

Abstract
To advance cancer research in a transformative way, we must redefine the problem. Although epithelial cancers, such as breast cancer, may be caused by random somatic gene mutations, the reality is that this is only one of many ways to induce tumor formation. Cancers also can be produced in experimental systems in vitro and in vivo, for example, by inducing sustained alterations of extracellular matrix (ECM) structure. Moreover, certain epithelial cancers can be induced to ‘reboot’ and regenerate normal tissue morphology when combined with embryonic mesenchyme or exogenous ECM scaffolds that are produced through epithelial-stromal interactions. At the same time, work in the field of Mechanical Biology has revealed that many cell behaviors critical for cancer formation (e.g., growth, differentiation, motility, apoptosis) can be controlled by physical interactions between cells and their ECM adhesions that alter the mechanical force balance in the ECM, cell and cytoskeleton. Epithelial tumor progression also can be induced in vitro by changing ECM mechanics or altering cytoskeletal tension generation through manipulation of the Rho GTPase signaling pathway. Mechanical interactions between capillary cells and ECM that are mediated by Rho signaling similarly mediate control of capillary cell growth and angiogenesis, which are equally critical for cancer progression and metastasis. These findings question basic assumptions in the cancer field, and raise the intriguing possibility that cancer may be a reversible disease that results from progressive deregulation of tissue architecture, which leads to physical changes in cells and altered mechanical signaling. This perspective raises the possibility of developing a tissue engineering approach to cancer therapy in which biologically-inspired materials that mimic the embryonic microenvironment are used to induce cancers to revert into normal tissues.
Keywords: mechanical, extracellular matrix, stroma, cell traction, cytoskeleton, cancer therapy
Introduction
Cancers are commonly thought to result from progressive accumulation of random gene mutations, and most research in this area has therefore sought to identify critical oncogenic genes or proteins. This paradigm led the pharmaceutical industry to focus on development of drugs that target single molecular components encoded or regulated by these genes. It is now clear, however, that cell behaviors are not regulated by a linear series of commands, but rather by networks of molecular interactions that involve positive and negative reinforcement, as well as high levels of cross talk integrated at the whole system (genome-wide gene and protein regulatory network) level [1–4].
In this type of dynamic regulatory network, switching between different stable states or phenotypes requires that activities of signaling molecules in multiple pathways change in concert [1, 3, 5, 6]. For example, different mitogens commonly activate over sixty genes in common [7], and master switch genes (e.g., myoD, Snail) similarly control the implementation of complex behavioral programs required for cell fate switching by regulating the concerted expression of scores of downstream genes [8, 9]. Adult skin fibroblasts also can be induced to revert to pluripotent embryonic stem cells by simultaneously co-expressing a handful of different transcription factors that, in turn, activate multiple downstream genes [10–13].
If multiple signaling elements in the cellular regulatory network must be altered simultaneously to change cell phenotype, then various types of environmental stimuli that produce pleiotropic effects in cells (and hence are not commonly thought of as ‘specific’ bioregulators) may contribute to normal and malignant tissue development. This may explain why switching between different cell fates that are critical for cancer (e.g., growth, differentiation, apoptosis, motility) can be triggered in normal and transformed cells, as well as stem cells, by changes in extracellular matrix (ECM) structure and cell shape distortion [14–20], ‘non-specific’ chemical solvents [6] and electrical ion flows [21–24] that influence multiple gene activities, as well as by distinct molecular factors or specific gene mutations in epithelial or mesenchymal cells. It also could explain why although a combination of four genes was recently found to be sufficient to induce fibroblasts to revert to embryonic stem cells in two different experimental reports, the four genes utilized were different in each study (only two genes were shared in common) [10,11,25]. Perhaps it is for this reason that conventional strategies for the development of anti-cancer therapeutics have been suboptimal, and why many active ‘single target’ drugs (e.g., Glivec) are later discovered to influence multiple signaling pathways simultaneously [26]. Thus, a key obstacle for future progress in the cancer therapeutics field is to develop experimental, theoretical and therapeutic strategies that take into account the structural complexity and system-level nature of cellular regulation [5, 27–29].
Another major problem restricting forward advance is that cancer is often defined as a disease of cell proliferation. But deregulated growth is not sufficient to make a tissue cancerous; a wart is a simple example. What makes a growing cancer malignant is its ability to break down tissue architecture, invade through disrupted tissue boundaries, and metastasize to distant organ sites. In simplest terms, cancer is a disease of development: it results from loss of the normal controls that direct cells to assemble into tissues, and that hold tissues within their organ confines [30–35]. This is consistent with the increasing appreciation of the importance of cancer stem cells [36], epithelial-mesenchymal transitions [37], and angiogenesis [38] for tumor formation and metastatic progression. In addition, provocative experiments first carried out over thirty five years ago show that certain cancers differentiate and normalize their growth when combined with normal mesenchyme, other embryonic tissues, or with ECMs that are deposited as a result of interactions between these tissues [30,39–48]. Some human malignant carcinomas also induce host stroma to be tumorigenic in nude mice [49]. Thus, cancer is a developmental disease that involves dysfunction of multi-cellular inductive interactions; it does not result from unregulated growth of a single cell type.
Interest in work pursuing developmental contributions and non-genetic causes of cancer waned when the molecular biology revolution surged and the focus shifted almost entirely to genetic causation. But there is now renewed interest in developmental and environmental contributions to cancer growth because of more recent studies that confirm the central role that the tissue microenvironment plays during tumor formation [35,50–52]. These experiments show, for example, that carcinogens must act on the connective tissue stroma as well as the epithelium to produce a cancer [53]; mechanical interactions between cancer cells and ECM can accelerate neoplastic transformation [18,54]; normal tissues can be induced to become cancerous in vivo by altering ECM structure [55]; and stroma from healthy adult animals can prevent neoplastic transformation and encourage normal growth of grafted epithelial cancer cells [48]. The recent clinical approval and use of angiogenesis inhibitors that target the vasculature in the stromal compartment of the tumor, and not the cancer cells themselves, provide additional evidence supporting the potential value of this unconventional view of cancer formation and progression.
Thus, the challenge is to retrace our steps, and to re-explore this old path of investigation in cancer research that was left by the wayside years ago. Specifically, these observations raise the possibility that the production of cancer stem cells, epithelial-mesenchymal transitions, increased angiogenesis and unrestrained cell growth that drive cancer formation may result from deregulation of the tissue microenvironment. Conversely, embryonic tissues may reverse cancerous growth by restoring these normal microenvironmental cues. In this article, we explore these possibilities in greater detail, and briefly discuss the central role that the Rho family of small GTPases plays in this micromechanical control system. The possibility of developing a tissue engineering approach to cancer therapy that involves creation of biomimetic materials that mimic the inductive, cancer-reversing properties of embryonic tissues, is also discussed.
Structural determinants of cancer formation
Epigenetic (non-genetic) factors play an important role in cancer formation. Although the term ‘epigenetic’ has come to be used in a very narrow way (i.e., to refer to stable chromatin modifications), the reality is that there are many other non-genetic contributors to cancer development. For example, while constitutive expression of an oncogene in the beta cells of pancreatic islets stimulates growth and produces pancreatic tumor formation in transgenic mice, these tumors remain in a benign hyperplastic state and do not progress to form cancerous lesions unless angiogenesis (new capillary blood vessel growth) is also stimulated in the neighboring stroma [56]. The deregulation of cell growth and loss of cell-cell relations that characterize early stages of pancreatic carcinoma formation also correlate with compromise of the structural integrity of the epithelial ECM (basement membrane) [30,57]. Moreover, these disorganized tumor cells suppress their growth and reform into a polarized epithelium when they come in direct contact with connective tissue stroma that induces them to accumulate an intact basement membrane in vivo, or when they are cultured on exogenous, intact basement membrane in vitro [30,43,44,57]. In addition, recent studies with mammary epithelial tumor cell show that transformation of these cells can be enhanced based on physical interactions with the ECM that increase cell contractility [18], and that their growth and differentiation can be normalized by modulating cell adhesion to the ECM [58].
