

Abstract
Oxidative stress caused by glutathione depletion after prolonged exposure to extracellular glutamate leads to a form of neuronal cell death that exhibits morphologically mixed features of both apoptosis and necrosis. However, specific downstream executioners involved in this form of cell death have yet to be identified. We report here that glutamate exposure does not activate caspase-3 in the HT22 neuronal cell line. Furthermore, no cytoprotection was achieved with either the pan-caspase inhibitor Z-VAD-fmk or the caspase-3-specific inhibitor DEVD-CHO. In contrast, inhibition of the proteasome by lactacystin protected both HT22 cells and rat primary neuronal cells against cell lysis. In parallel, oxidatively altered and ubiquitinated proteins accumulated in the mitochondrial fraction of cells after proteasome inhibition. These findings suggest that caspases can be decoupled from oxidative stress under some conditions, and implicate the ubiquitin/proteasome pathway in neuronal cell death caused by oxidative glutamate toxicity.
Keywords: calpain, caspase, epoxomicin, lactacystin, MG132, oxytosis
Oxidative stress is one of the major causes of delayed neuronal cell death after stroke, brain trauma and a variety of neurodegenerative diseases. One of its hallmarks is a decrease in the reduced form of the major intracellular antioxidant, glutathione. Experimentally, this can be induced in cell lines or in primary neuronal culture by interfering with the cell's capacity to synthesize glutathione (Murphy et al. 1990; Ratan et al. 2002). Extracellular glutamate can be utilized to this effect in the neuronal cell line HT22 or in freshly cultured primary neurons, both of which lack functional glutamate receptors and are thus not subject to excitotoxic cell death. Glutathione synthesis is disrupted because extracellular glutamate blocks a glutamate-cystine anti-porter in the plasma membrane, leading to a deficit in one of the building blocks for glutathione synthesis. This form of glutamate-induced oxidative injury leads to rapid neuronal cell lysis.
Extensive efforts have been made to define a cell death cascade for this oxidative stress-induced cell death. Morphologically, features of both apoptosis and necrosis can be detected in various types of neuronal cell lines including HT22 and HT4 cells (Tan et al. 1998; Tirosh et al. 2000). On a molecular level, a central role has been ascribed to activation of 12-lipoxygenase following the depletion of glutathione (Li et al. 1997a; Canals et al. 2003; Khanna et al. 2003). In addition, a requirement for de novo protein synthesis, activation of the extracellular signal-regulated kinase (ERK) MAPK and of soluble guanylyl cyclase have been reported (Li et al. 1997a,b; Satoh et al. 2000; Stanciu et al. 2000). Nevertheless, the downstream proteases that execute this form of neuronal cell death have yet to be defined.
In this study, we investigated candidate proteases that may be activated in the course of oxidative glutamate toxicity. In both HT22 cells and primary neuron cultures, we report that neither caspase nor calpain inhibition decreased cell death. In contrast, inhibition of the proteasome was significantly neuroprotective. These data suggest that the ubiquitin-proteasome pathway is a central part of the cell death cascade following prolonged exposure to extracellular glutamate.
Materials and methods
Inhibitor studies
HT22 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum and penicillin/streptomycin (all media from Invitrogen, Carlsbad, CA, USA). For viability experiments, cells were seeded at 5 × 105 cells/well in 24-well plates (Corning, Acton, MA, USA) and treated when 70% confluent. Primary neurons were isolated from E17 rats as described elsewhere (Ratan et al. 2002) and treated after 18 h in culture. Treatment consisted of exchanging the medium to 1 mL fresh culturing medium and adding 5 mm glutamate [stock solution 400 mm in phosphate-buffered saline (PBS)] in the presence or absence of dimethylsulfoxide (DMSO) (maximum 0.1% final concentration) as control or the indicated concentrations of inhibitors: Baicalein (Cayman Chemicals), ZVAD-fmk (Caspase Inhibitor I, Calbiochem, San Diego, CA, USA), DEVD-CHO (Calbiochem), Calpain Inhibitor III (Calbiochem), Lactacystin (Sigma, St. Louis, MO, USA), MG132 (Sigma), Proteasome Inhibitor I (Calbiochem), Epoxomicin (Calbiochem). After 24 h treatment, medium was collected and the cells were lysed in 1 mL 0.4% Triton-X-100, by incubating for 30 min at 37°C. Lactate dehydrogenase (LDH) content was determined separately for the cell extracts and corresponding media using a Cytotoxicity Detection Kit (Roche Molecular Biochemicals, Indianapolis, IN, USA), and the percentage of LDH released to the medium calculated after subtracting the corresponding background value. Results are given as mean ± SD. Multigroup comparisons were carried out by one-way anova followed by Tukey–Kramer HSD comparison.
