the apelin-LacZ reporter within the heart (left), quadriceps (middle), and lung (right column), reveals lacZ expression (blue) by vessels within the tissues. Endothelial cells of capillaries (black arrowheads) and veins (“V”) universally express the reporter, but arteries (“A”) do not. Bottom row: co-staining for LacZ reporter (blue) and the endothelial marker, CD31 (brown), confirms endothelial phenotype of apelin reporter-expressing cells (scale bars=25 µm). Three dimensional reconstruction (by serial confocal microscopy) of the same sections co-stained with fluorescent antibodies against LacZ and CD31 confirmed lacZ nuclei to be contained by endothelial cytosol (see Supplemental Videos 1 and 2).
To confirm that apelin reporter gene expression was limited to the endothelium in the lung (where histological distinction of endothelium may be difficult), we also carried out immunohistochemical staining for RAGE and surfactant protein C, targeting type I and II pneumocytes, respectively. Both of these cytosolic proteins failed to colocalize with nuclear lacZ, while CD31 staining confirmed that lacZ expression was restricted to pulmonary endothelial cells (Supplemental Figure 2). Integration along the Z-axis of multiple, consecutive confocal images further confirmed localization of the Xgal stained nuclei within the endothelium (Supplemental Video 1). Similar co-staining of the skeletal muscle for both nuclear lacZ and CD31 followed by Z-stack image analysis also revealed colocalization of the apelin reporter with capillary endothelium (Supplemental Video 2).
Endothelial apelin response to ischemic cardiac failure
We next carried out LAD-ligations in apelin+/lacZ mice to evaluate cell-specific changes in apelin expression following myocardial insult. Histologic assessment of reporter expression in the infarcted hearts of the apelin+/lacZ reporter mice revealed increased numbers of apelin reporter expressing endothelial cells in the healing infarct zone by 8 weeks, representing neovascularization associated with the healing process (Figure 3A). In addition, within regions distant from the infarct zone, we observed an increase in number of Xgal staining cells (Figure 3A and B). When serial sections of left ventricular, septal, and right ventricular tissues were evaluated, there was a significant average 2-fold increase in number of lacZ positive cells (P<0.001) with LAD ligation. Overall, apelin reporter expression remained restricted to capillary and venous endothelial cells, suggesting increased expression by those vessels, and not recruitment of arterial endothelium or de novo expression by other cell types such as cardiomyocytes. Similarly, in the skeletal muscle, LAD-ligation resulted in an increased number of apelin reporter-expressing capillary and venous endothelial cells, but no evidence of reporter protein production by other cell types was observed.
Figure 3.
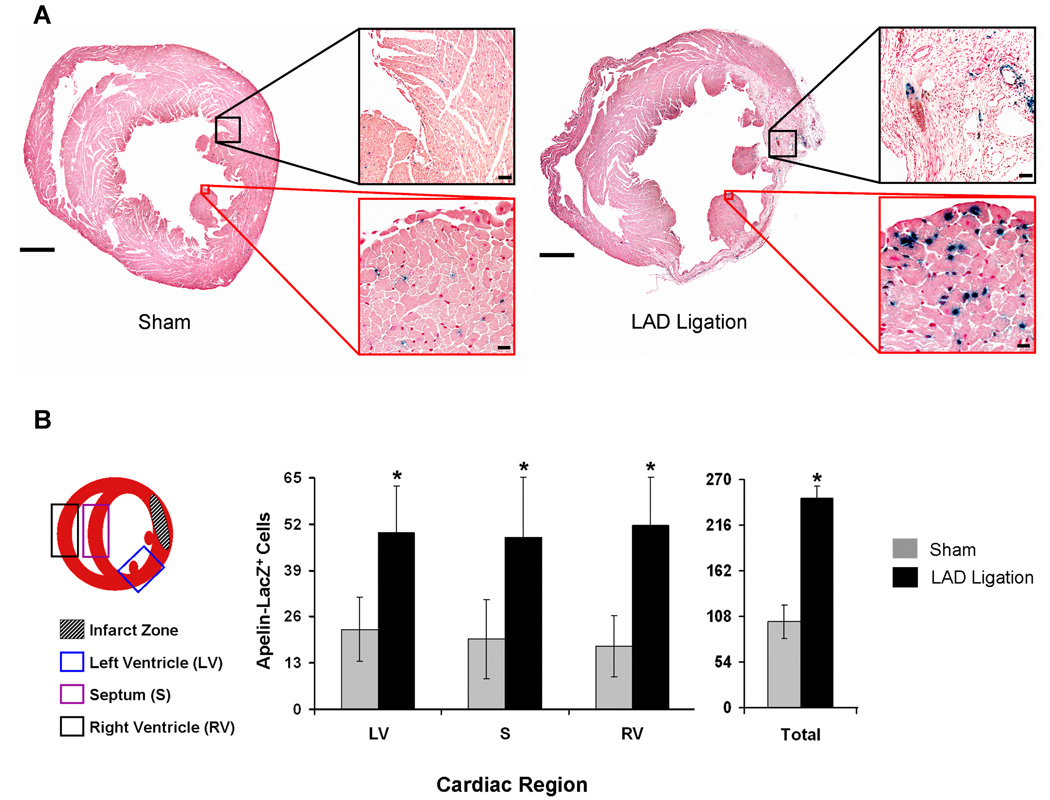
Ischemic myocardial failure induced by LAD-ligation results in upregulation of the apelin reporter by coronary endothelium. A, Images show representative whole heart sections from transgenic apelin lacZ reporter mice 8 weeks following sham (left) and LAD ligation (right), scale bars=1 mm. Apelin reporter expression can be seen in surviving endothelium as well as neoangiogenic cells within the areas of myocardial infarct following LAD ligation (black-bordered inlays, scale bars=175µm). Intense Xgal staining is observed in endothelium remote from the infarct, suggesting increased apelin expression by capillary endothelial cells in the spared portions of the myocardium (red-bordered inlays, scale bars=25µm). B, LAD ligation leads to increased number of apelin reporter protein producing cells at sites remote from the infarction region. The graph depicts mean number of cells±SD over three general areas of interest: left ventricle (LV), septum (S), right ventricle (RV). Mean total cell counts from all areas (LV+S+RV) are given on the right. Cells were counted in a blinded fashion over 4–6 high power fields (20x) in each region of interest (n=2 hearts per group, *P<0.001).
