

Abstract
Most homeostatic processes including gene transcription occur as a result of deviations in physiological tone that threatens the survival of the organism. A prototypical homeostatic stress response includes changes in gene expression following alterations in oxygen, iron or 2-oxoglutarate levels. Each of these cofactors plays an important role in cellular metabolism. Accordingly, a family of enzymes known as the Prolyl 4-hydroxylase (PHD) enzymes are a group of dioxygenases that have evolved to sense changes in 2-oxoglutarate, oxygen and iron via changes in enzyme activity. Indeed, PHDs are a part of an established oxygen sensor system that regulates transcriptional regulation of hypoxia/stress-regulated genes and thus are an important component of events leading to cellular rescue from oxygen, iron or 2-oxoglutarate deprivations. The ability of PHD activity to regulate homeostatic responses to oxygen, iron or 2-oxoglutarate metabolism has led to the development of small molecule inhibitors of the PHDs as a strategy for activating or augmenting cellular stress responses. These small molecules are proving effective in preclinical models of stroke and Parkinson's disease. However the precise protective pathways engaged by PHD inhibition are only beginning to be defined. In the current review, we summarize the role of iron, 2-oxoglutarate and oxygen in the PHD catalyzed hydroxylation reaction and provide a brief discussion of some of the transcription factors that play an effective role in neuroprotection against oxidative stress as a result of changes in PHD activity.
Keywords: Prolyl Hydroxylase, Dioxygenases, Iron, 2-Oxoglutarate, Oxygen, Transcription Factor, Camp Response Element Binding Protein, CREB, Nuclear Factor Kappa B, NF-kB, Activating protein, AP-1, specific protein 1, Sp1, Sp3, Hypoxia Inducible factor, HIF, Ischemia, Stroke, Oxidative Stress, Neuroprotection, Review
2. PROLYL HYDROXYLASE ACTIVITY AND REGULATION OF GENE TRANSCRIPTION
Hydroxylation of specific amino acids in proteins is as an enzyme catalyzed posttranslational modification that can lead to changes in interactions between proteins. Indeed, hydroxylation of proline residues has been shown to play an important role in the stability of proteins such as collagen (1), elastin (2), and prion protein (3). Hydroxylation of specific prolines (position 402 or 564) within the transcriptional activator “Hypoxia Inducible Factor-1” (HIF-1) (4, 5) regulates its transcriptional activity. HIF-1 hydroxylation is a prototypic example of a posttranslational modification capable of regulating gene transcription. As HIF is at the center of adaptive responses to ischemic and oxidative stress, the regulation of HIF hydroxylation becomes a viable strategy for engaging its homeostatic functions in a host of tissues, particularly the brain. The group of enzymes that can catalyze the hydroxylation reaction of HIF-1 are prolyl 4-hydroxylases (PHDs). These enzymes belong to a super family of 2-oxoglutarate dependent hydroxylases, that require iron in the catalytic moiety (6). They also utilize oxygen in the form of dioxygen, incorporating one oxygen atom into the proline residue, and the other into 2-oxoglutarate, yielding succinate and CO2. PHD activity negatively regulates the stability of hypoxia inducible factor-1 alpha (HIF-1α). The well-established strategic role HIF-1 plays in the regulation of adaptive mechanisms in response to hypoxia, the requirement of oxygen for the activity of PHDs and the inverse relationship between PHD catalytic activity and HIF-1 transcriptional activity has led researchers to appropriately designate PHDs as key players in the oxygen sensing transcriptional mechanisms regulated by HIF-1. Interestingly, oxygen is not the only co-factor required for PHD activity. The transition metal, iron is also required as is 2-oxoglutarate. The ability of both of these cofactors to regulate PHD activity suggests that PHD activity can be modulated under conditions of iron or 2-oxoglutarate deficiency. As iron and 2-oxoglutarate are critical for the optimal function of mitochondrial respiration via their functions on Fe/S cluster proteins and production of reducing equivalents (NADH and FADH2), it is not surprising that deficiency in either of these two co-substrates would trigger transcriptional responses that decrease the dependence on mitochondrial respiration and increase the dependence on glycolytic metabolism. In this review, we provide an overview of the enzymology of the PHDs and the transcriptional responses modified by changes in enzyme activity. We also discuss other transcriptional activators that are induced by oxidative stress in the central nervous system.
3. REGULATION OF PROLYL 4-HYDROXYLASE (PHD) ENZYME ACTIVITY VIA IRON, 2-OXOGLUTARATE AND OXYGEN BINDING SITES
The hydroxylation reaction catalyzed by the PHDs comprises of an iron-mediated incorporation of a hydroxyl group into the conserved proline residue with the consumption of a dioxygen molecule and release of carbon dioxide, whereas 2-oxoglutarate is converted to succinate (7) (for review, see Siddiq et al., 2007) (Figure 1). PHDs that selectively catalyze the formation of hydroxyproline in the HIF-1 molecule by the hydroxylation of conserved proline residues belong to a sub-group of the dioxygenases. The most extensively studied prolyl hydroxylases are the ones that hydroxylate a proline residues in collagen molecules (8-13). These studies reveal that the enzyme, isolated as a homogeneous protein by affinity chromatography from three different sources (8-10, 12, 14, 15), occurs as a tetramer with a molecular weight of about 240,000 (11, 16-18). The enzyme does not hydroxylate free proline, and recognizes a conserved motif (LXXLAP in the HIF-1 molecule; X indicates any amino acid and P indicates the hydroxyl acceptor proline) in the primary substrate for hydroxylation (5, 11, 19-22). The hydroxylation of prolyl residues in this sequence is influenced by the nature of the amino acid in the X position, the nature of the amino acids in the adjacent sequences, the chain length and conformation of the primary substrate (11, 16, 23).
Figure 1.
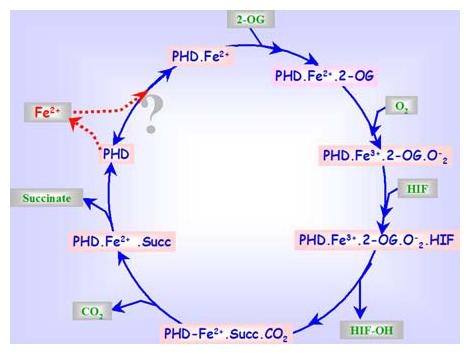
Schematic presentation of the proposed sequential mechanism for the prolyl hydroxylase reaction. The 2-oxoglutarate is stoichiometrically decarboxylated during the hydroxylation of HIF in the presence of dioxygen and iron, resulting in the generation of carbon dioxide (CO2) and succinate. The dashed lines between the enzyme and Fe2+ indicate that the enzyme may exist with or without Fe2+ in the catalytic moiety after each catalytic cycle.