Isolation of mitochondrial fractions
For isolation of a mitochondrially enriched fraction, HT22 cells were grown on 10 cm dishes (Corning) and treated when 70% confluent. After 11 h, cells were trypsinized, collected, and washed with PBS. Cells were then resuspended in 200 μL of a lysis buffer containing 20 mm HEPES–KOH pH 7.4, 10 mm NaCl, 1.5 mm MgCl2, 1 mm EDTA, 1 mm EGTA, 250 mm sucrose and freshly added 1 mm dithiothreitol (DTT), 2 μg/mL aprotinin, and 0.1 mm phenylmethylsulfonyl fluoride (PMSF). To disrupt the plasma membrane, cells were resuspended 25 times with a yellow pipette tip, followed by grinding eight times with a Disposable Pellet Pestle (Kontes Glass Co., Vineland, NJ, USA). Unbroken cells and nuclei were removed by centrifugation for 15 min at 1025 g, followed by 15 min centrifugation at 10 000 g to pellet mitochondria. For derivatization of oxidized proteins using the Oxyblot kit (Chemicon, Temecula, CA, USA), mitochondria were isolated as above, but the buffer contained 50 mm DTT and no PMSF. Twenty micrograms of protein were derivatized and analyzed as recommended by the manufacturer.
Proteasome activity assay
For proteasome activity assays, HT22 cells were grown on 10 cm plates and lysed after 6 h of treatment, prior to any effects on cell viability, in cell membrane lysis buffer (see above) containing 4 mm DTT and 2 mm ATP. After centrifugation for 10 min at 20 800 g, supernatants (20 μg protein) were incubated with 30 μm Proteasome Substrate I (Calbiochem) in a total volume of 100 μL for 60 min at 37°C. Fluorescence was measured in a 96-well plate reader (Biotek FL600) using a 360/40 nm excitation filter and a 460/40 nm emission filter.
Western blotting
For western blots, cells were incubated for the times indicated with or without 5 mm glutamate in the presence or absence of lactacystin. After washing once with PBS, cells were trypsinized, washed, and then either processed for mitochondrial isolation (see above) or extracted using cell lysis buffer (Cell Signaling Technology, Beverly, MA, USA) containing 0.1 mm PMSF (Sigma). Extracts, representing 20 μg protein per lane, were separated on 4–12% NuPAGE gels (Invitrogen), blotted to nitrocellulose membranes and probed with antibodies to total caspase-3 and to cleaved caspase-3 (both from Cell Signaling), or with an antibody to ubiquitin (Sigma).
FACScan analysis
To analyze glutamate-induced cell death by FACScan analysis, HT22 cells were incubated for 24 h in the presence or absence of 5 mm glutamate and either 10 μm lactacystin, 50 nm epoxomicin, or 1 μm Proteasome Inhibitor I. Cells were then trypsinized, washed twice with PBS and stained with the Annexin V-FITC Apoptosis Detection Kit II (BD Biosciences, Billerica, MA, USA) according to the manufacturer's protocol. A minimum of 5000 cells per sample was counted.
Results
To study the involvement of proteases in oxidative glutamate toxicity, we initially measured the impact of caspase and calpain inhibitors on viability of glutamate-treated HT22 cells. Neither the pan-caspase inhibitor ZVAD-fmk (10 μm) nor the more specific caspase-3 inhibitor DEVD-CHO (20 μm) had a significant impact on glutamate-induced cell death (Fig. 1a). Pre-treatment with an inhibitor of calpain (calpain inhibitor III, 10 μm) also did not yield cytoprotection, suggesting that this other major executioner protease was also not involved. In contrast, the 12-lipoxygenase inhibitor baicalein was fully protective, as previously reported (Li et al. 1997a).
Fig. 1.
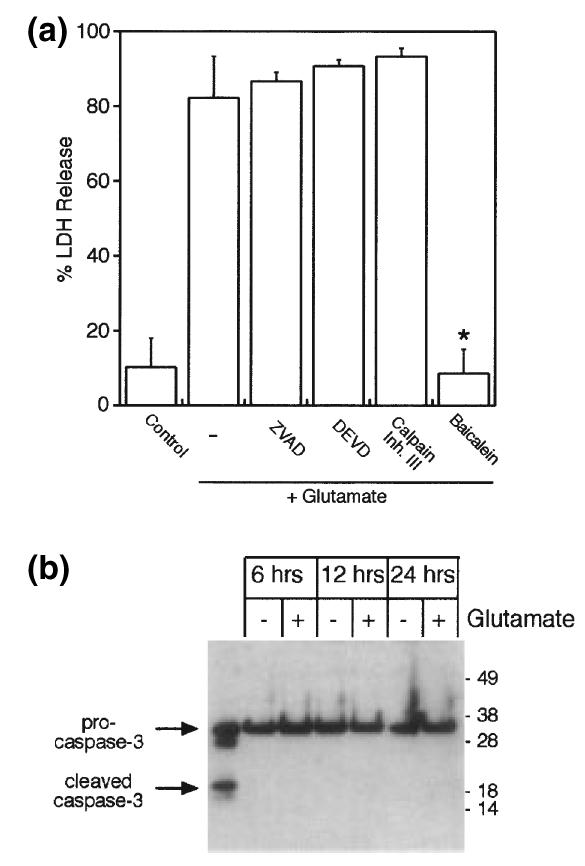
Caspase-3 is not activated by oxidative glutamate toxicity. (a) LDH release was measured after 24 h exposure of HT22 cells to 5 mm glutamate in the presence of 10 μm Z-VAD-fmk, the specific caspase-3 inhibitor DEVD-CHO (20 μm), 10 μm calpain inhibitor III, or the 12-lipoxygenase inhibitor baicalein (10 μm). *p < 0.05 versus glutamate alone. (b) Caspase-3 is not cleaved to the activated form in HT22 cells after glutamate challenge. First lane, hypoxia-treated rat primary neurons as control.