Changes in ECM structure also appear to play a central role in cancer formation in vivo. For example, normal breast epithelium can be induced to progress through hyperplasia and to transform into cancerous tissue by constitutively overexpressing the ECM-degrading enzyme, stromelysin, in transgenic mice [55]. Importantly, these cells that were transformed as a result of changes of tissue structure also exhibit genomic abnormalities [55,59,60]. This concept that structural or mechanical changes in the tissue microenvironment may actively contribute to tumor formation is supported by early experiments in the cancer research field, which showed that implanting a rigid piece of metal or plastic can trigger cancer formation in animals, whereas tumors do not form when the same material is introduced as a powder [61] Thus, changes in ECM and tissue structure appear to have the potential to be as carcinogenic as oncogenic chemicals, viruses, radiation and gene mutations.
Cancer as a disease of tissue development
Cancer is a developmental disease because it results from a breakdown of the fundamental rules that govern how cells stably organize within tissues, tissues within organs, and organs within the whole living organism. Uncontrolled cell growth is necessary for cancer formation, but it is not sufficient. It is only when growth becomes autonomous and leads to disorganization of normal tissue architecture that a pathologist can recognize that a normal tissue has undergone ‘neoplastic transformation’. In the case of epithelial tumors that make up over 90% of cancers, the tumor (tissue mass or swelling) is deemed malignant if there is a breakdown of boundaries between the epithelial and connective tissues that comprise the organ. Disruption of these tissue boundaries enables these cells to invade into nearby blood vessels or lymphatics, and thereby spread or ‘metastasize’ to distant organs resulting in multi-organ failure and death. It is therefore helpful to view cancer progression in the context of embryological development gone awry.
Because most cancer research is carried out using experimental tumor models, the fact that cancer results from dysregulation of a normal tissue and progressive loss of tissue architecture is often ignored. This is important because although tissue patterns are initially established in the embryo, all tissues are dynamic structures that undergo continual turnover. Thus, living organs must maintain functional signaling mechanisms that are necessary to ensure that the normal three-dimensional (3D) form of their tissues remain relatively constant throughout adult life.
The shapes of normal epithelial tissues arise in the embryo as a result of complex epithelial-mesenchymal interactions; however, the 3D pattern of many epithelial tissues is actually dictated by the underlying mesenchyme. For example, in studies in which epithelia isolated from mammary gland were recombined with mesenchyme from salivary gland, the final organ displayed the morphology of the salivary gland (i.e., the source of the mesenchyme); however, the cells secreted milk proteins into the duct instead of salivary enzymes [62]. Interestingly, mesenchyme from different tissues also differ in their ability to exert traction forces on their adhesions [63]. Thus, the mesenchyme apparently possesses all of the chemical and mechanical information necessary to guide 3D tissue and organ formation in a characteristic tissue pattern-specific manner.
Analysis of the mechanism of epithelial organ development has revealed that the mesenchyme sculpts epithelial tissue form by accelerating and slowing ECM (basement membrane) turnover at selective sites [34, 64, 65](Fig. 1). The mesenchyme produces high ECM-degrading activity (e.g., metalloproteinases) in selected regions, and the overlying epithelium responds by increasing ECM deposition to even greater levels so that net lateral extension and outward folding of the epithelial sheet-like basement membrane results at these sites. Epithelial cell proliferation also rises in these same regions that exhibit the highest ECM turnover [66–68], and thus every time the ECM area doubles in size it is precisely matched by cell division such that the thickness of the epithelium remains relatively stable during this dynamic remodeling process.
Fig. 1. Cancer as an Engineering Problem.
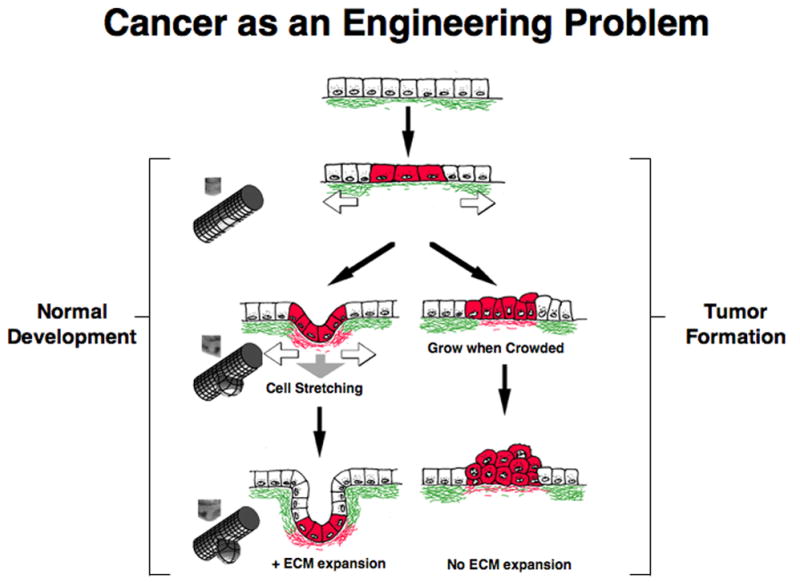
A theoretical model for mechanical control of tissue remodeling during normal epithelial development and tumor formation. Left) During normal development, regional increases in ECM turnover result in formation of local defects in the basement membrane (green), which stretches and thins due to the contraction and pulling of neighboring epithelium (white arrows) and underlying mesenchyme (gray arrow). Cells adherent to this region of the basement membrane will distort or experience increased stresses and thus, become preferentially sensitive to growth stimuli. Cell division is paralleled by deposition of new basement membrane (red) and thus, cell mass expansion and ECM extension are tightly coupled; this leads to bud formation in this localized region. Micrographs of normal epithelial bud formation during embryonic lung formation, and corresponding engineering depictions of the local mechanical strain distributions are shown at the far left. Right) During tumor formation in an adult epithelium, basement membrane thinning, changes in cell mechanics, and an increase in the sensitivity of adjacent cells to growth stimuli are also observed, much like during epithelial bud formation (left). But because cell division is not accompanied by basement membrane extension, piling up of epithelial cells and disorganization of normal tissue architecture result. If these changes in tissue structure and mechanics are sustained over time, then this continued growth stimulus could lead to selection of anchorage-independent cells, and development of a malignant carcinoma.
At the same time cell proliferation and basement membrane growth are accelerating at the tips of growing epithelial buds, the mesenchyme deposits fibrillar collagen that slows basement membrane degradation in the intervening regions (Fig. 1)[69]. In this manner, epithelial organs (e.g., glands, skin) are created that exhibit regular patterns containing multiple growing epithelial buds or branches separated by quiescent cleft regions. However, this is a delicate dynamic balance: if ECM degradation outpaces ECM synthesis, then basement membrane dissolution results, and this leads to involution of the entire developing tissue [70–72]. Cells within growth epithelium and endothelium regress because they lose their normal ECM anchoring points when basement membrane integrity is lost, and this causes the cells to detach, round and switch on the cell death (apoptosis) program [16,73].
Epithelial-mesenchymal interactions are generally discussed in the context of embryogenesis, and some assume that they end at birth. However, all adult tissues undergo turnover as their constituent cells and molecules are continually removed and replaced. Thus, in reality, it is the 3D architecture or pattern integrity of the tissue that is maintained over time, and not individual cellular or molecular subcomponents (e.g., normal intestinal epithelium ‘turns over’ every three days). Importantly, adult epithelium retains the ability to undergo normal morphogenesis when mixed with embryonic mesenchyme from different regions [42, 45, 74], and the source of the stromal tissue governs the final 3D form that epithelia will exhibit [75]. Moreover, studies of the growth of normal epithelium [67,76] and endothelium [77] show that local dissolution of basement membrane occurs before the onset of cell proliferation. Thus, ECM remodeling appears to drive growth differentials, and not the other way around.
These findings are relevant for cancer because tumor architecture also varies depending on the source of connective tissue [78], and chemical carcinogenesis of epidermis requires the presence of closely apposed carcinogen-treated dermis [79]. Moreover, different types of epithelial tumors produce distinct effects on production of stromal collagen by host fibroblasts [80], and grafted human epithelial tumors can recruit normal murine stromal cells to become tumorigenic in nude mice [49]. But perhaps the most interesting observation is that various epithelial cancers can be induced to ‘differentiate’ and regenerate normal epithelial organization and histodifferentiation by being mixed with normal embryonic mesenchyme [30,39–48].