In general, kinetic mechanisms for enzyme reactions fall into two major groups, sequential and substitutional. In sequential mechanisms, all the reactants must combine with the enzyme before the reaction can occur and the product is released; whereas in substitution mechanisms one or more products are released before all the substrates have become bound to the enzyme. Kinetics studies of PHDs suggest that the binding of the co-substrates Fe2+, 2-oxoglutarate, oxygen, and the proline containing primary substrate to the enzyme occurs by a sequential mechanism (23, 24) (Figure 1). However, several aspects of the co-substrate affinity, binding and requirements of prolyl hydroxylase have remained unresolved. For instance, while the absolute requirement of iron by PHDs and the mechanism by which iron mediates the transfer of a hydroxyl group in the proline residue are well established, there is still considerable disagreement concerning the affinity and binding strength of iron in the prolyl hydroxylase protein. Spectroscopic evidence has been reported for the firm binding of iron to the enzyme (25, 26), however a main concern about these studies is the impurity of the enzyme preparations. Some of the in vitro studies suggest that the enzyme was not completely inhibited by EDTA, 2,2′-dipyridyl and a variety of other chelating agents such as Desferrioxamine (DFO) (27, 28), but others have found complete inhibition with some of these compounds (e.g. (27, 29, 30). There are also observations that prolyl hydroxylase purified by affinity chromatography on its polypeptide substrate linked to agarose retained about 40% of its maximal activity without the addition of Fe2+ (22, 31). In vitro studies clearly indicate that the activity of pure prolyl hydroxylase is completely dependent on added Fe2+. Whether iron stays permanently bound to the enzyme in the tissues is not clear. There are reports suggesting that even when PHD is purified by an affinity column procedure, the enzyme does not retain sufficient quantities of iron to catalyze the reaction without the addition of this cation in vitro (32-34). Inhibition of the activity of PHDs in primary neurons by the treatment with iron chelators such as DFO, resulting in stabilization of HIF-1 and downstream target genes, indicates that these enzymes do rely on the labile pool of iron in the cells for their activity (35). Fe2+ is located in a pocket coordinated with the enzyme by three side-chains with two histidines and an aspartate forming the catalytic triad (36-38). However, the exact mode of the binding of iron to the enzyme molecule is not known, but it has been suggested in many previous reports that this binding may occur to one or more −SH groups present in the vicinity of the active site of the enzyme (33, 39). In agreement with this suggestion, in vitro studies show that sulphydryl reagents inhibit PHD activity (33), and this inhibition can be reversed with dithiothreitol (DTT) (33, 40, 41).
PHDs catalyze the uncoupled decarboxylation of 2-oxoglutarate in the absence of the polypeptide substrate (42-47). It thus seems that Fe2+, 2-oxoglutarate and oxygen, can bind to the enzyme in the absence of the polypeptide substrate. Studies using distinct structural analogs of 2-oxoglutarate such as dihydroxybenzoate (DHB) and dimethyl-oxalyl-glycine (DMOG) have been found to inhibit PHD activity (35). It is thus clear that the co-substrates 2-oxoglutarate and iron bind at separate sites on the enzyme molecule (30) and the inhibition of binding of either leads to inhibition of PHD activity and consequent activation of downstream pathways governed by PHDs. These sites are evidently also distinct from the binding site of the polypeptide substrate, as in vitro studies using oxaloacetate or Zn2+ do not affect the binding of the polypeptide substrate to the active site (27). This also clearly suggests that some of the citric acid cycle intermediates (27, 28) may act as physiological inhibitors of the enzymes.
The prolyl hydroxylase reaction is entirely dependent on O2, and during the reaction one atom of the O2 molecule becomes incorporated into the hydroxyl group of the formed hydroxyproline while the other is incorporated into the succinate (33). Mechanistic studies of the enzyme activity reveal that oxygen is activated before hydroxylation by the formation of a ferryl intermediate or hydroperoxide (32, 48-57). This occurs via interaction of molecular oxygen with Fe2+ leading to the oxidation of Fe2+ to Fe3+ (33, 36, 58-61). Compounds capable of inhibiting the formation of hydroperoxide, such as epinephrine and nitroblue tetrazolium, inhibit the activity of PHDs in vitro (62). A series of reports show that HIF-1 protein levels are generally low in rodent tissues under physiological conditions, however with organ or systemic hypoxia and diminished PHD activity, HIF-1 levels are significantly increased, and thus there is an increase in HIF-target gene transcription (35, 62-66).
4. PHD ACTIVITY-RESPONSIVE TRANSCRIPTION FACTORS
Most physiological processes including gene transcription occur as a result of changes in the tonic physiologic or biochemical state of the cells and tissues in order to either fulfill the basal endogenous needs to stay alive, or combat to and survive against an external stimuli such as stress. 2-oxoglutarate being a part of the TCA cycle and both Fe2+ and O2 being electronically-activated, radical-generating molecules play an important role in determining the ‘redox-state’ or ‘stress’ driven processes in living cells. The sensitivity of the PHD activities towards changing levels of fundamental molecules, like oxygen, 2-oxoglutarate and iron, presents a very complex but rather useful scheme that engages cellular adaptive responses leading to the activation of rescue mechanisms. These mechanisms help the cell survive by initiating the transcriptional upregulation of genes that enhance tissue perfusion and anaerobic ATP generation pathways. Gene array analysis has recently revealed significant information regarding global changes in the gene transcription pathways of cells in response to stress such as hypoxia/ischemia. These changes not only enhance the capability of the system to combat the hypoxic conditions but also contribute to hypoxia-induced phenotypic changes in the cells (67, 68). Studies have also demonstrated that stress as a result of changes in oxygen or iron has a cell-type and cell-state specific effect on the cellular transcriptome. Although the knowledge of the transcriptional mechanisms that are activated by the oxygen sensors is crucial to influence the downstream gene transcription events in favor of physiologic recovery, an effective therapeutic approach requires a global understanding of the upstream pathways. These stress-sensing mechanisms that communicate the stress signal to specific transcriptional regulators in the brain are areas of intense investigation in our own laboratory as well, and the current hypotheses include investigations regarding the direct role of oxygen, iron and 2-oxoglutarate dependent enzymes prolyl hydroxylases in neuroprotective transcriptional pathways. In the following section, we discuss the various transcription factors that play a established role in neuroprotection and are regulated in response to changes in oxygen, iron or 2-oxoglutarate and thus may be a potential PHD target or interactors.
4.1. The Hypoxia-Inducible-Factors (HIF)
The HIF transcription system including its homologs (e.g., HIF-1α, HIF-2α, HIF-3α) has emerged as a key regulator of responses to changes in PHD activity as a result of changes in oxygen and/or iron levels, both at local and systemic levels. So far, HIF is the most established PHD-regulated transcription factor. The degradation of HIF occurs in the presence of molecular oxygen by modification of oxygen-dependent degradation domains within the HIF protein carried out by the PHDs. These enzymes add a hydroxyl group in conserved proline residues (402 and 564 of the alpha subunit of HIF-1) thus facilitating interaction with the von Hippel-Lindau tumor suppressor, which targets HIF-1α for proteasomal degradation (Figure 2). Treatment of both isolated neurons or animals with PHD inhibitors leads to stabilization of HIF and an increase in the levels of its downstream target genes (e.g.erythropoietin, vascular endothelial growth factor, glycolytic enzymes) (35).
Figure 2.