Hypoxia-regulated genes are upregulated in a pattern similar to that of apelin-APJ in response to progressive heart failure
Given the gene expression changes in the murine infarct model as well as the predominance of apelin expression by endothelial cells, we explored possible common endothelial mechanisms for upregulation of apelin and APJ in the heart and peripheral muscle. One regulatory mechanism of interest was the hypoxia-regulated pathway, as previously suggested (9, 29). To confirm that hypoxia was in fact a stimulus associated with the LAD-ligation model, we measured blood oxygen saturation in the vessels feeding the quadriceps by pulse oximetry, a methodology shown to detect decreased blood oxygen content in humans with heart failure (Figure 4A) (28, 37). Preoperative oxygen saturations were determined to be 100 % in all animals and remained at this level following the sham procedure. By comparison, LAD ligation produced a significant drop in oxygen saturation by 4 weeks (96±2.1%), with further decline by week 8 (92±3.4%). To investigate the significance of these changes in oxygen tension, we assayed expression levels of genes known to be regulated by hypoxia in quadriceps tissue from the LAD-ligated animals. Hypoxia responsive genes included tumor necrosis factor alpha (TNF-α), vascular endothelial cell growth factor-A (VEGF-A), and hypoxia inducible factor 1alpha (HIF-1α). We found these validated target genes to be upregulated in a pattern similar to apelin and APJ (Figure 4B).
Figure 4.

LAD ligation-induced cardiac failure induces hypoxia of the skeletal muscle and upregulation of hypoxia-responsive genes. A, Oximetry measurements in quadriceps muscle demonstrate progressive hypoxia in murine peripheral skeletal muscle following LAD ligation compared to animals receiving sham procedure (n=5–7 animals per group, bars represent means±95%CI, *P<0.001). B, TNF-α, VEGF, and HIF-1α are significantly upregulated in the quadriceps muscle as heart failure progresses (n=11 per group; bars represent means ± 95% CI, P-values: *<0.045, **<0.0001, †<0.003, ††<0.009).
To better delineate the relationships between the hypoxic genes assayed, apelin and APJ, and other physiologic parameters measured in our LAD-ligation animals, we performed a multivariate correlation analysis and represented the data with an association matrix heatmap (Figure 5). Mapping data in this fashion revealed that both apelin and APJ expression changes occurred in concert with the panel of hypoxic genes and that there were significant, independent positive correlations between each of the known hypoxia genes, apelin, and APJ (Figure 5, r-values given in blue). Specifically, there was a strong positive correlation between apelin and APJ and HIF-1α in the skeletal muscle (p<0.01) similar to the relationship between HIF-1α and VEGF in the same tissue, suggesting a possible regulation of the apelin-APJ pathway by HIF-1α.
Figure 5.

”Expression heat map” of selected parameters and gene expression changes measured in the 44 animal cohort undergoing LAD-ligation or sham procedure. The map represents correlations between the expression patterns of genes as well as cardiac function (FS). Notable positive correlations include concerted upregulation of hypoxia responsive genes (VEGF, HIF-1α, TNF-α) and apelin/APJ. Cardiac β-natriuretic peptide (BNP) is inversely correlated to cardiac function as is apelin. Color scale for degree of correlation is given beneath the table, with green representing a negative correlation and red positive. The lower half of the table presents the same information in numerical format, with significant correlations (r-values) depicted in blue (FS= fractional shortening; *P<0.05, **P<0.01).
Hypoxia is a potent regulator of cardiac and pulmonary apelin-APJ expression in vivo
The previous data provides evidence that experimental CHF creates tissue hypoxia and simulates genetic upregulation of apelin and AJP gene expression, thus providing an association between tissue hypoxia and apelin-APJ gene expression. To fundamentally establish that apelin and APJ genes are regulated in vivo by hypoxia, we utilized a model of isolated hypoxic insult, avoiding confounding accompanying stimuli such as those found in the LAD-ligation myocardial failure model. WT and apelin+/lacZ reporter mice were exposed to systemic hypoxia (10 % FiO2) or kept at ambient oxygen conditions. Quantitative RT-PCR of cardiac and lung RNA revealed significant, near 8- and 4-fold hypoxia-induced upregulation of cardiac and pulmonary apelin, respectively (Figure 6A). Hypoxia also stimulated significant upregulation of APJ in these tissues, 2.2-fold in the heart and 3.2-fold in the lung. There was a 2-fold increase in mean apelin expression in skeletal muscle, however, inter-animal variation precluded these data achieving statistical significance. Interestingly, the cell-specific pattern of lacZ reporter gene expression in each of the three tissues examined remained constant, suggesting that apelin expression was limited to the endothelium. Quantitative analysis of the number of β-galactosidase expressing cells in the heart of apelin+/lacZ reporter mice revealed at least a 2-fold increase in the total number of staining nuclei in sections from the LV, RV and septum, suggesting the activation of apelin gene expression in additional endothelial cells under hypoxic conditions (data not shown).
Figure 6.