Localized differentials of cell growth and ECM turnover similar to those observed during epithelial-mesenchymal interactions in the embryo occur in tissues that retain their ability to undergo morphogenesis in the adult (e.g., breast epithelium during pregnancy, growing blood capillaries during wound healing and tumor angiogenesis). Interestingly, although stable adult epithelial tissues do not normally exhibit major changes in ECM structure or undergo active changes in form, ultrastructural changes in the basement membrane do occur during early phases of cancer formation, prior to development of a palpable tumor [81] [82, 83]. These structural alterations include the appearance of basement membrane gaps, thickening and reduplication, as well as loosening of basal cells from one another and from neighboring connective tissue [81]. Experimental treatment of thyroid gland with carcinogens similarly causes the epithelial basement membrane to become discontinuous (as seen in the tips of growing epithelial buds in the embryo), and then to completely dissolve in certain regions as the lesions progress from pre-nodular to nodular forms, and finally to overt invasive carcinomas [82].
Formation of breaks in the epithelial tumor basement membrane is a hallmark of malignant invasion because cells in non-malignant tumors surrounded by an intact basement membrane generally do not penetrate into surrounding tissues [84–86]. But ECM remodeling during tumor progression is likely more dynamic than is generally appreciated, and microenvironmental cues may govern whether basement membrane degradation or synthesis is induced locally. For example, different tumor lesions in a spontaneously metastatic murine mammary cancer vary greatly in terms of the number and size of basement membrane discontinuities they exhibit [87]. Most interesting is the observation that well developed, continuous basement membranes sometimes appear surrounding metastatic tumors at distant sites, even though the primary tumors have detectable breaks in their basement membranes that permit these lesions to form [81]. Taken together, these findings strongly suggest that the most common forms of cancer (i.e., carcinomas) may result from aberrant epithelial-mesenchymal interactions. This also may explain why tumors behave so differently in different stromal microenvironments.
A microstructural view of cancer formation
The early observations described above led to the proposal over twenty five years ago that local changes in the physical properties of the ECM in the tissue microenvironment might potentially lead to cancer formation [30–32,34,44,57]. This engineering view of cancer formation was based on the idea that tumors may result from progressive deregulation of normal epithelial-mesenchymal interactions that are required to maintain stable tissue form throughout adult life (Fig. 1). For example, although the spatial coupling between changes in ECM structure (e.g., basement membrane thinning) and increased cell proliferation is similar in premalignant epithelial lesions and embryonic tissues, there is a difference. In the embryo, the basement membrane grows rapidly (over hours to days) and expands laterally through folding because the net amount of ECM synthesis is greater than ECM degradation. In contrast, there is neither significant lateral expansion nor rapid dissolution of the basement membrane in adult tissues, and thus, the increased ECM breakdown observed in these adult tissues is not overcome (or even fully matched) by new basement membrane synthesis. If there is increased cell division in these regions of enhanced ECM remodeling without a commensurate expansion of basement membrane area, then these cells will pile on top of each other (Fig. 1).
When this ‘hyperplastic’ tissue mass enlarges enough to become palpable, it is recognized as a ‘tumor’. However, if the growth stimulus (e.g., ECM thinning) ceases, then the overlying cells will die because they lose adhesion to the underlying basement membrane that they require to survive, and hence hyperplasia is a reversible process. But hyperplasia represents only one point along a spectrum of tissue deregulation. If the growth stimulation is sustained long enough and cells do not terminally differentiate or undergo rapid apoptosis, it may lead to spontaneous mutations or selection of subpopulations of cells that can survive and proliferate autonomously of anchorage to basement membrane, much like when cells ‘spontaneously transform’ through continued culturing (and cell doublings) in vitro. These tumors will survive and continue to grow, even when the initiating stimulus is removed. Because these abnormal cells also become physically separated from the underlying stromal cells, they may progressively lose their ability to maintain basement membrane integrity over time (i.e., given that continued epithelial-mesenchymal interactions is required for basement membrane production in the embryo). Now when complete dissolution of the basement membrane occurs, the tissue does not undergo involution because the epithelial tumor cells survive in the absence of ECM adhesions. Instead, these cells are now free to invade into the neighboring stroma, blood vessels and lymphatics, and thus, to metastasize to other organs where they can implant and grow. This is a malignant cancer.
What is perhaps most novel about this microstructural model of cancer formation, however, is the idea that normal and malignant tissue differentiation are controlled mechanically [30–32,34]. This hypothesis was based on the observation that all adherent cells generate tensional forces in their contractile cytoskeleton, and exert traction on their adhesions to ECM and to other cells. Thus, when tissues appear stable in form, they are, in reality, in a state of isometric tension in which cell-generated stresses are balanced by other forces and structures in neighboring cells and their linked ECM scaffolds; these types of tensionally prestressed structures are called ‘tensegrity’ structures [28].
If living tissues are stabilized in this manner, then a local thinning of the basement membrane will cause it to stretch out more than its thicker (and stiffer) surrounding regions, much like a run in a woman’s stocking (Fig. 1). This local change in ECM structure will increase the mechanical stresses exerted locally on the cells that are adherent to the thinned portion of the basement membrane. Tensed or stretched cells exhibit increased sensitivity to soluble mitogens [15,16,88]; thus, this mechanical distortion could be responsible for the local changes in cell growth that drive embryonic morphogenesis, as well as tumor formation (Fig. 1) [30,32,34]. In fact, in vitro studies confirm that cell growth rates increase within regions of endothelial monolayers preferentially at sites where mechanical stresses concentrate [89], and that tissue geometry feeds back to determine sites of mammary epithelial branching morphogenesis [90]. Local traction forces exerted by epithelial tumor cells on their ECM adhesions also appear to control basement membrane turnover during cancer invasion [91]. Thus, local mechanical interactions between cells and their ECM may actively contribute to the carcinogenic process.
Mechanochemical control of cell fate switching by ECM
Studies carried out in the field of Mechanical Biology over the past twenty years have confirmed that cells can be switched between phenotypes critical for neoplastic transformation, including growth, differentiation, motility and apoptosis in the presence of soluble mitogens by mechanically distorting cells and altering cytoskeletal structure. For example, epithelial and endothelial cells generally proliferate on ECM substrates that resist cell traction forces generated in the actin cytoskeleton and promote cell distortion (spreading) (Fig. 2) [15,16,88]. In contrast, these cells round and undergo apoptosis on ECM substrates that fail to bear these loads, whereas they differentiate and express tissue-specific functions when cultured on substrates that maintain an intermediate degree of shape distortion [15,16,92]. Directional cell motility also can be controlled by altering physical interactions between cells and their ECM adhesions [17,93,94]. In addition, substrates that mimic the flexibility of the ECM of a particular tissue type best support the expression of the differentiated phenotype normally exhibited cells from that tissue [95,96]. Even human mesenchymal stem cell lineage switching can be controlled by modulating mechanical interactions between cells and their ECM adhesions, which alter cell shape and cytoskeletal organization [19,20].
Fig. 2. Mechanical Control of Cell Growth & Function.
