

Abstract
GABAB receptors are heterodimeric G-protein coupled receptors composed of R1 and R2 subunits that mediate slow synaptic inhibition in the brain by activating inwardly-rectifying K+ channels (GIRKs) and inhibiting Ca2+ channels. We demonstrate here that GABAB receptors are intimately associated with 5’AMP-dependent protein kinase (AMPK). AMPK acts as a metabolic sensor that is potently activated by increases in 5’AMP concentration caused by enhanced metabolic activity, anoxia or ischemia. AMPK binds the R1 subunit and directly phosphorylates S783 in the R2 subunit to enhance GABAB receptor activation of GIRKs. Phosphorylation of S783 is evident in many brain regions, and is increased dramatically after ischemic injury. Finally we also reveal that S783 plays a critical role in enhancing neuronal survival after ischemia. Together our results provide evidence of a novel neuroprotective mechanism, which under conditions of metabolic stress or after ischemia increases GABAB receptor function to reduce excitotoxicity and thereby promoting neuronal survival.
Introduction
GABAB receptors mediate slow prolonged inhibition in the brain by activating postsynaptic inwardly-rectifying K+ channels (GIRKs) and inactivating presynaptic voltage-gated Ca2+ channels. GABAB receptors also inhibit adenylate cyclase, leading to diminished activity of PKA signaling pathways (Bowery, 2006). Structurally GABAB receptors are members of the class C family of G-protein coupled receptors (GPCR) and are encoded in vertebrates by two genes - GABABR1 and GABABR2 respectively (Bettler et al., 2004; Couve et al., 2004a). In contrast to the majority of other GPCRs, heterodimerization between GABABR1 and R2 subunits is required for the formation of functional receptors that reproduce most of the pharmacological and physiological properties of their native counterparts (Bettler et al., 2004; Couve et al., 2004a).
Synaptic activity is critically dependent on cellular energy levels, which are in part determined by the metabolic sensor 5’AMP-activated protein kinase (AMPK). AMPK is rapidly activated when cellular levels of ATP coincident with an increase in AMP concentration due to high metabolic activity and in the pathological states of anoxia and ischaemia (Carling, 2005) (Kahn et al., 2005). AMPK is phosphorylated by the upstream kinases LKB on threonine 172 (T172) in a process that is stimulated by AMP. Calmodulin-dependent protein kinase kinase-β (CamKK) also phosphorylates T172 and phosphorylation of this residue by either of these enzyme enhances AMPK catalytic activity (Hawley et al., 2003); 2005; (Woods et al., 2003a); 2005; Hurley et al., 2005). AMPK principally mediates its effect on cellular energy levels by stimulating catabolic metabolism and simultaneously inhibiting anabolic pathways (Kahn et al., 2005). Accordingly, to date the majority of AMPK substrates identified are either enzymes or transcription factors that control carbohydrate and lipid metabolism (Kahn et al., 2005).
Here we reveal that AMPK binds directly to GABAB receptors and phosphorylates S783 in the cytoplasmic tail of the R2 subunit, a process that enhances receptor coupling to GIRKs. S783 is basally phosphorylated by AMPK activity in neurons and mutation of S783 reduces their survival after anoxia. Finally, ischemic injury to the brain produces large increases in S783 phosphorylation within the R2 subunit. In cultured neurones this phosphorylation is important for enhanced neuronal survival post- hypoxic insult. Together our results provide evidence of a novel neuroprotective mechanism to increase GABAB receptor function after ischemia to reduce excitotoxicity.
Results
AMPK binds directly to GABAB receptors
To identify signaling molecules that regulate GABAB receptor functional expression we searched for novel cytoplasmic binding partners for these proteins. To this end, the cytoplasmic tail of the GABABR1 subunit was used as a bait in a 2-hybrid screen of a rat brain cDNA library expressed in yeast (Couve et al., 2001; Restituito et al., 2005). This resulted in the identification of residues 289-550 of the α1 subunit of AMPK as a binding partner for the cytoplasmic tail of the GABABR1 subunit, but not to the corresponding domain of R2 (Figs. 1A and B). AMPK holoenzyme is found ubiquitously expressed in all cell types and is composed of catalytic α1 or α2 subunits together with regulatory β and γ subunits. Residues 289-550 of AMPK α1/2 subunits are not required for catalytic activity but this domain has been suggested to act as a scaffold mediating interaction with β and γ subunits (Carling, 2005) (Kahn et al., 2005).
Figure 1. Association of AMPK with GABAB receptors.
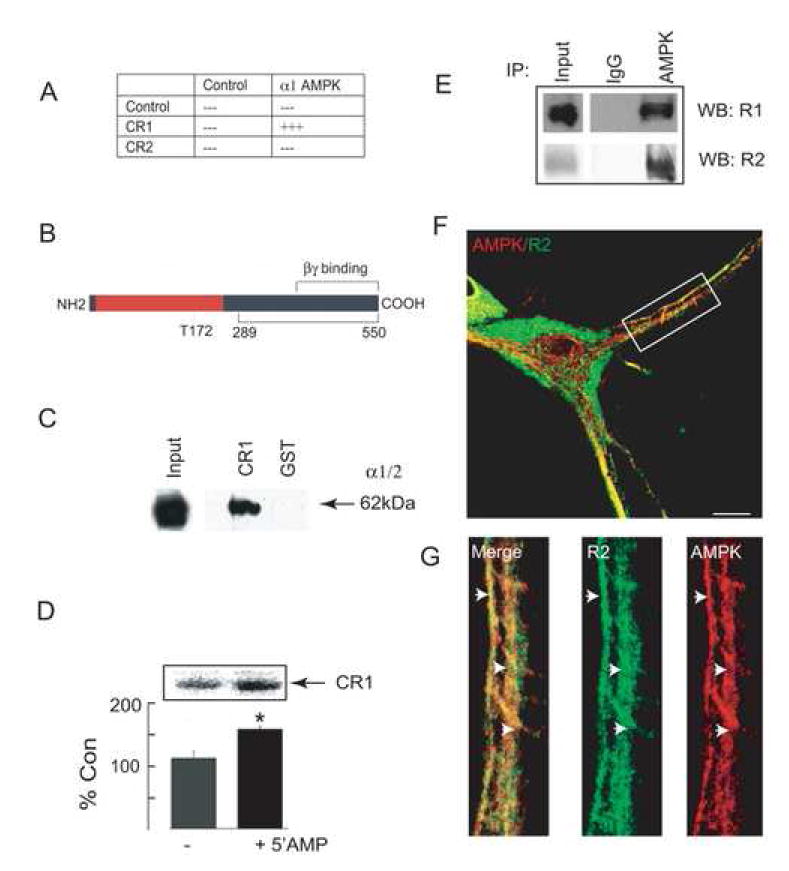
A. Interaction of CR1 with AMPK in yeast. A bait encoding the cytoplasmic tail of GABABR1 (CR1) was used to screen a rat brain library, resulting in the identification of clones encoding AMPK α1 subunit. The smallest clone-encoding amino acids 289-550 of the α1 subunit were re-transformed into yeast with either empty vector (control), CR1 or CR2. Resulting clones were then tested for β-gal activity; +++ = +ve, --- = –ve. B. A schematic of the major functional domains within the α1/2 subunits of AMPK. The catalytic domain (red) with the autophosphorylation site T172 and the domains for binding β/γ subunits in addition to GABAB receptors (black) are shown. C. In vitro binding of GST-CR1 and AMPK. Fusion proteins were immobilized on GST-agarose and exposed to detergent-solubilized brain extracts. Bound material was then immunoblotted with an antibody for α1/2 subunits of AMPK. The input lane represents 10% of the material used in each assay. D. Phosphorylation of GST-CR1 by AMPK from brain lysates. GST-CR1 was exposed to brain lysates and bound material was exposed to 32Pγ-ATP in the presence and absence of 100 μM 5’AMP for 10 min at 37°C. Phosphorylation was then assessed by SDS-PAGE (upper panel). The level of 32P incorporation was measured against controls in the absence of 5’AMP (= 100%; n = 3), * = significantly different from control (p<0.01; students-t test). E. Immunoprecipitation of AMPK and GABAB receptors from brain extracts. Crude brain membranes were immunoprecipitated with antibodies that recognize the α1/2 subunits of AMPK. Precipitated material was then immunoblotted with antibodies against the GABAB R1 and R2 subunits as indicated. Input represents 10% of the material used for each experiment. F. Co-localization of AMPK and GABAB receptors in hippocampal neurons at 12-16 DIV. Neurons were fixed, permeabilized and stained with antibodies against GABAB receptors and AMPK α1/2 subunits with secondary antibodies conjugated to FITC and rhodamine, respectively (scale bar represents 5 μm). G. Represents an enlargement of the boxed area shown in F.
To confirm our observations in yeast we first used in vitro binding assays. AMPK α1/2 subunits were detected binding to glutathione-S-transferase fusion proteins (GST) encoding the C-tail of the R1 subunit (GST-CR1) (Couve et al., 2001; Couve et al., 2004b) but not to GST alone from detergent solubilized brain extracts (Fig. 1C) as measured by immunoblotting with antibodies that recognize the α1/2 subunits of AMPK (Fryer et al., 2002). Using deletion analysis it was evident that residues 910-925 within the coiled-coil domain of the GABABR1 subunit (Bettler et al., 2004; Couve et al., 2004a) are essential in mediating the binding of CR1 to AMPK (data not shown; (Bettler et al., 2004; Couve et al., 2004a). Bound material was also subject to in vitro kinase assays. Phosphorylation of GST-CR1 was evident, which could be significantly enhanced by 5’AMP (Fig. 1D). Phosphorylation occurred exclusively on serine residues within a single major phospho-peptide (Supplementary Data Fig. 1A, B). No phosphorylation of GST alone was seen in these assays, a finding consistent with our previously published studies (Brandon et al., 1999b). Immunoprecipitation was also used to measure the association of GABAB receptors with AMPK. Both R1 and R2 subunits were detected co-immunoprecipitating with antibodies against the α1/2 subunits of AMPK from detergent solubilized brain extracts but not with control IgG (Fig.1E). Finally we examined the subcellular distribution of AMPK α1/2 subunits and GABAB receptors using immunofluorescence in 10-14 Div cultured hippocampal neurons. Significantly AMPK staining was evident in neuronal processes and was also found in the nucleus, consistent with previous studies on the distribution of this multifunctional signaling molecule (Fig. 1F) (Carling, 2005) (Kahn et al., 2005). GABAB receptor immunoreactivity was evident in neuronal cell bodies and processes (Fig. 1F) as detailed previously in cultured neurons (Couve et al., 2001; Restituito et al., 2005). Strikingly, co-localization of GABAB receptors with AMPK was evident for receptor populations close to or on the cell surface (Fig. 1G).
Collectively our experiments reveal that GABAB receptors can interact directly with AMPK and that these proteins are intimately associated in neurons.