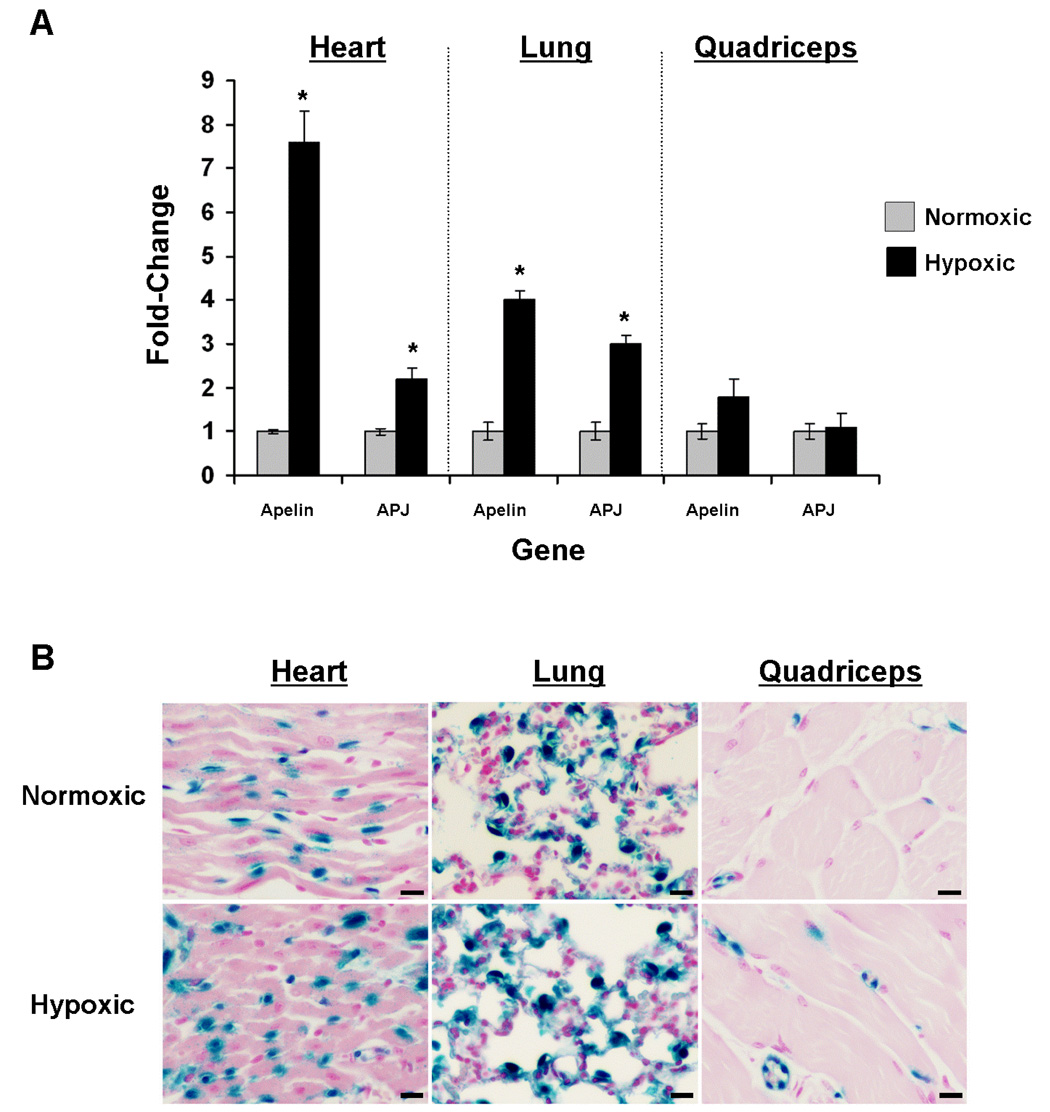
The apelin-APJ pathway is upregulated in endothelial cells following systemic hypoxia in vivo. A, Apelin and APJ expression are significantly increased in the heart and lung following continual, systemic hypoxia (10% FiO2) for 1 week (n=11 per group; bars represent means ± 95% CI; *P<0.001). In quadriceps tissue, apelin mRNA levels showed a nearly two-fold mean increase, but this difference did not reaching statistical significance. B, Representative histological sections of heart, lung, and quadriceps from apelin+/lacZ reporter mice kept under ambient normoxia (21% FiO2, top row) or hypoxia (10% FiO2) for 1 week, demonstrating that apelin reporter expression remains restricted to the endothelium and suggesting that increased tissue mRNA levels correlate to increased apelin expression by endothelial cells (scale bars=20 µm).
Hypoxia stimulates apelin expression in vitro and is regulated by HIF pathways
We next investigated hypoxia-induced apelin regulation in cultured human endothelial cells from the coronary artery (HCAEC), dermal microvasculature (HMVEC-D), and pulmonary artery (HPAEC). Hypoxia exposure induced 2.5-, 1.4, and 1.9-fold increases in apelin expression by HCAEC, HMVEC-D, and HPAEC, respectively (Figure 7A). We also tested hypoxia as a stimulus for apelin expression in a human-derived cell line (ECV-304) known to have responses similar to endothelial cells and found hypoxia induced a robust, near 23-fold increase in apelin expression in these cells.
Figure 7.