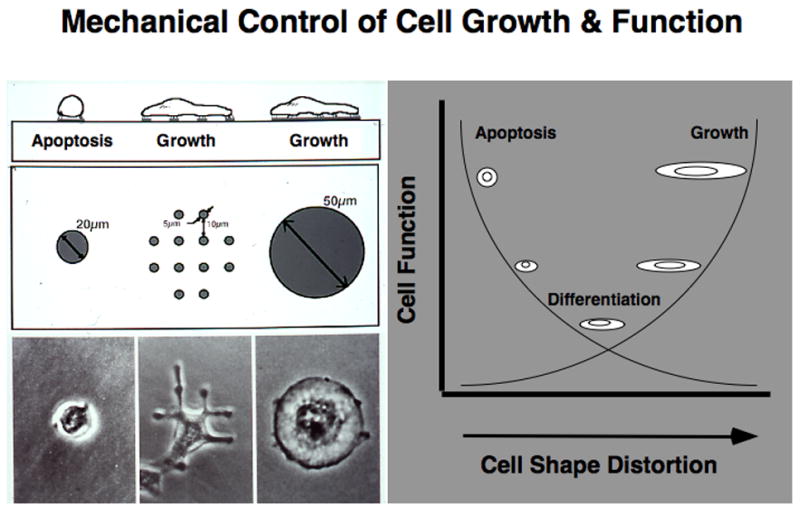
Left) Diagrammatic side-view and top-view representations of cells adherent to microfabricated circular ECM islands on the micrometer scale that constrain cell spreading are shown at the top. Phase contrast microscopic images of capillary endothelial cells cultured on these micropatterned substrates are displayed at the bottom. The effects of cell spreading (distortion) can be distinguished from those due to ECM contact formation by using many smaller, focal adhesion-sized islands (5 μm) ECM islands that cause the cells to spread from island to island; these cells spread as much as cells on the large island, but contact the total amount of ECM as on the small island. Right) Diagram depicting general effects of cell spreading on the growth, differentiation and apoptosis of epithelial and endothelial cells, when manipulated using the micropatterned substrates shown at left (see refs ???, for details). Note that, in general, cell growth increases with spreading, whereas apoptosis is switched on in round cells, and cells preferentially undergo differentiation in a moderately spread state.
A hallmark of malignant transformation is loss of “anchorage–dependent growth”, which enables cells to survive and continue proliferating when they pile up and lose adhesion to their underlying ECM. Past studies have shown that anchorage-dependence is largely due to ‘cell shape-dependence’: these cells must both bind ECM and physically distort their cytoskeleton in order to proliferate and prevent apoptosis (Fig. 2) [16, 97]. Furthermore, as normal cells become progressively more transformed, they exhibit a concomitant decrease in their sensitivity to growth inhibition by cell rounding, which manifests itself as a progressive increase in cell packing densities [98]. Continuous mechanical perturbation also can induce tumors in rodents [61], and there are many anecdotal accounts of cancer forming at sites of repeated physical injury.
The ability of changes in the mechanical microenvironment to promote tumor formation may, in part, be explained by the recent demonstration that increases of ECM stiffness can establish a mechanical autocrine loop (positive feedback loop). The small GTPase Rho is activated by tension application to cell surface integrin receptors that mediate ECM adhesion, and it is suppressed by tension dissipation in the cytoskeleton [99]. The malignant phenotype in mammary epithelial cells can be switched on by increases in ECM rigidity that enhance cell contractility through activation of RhoA, which feeds back to further increase ECM stiffness (as well as cell spreading and growth) when cells pull on their ECM adhesions and promote ECM fibril alignment [18]. Interestingly, RhoA signaling also mediates shape-dependent growth control in normal capillary endothelial cells during angiogenesis [100]. Furthermore, recent work suggests that tumor endothelial cells differ from their counterparts in that their Rho signaling is hyperactivated (Ghosh et al., unpublished observation), much like observedin tumor epithelial cells [18]. Rho signaling also contributes to the epithelial-mesenchymal transition in tumor cells as colon carcinoma cells exhibit a large increase in expression of the related isoform RhoC, as well as concomitant suppression of RhoA activity, when they undergo this transition [101]. Thus, the Rho family of small GTPases apparently represents a critical control element in normal tissue development, which can contribute to tumor formation when constitutively activated or aberrantly regulated in the appropriate tissue microenvironment.
Integration of structural and information processing networks
How can mechanical distortion of the cell influence its growth and function? Mechanical strain of the ECM that alters forces transmitted across cell surface integrin receptors can activate various intracellular signaling pathways (e.g., ERK, MAP, Ca++, src, G proteins, etc.) that are also triggered by soluble cytokines when their bind their surface receptors [28,102,103]. But cell fate switching cannot be explained in terms of changes in a single signaling pathway. For example, the angiogenic factor FGF actively signals through its receptors in endothelial cells regardless of their shape, yet spread cells respond by surviving and proliferating, whereas round cells switch on apoptosis and die [16,34]. Moreover, application of mechanical stress to cell surface integrins produces similar transmembrane signals in round and spread cells [104], yet the phenotypic switch is again governed by the degree to which the cell spreads. Thus, the overall degree of cell shape (and cytoskeletal) distortion governs how cells will respond at the level of the whole cell, and the entire genome-wide regulatory network.
Mechanical changes in cytoskeletal structure due to alterations of physical cues (e.g., ECM mechanics, tissue distortion) may simultaneously alter multiple signaling pathways throughout the cell because numerous signal transduction molecules and gene regulatory proteins normally function when immobilized on insoluble scaffolds that form the physically linked cytoskeleton-nuclear matrix lattice that bears mechanical loads inside the cell [5,105]. Mechanical stresses can influence the biochemical activities of these molecules by producing load-dependent changes in molecular shape that can alter binding kinetics and enzyme reaction rates [28,106,107].
This multiplexed signaling response is important because recent work suggests that cell fate switching requires multiple gene activities to change simultaneously [1–6, 29,108]. This is, in part, based on mathematical models of dynamic networks from the field of Complexity that exhibit regulatory circuits with stimulatory and inhibitory interactions between elements analogous to those observed in biological regulatory networks [1,3,29]. This physics-based approach to cell regulation suggests that multiple changes must occur at the same time within the genome-wide gene regulatory network to switch the entire network from one stable ‘attractor’ state or phenotype to another. Thus, cell and nuclear structure appear to have evolved to produce concerted transitions between distinct cell (and tissue) phenotypes by triggering these phenotypic switches through multiplexed signal transduction [5]. These switches may include transitions between quiescence and growth, stationary and motile states, and between different stable cell shapes (e.g., during epithelial-mesenchymal transition) that are central to tumor progression. The existence of self-reinforcing attractor states of this type based on complex dynamic signaling networks may explain the robustness of epithelial-mesenchymal transitions that appear to be nearly identical in various types of cancers, regardless of species and organ source. Similar self-propelling processes and self-stabilizing states likely contribute to tumor progression and metastatic conversion as well [29].
Can we devise new therapies that ‘reboot’ cancers to revert to normal tissues?
The studies and concepts reviewed above bring many of the existing assumptions in the Cancer Biology field into question. All scientific fields face conflicting hypotheses and mechanistic explanations during their formative stages, but then one or two theories win out and dominate thereafter. Often the losing theories are incorrect or significantly flawed; however, sometimes they are valid, but the tools do not yet exist to test them properly. The importance of the physicality of the tumor microenvironment for cancer formation is one of these theories that has awaited development of appropriate methods to experimentally determine its value.
It is widely accepted that cancer is an irreversible and deadly disease that results from accumulated gene mutations and chromosomal abnormalities. If cancers are caused by irreversible genetic alterations, then successful treatment will require that these cells must be killed or surgically removed to prevent loss of life; this perspective also validates the use of toxic therapies that often cause high morbidity in patients that are already horribly comprised by their disease.
But if cancer is a reversible process, then the entire anti-cancer paradigm will change. This is a highly provocative idea and cancer will remain a tough problem to solve; however, as described above, there are many studies in experimental systems to suggest that cancers can be induced to become quiescent, differentiate, die or form completely normal tissues, if provided with the correct set of complex signals, as conveyed by embryonic tissues or other microenvironmental cues (Fig. 3) [30,39–48]. Thus, the potential for cancer reversibility exists; however, rigorous experimental characterization of how this process proceeds is necessary before the existing research establishment will (or should) take notice and change its direction.
Fig. 3. An Engineering Solution to Cancer?