Identification of AMPK-dependent phosphorylation sites within the GABAB receptors
We used in vitro phosphorylation to further assess the significance ofAMPK binding to GABAB receptors. Consistent with our observations from brain extracts (Fig. 1D), purified AMPK phosphorylated GST-CR1 exclusively on serine residues, while GST alone was not significantly phosphorylated under the same conditions (Fig. 2A). GST-CR1 was phosphorylated solely on serine residues (Fig. 2B) within a single major tryptic peptide (supplementary data; Figs 1A and B). GST-32P-CR1 phosphorylated to a stoichiometry of 0.3 mol/mol by AMPK, was then subject to trypsin digestion and the resulting peptides fractionated by HPLC (supplementary data; Fig. 2A. This revealed that the majority of the radiolabel eluted as a single peak, which was subject to Edman degradation. A single major peptide was found in these fractions (RHQLQSRQQ) which contained a single serine residue corresponding to S917 in CR1 (Fig. 2C). To confirm the identity of this site we compared the phosphorylation of GST-CR1 and a mutant in which S917 had been converted to alanine (GST-CR1S917A). Mutation of this residue significantly decreased phosphorylation by AMPK to 70 ± 5.2 % of that seen with GST-CR1 (Fig. 2D; p<0.01). We next compared the patterns of phosphorylation of GST-32P-CR1 and GST-32P-CR1S917A by tryptic peptide mapping. For GST-32P-CR1 phosphorylation occurred within a single major peptide that was strikingly similar to that seen on the phosphorylation of GST-CR1 with brain extracts (peptide 1; supplementary Data; Figs. 1B, C). Mutation of S917 eliminated phospho-peptide 1; however a number of new phospho-peptides became evident, suggesting that mutation of S917 unmasks other AMPK substrates within CR1 (Supplementary Data; Fig. 1C). Consistent with this, we found that co-mutation of S923 with S917 (GST-CR1S917/923A) further reduced AMPK phosphorylation to 10 ± 5.5% of the level seen with GST-CR1 (Fig. 2D; p<0.01; n= 4) and eliminated the majority of the new phospho-peptides seen with GST-32P-CR1S917A.
Figure 2. Identification of AMPK phosphorylation sites in the cytoplasmic tail of GABAB receptor subunits.
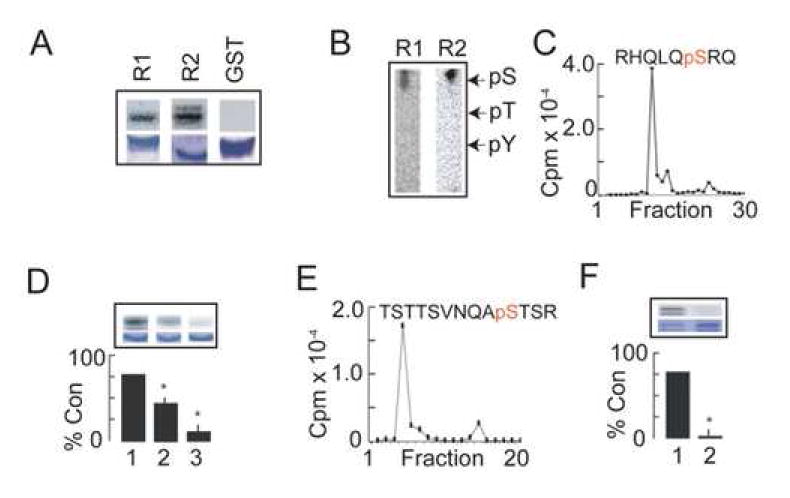
A. GABAB receptor cytoplasmic tails are phosphorylated by AMPK. GST-CR1 (R1), GST-CR2 (R2) and GST were phosphorylated in vitro by purified AMPK for 10 min at 30°C. Phosphorylation was then evaluated by autoradiography (upper panels) and coommassie staining (lower panels). B. GST-CR1 and GST-CR2 are phosphorylated on serine residues by AMPK. The migration of phospho-amino acid standards are indicated. C. S917 is a major phosphorylation site for AMPK in GST-CR1. A major peptide that contained a single phospho-serine residue corresponding to S917 in the GABABR1 subunit was identified using HPLC. D. Analysis of GST-CR1 phosphorylation using site-specific mutagenesis. GST-CR1 (1), GST-CR1S917A (2) and GST-CR1S917/923A (3) were phosphorylated in vitro using purified AMPK in the presence of 100 μM 5’AMP for 10 min at 37°C followed by SDS-PAGE and subject to autoradiography (upper panels) or coommassie staining (lower panels). The level of phosphorylation for each mutant was then compared to that seen for GST-CR1, which was given a value of 100%. * = significantly different from control (p<0.01; students-t test; n=4). E. S783 is the major site of phosphorylation for AMPK in GST-CR2. 32P-GST-CR2 was digested with trypsin and the resulting peptides then separated by HPLC. The level of 32P in the respective fractions was then quantified. The majority of the recovered counts eluted as a single peak, which was subject to mass spectroscopy (supplementary data; Fig. 2). This revealed the presence of a major peptide that contained a single phospho-serine residue corresponding to S783 in GABABR2. F. Analysis of GST-CR2 phosphorylation using site-specific mutagenesis. GST-CR2 (1) and GST-CR2S783A (2) were phosphorylated in vitro using purified AMPK for 10 min at 37°C followed by SDS-PAGE and subject to autoradiography (upper panels) or coommassie staining (lower panels). The level of phosphorylation for the S783A mutant was then compared to that seen for GST-CR2, which was given a value of 100%. * = significantly different from control (p<0.01; students-t test; n=5).
We also analyzed whether the C-tail of GABABR2 (GST-CR2) is a substrate of AMPK. GST-CR2 was phosphorylated by AMPK (Fig. 2A) and GST-32P-CR2 migrated as a doublet consistent with the instability of this purified protein as documented previously (Couve et al., 2002). Phospho-amino acid analysis and peptide mapping revealed that phosphorylation of GST-CR2 occurred exclusively on serine residues within a single major phospho-peptide (Fig. 2B; supplementary data; Fig. 1C). GST-32P-CR2 labeled to a stoichiometry of 0.15 mol/mol by AMPK was digested with trypsin and the resulting peptides then subject to fractionation by HPLC (Fig. 2E). The majority of the label counts eluted as a single peak and was further analyzed by HPLC-tandem mass spectroscopy (Supplementary Data; Fig. 2). Two peptides of 1546.65 Da and 1606.52 Da were detected. Analysis of CID (collisional induced dissociation) spectra revealed that these peptides represented the unphosphorylated peptide TSTSVTSVNQAS783TSR derived from the GABABR2 C-tail, and that the same peptide singly phosphorylated at S783 (supplementary data; Fig. 2B). To confirm the identity of S783 as an AMPK substrate we prepared a mutant form of GST-CR2 in which S783 was converted to an alanine residue (GST-CR2S783A). This mutation totally abolished AMPK-dependent phosphorylation of GST-CR2 (Fig. 2F).
Thus our in vitro studies suggest that GABAB receptors are substrates of AMPK with major phosphorylation sites S917 and S783 in the cytoplasmic tails of the R1 and R2 subunits respectively. Interestingly neither S917 nor S923 conform to the consensus for phosphorylation by AMPK previously determined on the basis experiments using synthetic peptides (Scott et al., 2002). Presumably the intimate association of GABAB receptors with AMPK facilitates the phosphorylation of non-consensus sites within these receptors.
Phosphorylation of S783 by AMPK activity decreases GABAB receptor run-down
Phosphorylation of the intracellular domains of GPCRs is critical in regulating their function. Therefore we measured whether the activation of AMPK regulates GABAB receptor effector coupling. To do so we investigated the activation of K+ channels by recombinant GABAB receptors using HEK-293 cells stably expressing Kir 3.1 and 3.2 channels (GIRK cells) and transiently expressing R1a and R2 GABAB receptor subunits. We conducted time-course studies for membrane currents activated with a sub-maximal (10 μM) GABA concentration. Consistent with previous studies (Couve et al., 2002), internal perfusion with a patch pipette electrolyte containing ATP and GTP initiated a time-dependent run-down in the GABA-activated K+ current to 33 ± 3 % of control after 22 minutes (Fig. 3A). Interestingly, inclusion of 1mM AMP in the patch pipette solution, in the presence of ATP and GTP, partially prevented the run-down of the GABA-activated K+ current to 52 ± 4 % after 22 min (Fig. 3A) compared with a patch pipette solution containing just ATP and GTP. The rundown in the GABAB receptor-activated K+ currents were all described by monoexponential decays, with time constants that appeared independent of the pipette composition and receptor subunit composition (supplementary data; Fig. 3).
Figure 3. 5’ AMP reduces the attenuation of GABA-activated K+ currents in GIRK cells via phosphorylation of S783 in R2.
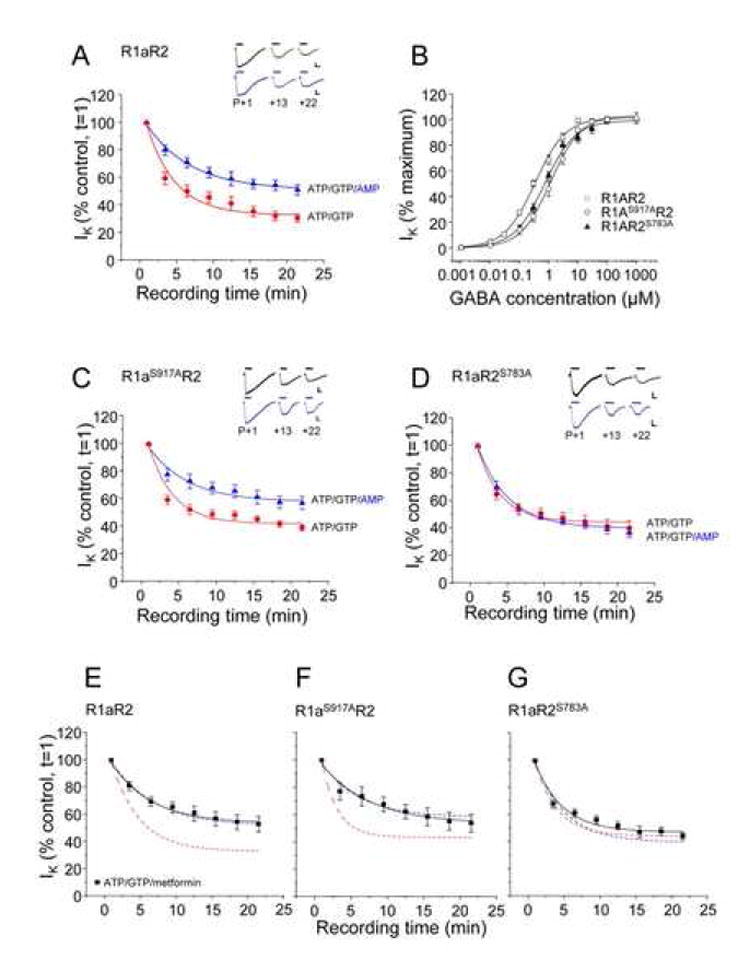
A. GIRK cells transfected with wild-type R1aR2 GABAB subunits were exposed to 10μM GABA, at the time points indicated, with the internal patch pipette solutions containing either 2mM ATP and 1mM GTP (●) or 1mM AMP, 2mM ATP and 1mM GTP (▲) from n = 5-13 cells. All Insets show sample membrane currents (calibration bars 200pA, 2s). B. Concentration response relationships for R1aR2 (□), R1aS917AR2(○) and R1aR2S783A (▲) GABAB receptors (n = 5-12 cells) with an internal solution containing 14mM CP, 2mM ATP and 1mM GTP normalized to the maximum GABA peak current (1mM) and fitted with the Hill equation (see methods). C and D. GIRK cells transfected with R1aS917AR2 (C) or R1aR2S783A (D) GABAB subunits were exposed to 10μM GABA, at the time points indicated, with the internal patch pipette solutions containing either 2mM ATP and 1mM GTP (●) or 1mM AMP, 2mM ATP and 1mM GTP (▲) from n=5-16 cells. E,F and G. Metformin reduces the attenuation of GABA-activated K+ currents of GABAB receptors. GIRK cells transfected with wild-type R1aR2 (E), R1aS917AR2 (F) and R1aR2S783A (G) GABAB subunits were exposed to 10μM GABA, at the time points indicated, with the internal patch pipette solution containing, 2mM ATP, 1mM GTP and 1mM metformin (■). Fitted lines taken from panels A, C and D are shown for comparison. Data from n=4-12 cells. All responses have been normalized to the response recorded just after the formation of the whole-cell recording mode (t=1). Data are mean ± s.e.m. Curves are monoexponential fits to the data points.