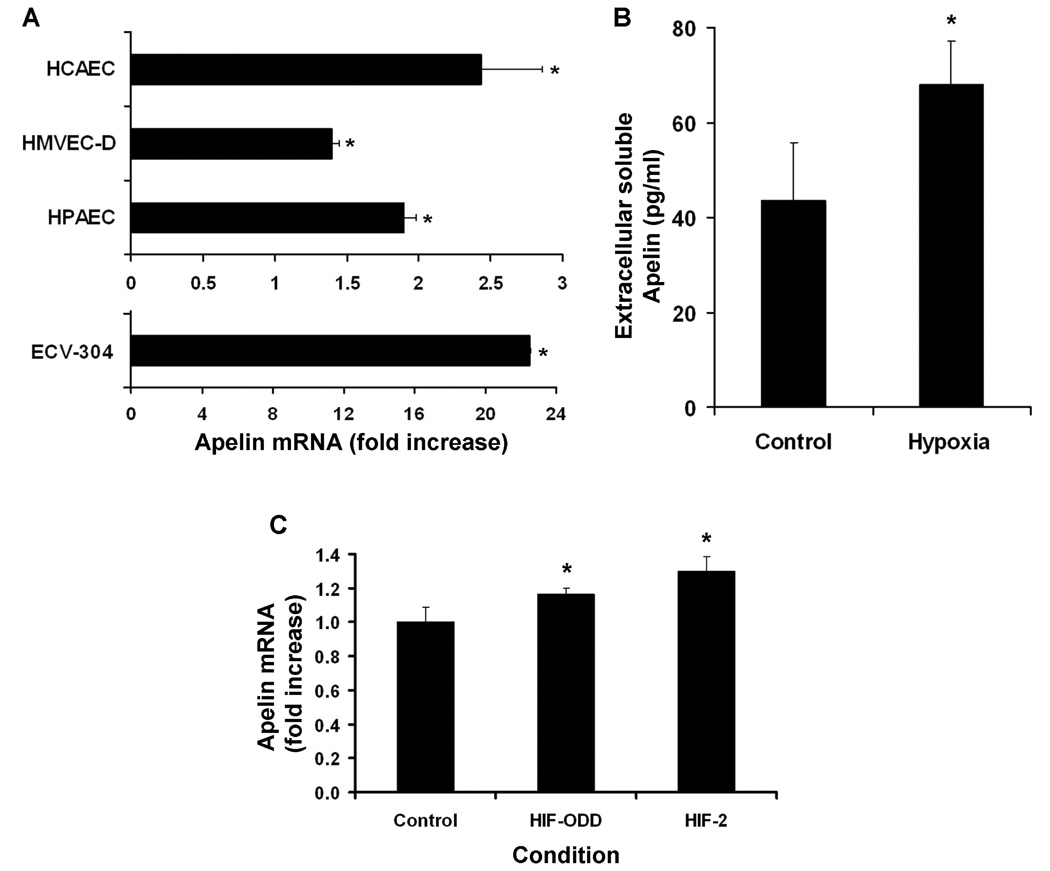
Hypoxia-induced apelin upregulation in human cultured human endothelial cells. A, Twenty-four hour hypoxic exposure (1%FiO2) resulted in significant upregulation of apelin by coronary artery endothelial cells (HCAEC), dermal microvascular endothelial cells (HMVEC-D), pulmonary artery endothelial cells (HPAEC) and ECV-304 cells in vitro (*P<0.005). B, Soluble apelin production (as determined by ELISA) by HCAEC is significantly increased following hypoxic exposure. C, Upregulation of apelin mRNA in cultured HEK293 cells transfected with constitutively active HIF-1α (HIF-ODD) and HIF-2α (*P<0.04, **P<0.003). In all panels, bars represent mean±SD from 2–5 representative experiments.
To confirm that hypoxia-induced upregulation of apelin transcript translated into increased apelin protein levels, media from HCAEC was assayed by ELISA for the biologically active apelin-12 isoform. Hypoxic exposure significantly increased extracellular soluble apelin from a baseline of 43.4±12.4 to 68.0±9.1 pg/ml (Figure 7B).
In order to evaluate the possibility of HIF-mediated downstream transcriptional regulation, we searched the human and mouse apelin promoter regions for known HIF binding domains using the NCBI-Entrez genome database and identified numerous putative binding regions, as previously described (Supplemental Table) (9). We then transfected HEK-293 cells with plasmids encoding for either the constitutively active form of HIF-1α (HIF-ODD) or HIF-2α. Both HIF-ODD and HIF-2α produced a significant upregulation of endogenous apelin gene expression, however, a greater induction was observed in HIF-2α transfected cells (Figure 7C).
Discussion
A growing number of studies have identified an association between the apelin-APJ pathway and cardiovascular disease states such as congestive heart failure (CHF). Initial work in this laboratory demonstrated that APJ was the most highly differentially expressed gene in the human failing heart among 12,000 screened, and that APJ and apelin expression increased with the compensation that occurs following left ventricular device placement and offloading of the failing ventricle (6). Studies of circulating apelin levels in subjects with varying degrees of heart failure suggested that downregulation of the apelin-APJ pathway is a feature of decompensated heart failure, but that in early and compensated heart failure the apelin-APJ pathway is upregulated. Other studies have found circulating apelin levels to be unchanged or decreased in various stages of human heart failure (8, 14, 36). These conflicting results may be due in part to heterogeneity of the study populations in terms of disease process, disease duration prior to enrolment, and co-existing medical conditions, but deserves further study.
Because of the disparity in human studies seeking to correlate circulating apelin levels with disease severity, and serious concern regarding the specificity and reproducibility of currently available immunoassays with blood measurements, we have undertaken studies at the genetic level in validated mouse models to gain insights regarding physiological regulators of gene expression as a guide for better understanding the biology of this pathway. In addition to quantitative RT-PCR studies of whole tissue mRNA levels, we have also utilized a gene targeted mouse with the bacterial lacZ reporter gene in the apelin locus, to study the cell-specificity of apelin expression in vivo in healthy and diseased animals. Overall, these studies strongly suggest that expression of the apelin-APJ pathway is upregulated at an early or compensated phase in heart failure, and correlates in a linear fashion with the degree of heart failure as assessed by echocardiographic indices and BNP expression. Indeed, the genetic response of both apelin and APJ at 4 weeks in skeletal muscle preceded the increase in cardiac BNP mRNA levels first detected at 8 weeks. Mice in the LAD ligation groups were considered to be in compensated failure, as they were not dying or exhibiting failure to thrive. Unfortunately, we were not able to correlate tissue expression levels of apelin to circulating peptide levels, as available immunoassays for rat and human apelin identify a cross-reacting antigen in mouse serum. However, a recent study in a rat model of ischemic heart failure provides evidence for increased APJ and apelin protein levels in failing heart, supporting our genetic studies (3). Also, increased apelin mRNA levels have been documented in human heart failure (14). Thus, there is genetic upregulation of the apelin-APJ pathway that is likely reflected in protein levels, and may serve to support cardiovascular homeostasis and systemic perfusion.