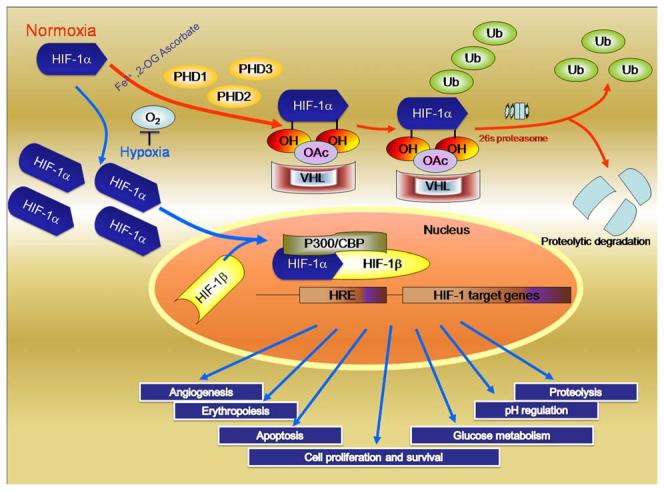
Cellular response to hypoxia: Levels of Hypoxia Inducible Factor-1 (HIF-1) are regulated by cellular oxygen by proline hydroxylation. The reaction is catalyzed by the enzymes prolyl 4-hydroxylases. Under normoxia (blue arrows), the intracellular level of HIF-1α is kept low by rapid ubiquitination and subsequent proteasomal degradation via recruitment of von Hippel–Lindau protein (pVHL), which depend on the hydroxylation of proline residues. In contrast, under hypoxia (orange arrows), both the intracellular level and the transcriptional activity of HIF-1α increase as a result of suppressed PHD activities. Consequently, HIF-1α forms a heterodimer with HIF-1β and changes the transcriptional rates of HIF-1-regulated genes under hypoxia; Reproduced with permission from # 7.
It is believed that approximately 1–1.5% of the genome is transcriptionally regulated by hypoxia. Many of these genes are known to be regulated by HIF-1α regulate biological changes (e.g. increased O2 delivery, increased angiogenesis, increased anaerobic glycolysis) that facilitate adaptation to hypoxia and associated metabolic compromise. In addition to its role in combating hypoxia, HIF-dependent gene expression provides resistance to oxidative stress, since many of the genes regulated by HIF-1 or HIF-2 (e.g. erythropoietin, VEGF, MnSOD) prevent oxidative stress-induced death by themselves (69-73). Recent evidence has pointed out that reactive oxygen species (ROS) generation occurs at multiple time points after stroke (74-76). Studies on the expression of HIF-1 and its target genes in the adult rat brain have shown that after focal cerebral ischemia, mRNAs encoding HIF-1α, glucose transporter-1 and several glycolytic enzymes including lactate dehydrogenase were up-regulated in the areas around the infarction (77-80).
Pharmacological and genetic strategies that reduce oxidative injury and decrease brain damage are now considered to be an effective approach for drug development in stroke. The well-established role of PHDs in the scheme of HIF regulated gene transcription presents researchers a distinct therapeutic target for activation of HIF by small molecule “drugs” against oxidative stress. The advantage of this strategy as compared to prior “antioxidants” is the ability of a single drug to selectively target a single molecule (i.e. PHD) that will activate more than seventy genes providing adaptation to ischemia and oxidative stress (35, 81, 82) (Figure 2).
4.2. Cyclic AMP response element binding protein (CREB)
The cyclic AMP response element binding protein (CREB) is one of a family of leucine zipper transcription factors regulated by intracellular signaling mechanisms such as cAMP and Ca2+. CREB family members contain a C-terminal basic domain that mediates DNA binding and a leucine zipper domain that facilitates dimerization. These two domains are separated by the kinase inducible domain (KID). Ser-133, present in the KID is phosphorylated in a stimulus-inducible manner leading to the binding of the KID domain to the transcriptional coactivator, CREB binding protein (CBP) (83, 84). This phosphorylation-dependent interaction between CREB and CBP is believed to function as the trigger for inducible gene expression. Since the identification and cloning of CREB, the molecular mechanisms by which it functions as an inducible regulator of transcription have been the focus of much investigation. Clear details of the upstream signaling mechanisms that convert extracellular stimuli into CREB activation, by Ser-133 phosphorylation, in order for CREB to function as a stimulus-dependent transcriptional activator are not known. In neurons, CREB phosphorylation occurs under a wide variety of cellular circumstances. These include responses to growth factors during the development of the nervous system, depolarization and synaptic activity during normal neuronal function, and hypoxia and stress responses during stroke or neural injury.
Acute mild hypoxia in neuronal cells activates CREB through phosphorylation at serine 133 (85) and CREB and HIF-1 may act together at promoters of genes involved in hypoxic compensation (e.g. LDH). In an intestinal epithelial cell model, more severe hypoxia results in the CREB degradation, an event mediated through decreased activity of protein phosphatase 1γ (86). CREB degradation leads to a derepression of inflammatory gene expression and thus contributes to hypoxia activated inflammatory processes. Interestingly, more prolonged exposure to severe hypoxia results in CREB stabilization and a resolution of inflammatory gene expression through the transcriptional upregulation of small ubiquitin-related modifier-1 (SUMO-1) modification (87). Thus the hypoxia regulated CREB-dependent gene expression is dependent upon the extent and degree of the stimulus.
There are also reports that show treatment of primary neurons with hypoxia mimics, such as iron chelators, increases CREB binding to DNA (88). Recent unpublished data from our lab shows that treatment of primary neurons with structurally distinct inhibitors of PHD activity increase CREB protein as well as mRNA levels (Siddiq et al., unpublished data). Although the molecular components by which hypoxia or hypoxia mimics alter signaling cascades culminating in CREB inhibition/activation have not been as precisely defined, some attractive candidates, such as SAPK2/p38MAPK exist. As a hypoxia-activated kinase, SAPK2 has at least three downstream targets, MAPKAP K2, MSK1, and MSK2. All of which are CREB kinases (89, 90). These appear to be critical regulators of CREB because in mouse fibroblasts lacking MSK1 and MSK2, CREB phosphorylation in response to stress is eliminated almost entirely (91). There are various reports showing a direct effect of hypoxia on the activity of these kinases. For instance, the activation of p38 MAPK requires dual phosphorylation of threonine 180 (Thr180) and tyrosine 182 (Tyr182) residues within the conserved threonine-glycine-tyrosine (TGY) motif. This is reported to be involved in conveying extracellular stress to cellular response such as inflammation and the processes of cell differentiation, growth, and death (92). However, reports suggest that under hypoxic conditions enhanced phosphorylation of CREB is accompanied by the decrease of ERK1/2 phosphorylation in mice brains (90, 93-96).
There are also reports that suggest inhibition of kinase activities by prolyl hydroxylases, such as the ones that regulate NF-κB activation by a hydroxylation mechanism (97) (see next section for details). It is possible that under normoxia inhibition of one or more of the phosphorylating kinases in the ERK/MAPK signaling pathway occurs by the addition of a hydroxyl group by the PHDs, whereas under hypoxic conditions, inhibition of PHD activity allows for the activation of the CREB by phosphorylation. Decrease in ERK phosphorylation under hypoxic conditions may be a result of feedback mechanisms once the survival machinery is active. In summary, there seems to be a cross talk between the CREB phosphorylation and activation under stress conditions and PHD activity under hypoxic conditions, but a cogent model of how these pathways intersect is just being established (Siddiq et al., unpublished observations).