Because most pathways of caspase-mediated cell death converge in activation of caspase-3, we examined cleavage of caspase-3 to the active form by western blotting. The 17/19 kDa cleaved forms of caspase-3 were not detected at all time points measured between 6 and 24 h of exposure to 5 mm glutamate (Fig. 1b), indicating that even at late time points caspase-3 is not activated.
In contrast, the proteasome played a central role in this form of neuronal cell death in our model systems. The potent proteasome inhibitor lactacystin significantly reduced cytoxicity in HT22 cells after glutamate-induced toxicity (Fig. 2a). To confirm the relevance of this pathway, experiments were also conducted in primary neuronal cells freshly isolated from E17 rat brains. Similarly, lactacystin significantly reduced glutamate-induced neurotoxicity (Fig. 2b). These results may seem somewhat surprising, because another proteasome inhibitor MG132, has previously been shown to induce apoptosis in HT22 cells (Stanciu and DeFranco 2002). We thus decided to investigate toxicity of these proteasome inhibitors themselves, in the absence of glutamate. As previously reported, MG132 was cytotoxic in HT22 cells in a dose-dependent manner, whereas lactacystin showed relatively little toxicity up to 30 μm concentration (Fig. 2c).
Fig. 2.
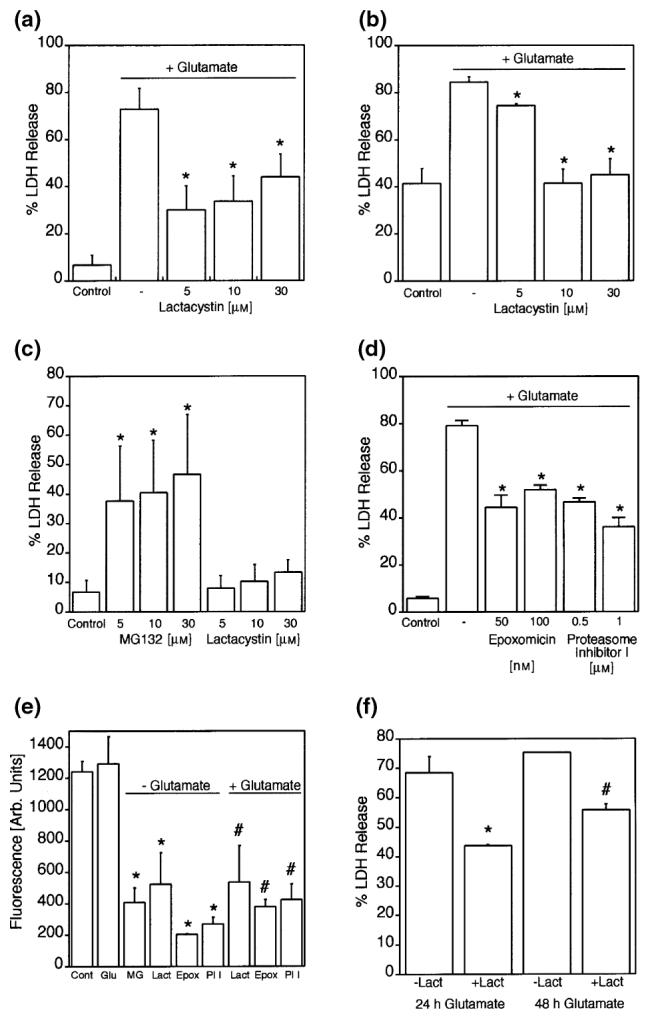
Inhibition of the proteasome with lactacystin protects against oxidative glutamate toxicity. (a) The proteasome inhibitor lactacystin protects HT22 cells against glutamate challenge. *p < 0.05 versus glutamate alone. (b) Lactacystin is also protective in freshly cultured primary neuronal cells. *p < 0.05 versus glutamate alone. (c) In contrast to MG132, lactacystin is not toxic to HT22 cells. *p < 0.05 versus control. (d) Two additional proteasome inhibitors, epoxomicin and Proteasome Inhibitor I, also protect HT22 cells against oxidative glutamate toxicity. *p < 0.05 versus glutamate alone. (e) Proteasome activity against a synthetic substrate in HT22 lysates inhibited by various proteasome inhibitors. Cont = control, Glu = 5 mm glutamate, MG = 10 μm MG132, Lact = 10 μm lactacystin, Epox = 50 nm epoxomicin, PI I = 1 μm Proteasome Inhibitor I. *p < 0.05 versus control, #p < 0.05 versus glutamate alone. (f) Long-term protection against glutamate by lactacystin. Cell death is increased after 48 h in the presence of glutamate and lactacystin, but less than with glutamate alone. Lact = lactacystin, *p < 0.05 versus 24 h glutamate alone, #p < 0.05 versus 48 h glutamate alone.