Localization of apelin expression to the endothelium as demonstrated by study of the apelin+/lacZ reporter mouse confirms observations by other investigators and provides further insight into the physiology of the pathway (20). Although in vitro studies have shown cultured myocytes to express apelin (29, 32), our studies suggest that in vivo, it is the endothelium that is responsible for the production of apelin peptide, even in the response to pathophysiologic stimuli such as cardiac injury and hypoxia. The observation that apelin expression is restricted to post-arterial (i.e., capillary and venous) endothelial cells has important implications regarding how the apelin pathway might “sense” and attempt to compensate for the decreased cardiac output characteristic of heart failure. Specifically, capillary endothelial cells are optimally situated to detect mismatches between blood and oxygen supply and demand at the tissue level. In addition, increased apelin produced by capillary endothelial cells would have little local function, arguing that apelin is released into the circulation to improve cardiac contractility and output and thus eventual oxygen delivery to tissues. Perhaps the most striking feature of this model is the unique opportunity for peripheral endothelial cells to respond to decreased systemic perfusion, and regulate through release of the apelin peptide, a spectrum of cardiovascular and fluid balance related functions that appear aimed at maintaining circulatory homeostasis.
Data from the in vivo systemic hypoxia study as well as that from cultured endothelial cells demonstrate the responsiveness of the apelin-APJ pathway to hypoxia in this cell type. With isolated hypoxic exposure alone, healthy mice exhibited a robust upregulation of both apelin and APJ in cardiac and pulmonary tissues. However, comparing the hypoxia-induced in vivo changes in gene expression to those observed in the LAD-ligation model highlights the complexity of regulation of this pathway in heart failure, and suggests that multiple stimuli are active in the clinical setting of CHF. For example, although we demonstrated a marked upregulation of apelin within hypoxic skeletal muscle following LAD ligation, systemic hypoxia produced only a modest, non-statisitcally significant upregulation within the same tissue. This would suggest that other stimui elicited by ischemic heart failure (e.g., changes in endothelial shear stress or flow, oxidative stress, inflammatory change, etc.) likely contributed to the observed response. Another striking comparison is that of the pulmonary response to ischemic heart failure versus systemic hypoxia alone. Whereas LAD-ligation resulted in no appreciable change in apelin expression in this tissue, hypoxia produced a robust upregulation of the protein and receptor in endothelial cells, both in vivo and in vitro. These findings underscore the sensitivity of pulmonary apelin-producing endothelium to hypoxia. Following LAD ligation, alveolar endothelium remained normoxic due to inhaled ambient oxygen. By contrast, animals in the systemic hypoxia experiment experienced pulmonary hypoxia as ambient oxygen concentrations were low and responded, in part, by the robust upregulation of apelin. These data suggest that for pulmonary endothelium, unlike skeletal muscle vasculature, hypoxia is a more potent trigger for apelin upregulation than other stimuli which might be induced by chronic heart failure.
Although further work is required to delineate the pathways by which hypoxia-mediated apelin regulation may occur, recent studies have clearly demonstrated that HIF-1α plays a role (9, 29). Our findings both confirm these observations and expand upon them by providing data to support the role of HIF-2α signaling as well. While the two HIF isoforms are closely related and bind the same DNA motif, they are differentially expressed in the adult (13, 35). Specifically, HIF-1α expression is ubiquitous, whereas the 2α isoform is predominantly expressed by the endothelium, re-enforcing its specific importance in the regulation of apelin (35). This initial observation is promising, and additional studies are warranted to better understand the regulation of this pathway in diseases associated with hypoxia.
In conclusion, our data present a longitudinal evaluation of apelin and APJ expression changes in the setting of heart failure and reveal the pathway is regulated both within the coronary and peripheral skeletal muscle vasculature following ischemic heart injury, presumably to modulate cardiac inotrpy and systemic vascular resistance. Moreover, we have provided evidence that apelin is primarily expressed by the endothelium and may be induced by hypoxia via the endothelial specific HIF-2α pathway. Taken together, these data suggest that apelin expression is predominantly governed by endothelial cells and may be modulated in response to disease-induced stimuli such as hypoxia to optimize cardiac inotropy and vasomotor tone, thereby maintaining cardiovascular homeostasis. The apelin-APJ pathway may thus provide a mechanism for systemic endothelial monitoring of tissue perfusion and adaptive regulation of cardiovascular function.
Supplementary Material
Supplemental Figure 1. LAD ligation heart failure model. A, Representative histological samples from LAD ligation and sham animals stained with Masson-Trichrome revealing aneurysmal, dilated left ventricle following LAD ligation at 12 weeks (right, scale bars=2mm). B, Corresponding echocardiographic recordings 12 weeks following sham operation (left) and LAD ligation (right; scale bars=2mm). C, Mean fractional shortening (FS) decreased significantly 4, 8, and 12 weeks following LAD ligation (n=11 per group; bars represent means ± 95% CI, *P%0.0001) D, In the same cohort, β-natruetic peptide (BNP) mRNA expression in the heart increases as failure progresses (*P<0.0001). Cardiac apelin mRNA expression also increased in a similar pattern (Figure 1).
Supplemental Figure 2. Immunofluorescent localization of LacZ in endothelium of mouse lung. LacZ is stained green while red staining shows cell-specific markers for Type II epithelial cells (anti-surfactant protein c), Type I epithelial cell (anti-RAGE), and endothelial cells (anti-CD31). LacZ localization is distinct from Type II epithelial cells, as demonstrated by cytoplasmic staining for surfactant protein c (left panel) and close to, but not contained within the overlying thin Type I epithelial cells (middle panel). By contrast, the CD31 endothelial marker completely encircles LacZ-positive nuclei (right panel, bars=10 µm). Three dimensional reconstruction of the same section along the Z-axis by serial laser confocal microscopy demonstrates co-localization of the apelin-LacZ nuclei and CD31 (+) cytosol (Supplemental Video 1).
Supplemental Table. Apelin promoter HIF binding sites. Putative binding sites for HIF-transcription factor (A/G CGTG) within the human and mouse apelin promoter regions. +1 designates transcription start site for human (above) and Exon 1 is labeled for mouse (below). HIF binding domains are highlighted.