Given that decreasing the ATP:AMP ratio results in the activation of AMPK, (Carling 2005; (Kahn et al., 2005) we tested if the effects of AMP on GABA-activated K+ currents were dependent upon the AMPK phosphorylation sites, S917 and S783, in the C-tails of the R1 and R2 subunits, respectively (Fig. 2). GABA concentration response curves for R1aS917AR2, R1aR2S783A and wild-type R1aR2 GABAB receptors (Fig. 3B) revealed that the mutations caused only minor rightward lateral shifts. These shifts were in accord with the 2 – 4 fold reduction in GABA potency compared to that measured on the wild-type receptor. The corresponding GABA EC50s were: R1aR2, 0.3 ± 0.03 μM; R1aS917AR2, 1.2 ± 0.1 μM; and R1aR2S783A, 0.7 ± 0.1 μM. The stability of the GABA-activated K+ currents was also assessed for both of the mutant receptors, using a sub-maximal GABA concentration (10μM). Cells expressing R1aS917AR2 subunits exhibited significant run-down when dialyzed with ATP and GTP alone (40 ± 2 % of control; Fig. 3C). After the addition of 1 mM AMP, to the ATP and GTP pipette solution, GABA responses declined to 57 ± 5 % after 22 min (Fig. 3C), which was similar in profile to the run-down noted for the wild-type receptor (Fig. 3A).
By contrast with wild-type or R1aS917AR2 GABAB receptors, GABA-activated K+ currents evoked on R1aR2S783A displayed a different time course profile when the patch pipette solution was supplemented with 1 mM AMP (Fig. 3D). Initially, using internal solutions containing ATP and GTP, the current run-downs were quite similar in profile to those recorded with wild-type receptors, (ATP and GTP alone; 42 ± 6 %; Fig. 3D). However, the addition of AMP, to the ATP and GTP internal solution, had little effect on the GABA-activated K+ current run-down (37 ± 4 %; Fig. 3D) compared with ATP and GTP alone. Similar effects of AMP were seen in cells expressing GABAB receptors containing both R1aS917A and R2S783A (data not shown). These data suggest that S783 on the R2 subunit of the GABAB receptor was important for the activity of AMP in reducing GABA current rundown, possibly involving the direct phosphorylation of the R2 subunit by AMPK.
To ensure that AMP was not having an indirect effect on the GABA-activated K+ current via GABAB receptors, an AMPK activator, metformin, was used. Metformin activates the AMPK complex independently of the ATP:AMP ratio inside the cell (Fryer et al., 2002; Hawley et al., 2002; Woollhead et al., 2005). Time course profiles for 10 μM GABA-activated K+ currents were constructed for all three GABAB receptors: R1aR2, R1aS917AR2 and R1aR2S783A. AMPK was activated by internally perfusing cells with 1 mM metformin (including ATP and GTP) in the patch pipette solution. The wild-type receptor and mutants R1aS917AR2 and R1aR2S783A exhibited similar time course profiles for the GABA-activated K+ currents in the presence of metformin as they did following the addition of internal AMP (Figs. 3E, F and G).
These results suggest that the activation of AMPK, by either AMP or metformin, and the subsequent phosphorylation of S783 on the GABAB R2 subunit, were responsible for ameliorating the run-down of GABA-activated K+ currents in GIRK cells.
Production of a phospho-specific antibody against S783 in GABAB R2
Our functional studies have illustrated that AMPK modulation of recombinant GABAB receptors is critically reliant upon S783 in the R2 subunit (Fig. 3), an in vitro substrate of this kinase (Fig. 2). To further analyze the phosphorylation of this site, an antiserum was raised against a synthetic peptide chemically phosphorylated on the residue corresponding to S783 (VNQAS-P04TSRL). Affinity-purified anti-pS783 recognized GST-CR2 only when the latter had been pre-phosphorylated by AMPK, which was abolished following pre-adsorption with the phospho- but not dephosphorylated antigen (Fig. 4A). We further analyzed the specificity of pS783 using recombinant expression GABAB receptors expressed in HEK-293 cells. For these experiments transfected HEK-293 cells expressing GABAB receptors were labeled with NHS-Biotin. Cell surface receptor populations were purified on immobilized avidin and subject to immunoblotting. A band of 102 kDa corresponding to the R2 subunit was detected in cells expressing either R2 or R2S783A subunits using anti-R2 antibodies (Fig. 4B; (Couve et al., 2004b; Couve et al., 2002). A band of identical size was evident only in cells expressing R2 subunits, which was specifically blocked by phosphorylated but not dephosphorylated antigen (Fig. 4B). Using R2 antibodies we compared the cell surface expression levels of GABAB receptors incorporating wild-type or R2S783A subunits and it was evident that mutation of S783 did not alter cell surface stability of the R2 subunit (Fig. 4D; 105 ± 9.4% of control; n = 4). The effects of phenformin, a more potent and cell permeable activator of AMPK than metformin, on S783 phosphorylation were assessed (Fryer et al., 2002; Hawley et al., 2002; Woollhead et al., 2005). This agent produced a large and highly significant increase in AMPK activity as measured by enhanced phosphorylation of T172 in AMPK α1/2 subunits (Fig 4E; Carling 2005; (Kahn et al., 2005). The effects of phenformin on S783 phosphorylation for cell surface GABAB receptors was assessed by immunoblotting of biotinylated cell surface fractions. Phenformin treatment produced a large increase in S783 phosphorylation without modifying R2 cell surface expression levels (Fig. 4E). In these experiments using Triton-soluble extracts, R2 often migrated as a doublet when blotted with anti-p783, which may either reflect degradation upon sample preparation or a specific phosphorylation dependent shift in mobility of the R2 subunit.
Figure 4. Analyzing S783 phosphorylation in expression systems, cultured neurons and the brain.
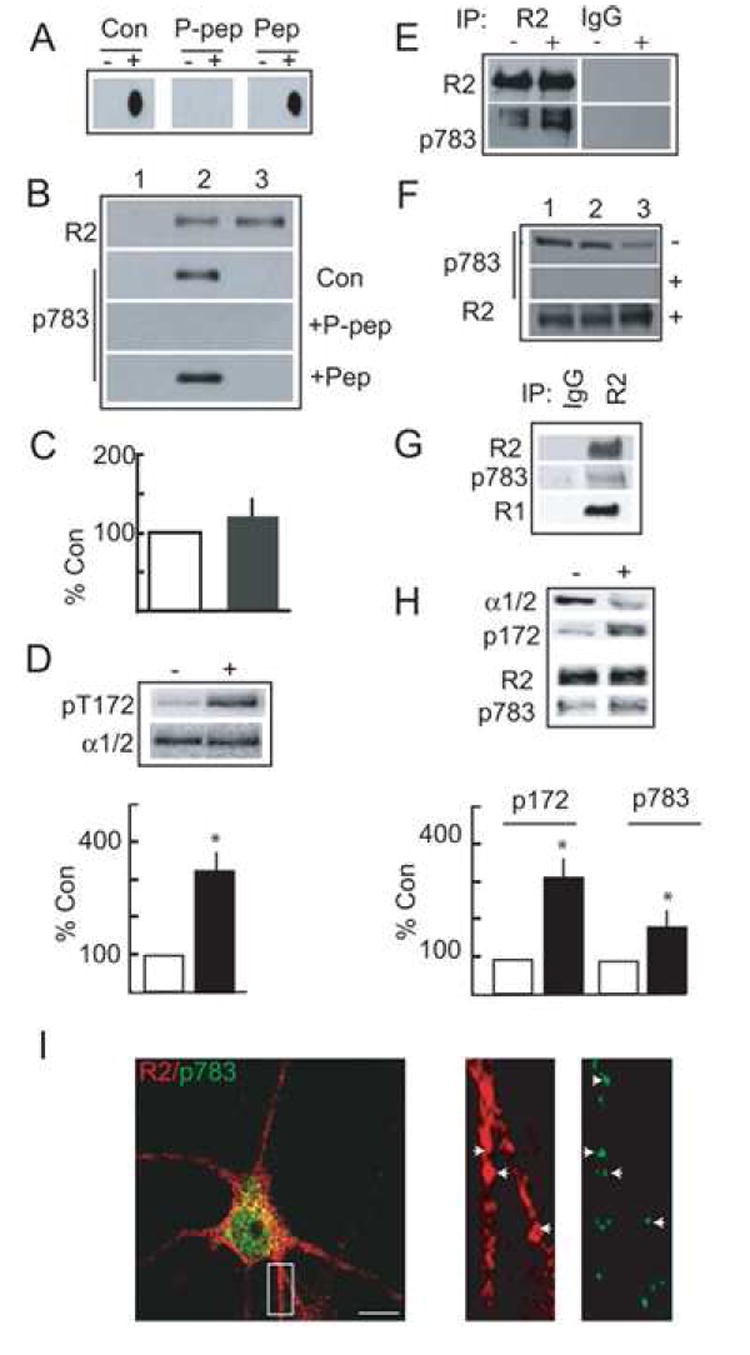
A. Specificity of pS783 antibody. GST-CR2 was transferred to a membrane with (+), or without (-) prior in vitro phosphorylation by AMPK and probed with affinity purified pS783 antibody alone, or with antibody preabsorbed with a 500-fold molar excess of phospho- (P-pep) or dephosphoantigens (Pep). B. Cell surface GABAB receptors are phosphorylated on S783. Mock transfected HEK-293 cells (lane 1) and those expressing GABAB receptors composed of R1R2 (2) or R1R2S783A (3) subunits were labeled with NHS-biotin and cell surface proteins and immunoblotted with antibodies against R2 subunits or pS783. Blots with pS783 were also performed with antibody that had been preabsorbed with a 500-fold molar excess of phospho (P-pep) and dephosphoantigens (Pep). C. Cell surface expression levels of GABABR2 (open bar, set to 100%) and R2S783A (filled bar) subunits when co-expressed with R1 subunits, measured by immunoblotting with antibodies against the R2 subunit (n = 4). D. Phenformin activates AMPK activity in HEK-293 cells. Cells expressing R1R2 GABAB receptors were treated with 10 mM phenformin for 1 h then immunoblotted with antibodies against the AMPK α1/2 subunits and pT172. The ratio of α1/2/p172 signals were then calculated from control cells (open) and from those treated with phenformin (filled). (n = 4). E. Phenformin treatment enhances S783 phosphorylation. HEK-293 cells were treated with (+) or without (-) 10 mM phenformin and labeled with NHS-biotin. Cell surface proteins were then denatured and subject to immunoprecipitation with R2 antibodies or control IgG. Precipitated material was immunoblotted with R2 or pS783 antibodies. In the experiment shown phenformin produced a 2.5-fold increase in R2S783 phosphorylation without altering cell surface expression levels of this receptor subunit. Similar results were seen in 3 other independent experiments. F. S783 is basally phosphorylated in the brain. Membrane protein prepared from rat cortex (1), hippocampus (2) and cerebellum (3) were immunoblotted with p-783 and R2 antibodies as indicated with (+) or without (-) prior treatment of the membrane with λ phosphatase. G. Analysis of S783 phosphorylation via immunoprecipitation. Hippocampal extracts were immunoprecipitated with anti-R2 antibody or control IgG. Precipitated material was then immunoblotted with antibodies for p783, R2 and R1 as indicated. H. Phenformin activates AMPK and enhances S783 phosphorylation in neurons. 12-14 DIV hippocampal neurons were treated with 10 mM phenformin for 1 h, and treated with NHS-Biotin. Cell surface fractions were then immunoblotted with AMPK α1/2, p172, anti-R2 and anti-p783 antibodies as indicated. The ratios of α1/2/p172 and R2/p783 were then calculated from control cells (open) and those treated with phenformin (filled). * = significantly different from control (p<0.01; students-t test; n = 5). The levels of cell surface GABABR2 subunits were unaltered by phenformin (110 ± 11.2%; n = 3; data not shown). I. Confocal analysis of p783 phosphorylation. 12-14 DIV hippocampal neurons were stained with R2 and pS783 antibodies coupled to rhodamine and FITC-conjugated secondaries, respectively (left hand panel, Scale bar 10 μm). The right panels represent enlargements of the boxed area in the left hand panel.