Given these differential effects of lactacystin and MG132, we decided to study additional inhibitors of the proteasome. Epoxomicin and Proteasome Inhibitor I are structurally unrelated to lactacystin and known to be specific proteasome inhibitors. Both protected HT22 cells against oxidative glutamate toxicity with similar efficacy as lactacystin, suggesting that the protection against oxidative stress afforded by lactacystin is indeed mediated by inhibition of the proteasome (Fig. 2d).
One possible explanation for this apparent discrepancy would be a differential effect of the inhibitors on proteasome activity. We therefore used a fluorogenic substrate to measure proteasome activity in HT22 cell lysates. After 6 h of treatment, prior to any effects on cell viability, proteasome activity was significantly reduced by all of the inhibitors tested (Fig. 2e). These results indicate that the proteasome was inhibited to a similar extent by all inhibitors at the concentrations used for the viability experiments. Furthermore, proteasome activity against the synthetic substrate was not significantly changed by glutamate treatment. Similar levels of reduction of proteasome activity were seen after 18 h of treatment with lactacystin or MG132 (data not shown). Thus, despite similar inhibitory activity towards the chymotrypsin-like proteasome activity in vitro, MG132 is cytotoxic for HT22 cells, whereas lactacystin, epoxomicin and Proteasome Inhibitor I are protective against oxidative glutamate toxicity.
To assess whether the protection afforded by lactacystin lasts beyond 24 h, we extended the incubation time with glutamate in the presence or absence of lactacystin to 48 h. Although cytotoxicity was slightly increased over time, reduction of glutamate-induced LDH release by lactacystin after 48 h remained statistically significant (Fig. 2f).
Although LDH release is a good measure of cell death as the final outcome, its reduction by lactacystin treatment does not necessarily imply that the remaining cells are healthy. To investigate a possible activation of an apoptotic program by treatment with the proteasome inhibitors, we analyzed HT22 cells using FACScan analysis, after 24 h incubation. Control cells were overwhelmingly negative for staining with both propidium iodide and FITC-labeled annexin V (Fig. 3a), while glutamate treatment shifted most of the cells to an apoptotic/necrotic phenotype, characterized as either annexin positive or both annexin and propidium iodide positive (Fig. 3b). In contrast, all three protective proteasome inhibitors shifted the majority of cells back to the annexin-negative field (Fig. 3c–e), arguing against an apoptotic state of these cells.
Fig. 3.

FACScan analysis of HT22 cells subjected to glutamate and proteasome inhibitor treatment. HT22 cells were incubated for 24 h in the presence or absence of 5 mm glutamate and either 10 μm lactacystin, 50 nm epoxomicin, or 1 μm Proteasome Inhibitor I and stained with annexin V–FITC and propidium iodide.
Nonetheless, because the proteasome is involved in the turnover of cellular proteins, we decided to examine the effects of lactacystin on levels of ubiquitinated proteins. Large amounts of ubiquitinated proteins of high molecular mass accumulated in the Triton-insoluble fraction of cells treated with both glutamate and the proteasome inhibitor lactacystin (Fig. 4a). To determine whether this type of lactacystin-dependent accumulation of ubiquitinated high molecular mass proteins was specific to an intracellular compartment, mitochondria-enriched fractions were isolated from HT22 cells. Indeed, the mitochondrial fraction of the lactacystin-treated cells, but not of those treated with glutamate alone or untreated, was enriched for ubiquitinated high molecular mass proteins (Fig. 4b). This finding underscored the efficacy of lactacystin in inhibiting the proteasome and indicated that the inhibition may be particularly relevant for the mitochondria. Finally, protein turnover may be impacted by oxidative glutamate toxicity because of oxidative damage to cellular proteins. These oxidatively modified proteins with carbonyl groups, known to be favored substrates specifically of the 20S proteasome (Weih et al. 2001), can be identified after derivatization with dinitrophenylhydrazine (Singhal et al. 2002). Lactacystin treatment led to an accumulation of oxidized proteins in the mitochondrial fraction (Fig. 4c).
Fig. 4.