Supplemental Video 1. Three dimensional reconstruction of 5 µm-thick histological sections of apelin+/lacZ reporter mouse lung co-stained with fluorescent antibodies against both nuclear LacZ (green) and CD31 (red). Movie demonstrates LacZ nuclei to be contained within the endothelial cytosol.
Supplemental Video 2. Three dimensional reconstruction of 5 µm-thick histological sections of apelin+/lacZ reporter mouse heart co-stained with fluorescent antibodies against both nuclear LacZ (green) and CD31 (red). Movie demonstrates LacZ nuclei to be contained within the endothelial cytosol.
Acknowledgements
We thank Drs Nancy Boudreau (University of California, San Francisco, CA) and Steven McKnight (University of Texas Southwestern, Dallas, TX) for the generous donation of HIF-ODD and HIF-2α plasmids, respectively, as well as Grant Hoyt for his technical expertise.
Sources of funding
This work was supported by NIH-NHLBI grant R01 HL077676 (TQ). A.Y.S was supported by grants from the Massachusetts General Hospital, Stanford University School of Medicine, and NIH-NHLBI (F32 HL084982-01).
Footnotes
Disclosures - None
References
- 1.Ashley E, Chun HJ, Quertermous T. Opposing cardiovascular roles for the angiotensin and apelin signaling pathways. J Mol Cell Cardiol. 2006;41:778–781. doi: 10.1016/j.yjmcc.2006.08.013. [DOI] [PubMed] [Google Scholar]
- 2.Ashley EA, Powers J, Chen M, Kundu R, Finsterbach T, Caffarelli A, Deng A, Eichhorn J, Mahajan R, Agrawal R, Greve J, Robbins R, Patterson AJ, Bernstein D, Quertermous T. The endogenous peptide apelin potently improves cardiac contractility and reduces cardiac loading in vivo. Cardiovasc Res. 2005;65:73–82. doi: 10.1016/j.cardiores.2004.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Atluri P, Morine KJ, Liao GP, Panlilio CM, Berry MF, Hsu VM, Hiesinger W, Cohen JE, Joseph Woo Y. Ischemic heart failure enhances endogenous myocardial apelin and APJ receptor expression. Cell Mol Biol Lett. 2007;12:127–138. doi: 10.2478/s11658-006-0058-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bayat H, Swaney JS, Ander AN, Dalton N, Kennedy BP, Hammond HK, Roth DM. Progressive heart failure after myocardial infarction in mice. Basic Res Cardiol. 2002;97:206–213. doi: 10.1007/s003950200013. [DOI] [PubMed] [Google Scholar]
- 5.Berry MF, Pirolli TJ, Jayasankar V, Burdick J, Morine KJ, Gardner TJ, Woo YJ. Apelin has in vivo inotropic effects on normal and failing hearts. Circulation. 2004;110:II187–II193. doi: 10.1161/01.CIR.0000138382.57325.5c. [DOI] [PubMed] [Google Scholar]
- 6.Chen MM, Ashley EA, Deng DX, Tsalenko A, Deng A, Tabibiazar R, Ben-Dor A, Fenster B, Yang E, King JY, Fowler M, Robbins R, Johnson FL, Bruhn L, McDonagh T, Dargie H, Yakhini Z, Tsao PS, Quertermous T. Novel role for the potent endogenous inotrope apelin in human cardiac dysfunction. Circulation. 2003;108:1432–1439. doi: 10.1161/01.CIR.0000091235.94914.75. [DOI] [PubMed] [Google Scholar]
- 7.Cheng X, Cheng XS, Pang CC. Venous dilator effect of apelin, an endogenous peptide ligand for the orphan APJ receptor, in conscious rats. Eur J Pharmacol. 2003;470:171–175. doi: 10.1016/s0014-2999(03)01821-1. [DOI] [PubMed] [Google Scholar]
- 8.Chong KS, Gardner RS, Morton JJ, Ashley EA, McDonagh TA. Plasma concentrations of the novel peptide apelin are decreased in patients with chronic heart failure. Eur J Heart Fail. 2006;8:355–360. doi: 10.1016/j.ejheart.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 9.Cox CM, D'Agostino SL, Miller MK, Heimark RL, Krieg PA. Apelin, the ligand for the endothelial G-protein-coupled receptor, APJ, is a potent angiogenic factor required for normal vascular development of the frog embryo. Dev Biol. 2006;296:177–189. doi: 10.1016/j.ydbio.2006.04.452. [DOI] [PubMed] [Google Scholar]
- 10.De Mota N, Lenkei Z, Llorens-Cortes C. Cloning, pharmacological characterization and brain distribution of the rat apelin receptor. Neuroendocrinology. 2000;72:400–407. doi: 10.1159/000054609. [DOI] [PubMed] [Google Scholar]
- 11.De Mota N, Reaux-Le Goazigo A, El Messari S, Chartrel N, Roesch D, Dujardin C, Kordon C, Vaudry H, Moos F, Llorens-Cortes C. Apelin, a potent diuretic neuropeptide counteracting vasopressin actions through inhibition of vasopressin neuron activity and vasopressin release. Proc Natl Acad Sci U S A. 2004;101:10464–10469. doi: 10.1073/pnas.0403518101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Devic E, Rizzoti K, Bodin S, Knibiehler B, Audigier Y. Amino acid sequence and embryonic expression of msr/apj, the mouse homolog of Xenopus X-msr and human APJ. Mech Dev. 1999;84:199–203. doi: 10.1016/s0925-4773(99)00081-7. [DOI] [PubMed] [Google Scholar]