Together these results reveal that S783 is an AMPK substrate, and that the amelioration of GABAB receptor activation of GIRKs in HEK-293 cells (Fig. 3) was likely to be mediated by direct phosphorylation of this residue.
S783 is phosphorylated in the brain and cultured neurons
Immunoblotting with anti-pS783 was used to evaluate the phosphorylation of GABAB receptors in brain extracts. This antibody recognized a band of 102 kDa identical to that seen with anti-R2 antibody in membrane fractions prepared from cerebral cortex, hippocampus and cerebellum, which was abolished by pretreatment with λ phosphatase (Fig. 4F). We further analyzed phosphorylation of GABABR2 using immunoprecipitation of detergent solubilized hippocampal membranes followed by immunoblotting. Bands of 102kDa were seen with both anti-R2 and p783 antibodies immunoprecipitating with R2 antibody but not with control IgG (Fig. 4G). The R1 subunit was also detected co-immunoprecipitating with AMPK antibody but not control IgG (Fig. 4G).
We next evaluated the phosphorylation of S783 in 10-14 Div cultured hippocampal neurons that were treated with or without 10 mM phenformin. In agreement with our observations in HEK-293 cells phenformin produced a significant increase in AMPK activity as measured by immunoblotting of pT172 (325 ± 45.2% of control; Fig. 4H; p<0.01, n=4). To measure the effects on GABAB receptors neurons were labeled with NHS-Biotin and cell-surface proteins were isolated on immobilized avidin and immunoblotted with R2 and pS783 antibodies. This revealed that phenformin significantly enhanced surface receptor S783 phosphorylation to 185 ± 20.2% of control (Fig. 4H; p<0.01) without significantly altering total R2 expression levels (Fig. 4H; 110 ± 11%).
Immunofluorescence was used to further analyze the distribution of phosphorylated GABAB receptors in cultured hippocampal neurons. At 12-14 DIV neurons were fixed, permeabilized andstained with anti-pS783. This antibody labeled neurons by the soma and neuronal processes, which was blocked by the phosphorylated peptide antigen but not its non-phosphorylated equivalent (supplementary data; Fig. 4). We also compared the distribution of pS783 staining with that for total R2 subunits using double immunofluorescence. Immunoreactivity for pS783 and R2 was evident in the cell bodies and neuronal processes of cultured hippocampal neurons (Fig. 4I). We compared the co-localization of cR2 and p783 immunofluorescent puncta for pS783 aand R2 on neuronal processes. 84.74 ± 10.2% (n = 4) of pS783 puncta also contained immunoreactivity for R2. In contrast only 25 ± 4.5% (n = 5) of R2 puncta contained immunoreactivity for pS783. This discrepancy most likely reflects that not all GABAB receptors containing R2 subunits are phosphorylated on S783, consistent with this treatment of cultured neurons with the AMPK activator metformin robustly increased phosphorylation of this residue in cultured neruons (Fig. 4 H).
Together these biochemical studies demonstrate that GABAB receptors in the brain and cultured neurons, are phosphorylated on S783 in the R2 subunit and strongly suggest a role for AMPK activity in regulating the phosphorylation of this residue.
Phosphorylation of S783 in hippocampal neurons is dependent upon AMPK activity
Given the paucity of specific AMPK inhibitors we used a genetic approach to further elucidate the role of this kinase in regulating GABAB receptor phosphorylation. To do so we utilized a dominant negative α1 subunit expression construct, in which aspartate 157 had been mutated to an alanine residue and modified with a myc epitope (DNα1; Woods et al., 2000). This construct is catalytically inactive but can still assemble with kinase β and γ subunits and has been used to inhibit AMPK activity in many experimental systems (Carling, 2005; Kahn et al., 2005). 10-12 Div hippocampal neurons were transfected with a plasmid encoding DNα1 and effects on GABAB receptor phosphorylation were then evaluated 24-48 h later using triple immunofluorescence and high-resolution confocal microscopy. In neurons expressing DNα1, as revealed by high levels of myc staining, there appeared to be a reduction in S783 fluorescence compared to that of non-transfected neurons, while the levels of R2 staining appeared similar (Fig. 5A and B). To quantify these results we compared the number of R2 puncta containing pS783 immunoreactivity (R2/p783 co-localization) with values data being normalized to values seen in control neurons (=100%). The number of R2 puncta containing p783 immunoreactivity in DNα1-expressing neurons identified via their myc fluorescence decreased significantly to 21 ± 4.5% of the level seen in control non-expressing neurons (Fig. 6A). To control for possible non-specific effects of DNα1 expression, we analyzed the level of phosphorylation of S892, a substrate of PKA, using another pphospho-specific antibody against this site (anti-pS892; (Couve et al., 2002). In both control neurons and those expressing DNα1 very similar levels of co-localization between pS892 and R2 staining were evident (Figs. 5C, D and 6A). As a final control we assessed the effects of expressing GFP (rather than GFP), on the phosphorylation of both S783 and S892 revealing that expression of this protein did not alter the levels of either S783 or S892 phosphorylation (Fig 6A).
Figure 5. Analyzing the effects of modulating AMPK activity on GABAB receptor phosphorylation in hippocampal neurons.
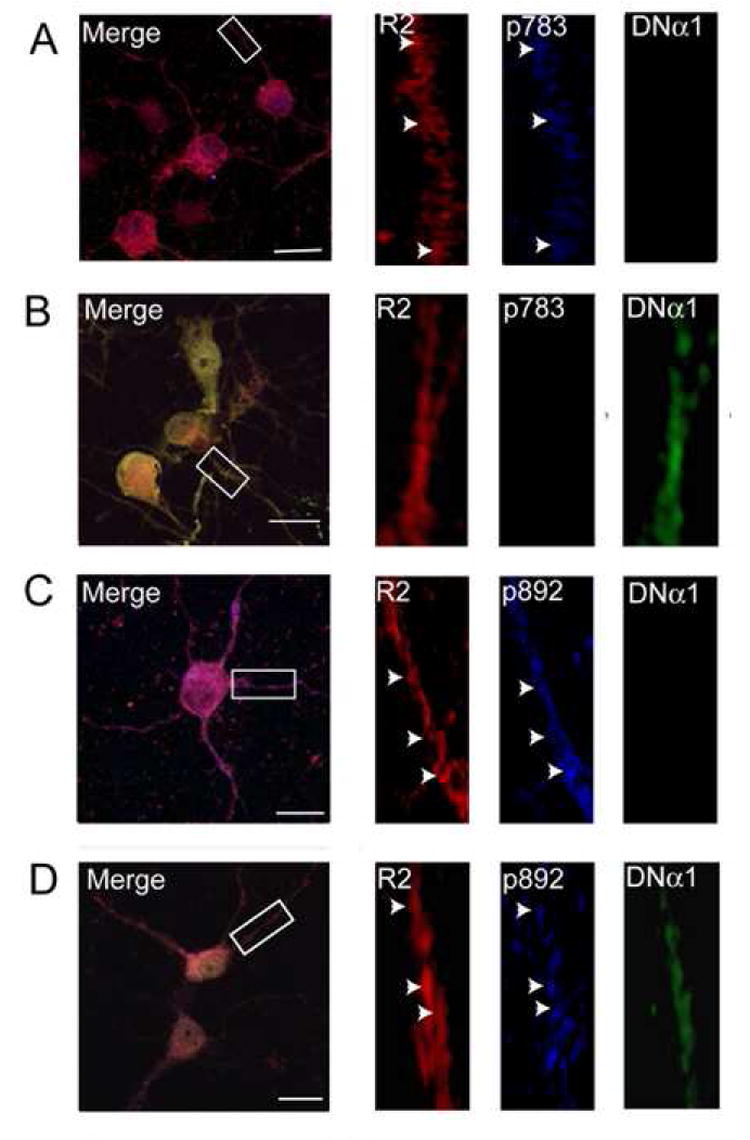
12-14 DIV cultured hippocampal neurons were transfected with a plasmid encoding myc-tagged DNα1. 24-48 h after transfection neurons were fixed, permeabilized and stained with antibodies specific for GABABR2 subunits (secondary antibody conjugated to rhodamine) and for myc (secondary antibody conjugated to FITC). Neurons were also stained with anti-pS783 antibodies (A-B) or pS892 (C-D) conjugated to Alexa 405. Images were recorded from both untransfected neurons (A and C) and those expressing DNα1 (B and D) using the same microscope settings. Scale bars represent 5 μm.
Figure 6. AMPK modulates GABAB receptor phosphorylation and function in hippocampal neurons.
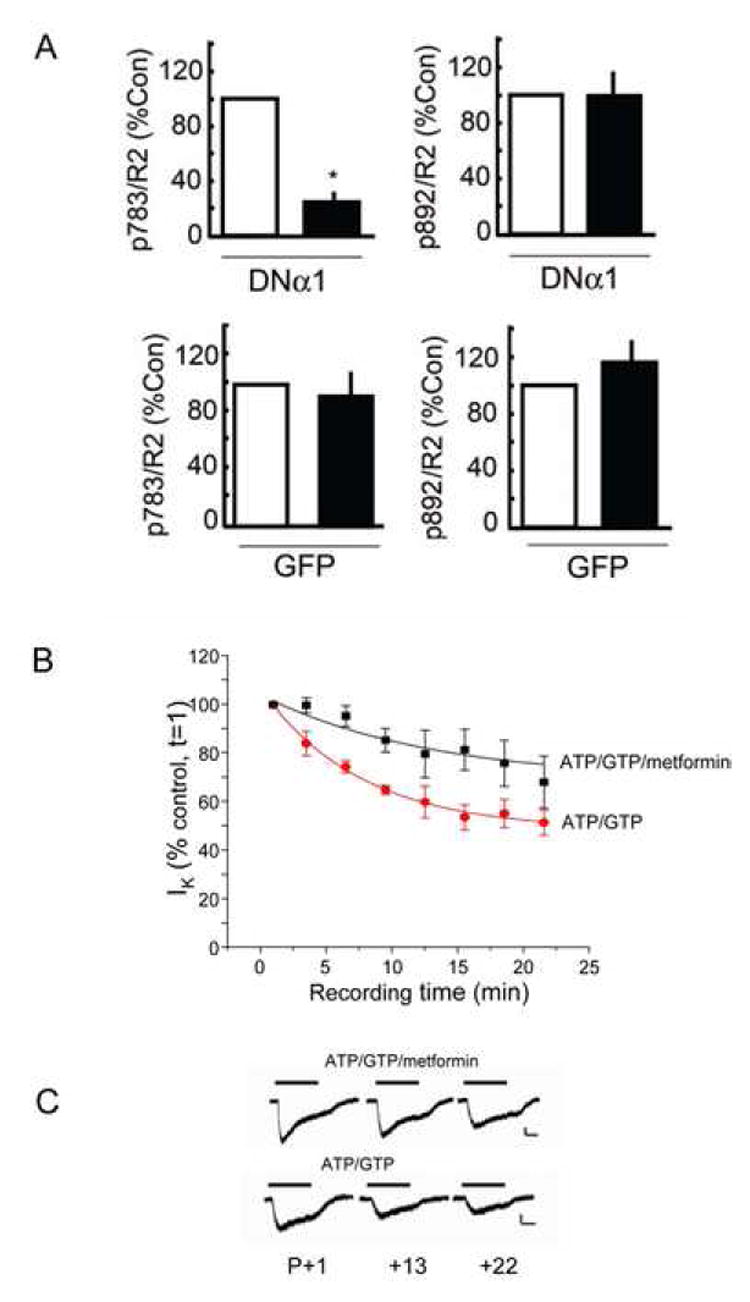
A. AMPK activity mediates basal phosphorylation of S783. Data for pS783, pS892 and R2 fluorescence levels were calculated from control non-transfected neurons and those expressing DNα1 (see Fig. 5). This data was then used to determine the co-localization of pS783/R2 and pS892/R2 signals in both non-transfected neurons (open) and those expressing either DNα1 or GFP (filled) as indicated (controls = 100%). * = significantly different from control (p<0.01; students-t test, n = 20 -35 neurons in 3 separate transfections). B. AMPK activity modulates GABAB receptor activity in hippocampal neurons. Hippocampal neurons (7-14 DIV) were exposed to 10μM baclofen, at the time points indicated, with the internal patch pipette solution containing either, 2mM ATP and 1mM GTP (●), or 2mM ATP, 1mM GTP and 1mM metformin (■) from n = 5 -12 cells. Response amplitudes have been normalized to the response recorded just after the formation of the whole-cell recording mode (t = 1). Data are mean ± s.e.m. Curve fits are monoexponential decays. All recordings were carried out in the presence of high K+ Krebs containing 20μM AP-5, 10μM CNQX, 500 nM TTX and 25μM bicuculline. C. Typical GABA-activated K+ currents recorded from hippocampal neurons using internal solutions as indicated (calibration bar 40pA, 1s).