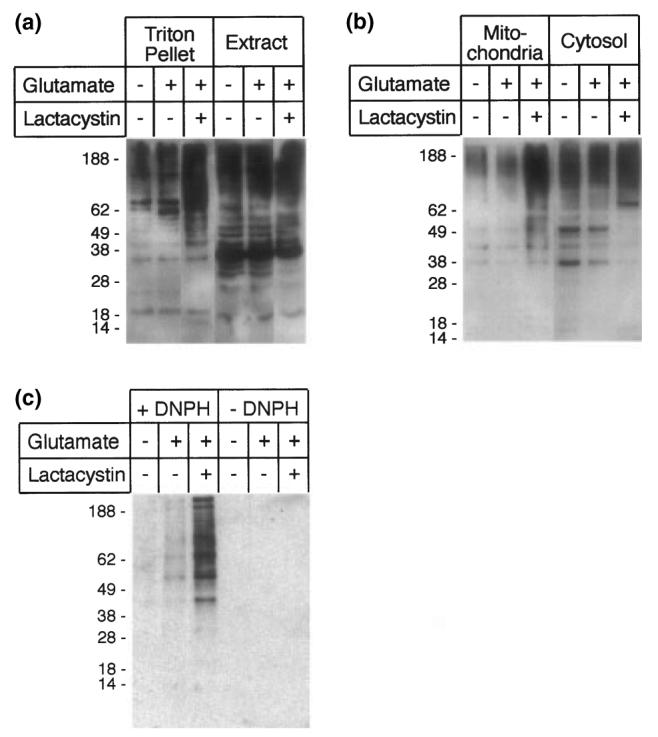
Consequences of proteasome inhibition in HT22 cells. (a) In the presence of lactacystin, ubiquitinated high molecular mass proteins accumulate in the Triton-X-100-insoluble fraction. (b) Ubiquitinated proteins accumulate specifically in the mitochondrial fraction of lactacystin-treated HT22 cells. (c) After inhibition with lactacystin, oxygenated proteins also accumulate specifically in the mitochondrial fraction. Mitochondrial proteins were derivatized with dinitrophenylhydrazine (+ DNPH) or sham-treated (– DNPH).
Discussion
In this study, we used the mouse hippocampal cell line HT22 and freshly isolated rat primary neurons to investigate pathways of oxidative toxicity that lead to neuronal cell death after glutamate injury. Our results identify the proteasome, the major catabolic protease in the cytosol, as part of the oxidative cell death cascade. Incubation of HT22 cells or freshly isolated rat primary neurons with glutamate in the presence of the specific proteasome inhibitor lactacystin leads to enhanced cell survival. Concomitantly, ubiquitinated proteins as well as oxidatively modified proteins accumulate specifically in the mitochondrial fraction of the cells. These findings also suggest a role for mitochondria, which, from ultrastructural studies, are known to be damaged prior to significant cell lysis (Tan et al. 1998; Tirosh et al. 2000) in the cell death pathway. Surprisingly, this proteasome involvement can be detected when using the highly specific inhibitor lactacystin but not the peptide inhibitor MG132, which is known to cause apoptosis in HT22 cells (Stanciu and DeFranco 2002). Because both inhibitors reduced proteasome activity in vitro to a similar extent (Fig. 2e), MG132 may act on an additional target besides the proteasome; alternatively, different activities of the multicatalytic proteasome may be affected to varying degrees. Concentration-dependent pro- or anti-apoptotic effects of MG132 have previously been demonstrated for Sindbis virus-induced cell death (Lin et al. 1998). That lactacystin-mediated inhibition of the proteasome is indeed responsible for protection against oxidative glutamate toxicity was verified by use of two additional, structurally unrelated proteasome inhibitors, epoxomicin and Proteasome Inhibitor I (Fig. 2d). In each case, we titrated the proteasome inhibitors over a wide concentration range in preliminary experiments; the concentrations shown here are all in a range commonly used for cultured cells. In contrast to the proteasome, neither caspases nor calpains appear to be involved in oxidative glutamate neurotoxicity.
Studies in various neuronal cells have found both toxic and protective effects of proteasome inhibitors (Sadoul et al. 1996; Canu et al. 2000; Phillips et al. 2000; Qiu et al. 2000; Weih et al. 2001). It is especially possible that prolonged proteasome inhibition may lead to perturbations in mitochondrial turnover and accumulation of lipofuscin inclusions in the cytoplasm, which may have profound clinical implications in terms of Parkinson's disease (Lang-Rollin et al. 2003; Sullivan et al. 2004). Although we did not see loss of neuroprotection at 48 h in our model, a major caveat in this study remains the lack of ultrastructural data that may tell us about organelle integrity over time. Ultimately, variable consequences of proteasome inhibition may be found even in somewhat similar neuronal cell paradigms (Hoglinger et al. 2003; Sawada et al. 2004), so that extrapolation into clinical applications should be undertaken with caution.