4.3. Nuclear factor kappa-B (NF-κB)
The transcription factor nuclear factor kappa-B (NF-κB) is known for its fundamental role in regulating immune and inflammatory responses. Originally discovered in B lymphocytes about 20 years ago (98), the NF-κB family members (p65 or RelA, RelB, c-Rel, p50/p105 or NF-κB 1, and p52/p100 or NF-κB 2) are also diffusely expressed in both neurons and non-neuronal cells (99). However, in certain regions of the brain, especially the cortex and hippocampus, constitutive nuclear NF-κB activity has been reported exclusively in neurons (100-103). Members of the NF-κB family share a conserved Rel-homology domain (RHD) responsible for DNA binding activity, protein dimerization, and nuclear translocation. NF-κB is bound to the repressor molecule inhibitory kappaB (IκB) in the cytosol in the absence of stimulus. This coupling of proteins masks the nuclear localization sequence (NLS) of NF-κB and sequesters the protein in the cytosolic compartment. Upon stimulation, IκB is targeted for ubiqutination and degradation by specific serine phosphorylation. The NLS of NF-κB is then exposed, and enables it to translocate to the nucleus where it carries out its transcriptional activity at specific κB sites within the promoter regions of target genes (104). NF-κB-responsive genes include those responsible for encoding inflammatory cytokines, chemokines and cell surface adhesion molecules and several hypoxia induced genes such as cyclooxygenase-2 (COX-2), tumor necrosis factor alpha (TNFα), interleukin-6 (IL-6) and macrophage inflammatory protein-2 (MIP-2). NF-κB plays a dynamic role in the survival and death of neuronal and non-neuronal cells under physiological and pathological conditions (105, 106). Research has established that the activation of NF-κB by cytokines enhances neuronal survival by preventing apoptosis and that the anti-apoptotic action of cytokines disappears in neurons that are treated with a super-repressor IkappaB-alpha protein, or lacking the RelA (p65) subunit of NF-κB (107). The inhibition of NF-κB renders various types of cells highly vulnerable to apoptosis (104, 108).
Until recently, the central event in NF-κB activation that is the removal of the IκB complex from the transcription factor was considered to be through a process involving phosphorylation and degradation in which the IκB degradation is preceded by phosphorylation of serine residues 32 and 36 mediated by the IκB kinase (IKK) complex. However more recent reports show that NF-κB activity is mainly regulated by the regulation of molecules further upstream of the IκB phosphorylation. Hypoxia and more importantly specific inhibition of PHD activity by pharmacological agents as well and siRNA stimulate NF-κB transcriptional activity (97). Mechanistic studies reveal that neither NF-κB nor IκB contain hydroxylation motif but two of the important upstream kinases namely, inhibitory kappaB kinase alpha (IKKα) and inhibitory kappaB kinase beta (IKKβ), contain the sequence LXXLAP, the conserved proline containing motif present in HIF-1 molecule making it a primary substrate for hydroxylation by PHDs. Mutation of this conserved proline residue to alanine resulted in the loss of hypoxic-inducibility of NF-κB activity. It is proposed that under normoxic conditions, hydroxylation of this conserved proline residue inhibits the phosphorylation activity of these kinases thereby inhibiting the phosphorylation and degradation of the IκB subunit. Under hypoxia, inhibition of PHD activity inhibits the hydroxylation of the IKKα and IKKβ, leading to the phosphorylation and degradation of IκB and subsequent activation of NF-κB transcriptional activities (Figure 3).
Figure 3.
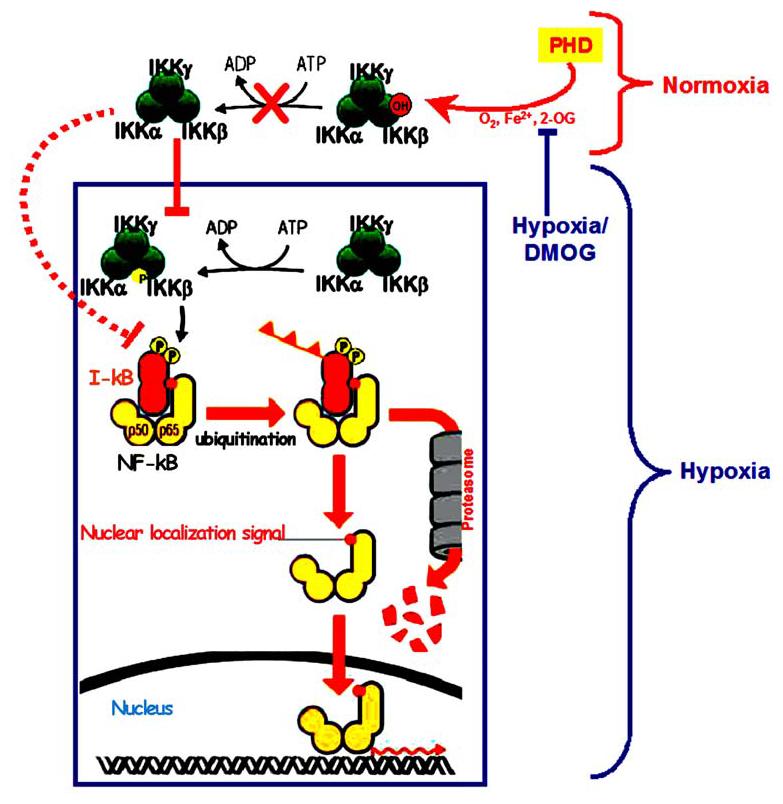
Nuclear factor kappaB regulation, NF-κB dimers are held in the inactive state by a family of inhibitors called I-κB. Diverse signaling mechanisms, such as inhibition of PHD activity by hypoxia or DMOG, leads to activation of a multisubunit kinase IKK complex which phosphorylates I-κB on two key serines. Phosphorylation of I-κB marks it for degradation by the ubiquitin pathway, NF-κB dimer is thus liberated to translocate to the nucleus, bind DNA and activate transcription. Under normoxia, a hydroxylation reaction, catalyzed by PHDs hydroxylates the proline on the IKK□ subunit, inhibits the downstream phosphorylation events thus inhibiting NF-κB activation.