Together these results in hippocampal neurons demonstrate that over expression of a dominant negative AMPK catalytic subunit can specifically decreases S783 phosphorylation, strongly suggesting the basal phosphorylation of this site in dependent upon AMPK activity.
AMPK modulates GABAB receptor function in hippocampal neurons
As neuronal AMPK was capable of phosphorylating GABAB receptors, we next determined whether such phosphorylation could affect native GABAB receptor function. We utilized different patch pipette solutions while whole-cell recording from cultured hippocampal neurons at 7-14 DIV and employed baclofen as the GABAB agonist to avoid the activation of native GABAA receptors. As noted previously in HEK-293 cells expressing recombinant GABAB receptors the activation of K+ current by 10μM baclofen exhibited significant rundown to 50 ± 5% of the level observed at zero time when dialyzed with electrolyte containing ATP and GTP (Fig. 7B). To assess the role of AMPK in modulation of GABAB receptor function, metformin (10mM) was added to the patch pipette solution (with ATP and GTP) to directly activate AMPK without altering the intracellular ATP:AMP ratio. Metformin significantly reduced rundown to 68 ± 11% of the level seen at zero time (Fig. 7B; p<0.01, n = 4-12 cells). Overall, these data suggest that the activation of AMPK, and hence phosphorylation of S783 in neurons, can maintain the stability of baclofen-activated K+ currents.
Figure 7. S783 phosphorylation is enhanced in the hippocampus after ischemic brain injury.
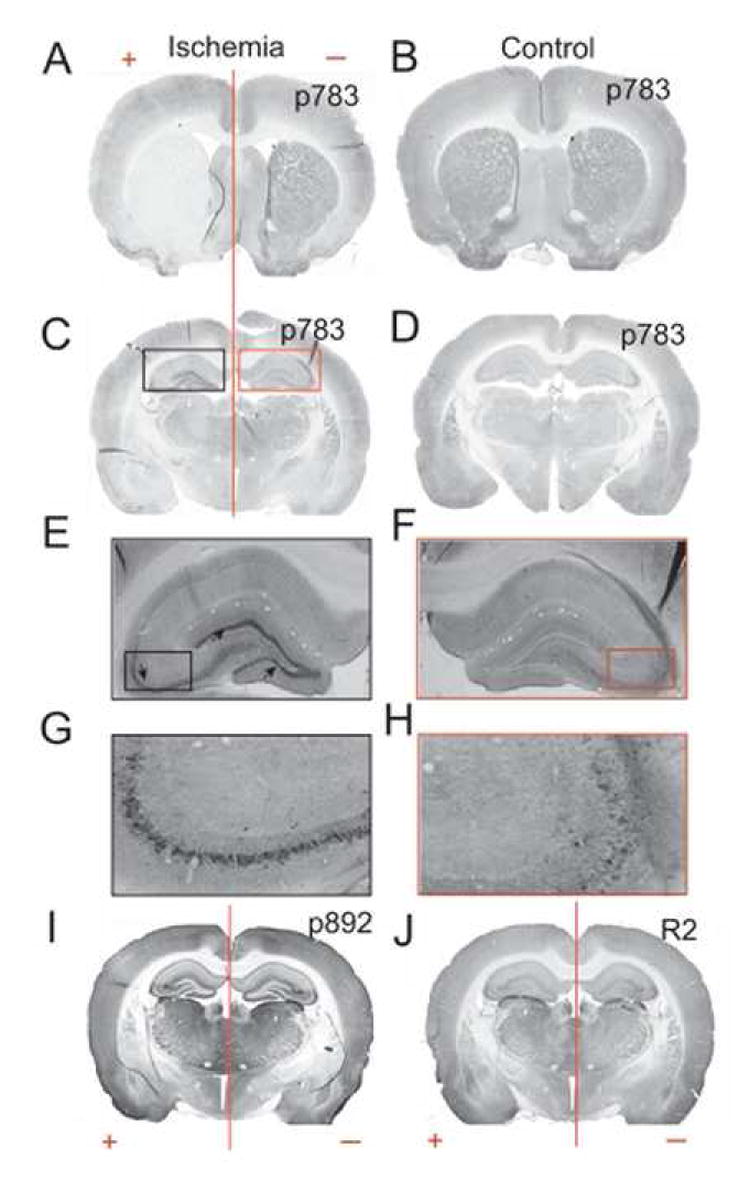
Rats subject to ischemic injury and aged match controls were cardiac perfused and 40 μm brain sections were prepared. These sections were then subject to immunohistochemistry using peroxidase/DAB staining for anti-pS783 (A to H), anti-pS892 (I) and anti-R2 (J). A. Coronal sections at the caudate putamen level showing ischemic tissue on the left hemisphere. B. Section through the caudate putamen in animals with no ischemic injury. C. Hippocampal level of ischemic brain showing the ischemic hemisphere on the left. D. Hippocampal level of control animal. E. Expansion of the black box in C showing the pyramidal cell layer of CA3 (arrows) and the granular layer of the dentate gyrus from ischemic brain. F. Expansion of the red box in C from uninjured tissue G. Magnification of the CA3 region showing intense staining in the pyramidal cell layer of the ischemic hemisphere. H. Magnification of CA3 of the non-ischemic hemisphere. I, J. coronal hippocampal sections of ischemic (+) and uninjured (-) rat brain.
S783 phosphorylation in the hippocampus is enhanced by ischemic brain injury
Anoxia and ischemia leads to rapid activation of AMPK (Carling, 2004; Kahn et al., 2005); (Gadalla et al., 2004; McCullough et al., 2005; Ramamurthy and Ronnett, 2006). We therefore sought to test whether ischemic brain injury after transient occlusion of the middle cerebral artery occlusion (MCAO) modifies GABAB phosphorylation in rats (Belayev et al., 1996; Longa et al., 1989). After injury rats were allowed to recover for 80 min before perfusion with 4% paraformaldehyde. It is well established MCAO produces a specific hemispheric injury (Belayev et al., 1996; Longa et al., 1989). To detect the location of the infarct in our experiments coronal brain sections from treated and untreated animals were first stained with anti-pS783 and then secondary antibodies conjugated to HRP before visualization with DAB followed by microscopy. In the animal shown in Fig. 7A, the area of ischemic damage (+) was localized to the left-hand side of the brain as measured by the drastic loss of pS783 staining in a large infarct that centered on the caudate/putamen. In contrast the right side of the brain (Fig 7A (-)) remained undamaged and was similar to the same brain regions from control animals (Fig. 7B). After identifying the area of brain injury we proceeded to examine S783 phosphorylation in the hippocampus for comparison with our studies in cultured neurons. Strikingly, enhanced immunoreactivity in the CA3 and DG was seen for the pS783 antibody only in the injured side of the brain (Fig. 7C, E and G black box) while the uninjured side of the brain (Fig. 7C, F, and H red box) exhibited levels of staining similar to those of control animals (Fig. 7D). However the levels of pS892 and R2 staining appeared unaltered in both the ischemic and uninjured sides of the brain (Fig. 7I and J). To further analyze the level of GABAB receptor phosphorylation in these experiments we directly compared optical density measurements of immunoreactivity between the varying regions of injured and control hippocampi and the corresponding areas of untreated animals using MetaMorph analysis. This revealed that ischemic injury produced large and highly significant increases in S783 phosphorylation within the CA3 and DG areas whilst the levels of R2 subunit immunoreactivity were unaltered (supplementary data; Fig. 4).
To examine the subcellular sites of S783 phosphorylation we stained hippocampal sections from ischemic brain with antibodies against p783 and parvalbumin, a marker for some interneurons (Celio, 1990). Immunofluorescent. images were then collected by confocal microscopy revealing that p783 staining was found in parvalbumin-positive interneurons (Fig. 8A). Strong p783 immunoreactivity was also evident as distinct puncta within the pyramidal layer of CA3 (Fig. 8A). These results demonstrate that S783 is phosphorylated in GABAergic interneurons and strongly suggest that this site is also phosphorylated in glutamatergic pyramidal neurons.
Figure 8. S783 phosphorylation promotes neuronal survival after anoxia insult.
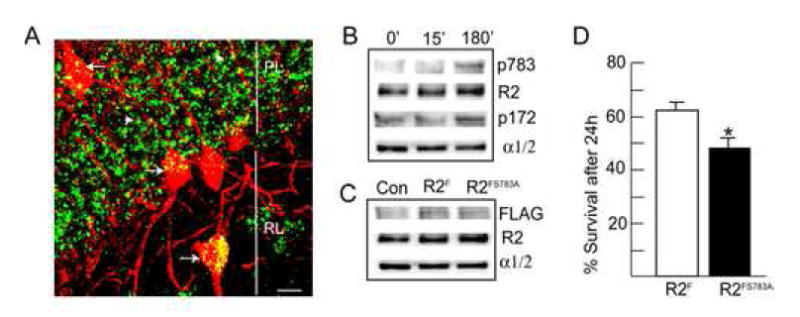
A. Analyzing the subcellular sites of S783 phosphorylation in the hippocampus using confocal microscopy. Hippocampal sections from MCAO animals were stained with antibodies against S783 and parvalbumin, a marker for subsets of interneurons, with secondary antibodies FITC and rhodamine respectively. Arrows indicate parvalbumin positive interneurons, white arrowheads represent parvalbumin negative interneurons, PL = pyramidal cell layer, RL = Stratum radiatum, scale bar = 10 μm. B. Anoxia increases AMPK activity and S783 phosphorylation. 12-14 DIV hippocampal neurons were given a 5 min anoxic insult, re-fed and incubated at 37°C for either 15 or 180 min as indicated. Neurons were then lysed and immunoblotted with antibodies against p783, GABABR2 (R2), p173 and AMPK α1/2 subunits (n=4-5). C. Expression of recombinant GABAB receptors in hippocampal neurons. 10-14 DIV hippocampal neurons expressing either GABABR2F, or GABABR2FS783A subunits were lysed and immunoblotted with FLAG antibodies. D. Mutation of S783 in GABABR2 decreases neuronal survival 24 hr after anoxia. Neurons were stained with trypan blue and viability was calculated as a percentage of unstained cells (viable cells) related to the total number of cells counted viable cells plus nonviable cells). At least 300 cells were counted on each coverslip. * = significantly different from control (p<0.01; students-t test, n=3)
S783 promotes neuronal survival after chemical anoxia
To further analyze the role of S783 in ischemia we used cultured hippocampal neurons (10-14 Div) and evaluated whether transient anoxic insult (10 mM deoxyglucose and 10 mM azide (Imura et al., 1999) (Kume et al., 2002) would activate AMPK and enhance GABAB receptor phosphorylation. In agreement with our studies in whole animals anoxic insult significantly increased the phosphorylation of S783 in GABABR2, and T172 in the α1/2 subunits of AMPK after 180 min by 330.20 ± 32% and 350.40 ± 45% h (p<0.01), without significantly altering the total cellular levels of these proteins (Fig. 8B).