At this point, it is not known whether there is a specific protein target of proteasomal degradation that functions as a ‘death switch’ in the oxidatively challenged cell, or whether bulk degradation of oxidized proteins by the proteasome pushes the cell so far out of equilibrium that recovery is not possible. Proteasome activity against a synthetic substrate was not significantly elevated in lysates of HT22 cells challenged with glutamate (Fig. 2e), but oxidation of proteins generates a larger pool of substrates available for degradation. Likewise, it is unclear whether transcription factors regulated by the proteasome such as p53, HIF1α, and NFκB are involved (Jaakkola et al. 2001; Jesenberger and Jentsch 2002). Our attempts to prevent nuclear uptake of activated NFκB with the inhibitory peptide SN50 did not protect glutamate-treated cells against cell death (data not shown), but this is certainly not a definitive test for NFκB involvement. Thus, at this point we cannot determine if the proteasome is an upstream mediator or if it functions downstream of 12-lipoxygenase activation, which has been shown to be involved in oxidative neuronal death. The effects of proteasome inhibition include the accumulation of both ubiquitinated and oxygenated proteins in the mitochondria of the cells, suggesting involvement of this organelle in the cell death process. Interestingly, alterations in mitochondrial turnover can be detected in neuronal cells after exposure to low levels of proteasome inhibitor (Sullivan et al. 2004). Quite possibly, the proteasome contributes to the mitochondrial damage seen during ultrastructural studies in both HT22 cells and the related HT4 cell line after glutamate challenge (Tan et al. 1998; Tirosh et al. 2000). One implication of these findings is that protection against oxidative glutamate toxicity by proteasome inhibition may only be transient; eventually, damage to mitochondrial proteins may be toxic to these oxidatively stressed cells even though the oxidized proteins are not degraded. After 48-h of glutamate treatment in the presence of lactacystin, cell death as measured by LDH release into the medium is increased compared with the 24 h time point, but is still significantly lower than with glutamate treatment alone (Fig. 2f).
In contrast to the ubiquitin/proteasome pathway, we have not found evidence for caspase-3 or calpain involvement in our models. Most apoptotic pathways involving caspases ultimately converge in the activation of caspase-3. However, we cannot unequivocally rule out the involvement of other members of the caspase family. Both the caspase-1 inhibitor Ac-YVAD-cmk (Tan et al. 1998; Dargusch and Schubert 2002) and the pan-caspase inhibitor Z-VAD-fmk used at 50 μm (Stanciu and DeFranco 2002) have been shown to protect against glutamate toxicity, although other proteases may also be inhibited at that concentration (Schotte et al. 1999; Caserta et al. 2003). Ultimately, the role of different proteases may be critically dependent on the model systems used. Notably, the proteasome is also implicated in interferon gamma (IFNγ)-induced apoptosis in murine lens epithelial cells, also without caspase-3 activation (Awasthi and Wagner 2004). Overall, our findings here suggest that under some conditions, oxidative stress can be decoupled from caspase activation.
Processes of oxidative glutamate toxicity have been previously found to involve, among others, activation of soluble guanylyl cyclase and the ERK MAPK pathway (Li et al. 1997a,b; Satoh et al. 2000; Stanciu et al. 2000). Our study now finds an additional component in the proteasome, although its place in the sequence of events has yet to be determined. Clearly, one of the first steps is the depletion of intracellular glutathione, which is also known to be one of the major factors in the delayed brain damage following stroke and traumatic injury, indicating that oxidative toxicity may be a major pathway by which neuronal cells die in these diseases. Ultimately, understanding oxidative glutamate toxicity may thus lead to novel therapeutic approaches to ameliorate brain damage. Regarding the proteasome, it has already been determined that a proteasome inhibitor closely related to lactacystin, MLN519, is neuroprotective after ischemia and reperfusion in rats (Phillips et al. 2000; Zhang et al. 2001; Williams et al. 2003), and clinical trials for this inhibitor as stroke treatment are underway (Shah et al. 2002). Our data here suggest that reduction of oxidative stress-mediated cell death may partially account for the neuroprotective effect of this proteasome inhibitor.
Acknowledgements
This work was supported in part by grants from the NIH: R01-NS40529, R01-NS37074, R01-NS38731, P01-NS10828.