4.4. Specific protein 1 and 3 (Sp1 and Sp3)
Sp1 and Sp3 are ubiquitous transcription factors of the Sp/XKLF transcription factor family that are involved in basal transcription and housekeeping gene expression (109-112). This family includes members such as Sp2–Sp4 that contain identical sequence binding motifs, but can display differential activity, depending on the stimuli. For instance, polyglutamine expansions in the huntingtin protein can induce neuronal toxicity, in part, by sequestering Sp1 and one of its coactivators, TATA binding protein-associated factor (TAF)II130, suggesting a role for Sp1 in neuronal survival (113, 114). Sp1 has also been shown to regulate prosurvival proteins e.g., the inhibitor of apoptosis (IAP) protein, survivin (115), and manganese superoxide dismutase (116) as well as prodeath proteins e.g., Fas ligand (117, 118) and 12-lipoxygenase (119). Like other transcription factors, the role of Sp1 in regulating cell death may depend on a number of factors, such as the cell type and the death stimulus (120). Levels of Sp1 are regulated to an extent by mRNA expression, but further regulation can be imposed by proteasomal degradation, for example, in response to nutrient starvation and adenylate cyclase stimulation (121). Reports suggest that in primary neurons, oxidative stress increases the levels as well as DNA binding of both Sp1 and Sp3 in neurons (122). Similarly, several classically hypoxia-responsive genes such as EPO and VEGF have Sp1/Sp3 binding sites within promoter regions that are thought to facilitate transcriptional activation (123, 124). Sp1 and Sp3 have also shown to be involved in COX-2 expression in a hypoxia independent manner. Both hypoxia and oxidative stress increase nuclear localization of Sp1 and Sp3 levels (122, 125). Forced expression of Sp1 and Sp3 enhances neuronal survival under oxidative stress conditions. Sp1 and Sp3 activation appear to be temporally related to the onset of oxidative stress in cortical neurons and not a late event that is a consequence of oxidative stress-induced cell death. Activation of Sp1 and Sp3 DNA binding occurs within the first 2 hr of glutamate or HCA exposure and is maximal by 5 hr. The kinetics of Sp1 and Sp3 activation demonstrate that induction of these factors is an “early” response to cell stress, and their activation is initiated 8–10 hr before the point at which neurons become irreversibly “committed” to the cell death pathway (88, 126-128). The close temporal relationship between oxidative stress and Sp1 and Sp3 activation is also supported by the observation that structurally diverse small molecules including inhibitors of the prolyl hydroxylases (e.g., DFO) that inhibit oxidative glutamate toxicity also block the activation of Sp1 and Sp3 by glutathione depletion, despite having no effect on glutathione depletion per se (88, 127). The direct effect of hypoxia or oxidative stress on Sp1 and Sp3 activities and presence of Sp1 and/or Sp3 binding sites in hypoxia-regulated genes such as VEGF, indicates a HIF-1 independent but redox-sensitive mechanism by which levels of survival genes may be regulated. Similarly, induction of VEGF may also occur via a p42/p44 MAP kinase-dependent mechanism (129). These studies reveal the presence of two Sp1-binding sites present in the MAP kinase responsive region. Wild type or mutant constructs for the Sp1 show that mutation of both Sp1-binding sites blocks the phosphorylation-dependent transcriptional activation and VEGF induction (130). There are also reports showing activation of the MAP kinase pathway by hypoxia /ischemia (93) which in turn affects Sp1 activation. The redox sensitive regulation of Sp1/Sp3 levels and transcriptional activity and their ability to induce neuroprotection and survival genes through diverse signaling mechanisms suggests the presence of a molecular framework connected via PHD activity.
4.5. Activating protein-1 (AP-1)
Activating protein-1 (AP-1) is a redox-sensitive transcription factor and it has been suggested that hypoxia and consequently the redox environment in the cell, initiates AP-1-mediated gene transcription (131). It comprises members of Fos, Jun, ATF (activating transcription factors) and MAF (musculoaponeurotic fibrosarcoma) (132-138) protein families that can homodimerize or heterodimerize to form the active AP-1 complex and modulate gene expression. The combinatorial interaction of these proteins provides multiple levels of gene expression control. In addition, cell type and the differentiation state can dictate the phenotypic outcome, accounting at least in part for how AP-1 can regulate apparently conflicting endpoints (137). AP-1 is involved in diverse cellular functions related to apoptosis, cell proliferation, cell differentiation, catecholamine biosynthesis, inflammation, xenobiotic metabolism, tumor invasion and angiogenesis (138). Genes regulated by AP-1 in hypoxia include tyrosine hydroxylase (139), VEGF (140), and endothelial NOS (eNOS) (131). AP-1 co-operates with other transcription factors such as HIF-1, GATA-2, NF-1 and NF-κB to complement the activation of hypoxia-sensitive genes (139-142). Thus, AP-1 may represent an important facilitator of stress-induced gene expression through interaction with other transcription factors. The mechanism by which AP-1 is activated in hypoxia has yet to be fully elucidated. This is likely to be a complex process, given AP-1's apparent activation by oxidants (131) and anti-oxidants alike (143). Another signaling mechanism proposed is the hypoxia-induced modulation of intracellular Ca2+ levels upstream of AP-1 activation (139, 140, 144, 145). This increase is thought to activate AP-1 independently of HIF. Other reports demonstrate a role for non-receptor tyrosine kinases in propagating the hypoxic signal from G protein-coupled receptors based on results implicating a role for Src (non-receptor tyrosine kinase) and Ras (145). Reports suggest that AP-1 activation under oxidative stress is mediated via a Jun N-terminal kinase (JNK)-dependent pathway (146). An interesting model of JunD induced gene expression via PHDs has been presented by Gerald et al (147). Accordingly, JunD, a member of the AP-1 family, regulates both genes involved in antioxidative defense and H2O2 production. Increased production of H2O2 by JunD inhibits the PHD enzyme activity by promoting iron oxidation i.e. by converting Fe2+ to Fe3+. An increased proportion of PHD in the Fe3+-inactivated state limits PHD activity, and therefore a decrease in HIF-1α hydroxylation, and degradation. Subsequently, HIF-1α accumulation enhances VEGF-A transcription. Reciprocally, JunD overexpression decreases intracellular H2O2 content, alleviates toxic effects of ROS, and efficiently counteracts Ras-induced angiogenesis in tumors.
5. PERSPECTIVES
Hypoxia is among the most fundamental of stresses for multicellular organisms that depend on oxygen as the terminal electron acceptor in efficient mitochondrial ATP production. The precise mechanism by which a change in oxygen tension leads to a complete reorganization of metabolic infrastructure is beginning to be elucidated. Central to these adaptive reorganization efforts is the change in activity of the oxygen, Fe2+ and 2-oxoglutarate dependent dioxygenases known as the HIF prolyl 4-hydroxylases. These enzymes possess a Km for oxygen that makes them ideal oxygen sensors (7). In response to changes in iron, oxygen or 2-oxoglutarate, PHD activity decreases. It is now clear that in addition to the canonical PHD substrate HIF, other transcription factor families such as CREB, NF-κB and AP-1 are also regulated by changes in PHD activity. The precise targets that must be hydroxylated to suppress adaptive hypoxia signaling are being defined. Moreover, an understanding of the tissue specific and subcellular mechanisms by which PHDs modify the tone of gene expression are only beginning to be defined. The current review summarizes our limited knowledge of what promises to be a very fruitful and therapeutically relevant field of investigation.
6. ACKNOWLEDGMENTS
We are thankful to Dr. Amir Siddiq, at the University of Sheffield, UK for help with figures and Dr. Renée Haskew-Layton at the Burke Medical Research Institute, NY, for help with the manuscript.