We next evaluated the effects over expressing FLAG tagged GABABR2F or R2FS783A subunits (Couve et al., 2002) on the survival of cultured neurons after anoxia. For these experiments we utilized nucleofection to express these differing transgenes in cultured neurons, a technique previously established to produce transfection efficiencies of approximately 35% (Kittler et al., 2004) (Couve et al., 2004b). To assess transgene expression, 10-14 Div nucleofected neurons were immunoblotted with FLAG antibodies. This revealed the expression of bands of 102 kDa in neurons nucelofected with GABABR2F or R2FS783A subunits but not in control cultures (Fig. 8C). Significantly, expression of recombinant R2 subunits did not alter the overall expression levels of R2 expression or the levels of AMPK α1/2 subunits (Fig. 8C). To measure the effect of S783 on survival, expressing neurons were exposed to a 15 min anoxic insult, cultured for a further 15h and stained with trypan blue to assess viability (Fig. 8D). This analysis revealed that survival for neurons expressing wild-type GABABR2F was 60.20 ± 2.3% while survival for neurons expressing R2FS783A subunits was significantly reduced by 11.08% to 49.12 ± 2.2% (p<0.01). After controlling for the efficiency of nucleofection, mutation of S783 decreases neuronal survival by approximately 31% after ischemia.
Together these in vitro provide the first direct evidence that S783 can modulate neuronal survival after anoxic injury.
Discussion
GABAB receptors are critical heterodimeric G-protein-coupled receptors which are important for the induction of slow and prolonged synaptic inhibition in the brain (Bowery, 2006). However, what has remained largely enigmatic is whether other signal transduction pathways are capable of exerting long-term regulation over GABAB receptor function. Here we reveal that functional heteromeric GABAB receptors composed of R1/R2 subunits directly bind to, and are phosphorylated by, AMPK, which then influences their function. The identification of the residue phosphorylated by AMPK, S783 on the R2 subunit, has subsequently allowed us to reveal a link between the induction of ischemia and increased phosphorylation of the GABAB receptor in the hippocampus, possibly by increased activity of AMPK. During ischemia intracellular ATP levels decrease and Ca2+ levels increase (Choi, 1998), which leads to the activation of kinases upstream of AMPK, including LKB1 and CaMKK (Hawley et al., 2005; Hurley et al., 2005; Woods et al., 2003a; Woods et al., 2003b). This increase in GABAB receptor phosphorylation, evident during ischemia, is predicted to enhance receptor effector coupling and provides a new and potentially neuroprotective mechanism that may limit neuronal exposure to excitotoxicity.
GABAB receptors as substrates for AMPK
By using Y2H, in vitro binding/kinase assays, immunoprecipitation and immunofluorescence assays it is clear that the interaction of GABAB receptors and AMPK is mediated initially by the direct binding of the kinase α1/2 subunits to the C-tail of the GABABR1 subunit. Interestingly, AMPK phosphorylated the C-tails of both the GABABR1a and R2 subunits in vitro and this phosphorylation occurred exclusively on serine residues. We subsequently identified two major sites of AMPK phosphorylation: S917 and S783 in the C-tails of GABABR1a and R2 subunits respectively.
Functional modulation of GABAB receptors by AMPK
A number of binding partners for GABAB receptors have been identified to date including 14-3-3 isoforms, ATF4/CREB2 and Marlin-1, but the precise role that these binding partners play in regulating GABAB receptor functional expression remains to be determined (Couve et al., 2004a). While AMPK has been established as an important regulator of the cellular levels of ATP, via inhibition of anabolic metabolism and activation catabolic pathways (Carling, 2004; Kahn et al., 2005), our results now suggest a novel role for this kinase in modulating inhibitory neurotransmitter receptors. Using patch clamp recording, the coupling of GABAB receptors to GIRKs in HEK-293 cells typically exhibited significant levels of rundown when activated by GABA despite the internal solution containing ATP and GTP. When this was supplemented by AMP or metformin, rundown was substantially ameliorated, suggesting that the activation of AMPK was instrumental in this modulation. By using mutagenesis we were able to determine whether both GABAB receptor subunits needed to be phosphorylated for AMPK to regulate receptor function. From this approcah it was evident that the functional effects of AMPK were mediated via the Ser783 on R2, rather than Ser917 on R1a. We therefore proposed that the C-tail of the R1a subunit was more likely to mediate the binding of AMPK to GABAB receptors rather than transduce any functional effect. Consistent with this role for R2, we have demonstrated that phosphorylation of this subunit by PKA on Ser892 also alleviates GABAB receptor rundown by enhancing its activation of GIRK channels (Fairfax et al., 2004) (Couve et al., 2002).
Direct phosphorylation of neuronal GABAB receptors
To directly measure the phosphorylation of S783 we raised a phospho-specific antibody against this residue. Using pS783 we were able to confirm that this residue was basally phosphorylated in HEK-293 cells, rat brain membranes and cultured hippocampal neurons. The level of phosphorylation of S783 was significantly enhanced after AMPK activation with phenformin. Phosphorylation of S783 did not modify the cell surface expression levels of GABAB receptors in these various cell systems. This supports a number of studies showing that neuronal GABAB receptors do not undergo agonist or PKA-induced internalization (Couve et al., 2004a).
To directly evaluate S783 phosphorylation in neurons, we employed a dominant negative α1 AMPK subunit construct because specific inhibitors for this kinase do not exist. In neurons expressing DNα1a large decrease in the phosphorylation of S783 was evident. However, phosphorylation of S892, a residue predicted to be adjacent to S783 in GABABR2 and previously identified as a PKA substrate, remained unaltered. These results demonstrate that neuronal GABAB receptors are indeed phosphorylated on S783 and that AMPK activity plays a critical role in regulating the phosphorylation of this residue. To examine the functional consequences of AMPK phosphorylation on the activity of neuronal GABAB receptors, electrophysiological analyses were utilized. In agreement with our recombinant data, the activation of GIRK channels by GABAB receptors again exhibited rundown under control conditions. This rundown was largely abrogated using a patch pipette solution supplemented with metformin. These results provide strong evidence of a role for AMPK activity in regulating GABAB receptor effector coupling in neurons.
Modulation of AMPK after ischemic insult
Given the critical role AMPK activity plays in conserving cellular levels of ATP, we considered that its ability to enhance GABAB receptor functional coupling may be significant under conditions of metabolic stress. This type of stress could be imposed on a cell by persistent neuronal depolarization or alternatively it can manifest during anoxic or ischemic injury, all of which conditions are known to decrease cellular levels of ATP or increase Ca2+ levels and lead to the robust activation of AMPK (Carling, 2004; Kahn et al., 2005) (Gadalla et al., 2004; McCullough et al., 2005; Ramamurthy and Ronnett, 2006). To test our hypothesis we analyzed the phosphorylation of GABAB receptors after ischemic injury to the brain induced by MCAO. This insult revealed a large increase in the phosphorylation of S783 in the CA3 and dentate gyrus, but not in the CA2 regions of the hippocampus. In contrast, the phosphorylation of S892 in GABABR2 remained unaltered, as did the overall expression levels of R2.
To provide further insight into the possible role of S783 phosphorylation in ischemia we examined the effects of anoxia on cultured hippocampal neurons. Anoxia significantly increased AMPK activity, and enhanced phosphorylation of S783, consistent with our studies in whole animals. Moreover, using overexpression of R2 we established that mutating S783 significantly decreased neuronal survival compared to neurons expressing wild type GABAB R2 subunits. Thus, our in vivo and in vitro results suggest that ischemic/anoxic injury leads to an increased phosphorylation of S783, which increases GABAB receptor activation of postsynaptic GIRK channels, leading to increased membrane conductance, thereby limiting neuronal activity. If such an effect was also manifest presynaptically, then we would predict that AMPK should also increase the inactivation of Ca2+ channels via GABAB receptor activation, leading to decreased release of glutamate, GABA and a range of other neurotransmitters, which may have varying effects on neuronal survival. However, it is interesting to note that GABAB receptor agonists are neuroprotective in animal models of ischemia, further highlighting a predominant role for these receptors in limiting neuronal activity after ischemic/anoxic injury (Kulinskii and Mikhel’son, 2000); (Dave et al., 2005) (Jackson-Friedman et al., 1997). Finally, and in agreement with these whole animal studies, we were able to establish that phosphorylation of S783 appears to play a neuroprotective role in cultured neurons after anoxia.
In conclusion, we have uncovered a novel role for AMPK activity binding to and phosphorylating GABAB receptors, a process which enhances their functional coupling to K+ channels. Given that AMPK has a unique ability to respond to decreased cellular levels of ATP, this mechanism, which relies on a single phosphorylatable serine residue in the R2 subunit, may act to decrease synaptic activity under conditions of intense metabolic stress or ischemic injury to conserve cellular energy levels in addition to limiting neuronal injury via excitotoxicity.
Methods and Materials
Antibodies
Rabbit antibodies against p172 in AMPK were purchased from Cell Signaling Systems. Rabbit polyclonal antibodies against the α1/α2 subunits have been detailed previously (Fryer et al., 2002). Anti-pS783 was raised in rabbits against the synthetic peptide: VNQApSTSRL, and the resulting antisera was sequentially purified on dephospho- and phospho-antigens (PhosphoSolutions, Colorado Bioscience Park, 12635 East Montview Blvd., # 213 Aurora, CO). Anti-pS892 in GABABR2 (UCL-72) was used as described previously (Couve et al., 2002). Antibodies were raised against GST-fusion proteins encoding the cytoplasmic tails of GABAB R1 and R2 in both guinea pigs and chickens. Sera were affinity purified on immobilized antigens before use as outlined previously (Calver et al., 2001; Couve et al., 2001).
Cell culture and transfection
Hippocampal cultures were prepared as described previously (Couve et al., 2004b; Couve et al., 2002). 10-12 DIV hippocampal neurons were transfected using Effectene (Qiagen, Valencia, CA, USA; (Jacob et al., 2005). HEK-293 cells expressing Kir 3.1 and 3.2 channels (GIRK cells; (Leaney et al., 2000) were transfected by calcium phosphate using 1:5:1 ratios of GABABR1a, R2 and GFP constructs, respectively (Couve et al., 2002).