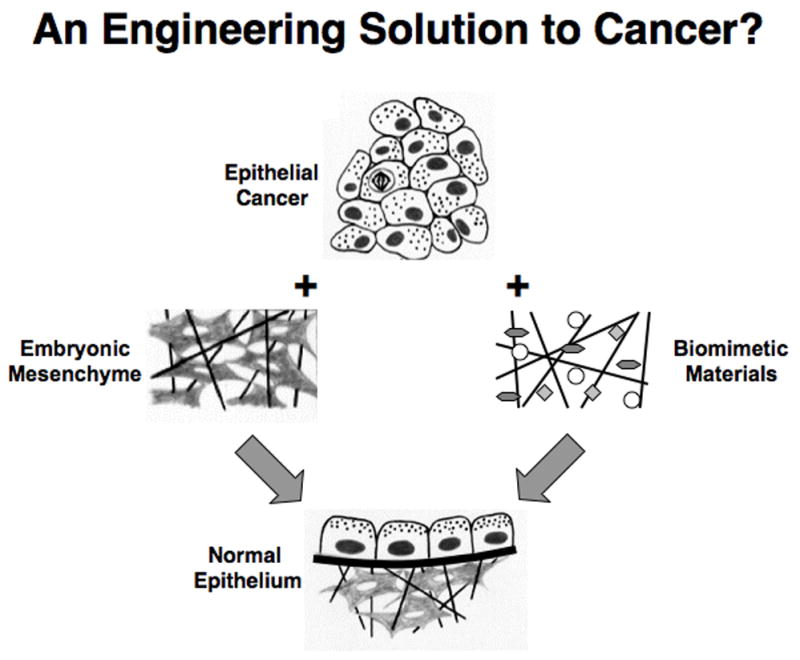
Left) Diagrams showing how combining epithelial tumor cells with embryonic mesenchyme induces the cancer cells to reverse their phenotype and restore normal tissue morphology. Right) Analogous diagrams describing a potential future form of cancer reversal therapy that utilizes synthetic biomimetic materials, which mimic the inductive behavior of embryonic mesenchyme.
For those cancer researchers who know of the ability of embryonic tissues to induce cancer reversion, most assume that it is due to production of some critical molecular morphogen or change in gene activity. However, the finding that cancer formation can be promoted or accelerated by altering the mechanics or structure of the ECM [18,30,54,55], or by chemically altering the connective tissue stroma [53], suggests that physical cues conveyed by stroma (mesenchyme) may be equally important. Similarly, few investigators are aware of the critical role that electrical potentials play in normal tissue and organ regeneration, that membrane potentials are altered in tumor cells, and that certain cancers can be induced to stop growing and differentiate by normalizing their bioelectrical properties [21–24,109,110]. Thus, demonstration that embryonic tissues harness these physical signals (in addition to chemical cues) could open entirely new avenues for therapeutic intervention in cancer.
There are currently three modalities of cancer therapy: molecular inhibitors (drugs), radiation and surgery. Nanobiotechnologies have recently gained interest as alternative multifunctional agents that may incorporate standard drug therapies, as well as targeting and imaging functions. However, there is another possibility: if physical factors contribute to cancer formation, then biomaterials and scaffolds used for medical devices and tissue engineering applications could provide yet another modality for cancer therapy (Fig. 3). These materials would need to be biomimetic in that they must mimic the structural, adhesive, molecular, chemical, mechanical, electrical cues that embryonic tissues convey when they induce cancer reversal.
In fact, engineers and cancer biologists are beginning to work together, and are finding that synthetic ECM scaffolds can induce cultured tumor cells to better mimic their in vivo phenotype [111]. Biologists interested in control of normal stem cell lineage switching also have reached out to tissue engineers because they now recognize the importance of regenerating a ‘stem cell niche’ with correct physical and chemical properties, and that providing cytokines alone is not sufficient [112,113]. Given the central role of cancer stem cells in driving cancer progression, it is possible that progressive deregulation of normal tissue architecture also may contribute to development of these aberrant stem cells by disrupting the structural determinants of the natural stem cell niche. Thus, the idea of using tissue engineering approaches to restore the normal niche and thereby, reverse cancer development is exciting. Similarly, if it were possible to engineer biomimetic materials or scaffolds that mimic the embryonic mesenchyme’s ability to revert carcinomas into normal epithelium, then these materials could be applied at tumor resection sites to prevent cancer recurrence. One also might envision injectable biomimetic materials that can target to primary and metastatic tumor sites, where they would integrate into the local stroma and self assemble into similar bioinspired scaffolds that act like mesenchyme to induce cancer reversal. These concepts have not yet been explored experimentally; however, I hope that this article stimulates young investigators who currently work in ostensibly unrelated fields (e.g., tissue engineering, materials science, physics) to join the battle on cancer, so that we can transform the way this disease is treated in the future.
Acknowledgments
This article reviews work that was supported by NIH. This article is based on a proposal that has been funded by a DoD Breast Cancer Innovator Award (to D.E.I.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kauffman SA. The Origins of Order. New York: Oxford Univ. Press; 1993. [Google Scholar]
- 2.Coffey DS. Self-organization, complexity and chaos: the new biology for medicine. Nat Med. 1998;4:882–885. doi: 10.1038/nm0898-882. [DOI] [PubMed] [Google Scholar]
- 3.Huang S. Gene expression profiling, genetic networks, and cellular states: an integrating concept for tumorigenesis and drug discovery. J Mol Med. 1999;77:469–480. doi: 10.1007/s001099900023. [DOI] [PubMed] [Google Scholar]
- 4.Ideker T, Galitski T, Hood L. A new approach to decoding life: systems biology. Annu Rev Genomics Hum Genet. 2001;2:343–372. doi: 10.1146/annurev.genom.2.1.343. [DOI] [PubMed] [Google Scholar]
- 5.Ingber DE. Tensegrity II. How structural networks influence cellular information processing networks. J Cell Sci. 2003;116:1397–1408. doi: 10.1242/jcs.00360. [DOI] [PubMed] [Google Scholar]
- 6.Huang S, Eichler G, Bar-Yam Y, Ingber DE. Cell fates as high-dimensional attractor states of a complex gene regulatory network. Phys Rev Lett. 2005;94:128701. doi: 10.1103/PhysRevLett.94.128701. [DOI] [PubMed] [Google Scholar]
- 7.Fambrough D, McClure K, Kazlauskas A, Lander ES. Diverse signaling pathways activated by growth factor receptors induce broadly overlapping, rather than independent, sets of genes. Cell. 1999;97:727–741. doi: 10.1016/s0092-8674(00)80785-0. [DOI] [PubMed] [Google Scholar]
- 8.Tapscott SJ. The circuitry of a master switch: Myod and the regulation of skeletal muscle gene transcription. Development. 2005;132:2685–2695. doi: 10.1242/dev.01874. [DOI] [PubMed] [Google Scholar]
- 9.Barrallo-Gimeno A, Nieto MA. The Snail genes as inducers of cell movement and survival: implications in development and cancer. Development. 2005;132:3151–3161. doi: 10.1242/dev.01907. [DOI] [PubMed] [Google Scholar]
- 10.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 11.Wernig M, Meissner A, Foreman R, Brambrink T, Ku M, et al. In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature. 2007;448:318–324. doi: 10.1038/nature05944. [DOI] [PubMed] [Google Scholar]
- 12.Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, et al. Induced Pluripotent Stem Cell Lines Derived from Human Somatic Cells. Science. 2007 doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 13.Meissner A, Wernig M, Jaenisch R. Direct reprogramming of genetically unmodified fibroblasts into pluripotent stem cells. Nat Biotechnol. 2007;25:1177–1181. doi: 10.1038/nbt1335. [DOI] [PubMed] [Google Scholar]
- 14.Lee EY, Lee WH, Kaetzel CS, Parry G, Bissell MJ. Interaction of mouse mammary epithelial cells with collagen substrata: regulation of casein gene expression and secretion. Proc Natl Acad Sci U S A. 1985;82:1419–1423. doi: 10.1073/pnas.82.5.1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Singhvi R, Kumar A, Lopez GP, Stephanopoulos GN, Wang DI, et al. Engineering cell shape and function. Science. 1994;264:696–698. doi: 10.1126/science.8171320. [DOI] [PubMed] [Google Scholar]
- 16.Chen CS, Mrksich M, Huang S, Whitesides GM, Ingber DE. Geometric control of cell life and death. Science. 1997;276:1425–1428. doi: 10.1126/science.276.5317.1425. [DOI] [PubMed] [Google Scholar]