Glossary
Abbreviations
- 2-OG
2-oxoglutarate
- AP-1
activator protein 1
- ATP
Adenosine triphosphate
- CO2
Carbon dioxide
- CBP
CREB binding protein
- CREB
camp response element binding protein
- DFO
Desferrioxamine
- DMOG
Dimethyl-oxalyl-glycine
- DTT
Dithiothreitol
- EDTA
ethylenediamine tetraacetic acid
- ERK
Extracellular signal-regulated kinase
- FADH
Flavin adenine dinucleotide
- Fe2+
Ferrous
- Fe3+
Ferric
- HIF
Hypoxia inducible factor
- HIF-1?
Hypoxia inducible factor-1 alpha
- HIF-1?
Hypoxia inducible factor-1 beta
- I-kB
Inhibitory kappaB
- IKK
Inhibitory kappaB kinase
- KID
Kinase inhibitory domain
- LDH
Lactate dehydrogenase
- MAPK
Mitogen-activated protein kinases
- MAPKAP K2
mitogen-activated protein kinase-activated protein kinase-2
- MnSOD
Manganese Superoxide Dismutase
- MSK
mitogen stimulated kinase
- NADH
Nicotinamide adenine dinucleotide
- NF-kB
nuclear factor kappa B
- NLS
Nuclear localization signal
- O2
Oxygen
- PHD
prolyl 4-hydroxylase domain
- pVHL
von Hippel-Lindau protein
- ROS
Reactive Oxygen Species
- RHD
Rel homology domain
- SAPK
stress-activated protein kinase
- SP1
specific protein1
- SP3
specific protein3
- SUMO-1
small ubiquitin-related modifier-1
- TCA
Tricarboxylic acid
- VEGF
Vascular endothelial growth factor
7. REFERENCES
- 1.Chopra RK, Ananthanarayanan VS. Conformational implications of enzymatic proline hydroxylation in collagen. Proc Natl Acad Sci U S A. 1982;79:7180–4. doi: 10.1073/pnas.79.23.7180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosenbloom J, Cywinski A. Inhibition of proline hydroxylation does not inhibit secretion of tropoelastin by chick aorta cells. FEBS Lett. 1976;65:246–50. doi: 10.1016/0014-5793(76)80490-5. [DOI] [PubMed] [Google Scholar]
- 3.Gill AC, Ritchie MA, Hunt LG, Steane SE, Davies KG, Bocking SP, Rhie AG, Bennett AD, Hope J. Post-translational hydroxylation at the N-terminus of the prion protein reveals presence of PPII structure in vivo. Embo J. 2000;19:5324–31. doi: 10.1093/emboj/19.20.5324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bruick RK, McKnight SL. A conserved family of prolyl-4-hydroxylases that modify HIF. Science. 2001;294:1337–40. doi: 10.1126/science.1066373. [DOI] [PubMed] [Google Scholar]
- 5.Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG., Jr. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292:464–8. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 6.Hewitson KS, McNeill LA, Riordan MV, Tian YM, Bullock AN, Welford RW, Elkins JM, Oldham NJ, Bhattacharya S, Gleadle JM, Ratcliffe PJ, Pugh CW, Schofield CJ. Hypoxia-inducible factor (HIF) asparagine hydroxylase is identical to factor inhibiting HIF (FIH) and is related to the cupin structural family. J Biol Chem. 2002;277:26351–5. doi: 10.1074/jbc.C200273200. [DOI] [PubMed] [Google Scholar]
- 7.Siddiq A, Aminova LR, Ratan RR. Hypoxia inducible factor prolyl 4-hydroxylase enzymes: center stage in the battle against hypoxia, metabolic compromise and oxidative stress. Neurochem Res. 2007;32:931–46. doi: 10.1007/s11064-006-9268-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kivirikko KI. Collagen biosynthesis: a mini-review cluster. Matrix Biol. 1998;16:355–6. doi: 10.1016/s0945-053x(98)90008-7. [DOI] [PubMed] [Google Scholar]
- 9.Kivirikko KI, Myllyharju J. Prolyl 4-hydroxylases and their protein disulfide isomerase subunit. Matrix Biol. 1998;16:357–68. doi: 10.1016/s0945-053x(98)90009-9. [DOI] [PubMed] [Google Scholar]
- 10.Kivirikko KI, Pihlajaniemi T. Collagen hydroxylases and the protein disulfide isomerase subunit of prolyl 4-hydroxylases. Adv Enzymol Relat Areas Mol Biol. 1998;72:325–98. doi: 10.1002/9780470123188.ch9. [DOI] [PubMed] [Google Scholar]
- 11.Kukkola L, Koivunen P, Pakkanen O, Page AP, Myllyharju J. Collagen prolyl 4-hydroxylase tetramers and dimers show identical decreases in Km values for peptide substrates with increasing chain length: mutation of one of the two catalytic sites in the tetramer inactivates the enzyme by more than half. J Biol Chem. 2004;279:18656–61. doi: 10.1074/jbc.M401514200. [DOI] [PubMed] [Google Scholar]
- 12.Tuderman L, Myllyla R, Kivirikko KI. Nutrition classics. European Journal of Biochemistry, Volume 80, 1977: Mechanism of the prolyl hydroxylase reaction. 1. Role of co-substrates. Nutr Rev. 1982;40:306–9. doi: 10.1111/j.1753-4887.1982.tb05237.x. [DOI] [PubMed] [Google Scholar]
- 13.Tuderman L, Prockop DJ. Procollagen N-proteinase. Properties of the enzyme purified from chick embryo tendons. Eur J Biochem. 1982;125:545–9. doi: 10.1111/j.1432-1033.1982.tb06716.x. [DOI] [PubMed] [Google Scholar]
- 14.Olsen BR, Berg RA, Kivirikko KT, Prockop DJ. Structure of protocollagen proline hydroxylase from chick embryos. Eur J Biochem. 1973;35:135–47. doi: 10.1111/j.1432-1033.1973.tb02819.x. [DOI] [PubMed] [Google Scholar]
- 15.Tuderman L. Developmental changes in prolyl hydroxylase activity and protein in chick embryo. Eur J Biochem. 1976;66:615–21. doi: 10.1111/j.1432-1033.1976.tb10589.x. [DOI] [PubMed] [Google Scholar]
- 16.Koivunen P, Hirsila M, Kivirikko KI, Myllyharju J. The length of peptide substrates has a marked effect on hydroxylation by the hypoxia-inducible factor prolyl 4-hydroxylases. J Biol Chem. 2006;281:28712–20. doi: 10.1074/jbc.M604628200. [DOI] [PubMed] [Google Scholar]
- 17.Kukkola L, Hieta R, Kivirikko KI, Myllyharju J. Identification and characterization of a third human, rat, and mouse collagen prolyl 4-hydroxylase isoenzyme. J Biol Chem. 2003;278:47685–93. doi: 10.1074/jbc.M306806200. [DOI] [PubMed] [Google Scholar]