- 13.Ema M, Taya S, Yokotani N, Sogawa K, Matsuda Y, Fujii-Kuriyama Y. A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1alpha regulates the VEGF expression and is potentially involved in lung and vascular development. Proc Natl Acad Sci U S A. 1997;94:4273–4278. doi: 10.1073/pnas.94.9.4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Foldes G, Horkay F, Szokodi I, Vuolteenaho O, Ilves M, Lindstedt KA, Mayranpaa M, Sarman B, Seres L, Skoumal R, Lako-Futo Z, deChatel R, Ruskoaho H, Toth M. Circulating and cardiac levels of apelin, the novel ligand of the orphan receptor APJ, in patients with heart failure. Biochem Biophys Res Commun. 2003;308:480–485. doi: 10.1016/s0006-291x(03)01424-4. [DOI] [PubMed] [Google Scholar]
- 15.Inui M, Fukui A, Ito Y, Asashima M. Xapelin and Xmsr are required for cardiovascular development in Xenopus laevis. Dev Biol. 2006;298:188–200. doi: 10.1016/j.ydbio.2006.06.028. [DOI] [PubMed] [Google Scholar]
- 16.Ishida J, Hashimoto T, Hashimoto Y, Nishiwaki S, Iguchi T, Harada S, Sugaya T, Matsuzaki H, Yamamoto R, Shiota N, Okunishi H, Kihara M, Umemura S, Sugiyama F, Yagami K, Kasuya Y, Mochizuki N, Fukamizu A. Regulatory roles for APJ, a seven-transmembrane receptor related to angiotensin-type 1 receptor in blood pressure in vivo. J Biol Chem. 2004;279:26274–26279. doi: 10.1074/jbc.M404149200. [DOI] [PubMed] [Google Scholar]
- 17.Iwanaga Y, Kihara Y, Takenaka H, Kita T. Down-regulation of cardiac apelin system in hypertrophied and failing hearts: Possible role of angiotensin II-angiotensin type 1 receptor system. J Mol Cell Cardiol. 2006 doi: 10.1016/j.yjmcc.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 18.Jia YX, Pan CS, Zhang J, Geng B, Zhao J, Gerns H, Yang J, Chang JK, Tang CS, Qi YF. Apelin protects myocardial injury induced by isoproterenol in rats. Regul Pept. 2006;133:147–154. doi: 10.1016/j.regpep.2005.09.033. [DOI] [PubMed] [Google Scholar]
- 19.Kasai A, Shintani N, Oda M, Kakuda M, Hashimoto H, Matsuda T, Hinuma S, Baba A. Apelin is a novel angiogenic factor in retinal endothelial cells. Biochem Biophys Res Commun. 2004;325:395–400. doi: 10.1016/j.bbrc.2004.10.042. [DOI] [PubMed] [Google Scholar]
- 20.Kleinz MJ, Davenport AP. Immunocytochemical localization of the endogenous vasoactive peptide apelin to human vascular and endocardial endothelial cells. Regul Pept. 2004;118:119–125. doi: 10.1016/j.regpep.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 21.Kleinz MJ, Skepper JN, Davenport AP. Immunocytochemical localisation of the apelin receptor, APJ, to human cardiomyocytes, vascular smooth muscle and endothelial cells. Regul Pept. 2005;126:233–240. doi: 10.1016/j.regpep.2004.10.019. [DOI] [PubMed] [Google Scholar]
- 22.Kuba K, Zhang L, Imai Y, Arab S, Chen M, Maekawa Y, Leschnik M, Leibbrandt A, Makovic M, Schwaighofer J, Beetz N, Musialek R, Neely GG, Komnenovic V, Kolm U, Metzler B, Ricci R, Hara H, Meixner A, Nghiem M, Chen X, Dawood F, Wong KM, Sarao R, Cukerman E, Kimura A, Hein L, Thalhammer J, Liu PP, Penninger JM. Impaired Heart Contractility in Apelin Gene Deficient Mice Associated With Aging and Pressure Overload. Circ Res. 2007 doi: 10.1161/CIRCRESAHA.107.158659. [DOI] [PubMed] [Google Scholar]
- 23.Lee DK, Cheng R, Nguyen T, Fan T, Kariyawasam AP, Liu Y, Osmond DH, George SR, O'Dowd BF. Characterization of apelin, the ligand for the APJ receptor. J Neurochem. 2000;74:34–41. doi: 10.1046/j.1471-4159.2000.0740034.x. [DOI] [PubMed] [Google Scholar]
- 24.O'Dowd BF, Heiber M, Chan A, Heng HH, Tsui LC, Kennedy JL, Shi X, Petronis A, George SR, Nguyen T. A human gene that shows identity with the gene encoding the angiotensin receptor is located on chromosome 11. Gene. 1993;136:355–360. doi: 10.1016/0378-1119(93)90495-o. [DOI] [PubMed] [Google Scholar]
- 25.Pereira T, Zheng X, Ruas JL, Tanimoto K, Poellinger L. Identification of residues critical for regulation of protein stability and the transactivation function of the hypoxia-inducible factor-1alpha by the von Hippel-Lindau tumor suppressor gene product. J Biol Chem. 2003;278:6816–6823. doi: 10.1074/jbc.M209297200. [DOI] [PubMed] [Google Scholar]
- 26.Reaux A, De Mota N, Skultetyova I, Lenkei Z, El Messari S, Gallatz K, Corvol P, Palkovits M, Llorens-Cortes C. Physiological role of a novel neuropeptide, apelin, and its receptor in the rat brain. J Neurochem. 2001;77:1085–1096. doi: 10.1046/j.1471-4159.2001.00320.x. [DOI] [PubMed] [Google Scholar]
- 27.Reaux-Le Goazigo A, Morinville A, Burlet A, Llorens-Cortes C, Beaudet A. Dehydration-induced cross-regulation of apelin and vasopressin immunoreactivity levels in magnocellular hypothalamic neurons. Endocrinology. 2004;145:4392–4400. doi: 10.1210/en.2004-0384. [DOI] [PubMed] [Google Scholar]
- 28.Resta O, Foschino-Barbaro MP, Bonfitto PG, Talamo S, Nocerino MC, Stefa no A, Biasco G. Nocturnal oxygen desaturation in patients with congestive heart failure. Boll Soc Ital Biol Sper. 1999;75:31–38. [PubMed] [Google Scholar]
- 29.Ronkainen VP, Ronkainen JJ, Hanninen SL, Leskinen H, Ruas JL, Pereira T, Poellinger L, Vuolteenaho O, Tavi P. Hypoxia inducible factor regulates the cardiac expression and secretion of apelin. Faseb J. 2007;21:1821–1830. doi: 10.1096/fj.06-7294com. [DOI] [PubMed] [Google Scholar]