- 17.Parker KK, Brock AL, Brangwynne C, Mannix RJ, Wang N, et al. Directional control of lamellipodia extension by constraining cell shape and orienting cell tractional forces. FASEB J. 2002;16:1195–1204. doi: 10.1096/fj.02-0038com. [DOI] [PubMed] [Google Scholar]
- 18.Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, et al. Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005;8:241–254. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 19.McBeath R, Pirone DM, Nelson CM, Bhadriraju K, Chen CS. Cell shape, cytoskeletal tension, and RhoA regulate stem cell lineage commitment. Dev Cell. 2004;6:483–495. doi: 10.1016/s1534-5807(04)00075-9. [DOI] [PubMed] [Google Scholar]
- 20.Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126:677–689. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 21.Hellmann P, Grummer R, Schirrmacher K, Rook M, Traub O, Winterhager E. Transfection with different connexin genes alters growth and differentiation of human choriocarcinoma cells. Exp Cell Res. 1999;246:480–490. doi: 10.1006/excr.1998.4332. [DOI] [PubMed] [Google Scholar]
- 22.Jones SM, Ribera AB. Overexpression of a potassium channel gene perturbs neural differentiation. J Neurosci. 1994;14:2789–2799. doi: 10.1523/JNEUROSCI.14-05-02789.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Strobl JS, Wonderlin WF, Flynn DC. Mitogenic signal transduction in human breast cancer cells. Gen Pharmacol. 1995;26:1643–1649. doi: 10.1016/0306-3623(95)00062-3. [DOI] [PubMed] [Google Scholar]
- 24.Rich IN, Worthington-White D, Garden OA, Musk P. Apoptosis of leukemic cells accompanies reduction in intracellular pH after targeted inhibition of the Na(+)/H(+) exchanger. Blood. 2000;95:1427–1434. [PubMed] [Google Scholar]
- 25.Okita K, Ichisaka T, Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature. 2007;448:313–317. doi: 10.1038/nature05934. [DOI] [PubMed] [Google Scholar]
- 26.Drucker L, Afensiev F, Radnay J, Shapira H, Lishner M. Co-administration of simvastatin and cytotoxic drugs is advantageous in myeloma cell lines. Anticancer Drugs. 2004;15:79–84. doi: 10.1097/00001813-200401000-00012. [DOI] [PubMed] [Google Scholar]
- 27.Ingber DE. Tensegrity I. Cell structure and hierarchical systems biology. J Cell Sci. 2003;116:1157–1173. doi: 10.1242/jcs.00359. [DOI] [PubMed] [Google Scholar]
- 28.Ingber DE. Cellular mechanotransduction: putting all the pieces together again. Faseb J. 2006;20:811–827. doi: 10.1096/fj.05-5424rev. [DOI] [PubMed] [Google Scholar]
- 29.Huang S, Ingber DE. A non-genetic basis for cancer progression and metastasis: self-organizing attractors in cell regulatory networks. Breast Disease. 2007;26:27–54. doi: 10.3233/bd-2007-26104. [DOI] [PubMed] [Google Scholar]
- 30.Ingber DE, Madri JA, Jamieson JD. Role of basal lamina in neoplastic disorganization of tissue architecture. Proc Natl Acad Sci U S A. 1981;78:3901–3905. doi: 10.1073/pnas.78.6.3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ingber DE, Jamieson JD. In: Tumor formation and malignant invasion: role of basal lamina. Liotta LA, Hart IR, editors. The Hague, Netherlands; Martinus Nijhoff: 1982. pp. 335–357. [Google Scholar]
- 32.Ingber D, Jamieson JD. Cells as tensegrity structures: architectural regulation of histodifferentiation by physical forces tranduced over basement membrane. Orlando: Academic Press; 1985. [Google Scholar]
- 33.Clark WH., Jr The nature of cancer: morphogenesis and progressive (self)-disorganization in neoplastic development and progression. Acta Oncol. 1995;34:3–21. doi: 10.3109/02841869509093632. [DOI] [PubMed] [Google Scholar]
- 34.Huang S, Ingber DE. The structural and mechanical complexity of cell-growth control. Nat Cell Biol. 1999;1:E131–138. doi: 10.1038/13043. [DOI] [PubMed] [Google Scholar]
- 35.Ingber DE. Cancer as a disease of epithelial-mesenchymal interactions and extracellular matrix regulation. Differentiation. 2002;70:547–560. doi: 10.1046/j.1432-0436.2002.700908.x. [DOI] [PubMed] [Google Scholar]
- 36.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 37.LaGamba D, Nawshad A, Hay ED. Microarray analysis of gene expression during epithelial-mesenchymal transformation. Dev Dyn. 2005;234:132–142. doi: 10.1002/dvdy.20489. [DOI] [PubMed] [Google Scholar]
- 38.Folkman J. Angiogenesis. Annu Rev Med. 2006;57:1–18. doi: 10.1146/annurev.med.57.121304.131306. [DOI] [PubMed] [Google Scholar]
- 39.Ellison ML, Ambrose EJ, Easty GC. Differentiation in a transplantable rat tumour maintained in organ culture. Exp Cell Res. 1969;55:198–204. doi: 10.1016/0014-4827(69)90481-9. [DOI] [PubMed] [Google Scholar]
- 40.Lakshmi MS, Sherbet GV. In: Embryonic and tumour cell interactions. Sherbet GV, editor. New York: S. Karger; 1974. [Google Scholar]
- 41.DeCosse JJ, Gossens CL, Kuzma JF, Unsworth BR. Breast cancer: induction of differentiation by embryonic tissue. Science. 1973;181:1057–1058. doi: 10.1126/science.181.4104.1057. [DOI] [PubMed] [Google Scholar]
- 42.Cunha GR, Fujii H, Neubauer BL, Shannon JM, Sawyer L, Reese BA. Epithelial-mesenchymal interactions in prostatic development. I. morphological observations of prostatic induction by urogenital sinus mesenchyme in epithelium of the adult rodent urinary bladder. J Cell Biol. 1983;96:1662–1670. doi: 10.1083/jcb.96.6.1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Watanabe TK, Hansen LJ, Reddy NK, Kanwar YS, Reddy JK. Differentiation of pancreatic acinar carcinoma cells cultured on rat testicular seminiferous tubular basement membranes. Cancer Res. 1984;44:5361–5368. [PubMed] [Google Scholar]
- 44.Ingber DE, Madri JA, Jamieson JD. Basement membrane as a spatial organizer of polarized epithelia. Exogenous basement membrane reorients pancreatic epithelial tumor cells in vitro. Am J Pathol. 1986;122:129–139. [PMC free article] [PubMed] [Google Scholar]
- 45.Chung LW, Zhau HE, Ro JY. Morphologic and biochemical alterations in rat prostatic tumors induced by fetal urogenital sinus mesenchyme. Prostate. 1990;17:165–174. doi: 10.1002/pros.2990170210. [DOI] [PubMed] [Google Scholar]
- 46.Wong YC, Cunha GR, Hayashi N. Effects of mesenchyme of the embryonic urogenital sinus and neonatal seminal vesicle on the cytodifferentiation of the Dunning tumor: ultrastructural study. Acta Anat (Basel) 1992;143:139–150. doi: 10.1159/000147240. [DOI] [PubMed] [Google Scholar]
- 47.Kenny PA, Bissell MJ. Tumor reversion: correction of malignant behavior by microenvironmental cues. Int J Cancer. 2003;107:688–695. doi: 10.1002/ijc.11491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maffini MV, Calabro JM, Soto AM, Sonnenschein C. Stromal regulation of neoplastic development: age-dependent normalization of neoplastic mammary cells by mammary stroma. Am J Pathol. 2005;167:1405–1410. doi: 10.1016/S0002-9440(10)61227-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Goldenberg DM, Pavia RA. Malignant potential of murine stromal cells after transplantation of human tumors into nude mice. Science. 1981;212:65–67. doi: 10.1126/science.7209521. [DOI] [PubMed] [Google Scholar]