- 18.Pekkala M, Hieta R, Kursula P, Kivirikko KI, Wierenga RK, Myllyharju J. Crystallization of the proline-rich-peptide binding domain of human type I collagen prolyl 4-hydroxylase. Acta Crystallogr D Biol Crystallogr. 2003;59:940–2. doi: 10.1107/s0907444903005420. [DOI] [PubMed] [Google Scholar]
- 19.Counts DF, Cardinale GJ, Udenfriend S. Prolyl hydroxylase half reaction: peptidyl prolyl-independent decarboxylation of alpha-ketoglutarate. Proc Natl Acad Sci U S A. 1978;75:2145–9. doi: 10.1073/pnas.75.5.2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hayaishi O. Enzymic studies on the mechanism of double hydroxylation. Pharmacol Rev. 1966;18:71–5. [PubMed] [Google Scholar]
- 21.Hayaishi O. Enzymic hydroxylation. Annu Rev Biochem. 1969;38:21–44. doi: 10.1146/annurev.bi.38.070169.000321. [DOI] [PubMed] [Google Scholar]
- 22.Prockop DJ, Juva K, Engel J. Enzymatic synthesis of hydroxyproline by the hydroxylation of poly (L-propyl-glycyl-L-prolyl) Hoppe Seylers Z Physiol Chem. 1967;348:553–60. doi: 10.1515/bchm2.1967.348.1.553. [DOI] [PubMed] [Google Scholar]
- 23.Tiainen P, Myllyharju J, Koivunen P. Characterization of a second Arabidopsis thaliana prolyl 4-hydroxylase with distinct substrate specificity. J Biol Chem. 2005;280:1142–8. doi: 10.1074/jbc.M411109200. [DOI] [PubMed] [Google Scholar]
- 24.Cleland LG, Betts WH, Vernon-Roberts B, Bielicki J. Role of iron and influence of antiinflammatory drugs on oxygen-derived free radical production and reactivity. J Rheumatol. 1982;9:885–92. [PubMed] [Google Scholar]
- 25.Tuderman L, Kuutti ER, Kivirikko KI. An affinity-column procedure using poly (L-proline) for the purification of prolyl hydroxylase. Purification of the enzyme from chick embryos. Eur J Biochem. 1975;52:9–16. doi: 10.1111/j.1432-1033.1975.tb03967.x. [DOI] [PubMed] [Google Scholar]
- 26.Tuderman L, Kuutti ER, Kivirikko KI. Radiommunoassay for human and chick prolyl hydroxylases. Eur J Biochem. 1975;60:399–405. doi: 10.1111/j.1432-1033.1975.tb21016.x. [DOI] [PubMed] [Google Scholar]
- 27.Hirsila M, Koivunen P, Xu L, Seeley T, Kivirikko KI, Myllyharju J. Effect of desferrioxamine and metals on the hydroxylases in the oxygen sensing pathway. Faseb J. 2005;19:1308–10. doi: 10.1096/fj.04-3399fje. [DOI] [PubMed] [Google Scholar]
- 28.Lu H, Dalgard CL, Mohyeldin A, McFate T, Tait AS, Verma A. Reversible inactivation of HIF-1 prolyl hydroxylases allows cell metabolism to control basal HIF-1. J Biol Chem. 2005;280:41928–39. doi: 10.1074/jbc.M508718200. [DOI] [PubMed] [Google Scholar]
- 29.Kivirikko KI, Ryhanen L, Anttinen H, Bornstein P, Prockop DJ. Further hydroxylation of lysyl residues in collagen by protocollagen lysyl hydroxylase in vitro. Biochemistry. 1973;12:4966–71. doi: 10.1021/bi00748a023. [DOI] [PubMed] [Google Scholar]
- 30.Tuderman L, Myllyla R, Kivirikko KI. Mechanism of the prolyl hydroxylase reaction. 1. Role of co-substrates. Eur J Biochem. 1977;80:341–8. doi: 10.1111/j.1432-1033.1977.tb11888.x. [DOI] [PubMed] [Google Scholar]
- 31.de Jong L, Albracht SP, Kemp A. Prolyl 4-hydroxylase activity in relation to the oxidation state of enzyme-bound iron. The role of ascorbate in peptidyl proline hydroxylation. Biochim Biophys Acta. 1982;704:326–32. doi: 10.1016/0167-4838(82)90162-5. [DOI] [PubMed] [Google Scholar]
- 32.Hirsila M, Koivunen P, Gunzler V, Kivirikko KI, Myllyharju J. Characterization of the human prolyl 4-hydroxylases that modify the hypoxia-inducible factor. J Biol Chem. 2003;278:30772–80. doi: 10.1074/jbc.M304982200. [DOI] [PubMed] [Google Scholar]
- 33.Myllyharju J, Kivirikko KI. Characterization of the iron- and 2-oxoglutarate-binding sites of human prolyl 4-hydroxylase. Embo J. 1997;16:1173–80. doi: 10.1093/emboj/16.6.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Myllyla R, Kuutti-Savolainen ER, Kivirikko KI. The role of ascorbate in the prolyl hydroxylase reaction. Biochem Biophys Res Commun. 1978;83:441–8. doi: 10.1016/0006-291x(78)91010-0. [DOI] [PubMed] [Google Scholar]
- 35.Siddiq A, Ayoub IA, Chavez JC, Aminova L, Shah S, LaManna JC, Patton SM, Connor JR, Cherny RA, Volitakis I, Bush AI, Langsetmo I, Seeley T, Gunzler V, Ratan RR. Hypoxia-inducible factor prolyl 4-hydroxylase inhibition. A target for neuroprotection in the central nervous system. J Biol Chem. 2005;280:41732–43. doi: 10.1074/jbc.M504963200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hanauske-Abel HM, Gunzler V. A stereochemical concept for the catalytic mechanism of prolylhydroxylase: applicability to classification and design of inhibitors. J Theor Biol. 1982;94:421–55. doi: 10.1016/0022-5193(82)90320-4. [DOI] [PubMed] [Google Scholar]
- 37.Roach PL, Clifton IJ, Fulop V, Harlos K, Barton GJ, Hajdu J, Andersson I, Schofield CJ, Baldwin JE. Crystal structure of isopenicillin N synthase is the first from a new structural family of enzymes. Nature. 1995;375:700–4. doi: 10.1038/375700a0. [DOI] [PubMed] [Google Scholar]
- 38.Roach PL, Schofield CJ, Baldwin JE, Clifton IJ, Hajdu J. Crystallization and preliminary X-ray diffraction studies on recombinant isopenicillin N synthase from Aspergillus nidulans. Protein Sci. 1995;4:1007–9. doi: 10.1002/pro.5560040521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Min SK, Kim SY, Kim CH, Woo JS, Jung JS, Kim YK. Role of lipid peroxidation and poly (ADP-ribose) polymerase activation in oxidant-induced membrane transport dysfunction in opossum kidney cells. Toxicol Appl Pharmacol. 2000;166:196–202. doi: 10.1006/taap.2000.8956. [DOI] [PubMed] [Google Scholar]