Abbreviations used
- DMEM
Dulbecco's modified Eagle's medium
- DMSO
dimethylsulfoxide
- DTT
dithiotreitol
- ERK
extracellular signal-regulated kinase
- FACS
fluorescence-activated cell sorter
- FBS
fetal bovine serum
- LDH
lactate dehydrogenase
- PBS
phosphate-buffered saline
- PMSF
phenylmethylsulfonyl fluoride
References
- Awasthi N, Wagner BJ. Interferon-gamma induces apoptosis of lens alphaTN4-1 cells and proteasome inhibition has an antiapoptotic effect. Invest. Ophthalmol. Vis. Sci. 2004;45:222–229. doi: 10.1167/iovs.03-0571. [DOI] [PubMed] [Google Scholar]
- Canals S, Casarejos MJ, de Bernardo S, Rodriguez-Martin E, Mena MA. Nitric oxide triggers the toxicity due to glutathione depletion in midbrain cultures through 12-lipoxygenase. J. Biol. Chem. 2003;278:21 542–21 549. doi: 10.1074/jbc.M213174200. [DOI] [PubMed] [Google Scholar]
- Canu N, Barbato C, Ciotti MT, Serafino A, Dus L, Calissano P. Proteasome involvement and accumulation of ubiquitinated proteins in cerebellar granule neurons undergoing apoptosis. J. Neurosci. 2000;20:589–599. doi: 10.1523/JNEUROSCI.20-02-00589.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caserta TM, Smith AN, Gultice AD, Reedy MA, Brown TL. Q-VD-OPh, a broad spectrum caspase inhibitor with potent antiapoptotic properties. Apoptosis. 2003;8:345–352. doi: 10.1023/a:1024116916932. [DOI] [PubMed] [Google Scholar]
- Dargusch R, Schubert D. Specificity of resistance to oxidative stress. J. Neurochem. 2002;81:1394–1400. doi: 10.1046/j.1471-4159.2002.00950.x. [DOI] [PubMed] [Google Scholar]
- Hoglinger GU, Carrard G, Michel PP, Medja F, Lombes A, Ruberg M, Friguet B, Hirsch EC. Dysfunction of mitochondrial complex-I and the proteasome: interactions between two biochemical deficits in a cellular model of Parkinson’s disease. J. Neurochem. 2003;86:1297–1307. doi: 10.1046/j.1471-4159.2003.01952.x. [DOI] [PubMed] [Google Scholar]
- Jaakkola P, Mole DR, Tian YM, et al. Targeting of HIF-alpha to the von Hippel–Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- Jesenberger V, Jentsch S. Deadly encounter: ubiquitin meets apoptosis. Nat. Rev. Mol. Cell Biol. 2002;3:112–121. doi: 10.1038/nrm731. [DOI] [PubMed] [Google Scholar]
- Khanna S, Roy S, Ryu H, Bahadduri P, Swaan PW, Ratan RR, Sen CK. Molecular basis of vitamin E action: tocotrienol modulates 12-lipoxygenase, a key mediator of glutamate-induced neurodegeneration. J. Biol. Chem. 2003;278:43 508–43 515. doi: 10.1074/jbc.M307075200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang-Rollin I, Rideout H, Stefanis L. Ubiquitinated inclusions and neuronal cell death. Histol. Histopathol. 2003;18:509–517. doi: 10.14670/HH-18.509. [DOI] [PubMed] [Google Scholar]
- Li Y, Maher P, Schubert D. A role for 12-lipoxygenase in nerve cell death caused by glutathione depletion. Neuron. 1997a;19:453–463. doi: 10.1016/s0896-6273(00)80953-8. [DOI] [PubMed] [Google Scholar]
- Li Y, Maher P, Schubert D. Requirement for cGMP in nerve cell death caused by glutathione depletion. J. Cell Biol. 1997b;139:1317–1324. doi: 10.1083/jcb.139.5.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin KI, Baraban JM, Ratan RR. Inhibition versus induction of apoptosis by proteasome inhibitors depends on concentration. Cell Death Differ. 1998;5:577–583. doi: 10.1038/sj.cdd.4400384. [DOI] [PubMed] [Google Scholar]
- Murphy TH, Schnaar RL, Coyle JT. Immature cortical neurons are uniquely sensitive to glutamate toxicity by inhibition of cystine uptake. FASEB J. 1990;4:1624–1633. [PubMed] [Google Scholar]
- Phillips JB, Williams AJ, Adams J, Elliott PJ, Tortella FC. Proteasome inhibitor PS519 reduces infarction and attenuates leukocyte infiltration in a rat model of focal cerebral ischemia. Stroke. 2000;31:1686–1693. doi: 10.1161/01.str.31.7.1686. [DOI] [PubMed] [Google Scholar]
- Qiu JH, Asai A, Chi S, Saito N, Hamada H, Kirino T. Proteasome inhibitors induce cytochrome-c-caspase-3-like protease-mediated apoptosis in cultured cortical neurons. J. Neurosci. 2000;20:259–265. doi: 10.1523/JNEUROSCI.20-01-00259.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratan RR, Ryu H, Lee J, Mwidau A, Neve RL. In vitro model of oxidative stress in cortical neurons. Methods Enzymol. 2002;352:183–190. doi: 10.1016/s0076-6879(02)52018-8. [DOI] [PubMed] [Google Scholar]