- 50.Wiseman BS, Werb Z. Stromal effects on mammary gland development and breast cancer. Science. 2002;296:1046–1049. doi: 10.1126/science.1067431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Laconi E. The evolving concept of tumor microenvironments. Bioessays. 2007;29:738–744. doi: 10.1002/bies.20606. [DOI] [PubMed] [Google Scholar]
- 52.Albini A, Sporn MB. The tumour microenvironment as a target for chemoprevention. Nat Rev Cancer. 2007;7:139–147. doi: 10.1038/nrc2067. [DOI] [PubMed] [Google Scholar]
- 53.Maffini MV, Soto AM, Calabro JM, Ucci AA, Sonnenschein C. The stroma as a crucial target in rat mammary gland carcinogenesis. J Cell Sci. 2004;117:1495–1502. doi: 10.1242/jcs.01000. [DOI] [PubMed] [Google Scholar]
- 54.Huang S, Ingber DE. Cell tension, matrix mechanics, and cancer development. Cancer Cell. 2005;8:175–176. doi: 10.1016/j.ccr.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 55.Sternlicht MD, Lochter A, Sympson CJ, Huey B, Rougier JP, et al. The stromal proteinase MMP3/stromelysin-1 promotes mammary carcinogenesis. Cell. 1999;98:137–146. doi: 10.1016/s0092-8674(00)81009-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Folkman J, Watson K, Ingber D, Hanahan D. Induction of angiogenesis during the transition from hyperplasia to neoplasia. Nature. 1989;339:58–61. doi: 10.1038/339058a0. [DOI] [PubMed] [Google Scholar]
- 57.Ingber DE, Madri JA, Jamieson JD. Neoplastic disorganization of pancreatic epithelial cell-cell relations. Role of basement membrane. Am J Pathol. 1985;121:248–260. [PMC free article] [PubMed] [Google Scholar]
- 58.Weaver VM, Petersen OW, Wang F, Larabell CA, Briand P, et al. Reversion of the malignant phenotype of human breast cells in three-dimensional culture and in vivo by integrin blocking antibodies. J Cell Biol. 1997;137:231–245. doi: 10.1083/jcb.137.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lochter A, Werb Z, Bissell MJ. Transcriptional regulation of stromelysin-1 gene expression is altered during progression of mouse mammary epithelial cells from functionally normal to malignant. Matrix Biol. 1999;18:455–467. doi: 10.1016/s0945-053x(99)00036-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sternlicht MD, Bissell MJ, Werb Z. The matrix metalloproteinase stromelysin-1 acts as a natural mammary tumor promoter. Oncogene. 2000;19:1102–1113. doi: 10.1038/sj.onc.1203347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bischoff F, Bryson G. Carcinogenesis through Solid State Surfaces. Prog Exp Tumor Res. 1964;14:85–133. doi: 10.1159/000385997. [DOI] [PubMed] [Google Scholar]
- 62.Sakakura T, Nishizuka Y, Dawe CJ. Mesenchyme-dependent morphogenesis and epithelium-specific cytodifferentiation in mouse mammary gland. Science. 1976;194:1439–1441. doi: 10.1126/science.827022. [DOI] [PubMed] [Google Scholar]
- 63.Nogawa H, Nakanishi Y. Mechanical aspects of the mesenchymal influence on epithelial branching morphogenesis of mouse salivary gland. Development. 1987;101:491–500. [Google Scholar]
- 64.Banerjee SD, Cohn RH, Bernfield MR. Basal lamina of embryonic salivary epithelia. Production by the epithelium and role in maintaining lobular morphology. J Cell Biol. 1977;73:445–463. doi: 10.1083/jcb.73.2.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bernfield MR, Banerjee SD. The basal lamina in epithelial-mesenchymal interactions. In: Kefalides N, editor. Biology and Chemistry of Basement Membranes. New York: Academic Press; 1978. pp. 137–148. [Google Scholar]
- 66.Bernfield MR, Banerjee SD, Cohn RH. Dependence of salivary epithelial morphology and branching morphogenesis upon acid mucopolysaccharide-protein (proteoglycan) at the epithelial surface. J Cell Biol. 1972;52:674–689. doi: 10.1083/jcb.52.3.674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mollard R, Dziadek M. A correlation between epithelial proliferation rates, basement membrane component localization patterns, and morphogenetic potential in the embryonic mouse lung. Am J Respir Cell Mol Biol. 1998;19:71–82. doi: 10.1165/ajrcmb.19.1.3158. [DOI] [PubMed] [Google Scholar]
- 68.Nogawa H, Morita K, Cardoso WV. Bud formation precedes the appearance of differential cell proliferation during branching morphogenesis of mouse lung epithelium in vitro. Dev Dyn. 1998;213:228–235. doi: 10.1002/(SICI)1097-0177(199810)213:2<228::AID-AJA8>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 69.David G, Bernfield MR. Collagen reduces glycosaminoglycan degradation by cultured mammary epithelial cells: possible mechanism for basal lamina formation. Proc Natl Acad Sci U S A. 1979;76:786–790. doi: 10.1073/pnas.76.2.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wicha MS, Liotta LA, Vonderhaar BK, Kidwell WR. Effects of inhibition of basement membrane collagen deposition on rat mammary gland development. Dev Biol. 1980;80:253–256. doi: 10.1016/0012-1606(80)90402-9. [DOI] [PubMed] [Google Scholar]
- 71.Trelstad RL, Hayashi A, Hayashi K, Donahoe PK. The epithelial-mesenchymal interface of the male rate Mullerian duct: loss of basement membrane integrity and ductal regression. Dev Biol. 1982;92:27–40. doi: 10.1016/0012-1606(82)90147-6. [DOI] [PubMed] [Google Scholar]
- 72.Ingber DE, Madri JA, Folkman J. A possible mechanism for inhibition of angiogenesis by angiostatic steroids: induction of capillary basement membrane dissolution. Endocrinology. 1986;119:1768–1775. doi: 10.1210/endo-119-4-1768. [DOI] [PubMed] [Google Scholar]
- 73.Frisch SM, Francis H. Disruption of epithelial cell-matrix interactions induces apoptosis. J Cell Biol. 1994;124:619–626. doi: 10.1083/jcb.124.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sakakura T, Sakagami Y, Nishizuka Y. Persistence of responsiveness of adult mouse mammary gland to induction by embryonic mesenchyme. Dev Biol. 1979;72:201–210. doi: 10.1016/0012-1606(79)90111-8. [DOI] [PubMed] [Google Scholar]
- 75.Tarin D. Tissue interactions in morphogenesis, morphostasis and carcinogenesis. J Theor Biol. 1972;34:61–72. doi: 10.1016/0022-5193(72)90054-9. [DOI] [PubMed] [Google Scholar]
- 76.Vasiliev JM. The role of connective tissue proliferation in invasive growth of normal and malignant tissues: a review. Br J Cancer. 1958;12:524–536. doi: 10.1038/bjc.1958.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ausprunk DH, Folkman J. Migration and proliferation of endothelial cells in preformed and newly formed blood vessels during tumor angiogenesis. Microvasc Res. 1977;14:53–65. doi: 10.1016/0026-2862(77)90141-8. [DOI] [PubMed] [Google Scholar]
- 78.Foley JF, Aftonomos BT, Heidrick ML. Influence of fibroblastic collagen and mucopolysaccharides on HeLa cell colonial morphology. Life Sci. 1968;7:1003–1008. doi: 10.1016/0024-3205(68)90177-x. [DOI] [PubMed] [Google Scholar]
- 79.Orr JW, Spencer AT. Transplantation studies on the mechanism of carcinogenesis. New York: Academic Press; 1972. [Google Scholar]
- 80.Gullino PM. The internal milieu of tumors. Prog Exp Tumor Res. 1966;8:1–25. doi: 10.1159/000386002. [DOI] [PubMed] [Google Scholar]
- 81.Tarin D. Ultrastructural features of neural induction in Xenopus laevis. J Anat. 1972;111:1–28. [PMC free article] [PubMed] [Google Scholar]
- 82.Lu S, Huang M, Kobayashi Y, Komiyama A, Li X, et al. Alterations of basement membrane in di-isopropanolnitrosamine-induced carcinogenesis of the rat thyroid gland: an immunohistochemical study. Virchows Arch. 2000;436:595–601. doi: 10.1007/s004280000180. [DOI] [PubMed] [Google Scholar]