- 30.Saint-Geniez M, Argence CB, Knibiehler B, Audigier Y. The msr/apj gene encoding the apelin receptor is an early and specific marker of the venous phenotype in the retinal vasculature. Gene Expr Patterns. 2003;3:467–472. doi: 10.1016/s1567-133x(03)00062-0. [DOI] [PubMed] [Google Scholar]
- 31.Scott IC, Masri B, D'Amico LA, Jin S-W, Jungblut B, Wehman AM, Baier H, Audigier Y, Stanier DYR. The G-protein coupled receptor Agtrl1b regulates early development of myocardial progenitors. Developmental Cell. 2007 doi: 10.1016/j.devcel.2007.01.012. in press. [DOI] [PubMed] [Google Scholar]
- 32.Szokodi I, Tavi P, Foldes G, Voutilainen-Myllyla S, Ilves M, Tokola H, Pikkarainen S, Piuhola J, Rysa J, Toth M, Ruskoaho H. Apelin, the novel endogenous ligand of the orphan receptor APJ, regulates cardiac contractility. Circ Res. 2002;91:434–440. doi: 10.1161/01.res.0000033522.37861.69. [DOI] [PubMed] [Google Scholar]
- 33.Tatemoto K, Hosoya M, Habata Y, Fujii R, Kakegawa T, Zou MX, Kawamata Y, Fukusumi S, Hinuma S, Kitada C, Kurokawa T, Onda H, Fujino M. Isolation and characterization of a novel endogenous peptide ligand for the human APJ receptor. Biochem Biophys Res Commun. 1998;251:471–476. doi: 10.1006/bbrc.1998.9489. [DOI] [PubMed] [Google Scholar]
- 34.Tatemoto K, Takayama K, Zou MX, Kumaki I, Zhang W, Kumano K, Fujimiya M. The novel peptide apelin lowers blood pressure via a nitric oxide-dependent mechanism. Regul Pept. 2001;99:87–92. doi: 10.1016/s0167-0115(01)00236-1. [DOI] [PubMed] [Google Scholar]
- 35.Tian H, McKnight SL, Russell DW. Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev. 1997;11:72–82. doi: 10.1101/gad.11.1.72. [DOI] [PubMed] [Google Scholar]
- 36.van Kimmenade RR, Januzzi JL, Jr., Ellinor PT, Sharma UC, Bakker JA, Low AF, Martinez A, Crijns HJ, MacRae CA, Menheere PP, Pinto YM. Utility of amino-terminal pro-brain natriuretic peptide, galectin-3, and apelin for the evaluation of patients with acute heart failure. J Am Coll Cardiol. 2006;48:1217–1224. doi: 10.1016/j.jacc.2006.03.061. [DOI] [PubMed] [Google Scholar]
- 37.Yamaya Y, Bogaard HJ, Wagner PD, Niizeki K, Hopkins SR. Validity of pulse oximetry during maximal exercise in normoxia, hypoxia, and hyperoxia. J Appl Physiol. 2002;92:162–168. doi: 10.1152/japplphysiol.00409.2001. [DOI] [PubMed] [Google Scholar]
- 38.Zeng X-XI, Wilm TP, Sepich DS, Solnica-Krezel L. Apeliln and it s receptor control heart field formation during zebrafish gastrulation. Developmental Cell. 2007 doi: 10.1016/j.devcel.2007.01.011. in press. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. LAD ligation heart failure model. A, Representative histological samples from LAD ligation and sham animals stained with Masson-Trichrome revealing aneurysmal, dilated left ventricle following LAD ligation at 12 weeks (right, scale bars=2mm). B, Corresponding echocardiographic recordings 12 weeks following sham operation (left) and LAD ligation (right; scale bars=2mm). C, Mean fractional shortening (FS) decreased significantly 4, 8, and 12 weeks following LAD ligation (n=11 per group; bars represent means ± 95% CI, *P%0.0001) D, In the same cohort, β-natruetic peptide (BNP) mRNA expression in the heart increases as failure progresses (*P<0.0001). Cardiac apelin mRNA expression also increased in a similar pattern (Figure 1).
Supplemental Figure 2. Immunofluorescent localization of LacZ in endothelium of mouse lung. LacZ is stained green while red staining shows cell-specific markers for Type II epithelial cells (anti-surfactant protein c), Type I epithelial cell (anti-RAGE), and endothelial cells (anti-CD31). LacZ localization is distinct from Type II epithelial cells, as demonstrated by cytoplasmic staining for surfactant protein c (left panel) and close to, but not contained within the overlying thin Type I epithelial cells (middle panel). By contrast, the CD31 endothelial marker completely encircles LacZ-positive nuclei (right panel, bars=10 µm). Three dimensional reconstruction of the same section along the Z-axis by serial laser confocal microscopy demonstrates co-localization of the apelin-LacZ nuclei and CD31 (+) cytosol (Supplemental Video 1).
Supplemental Table. Apelin promoter HIF binding sites. Putative binding sites for HIF-transcription factor (A/G CGTG) within the human and mouse apelin promoter regions. +1 designates transcription start site for human (above) and Exon 1 is labeled for mouse (below). HIF binding domains are highlighted.
Supplemental Video 1. Three dimensional reconstruction of 5 µm-thick histological sections of apelin+/lacZ reporter mouse lung co-stained with fluorescent antibodies against both nuclear LacZ (green) and CD31 (red). Movie demonstrates LacZ nuclei to be contained within the endothelial cytosol.
Supplemental Video 2. Three dimensional reconstruction of 5 µm-thick histological sections of apelin+/lacZ reporter mouse heart co-stained with fluorescent antibodies against both nuclear LacZ (green) and CD31 (red). Movie demonstrates LacZ nuclei to be contained within the endothelial cytosol.