- Sadoul R, Fernandez PA, Quiquerez AL, Martinou I, Maki M, Schroter M, Becherer JD, Irmler M, Tschopp J, Martinou JC. Involvement of the proteasome in the programmed cell death of NGF-deprived sympathetic neurons. EMBO J. 1996;15:3845–3852. [PMC free article] [PubMed] [Google Scholar]
- Satoh T, Nakatsuka D, Watanabe Y, Nagata I, Kikuchi H, Namura S. Neuroprotection by MAPK/ERK kinase inhibition with U0126 against oxidative stress in a mouse neuronal cell line and rat primary cultured cortical neurons. Neurosci. Lett. 2000;288:163–166. doi: 10.1016/s0304-3940(00)01229-5. [DOI] [PubMed] [Google Scholar]
- Sawada H, Kohno R, Kihara T, et al. Proteasome mediates dopaminergic neuronal degeneration, and its inhibition causes alpha-synuclein inclusions. J. Biol. Chem. 2004;279:10 710–10 719. doi: 10.1074/jbc.M308434200. [DOI] [PubMed] [Google Scholar]
- Schotte P, Declercq W, Van Huffel S, Vandenabeele P, Beyaert R. Non-specific effects of methyl ketone peptide inhibitors of caspases. FEBS Lett. 1999;442:117–121. doi: 10.1016/s0014-5793(98)01640-8. [DOI] [PubMed] [Google Scholar]
- Shah IM, Lees KR, Pien CP, Elliott PJ. Early clinical experience with the novel proteasome inhibitor PS-519. Br. J. Clin. Pharmacol. 2002;54:269–276. doi: 10.1046/j.1365-2125.2002.01638.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singhal AB, Wang X, Sumii T, Mori T, Lo EH. Effects of normobaric hyperoxia in a rat model of focal cerebral ischemia–reperfusion. J. Cereb. Blood Flow Metab. 2002;22:861–868. doi: 10.1097/00004647-200207000-00011. [DOI] [PubMed] [Google Scholar]
- Stanciu M, DeFranco DB. Prolonged nuclear retention of activated extracellular signal-regulated protein kinase promotes cell death generated by oxidative toxicity or proteasome inhibition in a neuronal cell line. J. Biol. Chem. 2002;277:4010–4017. doi: 10.1074/jbc.M104479200. [DOI] [PubMed] [Google Scholar]
- Stanciu M, Wang Y, Kentor R, et al. Persistent activation of ERK contributes to glutamate-induced oxidative toxicity in a neuronal cell line and primary cortical neuron cultures. J. Biol. Chem. 2000;275:12 200–12 206. doi: 10.1074/jbc.275.16.12200. [DOI] [PubMed] [Google Scholar]
- Sullivan PG, Dragicevic NB, Deng JH, Bai Y, Dimayuga E, Ding Q, Chen Q, Bruce-Keller AJ, Keller JN. Proteasome inhibition alters neural mitochondrial homeostasis and mitochondria turnover. J. Biol. Chem. 2004;279:20 699–20 707. doi: 10.1074/jbc.M313579200. [DOI] [PubMed] [Google Scholar]
- Tan S, Wood M, Maher P. Oxidative stress induces a form of programmed cell death with characteristics of both apoptosis and necrosis in neuronal cells. J. Neurochem. 1998;71:95–105. doi: 10.1046/j.1471-4159.1998.71010095.x. [DOI] [PubMed] [Google Scholar]
- Tirosh O, Sen CK, Roy S, Packer L. Cellular and mitochondrial changes in glutamate-induced HT4 neuronal cell death. Neuroscience. 2000;97:531–541. doi: 10.1016/s0306-4522(00)00028-2. [DOI] [PubMed] [Google Scholar]
- Weih M, Schmitt M, Gieche J, Harms C, Ruscher K, Dirnagl U, Grune T. Proteolysis of oxidized proteins after oxygen–glucose deprivation in rat cortical neurons is mediated by the proteasome. J. Cereb. Blood Flow Metab. 2001;21:1090–1096. doi: 10.1097/00004647-200109000-00006. [DOI] [PubMed] [Google Scholar]
- Williams AJ, Hale SL, Moffett JR, Dave JR, Elliott PJ, Adams J, Tortella FC. Delayed treatment with MLN519 reduces infarction and associated neurologic deficit caused by focal ischemic brain injury in rats via antiinflammatory mechanisms involving nuclear factor-kappaB activation, gliosis, and leukocyte infiltration. J. Cereb. Blood Flow Metab. 2003;23:75–87. doi: 10.1097/01.WCB.0000039285.37737.C2. [DOI] [PubMed] [Google Scholar]
- Zhang L, Zhang ZG, Zhang RL, Lu M, Adams J, Elliott PJ, Chopp M. Postischemic (6-hour) treatment with recombinant human tissue plasminogen activator and proteasome inhibitor PS-519 reduces infarction in a rat model of embolic focal cerebral ischemia. Stroke. 2001;32:2926–2931. doi: 10.1161/hs1201.100207. [DOI] [PubMed] [Google Scholar]