Neuronal transfection and anoxia
Dissociated E18 hippocampal neurons were transfected with 3 μg of CMV based expression constructs encoding N-terminally FLAG tagged R2F (Couve et al., 2002), or R2FS783A using 3 μg of DNA per 5×106 neurons using the Rat Nucleofector™ kit (Amaxa). Cells were used after 10-15 DIV (Couve et al., 2004b; Kittler et al., 2004) (Jacob et al., 2005). For anoxia, neurons were exposed to media containing 10 mM Na+ azide and 1 mM glucose for 5 min at 37°C. Neurons were then re-fed with conditioned media and cultured for a further 24h. To measure neuronal survival after this time period live neurons were stained with trypan blue, then washed extensively before chilling on ice prior to immunohistochemistry with FLAG antibody. Neurons were then fixed and the number of FLAG positive surviving neurons (Trypan blue negative) were calculated for each expression construct.
Expression constructs
Expression constructs encoding the rat GABABR1, GABABR2 subunits modified with N-terminal extracellular FLAG epitopes, GFP, GST-R1, GST-R2 fusions and myc-tagged DNα1 expression constructs have all been described previously (Couve et al., 2001; Woods et al., 2000).
GST affinity purification assays
GST-fusion proteins encoding the C-tails of GABABR1 subunits were expressed and purified as outlined previously (Couve et al., 2001); 2004b). For affinity purification GST-fusion proteins were immobilized on agarose and exposed to crude brain membranes solubilized in a buffer containing 1% Triton X-100 and (in mM) 150 NaCl, 50 Tris , pH 7.6, 5 EGTA, 5 EDTA, 50 NaF, 1 Na orthovanadate, 100 PMSF, and 10 μg/ml leupeptin, pepstatin, antipain, and aprotinin. After extensive washing in a buffer supplemented with 0.5M NaCl bound material was subject to immunoblotting. Bound material was also subject to in vitro kinase assays in a buffer containing 10 mM Tris pH 7.5, 10 mM MgCl2 and 5μCi 32Pγ-ATP with or without 100 μM 5’AMP for 10 min at 37°C followed by SDS-PAGE (Brandon et al., 1999a; Brandon et al., 1999b).
Immunoprecipitation
Crude brain membranes were solubilized in a buffer containing 2% CHAPS and (in mM) 150 NaCl, 50 Tris, pH 7.6, 5 EGTA, 5 EDTA, 50 NaF, 10 Na pyrophosphate, 1 Na orthovanadate, 10 PMSF, and 10 μg/ml leupeptin, pepstatin, antipain, and aprotinin. Lysates were then subject to immunoprecipitation with antibodies immobilized on protein A sepharose (Couve et al., 2002) and precipitated material then subject to SDS-PAGE followed by immunoblotting with antibodies against GABAB receptors or AMPK.
Immunoblot analysis and biotinylation
Cell surface biotinylation of neurons using NHS-biotin and subsequent immunoblotting with GABAB antibodies using [125I]-coupled anti-rabbit IgG and quantified using a phosphoimager were performed as detailed previously (Couve et al., 2004b; Fairfax et al., 2004).
Immunofluorescence
Hippocampal neurons were processed for immunohistochemistry under both permeabilized (+ 0.05% Triton X-100) and non-permeabilized conditions using the following primary antibodies: Anti-pS892 1:1000; Anti-pS783 1:500; anti-R1 1:1000; and anti-R2 1: 5000 (Couve et al., 2002). Images were then collected on a Bio-Rad Radiance II microscope. All channels were first background subtracted and the threshold value determined for each culture and used for all images from that culture. Quantification of fluorescence levels was performed on neuronal processes extending from the cell body in the plane of focus. Pixel intensity was then determined and measured in 2-3 μm2 areas of interest using MetaMorph software (Universal Imaging).
Immunohistochemistry
Animals were deeply anesthetized and intracardially perfused with saline solution followed by 4% paraformaldehyde. Brains were removed, post-fixed overnight and cryoprotected in 30% sucrose. Free-floating sections were cut at 40 μm using a freezing microtome and stored at -20°C in cryoprotective solution (30% sucrose, 30% ethylenglycol, 1% polyvinylpyrrolidone in PBS) until processing. Sections were washed in PBS and incubated for 10 min in 0.1% H2O2 to block endogenous peroxidase activity. After washing in PBS sections were incubated for 2 h in blocking solution (2% normal horse serum, 0.5% BSA and 0.3% Triton X-100 in PBS) followed by incubation in blocking solution containing the primary antibody for 48 h at 4°C. After rinsing in PBS sections were incubated in blocking solution containing biotinylated anti-rabbit or guinea pig antibody for 2 h at room temperature, then rinsed in PBS and incubated for 1 h in ABC solution (Vectastain Elite ABC kit, Vectorlabs). After washing, sections were incubated for 10 min in diaminobenzidine (peroxidase substrate) containing nickel chloride as an enhancing reagent. The reaction was stopped by washing twice in water. Sections were mounted onto slides, air-dried, dehydrated through graded alcohols followed by xylenes and then mounted for viewing. Sections were visualized with an Olympus BX51 microscope (Olympus Optical) and the optical density obtained using MetaMorph software (Universal Imaging Corporation).
In vitro phosphorylation, peptide mapping and phospho-amino acid analysis
GST-fusion proteins were phosphorylated in vitro using 100-1000U of AMPK in the presence or absence of 100 μM 5’AMP using 5-50 μCi of 32P-γ-ATP and a final ATP concentration of 50 μM for 30 min at 30°C. Purified AMPK was purchased from Upstate Biotechnology (Lake Placid NY) and had an activity in excess of 4000U/μg in the presence of 100 μM 5’-AMP. Phosphorylation was then assessed by autoradiography and quantified using a phospho-imager. Tryptic peptide mapping and phospho-amino acid analysis were performed as described previously (Brandon et al., 1999a).
Off-line HPLC and on-line HPLC-tandem mass spectrometry
In parallel with samples used for TLC tryptic mapping, digests of in vitro GST-CR2 fusion proteins were analyzed using capillary RP-HPLC. Radiolabelled samples (estimated 5 pmol peptides) were loaded on a 150 μm × 10 cm Microsil C8 column (Ultra-Plus II Micro LC, Micro-Tech Scientific) and 5 min fractions were collected with a robotic micro fraction collector (Probot, Dionex). Fraction samples were spotted and quantitated using a phospho-imager (Typhoon, Molecular Dynamics). In parallel, in vitro digests and off-line fractionation was performed using fusion proteins phosphorylated in the absence of 32P. Digests of these non-radioactive samples (1 μl, estimated 1 pmol peptides) were analyzed by nanoflow reverse phase liquid chromatography (RPLC) (Ultimate LC Packings, Dionex) coupled on-line to a quadrupole-orthogonal time-of-flight (QTOF) tandem mass spectrometer (MS) (QSTAR XL, Applied Biosystems/MDS Sciex) using methods described previously (Huang et al., 2001). The QSTAR MS was first operated in an information-dependent mode in which each full MS scan (1 s acquisition) was followed by MS/MS scan (3 s acquisition for MS/MS scan), where the most abundant peptide molecular ions were dynamically selected for low energy collision-induced dissociation (CID). Tandem MS/MS spectra were analyzed using both Mascot and a development version of Protein Prospector (Huang et al., 2001).
Patch clamp electrophysiology
Membrane currents were recorded from single GIRK cells using the whole-cell patch clamp configuration in conjunction with an Axopatch 1-C amplifier. Patch pipettes (resistances 3-5 MΩ ) were pulled from thin-walled borosilicate glass and filled with a solution containing (mM): 120 KCl, 2 MgCl2, 11 EGTA, 30 KOH, 10 HEPES, 1 CaCl2, 1 guanine triphosphate, 2 adenosine triphosphate with the addition of components as listed in the text that includes either 14mM creatine phosphate, 1mM adenosine monophosphate, 1mM metformin and 1.25U/ml AMPK (Upstate cell signaling solutions, NY), pH 7.0. The cells were continuously perfused with Krebs solution containing (mM): 140 NaCl, 4.7 KCl, 1.2 MgCl2, 2.5 CaCl2, 11 Glucose and 5 HEPES, pH 7.4. Before application of GABA or baclofen, the K+ concentration of the Krebs solution was increased to 25mM and the Na+ concentration was reduced to 120mM. Cells were recorded from 20-48 h after transfection. Membrane currents were filtered at 5 kHz (-3dB, 6th pole Bessel, 36 dB/octave) and stored onto a Dell Pentium III computer for later analysis with Clampex 8. Any change of more than 10% in the resting membrane input conductance or series resistance resulted in the recording being discarded. Peak amplitude GABA-activated K+ currents were determined at -70mV holding potential. Drugs and solutions were rapidly applied to the cells using a modified Y-tube positioned approximately 300:m from the recorded cell. To construct concentration-response relationships for GABA, the current (I) was measured in the presence of each concentration of GABA applied at 3 min intervals. The currents were normalized to the maximum GABA response (Imax) and the concentration response relationship fitted with the Hill equation:
where the EC50 represents the concentration of GABA ([A]) inducing 50% of the maximal K+ current evoked by a saturating concentration of GABA and nH is the Hill coefficient. For the analysis of current run-down, GABA or baclofen-activated current amplitudes were measured at G3 min intervals and the resulting time stability relatiohsips fitted with a monoexponential function of the form:
where y is the GABA/baclofen current amplitude at time t, yo is the residual amplitude at the end of drug application, A represents the amplitude and τ is the decay time constant. The fit was determined using a Marquadt non-linear least squares routine.
Yeast two-hybrid screens
Yeast two-hybrid screens were performed as described previously (Restituito et al., 2005). The bait plasmid containing the sequence corresponding to the cytoplasmic tails of GABABR1 (pMW101-CR1) was used to screen a rat brain cDNA library in yeast vector pJG4-5. Potential interactions were analyzed by growth on plates lacking leucine, uracil, tryptophan and histidine and confirmed by galactose-dependency and β-galactosidase assays. All positive interactions were confirmed by retransformation into the EGY48 strain and also with empty vector and bait encoding the cytoplasmic tail of the R2 subunit.
Transient middle cerebral artery occlusion (MCAO) surgery
Adult male Wistar rats (290-310g) were anesthesized with 3% isoflurane in 70% nitrous oxide and 30% oxygen through a nose cone. Temperature was maintained at 37°C throughout the surgery using a heating lamp. Transient MCAO was induced for 90 min using the intraluminal suture method (Longa et al., 1989). Briefly, an 18 mm length of 4-0 monofilament nylon suture coated with poly-L-lysine (Belayev et al., 1996) and a flame-rounded tip was inserted into the external carotid artery and advanced through the internal carotid to occlude the origin of the middle cerebral artery (MCA). Ninety minutes later the rats were re-anesthetized and the suture was withdrawn to allow for reperfusion of cerebral blood flow. Sham operated controls were subject to the same surgery but without advancement of the suture into the MCA. 2 h following reperfusion, rats were deeply anesthetized and perfused transcardially with 50 mls of saline followed by 200 mls of freshly made 4% paraformaldehyde in PBS. Brains were extracted and post-fixed for 2 h in 4% paraformaldehyde then processed for immunohistochemistry.