- 83.Li SC, Chen GF, Chan PS, Choi HL, Ho SM, Chan FL. Altered expression of extracellular matrix and proteinases in Noble rat prostate gland after long-term treatment with sex steroids. Prostate. 2001;49:58–71. doi: 10.1002/pros.1118. [DOI] [PubMed] [Google Scholar]
- 84.Ozzello L. The behavior of basement membranes in intraductal carcinoma of the breast. Am J Pathol. 1959;35:887–899. [PMC free article] [PubMed] [Google Scholar]
- 85.Luibel FJ, Sanders E, Ashworth CT. An electron microscopic study of carcinoma in situ and invasive carcinoma of the cervix uteri. Cancer Res. 1960;20:357–361. [PubMed] [Google Scholar]
- 86.Rubio CA, Biberfeld P. The basement membrane in experimentally induced atypias and carcinoma of the uterine cervix in mice. An immunofluorescence study. Virchows Arch A Pathol Anat Histol. 1979;381:205–209. doi: 10.1007/BF01257885. [DOI] [PubMed] [Google Scholar]
- 87.Pitelka DR, Hamamoto ST, Taggart BN. Epithelial cell junctions in primary and metastatic mammary tumors of mice. Cancer Res. 1980;40:1588–1599. [PubMed] [Google Scholar]
- 88.Folkman J, Moscona A. Role of cell shape in growth control. Nature. 1978;273:345–349. doi: 10.1038/273345a0. [DOI] [PubMed] [Google Scholar]
- 89.Nelson CM, Jean RP, Tan JL, Liu WF, Sniadecki NJ, et al. Emergent patterns of growth controlled by multicellular form and mechanics. Proc Natl Acad Sci U S A. 2005;102:11594–11599. doi: 10.1073/pnas.0502575102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nelson CM, Vanduijn MM, Inman JL, Fletcher DA, Bissell MJ. Tissue geometry determines sites of mammary branching morphogenesis in organotypic cultures. Science. 2006;314:298–300. doi: 10.1126/science.1131000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rabinovitz I, Gipson IK, Mercurio AM. Traction forces mediated by alpha6beta4 integrin: implications for basement membrane organization and tumor invasion. Mol Biol Cell. 2001;12:4030–4043. doi: 10.1091/mbc.12.12.4030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Dike LE, Chen CS, Mrksich M, Tien J, Whitesides GM, Ingber DE. Geometric control of switching between growth, apoptosis, and differentiation during angiogenesis using micropatterned substrates. In Vitro Cell Dev Biol Anim. 1999;35:441–448. doi: 10.1007/s11626-999-0050-4. [DOI] [PubMed] [Google Scholar]
- 93.Lo CM, Wang HB, Dembo M, Wang YL. Cell movement is guided by the rigidity of the substrate. Biophys J. 2000;79:144–152. doi: 10.1016/S0006-3495(00)76279-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Xia N, Thodeti C, Hunt TM, Xu Q, Ho M, et al. FASEB J. 2008. Directional control of cell motility through focal adhesion positioning and spatial control of Rac activation. in press. [DOI] [PubMed] [Google Scholar]
- 95.Engler AJ, Griffin MA, Sen S, Bonnemann CG, Sweeney HL, Discher DE. Myotubes differentiate optimally on substrates with tissue-like stiffness: pathological implications for soft or stiff microenvironments. J Cell Biol. 2004;166:877–887. doi: 10.1083/jcb.200405004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Georges PC, Janmey PA. Cell type-specific response to growth on soft materials. J Appl Physiol. 2005;98:1547–1553. doi: 10.1152/japplphysiol.01121.2004. [DOI] [PubMed] [Google Scholar]
- 97.Numaguchi Y, Huang S, Polte TR, Eichler GS, Wang N, Ingber DE. Caldesmon-dependent switching between capillary endothelial cell growth and apoptosis through modulation of cell shape and contractility. Angiogenesis. 2003;6:55–64. doi: 10.1023/a:1025821517679. [DOI] [PubMed] [Google Scholar]
- 98.Wittelsberger SC, Kleene K, Penman S. Progressive loss of shape-responsive metabolic controls in cells with increasingly transformed phenotype. Cell. 1981;24:859–866. doi: 10.1016/0092-8674(81)90111-2. [DOI] [PubMed] [Google Scholar]
- 99.Riveline D, Zamir E, Balaban NQ, Schwarz US, Ishizaki T, et al. Focal contacts as mechanosensors: externally applied local mechanical force induces growth of focal contacts by an mDia1-dependent and ROCK-independent mechanism. J Cell Biol. 2001;153:1175–1186. doi: 10.1083/jcb.153.6.1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mammoto A, Huang S, Moore K, Oh P, Ingber DE. Role of RhoA, mDia, and ROCK in cell shape-dependent control of the Skp2-p27kip1 pathway and the G1/S transition. J Biol Chem. 2004;279:26323–26330. doi: 10.1074/jbc.M402725200. [DOI] [PubMed] [Google Scholar]
- 101.Bellovin DI, Simpson KJ, Danilov T, Maynard E, Rimm DL, et al. Reciprocal regulation of RhoA and RhoC characterizes the EMT and identifies RhoC as a prognostic marker of colon carcinoma. Oncogene. 2006;25:6959–6967. doi: 10.1038/sj.onc.1209682. [DOI] [PubMed] [Google Scholar]
- 102.Alenghat FJ, Ingber DE. Mechanotransduction: all signals point to cytoskeleton, matrix, and integrins. Sci STKE. 2002;2002:PE6. doi: 10.1126/stke.2002.119.pe6. [DOI] [PubMed] [Google Scholar]
- 103.Bershadsky AD, Balaban NQ, Geiger B. Adhesion-dependent cell mechanosensitivity. Annu Rev Cell Dev Biol. 2003;19:677–695. doi: 10.1146/annurev.cellbio.19.111301.153011. [DOI] [PubMed] [Google Scholar]
- 104.Meyer CJ, Alenghat FJ, Rim P, Fong JH, Fabry B, Ingber DE. Mechanical control of cyclic AMP signalling and gene transcription through integrins. Nat Cell Biol. 2000;2:666–668. doi: 10.1038/35023621. [DOI] [PubMed] [Google Scholar]
- 105.Ingber DE. The riddle of morphogenesis: a question of solution chemistry or molecular cell engineering? Cell. 1993;75:1249–1252. doi: 10.1016/0092-8674(93)90612-t. [DOI] [PubMed] [Google Scholar]
- 106.Sawada Y, Tamada M, Dubin-Thaler BJ, Cherniavskaya O, Sakai R, et al. Force sensing by mechanical extension of the Src family kinase substrate p130Cas. Cell. 2006;127:1015–1026. doi: 10.1016/j.cell.2006.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lele TP, Pendse J, Kumar S, Salanga M, Karavitis J, Ingber DE. Mechanical forces alter zyxin unbinding kinetics within focal adhesions of living cells. J Cell Physiol. 2006;207:187–194. doi: 10.1002/jcp.20550. [DOI] [PubMed] [Google Scholar]
- 108.Huang S, Ingber DE. Shape-dependent control of cell growth, differentiation, and apoptosis: switching between attractors in cell regulatory networks. Exp Cell Res. 2000;261:91–103. doi: 10.1006/excr.2000.5044. [DOI] [PubMed] [Google Scholar]
- 109.Levin M, Thorlin T, Robinson KR, Nogi T, Mercola M. Asymmetries in H+/K+-ATPase and cell membrane potentials comprise a very early step in left-right patterning. Cell. 2002;111:77–89. doi: 10.1016/s0092-8674(02)00939-x. [DOI] [PubMed] [Google Scholar]
- 110.Levin M. Bioelectromagnetics in morphogenesis. Bioelectromagnetics. 2003;24:295–315. doi: 10.1002/bem.10104. [DOI] [PubMed] [Google Scholar]
- 111.Fischbach C, Chen R, Matsumoto T, Schmelzle T, Brugge JS, et al. Engineering tumors with 3D scaffolds. Nat Methods. 2007;4:855–860. doi: 10.1038/nmeth1085. [DOI] [PubMed] [Google Scholar]
- 112.Scadden DT. The stem-cell niche as an entity of action. Nature. 2006;441:1075–1079. doi: 10.1038/nature04957. [DOI] [PubMed] [Google Scholar]
- 113.Mikos AG, Herring SW, Ochareon P, Elisseeff J, Lu HH, et al. Engineering complex tissues. Tissue Eng. 2006;12:3307–3339. doi: 10.1089/ten.2006.12.3307. [DOI] [PMC free article] [PubMed] [Google Scholar]