Supplementary Material
Acknowledgments
This work was supported by MRC (UK), Wellcome Trust grants to TGS and SJM and NIH grants NS 046478, NS 048045 and NS051195 to SJM. We are grateful to Yolande Haydon and Mansi Vithlani for their comments on this manuscript. In memory of Dr Frederic Flach, KHS MD 1927-2006. Correspondence should be addressed to SJM, Email: sjmoss@mail.med.upenn.edu.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Belayev L, Alonso OF, Busto R, Zhao W, Ginsberg MD. Middle cerebral artery occlusion in the rat by intraluminal suture. Neurological and pathological evaluation of an improved model. Stroke. 1996;27:1616–1622. doi: 10.1161/01.str.27.9.1616. discussion 1623. [DOI] [PubMed] [Google Scholar]
- Bettler B, Kaupmann K, Mosbacher J, Gassmann M. Molecular structure and physiological functions of GABA(B) receptors. Physiol Rev. 2004;84:835–867. doi: 10.1152/physrev.00036.2003. [DOI] [PubMed] [Google Scholar]
- Bowery NG. GABA(B) receptor: a site of therapeutic benefit. Curr Opin Pharmacol. 2006;6:37–43. doi: 10.1016/j.coph.2005.10.002. [DOI] [PubMed] [Google Scholar]
- Brandon NJ, Bedford FK, Connolly CN, Couve A, Kittler JT, Hanley JG, Jovanovic JN, Uren J, Taylor P, Thomas P, et al. Synaptic targeting and regulation of GABA(A) receptors. Biochem Soc Trans. 1999a;27:527–530. doi: 10.1042/bst0270527. [DOI] [PubMed] [Google Scholar]
- Brandon NJ, Uren JM, Kittler JT, Wang H, Olsen R, Parker PJ, Moss SJ. Subunit-specific association of protein kinase C and the receptor for activated C kinase with GABA type A receptors. J Neurosci. 1999b;19:9228–9234. doi: 10.1523/JNEUROSCI.19-21-09228.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calver AR, Robbins MJ, Cosio C, Rice SQ, Babbs AJ, Hirst WD, Boyfield I, Wood MD, Russell RB, Price GW, et al. The C-terminal domains of the GABA(b) receptor subunits mediate intracellular trafficking but are not required for receptor signaling. J Neurosci. 2001;21:1203–1210. doi: 10.1523/JNEUROSCI.21-04-01203.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carling D. The AMP-activated protein kinase cascade--a unifying system for energy control. Trends Biochem Sci. 2004;29:18–24. doi: 10.1016/j.tibs.2003.11.005. [DOI] [PubMed] [Google Scholar]
- Carling D. AMP-activated protein kinase: balancing the scales. Biochimie. 2005;87:87–91. doi: 10.1016/j.biochi.2004.10.017. [DOI] [PubMed] [Google Scholar]
- Celio MR. Calbindin D-28k and parvalbumin in the rat nervous system. Neuroscience. 1990;35:375–475. doi: 10.1016/0306-4522(90)90091-h. [DOI] [PubMed] [Google Scholar]
- Choi D. Antagonizing excitotoxicity: a therapeutic strategy for stroke? Mt Sinai J Med. 1998;65:133–138. [PubMed] [Google Scholar]
- Couve A, Calver AR, Fairfax B, Moss SJ, Pangalos MN. Unravelling the unusual signalling properties of the GABA(B) receptor. Biochem Pharmacol. 2004a;68:1527–1536. doi: 10.1016/j.bcp.2004.06.036. [DOI] [PubMed] [Google Scholar]
- Couve A, Kittler JT, Uren JM, Calver AR, Pangalos MN, Walsh FS, Moss SJ. Association of GABA(B) receptors and members of the 14-3-3 family of signaling proteins. Mol Cell Neurosci. 2001;17:317–328. doi: 10.1006/mcne.2000.0938. [DOI] [PubMed] [Google Scholar]
- Couve A, Restituito S, Brandon JM, Charles KJ, Bawagan H, Freeman KB, Pangalos MN, Calver AR, Moss SJ. Marlin-1, a novel RNA-binding protein associates with GABA receptors. J Biol Chem. 2004b;279:13934–13943. doi: 10.1074/jbc.M311737200. [DOI] [PubMed] [Google Scholar]
- Couve A, Thomas P, Calver AR, Hirst WD, Pangalos MN, Walsh FS, Smart TG, Moss SJ. Cyclic AMP-dependent protein kinase phosphorylation facilitates GABA(B) receptor-effector coupling. Nat Neurosci. 2002;5:415–424. doi: 10.1038/nn833. [DOI] [PubMed] [Google Scholar]
- Dave KR, Lange-Asschenfeldt C, Raval AP, Prado R, Busto R, Saul I, Perez-Pinzon MA. Ischemic preconditioning ameliorates excitotoxicity by shifting glutamate/gamma-aminobutyric acid release and biosynthesis. J Neurosci Res. 2005;82:665–673. doi: 10.1002/jnr.20674. [DOI] [PubMed] [Google Scholar]
- Fairfax BP, Pitcher JA, Scott MG, Calver AR, Pangalos MN, Moss SJ, Couve A. Phosphorylation and chronic agonist treatment atypically modulate GABAB receptor cell surface stability. J Biol Chem. 2004;279:12565–12573. doi: 10.1074/jbc.M311389200. [DOI] [PubMed] [Google Scholar]
- Fryer LG, Parbu-Patel A, Carling D. The Anti-diabetic drugs rosiglitazone and metformin stimulate AMP-activated protein kinase through distinct signaling pathways. J Biol Chem. 2002;277:25226–25232. doi: 10.1074/jbc.M202489200. [DOI] [PubMed] [Google Scholar]
- Gadalla AE, Pearson T, Currie AJ, Dale N, Hawley SA, Sheehan M, Hirst W, Michel AD, Randall A, Hardie DG, Frenguelli BG. AICA riboside both activates AMP-activated protein kinase and competes with adenosine for the nucleoside transporter in the CA1 region of the rat hippocampus. J Neurochem. 2004;88:1272–1282. doi: 10.1046/j.1471-4159.2003.02253.x. [DOI] [PubMed] [Google Scholar]
- Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Makela TP, Alessi DR, Hardie DG. Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J Biol. 2003;2:28. doi: 10.1186/1475-4924-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley SA, Gadalla AE, Olsen GS, Hardie DG. The antidiabetic drug metformin activates the AMP-activated protein kinase cascade via an adenine nucleotide-independent mechanism. Diabetes. 2002;51:2420–2425. doi: 10.2337/diabetes.51.8.2420. [DOI] [PubMed] [Google Scholar]
- Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, Frenguelli BG, Hardie DG. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005;2:9–19. doi: 10.1016/j.cmet.2005.05.009. [DOI] [PubMed] [Google Scholar]
- Huang L, Jacob RJ, Pegg SC, Baldwin MA, Wang CC, Burlingame AL, Babbitt PC. Functional assignment of the 20 S proteasome from Trypanosoma brucei using mass spectrometry and new bioinformatics approaches. J Biol Chem. 2001;276:28327–28339. doi: 10.1074/jbc.M008342200. [DOI] [PubMed] [Google Scholar]
- Hurley RL, Anderson KA, Franzone JM, Kemp BE, Means AR, Witters LA. The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J Biol Chem. 2005;280:29060–29066. doi: 10.1074/jbc.M503824200. [DOI] [PubMed] [Google Scholar]
- Imura T, Shimohama S, Sato M, Nishikawa H, Madono K, Akaike A, Kimura J. Differential expression of small heat shock proteins in reactive astrocytes after focal ischemia: possible role of beta-adrenergic receptor. J Neurosci. 1999;19:9768–9779. doi: 10.1523/JNEUROSCI.19-22-09768.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson-Friedman C, Lyden PD, Nunez S, Jin A, Zweifler R. High dose baclofen is neuroprotective but also causes intracerebral hemorrhage: a quantal bioassay study using the intraluminal suture occlusion method. Exp Neurol. 1997;147:346–352. doi: 10.1006/exnr.1997.6637. [DOI] [PubMed] [Google Scholar]
- Jacob TC, Bogdanov YD, Magnus C, Saliba RS, Kittler JT, Haydon PG, Moss SJ. Gephyrin regulates the cell surface dynamics of synaptic GABAA receptors. J Neurosci. 2005;25:10469–10478. doi: 10.1523/JNEUROSCI.2267-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005;1:15–25. doi: 10.1016/j.cmet.2004.12.003. [DOI] [PubMed] [Google Scholar]
- Kittler JT, Thomas P, Tretter V, Bogdanov YD, Haucke V, Smart TG, Moss SJ. Huntingtin-associated protein 1 regulates inhibitory synaptic transmission by modulating gamma-aminobutyric acid type A receptor membrane trafficking. Proc Natl Acad Sci U S A. 2004;101:12736–12741. doi: 10.1073/pnas.0401860101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulinskii VI, Mikhel’son GV. Additivity and independence of neuroprotective effects of GABAA and GABAB receptor agonists in complete global cerebral ischemia. Bull Exp Biol Med. 2000;130:772–774. doi: 10.1007/BF02766091. [DOI] [PubMed] [Google Scholar]
- Kume T, Nishikawa H, Taguchi R, Hashino A, Katsuki H, Kaneko S, Minami M, Satoh M, Akaike A. Antagonism of NMDA receptors by sigma receptor ligands attenuates chemical ischemia-induced neuronal death in vitro. Eur J Pharmacol. 2002;455:91–100. doi: 10.1016/s0014-2999(02)02582-7. [DOI] [PubMed] [Google Scholar]
- Leaney JL, Milligan G, Tinker A. The G protein alpha subunit has a key role in determining the specificity of coupling to, but not the activation of, G protein-gated inwardly rectifying K(+) channels. J Biol Chem. 2000;275:921–929. doi: 10.1074/jbc.275.2.921. [DOI] [PubMed] [Google Scholar]
- Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- McCullough LD, Zeng Z, Li H, Landree LE, McFadden J, Ronnett GV. Pharmacological inhibition of AMP-activated protein kinase provides neuroprotection in stroke. J Biol Chem. 2005;280:20493–20502. doi: 10.1074/jbc.M409985200. [DOI] [PubMed] [Google Scholar]
- Ramamurthy S, Ronnett GV. Developing a Head for Energy Sensing: AMP-activated Protein Kinase as a Multifunctional Metabolic Sensor in the Brain. J Physiol. 2006 doi: 10.1113/jphysiol.2006.110122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Restituito S, Couve A, Bawagan H, Jourdain S, Pangalos MN, Calver AR, Freeman KB, Moss SJ. Multiple motifs regulate the trafficking of GABA(B) receptors at distinct checkpoints within the secretory pathway. Mol Cell Neurosci. 2005;28:747–756. doi: 10.1016/j.mcn.2004.12.006. [DOI] [PubMed] [Google Scholar]
- Scott JW, Norman DG, Hawley SA, Kontogiannis L, Hardie DG. Protein kinase substrate recognition studied using the recombinant catalytic domain of AMP-activated protein kinase and a model substrate. J Mol Biol. 2002;317:309–323. doi: 10.1006/jmbi.2001.5316. [DOI] [PubMed] [Google Scholar]
- Woods A, Azzout-Marniche D, Foretz M, Stein SC, Lemarchand P, Ferre P, Foufelle F, Carling D. Characterization of the role of AMP-activated protein kinase in the regulation of glucose-activated gene expression using constitutively active and dominant negative forms of the kinase. Mol Cell Biol. 2000;20:6704–6711. doi: 10.1128/mcb.20.18.6704-6711.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LG, Neumann D, Schlattner U, Wallimann T, Carlson M, Carling D. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol. 2003a;13:2004–2008. doi: 10.1016/j.cub.2003.10.031. [DOI] [PubMed] [Google Scholar]
- Woods A, Vertommen D, Neumann D, Turk R, Bayliss J, Schlattner U, Wallimann T, Carling D, Rider MH. Identification of phosphorylation sites in AMP-activated protein kinase (AMPK) for upstream AMPK kinases and study of their roles by site-directed mutagenesis. J Biol Chem. 2003b;278:28434–28442. doi: 10.1074/jbc.M303946200. [DOI] [PubMed] [Google Scholar]
- Woollhead AM, Scott JW, Hardie DG, Baines DL. Phenformin and 5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside (AICAR) activation of AMP-activated protein kinase inhibits transepithelial Na+ transport across H441 lung cells. J Physiol. 2005;566:781–792. doi: 10.1113/jphysiol.2005.088674. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.