- 40.Halme J, Jaaskelainen M. Protocollagen proline hydroxylase of the mouse uterus during pregnancy and post-partum involution. Biochem J. 1970;116:367–9. doi: 10.1042/bj1160367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Halme J, Kivirikko KI, Simons K. Isolation and partial characterization of highly purified protocollagen proline hydroxylase. Biochim Biophys Acta. 1970;198:460–70. doi: 10.1016/0005-2744(70)90124-5. [DOI] [PubMed] [Google Scholar]
- 42.Schofield CJ, Zhang Z. Structural and mechanistic studies on 2-oxoglutarate-dependent oxygenases and related enzymes. Curr Opin Struct Biol. 1999;9:722–31. doi: 10.1016/s0959-440x(99)00036-6. [DOI] [PubMed] [Google Scholar]
- 43.Prescott AG, Lloyd MD. The iron (II) and 2-oxoacid-dependent dioxygenases and their role in metabolism. Nat Prod Rep. 2000;17:367–83. doi: 10.1039/a902197c. [DOI] [PubMed] [Google Scholar]
- 44.Wu M, Moon HS, Pirskanen A, Myllyharju J, Kivirikko KI, Begley TP. Mechanistic studies on prolyl-4-hydroxylase: the vitamin C requiring uncoupled oxidation. Bioorg Med Chem Lett. 2000;10:1511–4. doi: 10.1016/s0960-894x(00)00224-9. [DOI] [PubMed] [Google Scholar]
- 45.Costas M, Mehn MP, Jensen MP, Que L., Jr. Dioxygen activation at mononuclear nonheme iron active sites: enzymes, models, and intermediates. Chem Rev. 2004;104:939–86. doi: 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]
- 46.Hausinger RP. FeII/alpha-ketoglutarate-dependent hydroxylases and related enzymes. Crit Rev Biochem Mol Biol. 2004;39:21–68. doi: 10.1080/10409230490440541. [DOI] [PubMed] [Google Scholar]
- 47.Nietfeld JJ, de Jong L, Kemp A. The influence of 2-oxoglutarate on the activity of prolyl 4-hydroxylase. Biochim Biophys Acta. 1982;704:321–5. doi: 10.1016/0167-4838(82)90161-3. [DOI] [PubMed] [Google Scholar]
- 48.Li L, Narducci Sarjeant AA, Vance MA, Zakharov LN, Rheingold AL, Solomon EI, Karlin KD. Exogenous nitrile substrate hydroxylation by a new dicopper-hydroperoxide complex. J Am Chem Soc. 2005;127:15360–1. doi: 10.1021/ja054948a. [DOI] [PubMed] [Google Scholar]
- 49.Li QS, Ogawa J, Schmid RD, Shimizu S. Indole hydroxylation by bacterial cytochrome P450 BM-3 and modulation of activity by cumene hydroperoxide. Biosci Biotechnol Biochem. 2005;69:293–300. doi: 10.1271/bbb.69.293. [DOI] [PubMed] [Google Scholar]
- 50.Li SW, Lin TS, Minteer S, Burke WJ. 3,4-Dihydroxyphenylacetaldehyde and hydrogen peroxide generate a hydroxyl radical: possible role in Parkinson's disease pathogenesis. Brain Res Mol Brain Res. 2001;93:1–7. doi: 10.1016/s0169-328x(01)00120-6. [DOI] [PubMed] [Google Scholar]
- 51.Li QS, Ogawa J, Shimizu S. Critical role of the residue size at position 87 in H2O2- dependent substrate hydroxylation activity and H2O2 inactivation of cytochrome P450BM-3. Biochem Biophys Res Commun. 2001;280:1258–61. doi: 10.1006/bbrc.2001.4261. [DOI] [PubMed] [Google Scholar]
- 52.Pan Z, Zhang R, Newcomb M. Kinetic studies of reactions of iron (IV)-oxo porphyrin radical cations with organic reductants. J Inorg Biochem. 2006;100:524–32. doi: 10.1016/j.jinorgbio.2005.12.022. [DOI] [PubMed] [Google Scholar]
- 53.Shepherd RE, Isaacson Y, Chensny L, Zhang S, Kortes R, John K. Lactobionic and gluconic acid complexes of FeII and FeIII; control of oxidation pathways by an organ transplantation preservant. J Inorg Biochem. 1993;49:23–48. doi: 10.1016/0162-0134(93)80046-c. [DOI] [PubMed] [Google Scholar]
- 54.Varotsis C, Zhang Y, Appelman EH, Babcock GT. Resolution of the reaction sequence during the reduction of O2 by cytochrome oxidase. Proc Natl Acad Sci U S A. 1993;90:237–41. doi: 10.1073/pnas.90.1.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang Y, Oldfield E. Cytochrome P450: an investigation of the Mossbauer spectra of a reaction intermediate and an Fe (IV)[double bond]O model system. J Am Chem Soc. 2004;126:4470–1. doi: 10.1021/ja030664j. [DOI] [PubMed] [Google Scholar]
- 56.Zhang Z, Ren J, Stammers DK, Baldwin JE, Harlos K, Schofield CJ. Structural origins of the selectivity of the trifunctional oxygenase clavaminic acid synthase. Nat Struct Biol. 2000;7:127–33. doi: 10.1038/72398. [DOI] [PubMed] [Google Scholar]
- 57.Burzlaff NI, Rutledge PJ, Clifton IJ, Hensgens CM, Pickford M, Adlington RM, Roach PL, Baldwin JE. The reaction cycle of isopenicillin N synthase observed by X-ray diffraction. Nature. 1999;401:721–4. doi: 10.1038/44400. [DOI] [PubMed] [Google Scholar]
- 58.Hanauske-Abel HM. Prolyl 4-hydroxylase, a target enzyme for drug development. Design of suppressive agents and the in vitro effects of inhibitors and proinhibitors. J Hepatol. 1991;13(Suppl 3):S8–15. doi: 10.1016/0168-8278(91)90003-t. discussion S16. [DOI] [PubMed] [Google Scholar]
- 59.Hamilton GA, Giacin JR, Hellman TM, Snook ME, Weller JW. Oxenoid models for enzymic hydroxylations. Ann N Y Acad Sci. 1973;212:4–12. doi: 10.1111/j.1749-6632.1973.tb47582.x. [DOI] [PubMed] [Google Scholar]
- 60.Hobza P, Hurych J, Zahradnik R. Quantum chemical study of the mechanism of collagen proline hydroxylation. Biochim Biophys Acta. 1973;304:466–72. doi: 10.1016/0304-4165(73)90266-3. [DOI] [PubMed] [Google Scholar]
- 61.Juva K. Hydroxylation of proline in the biosynthesis of collagen. An experimental study with chick embryo and granulation tissue of rat. Acta Physiol Scand Suppl. 1968;308:1–73. [PubMed] [Google Scholar]
- 62.Liu MS, Siess M, Hoffmann PC. The effect of changes in functional activity on ubiquinone redox status in isolated atria. Eur J Biochem. 1973;37:259–69. doi: 10.1111/j.1432-1033.1973.tb02984.x. [DOI] [PubMed] [Google Scholar]
- 63.Chavez JC, LaManna JC. Hypoxia-inducible factor-1alpha accumulation in the rat brain in response to hypoxia and ischemia is attenuated during aging. Adv Exp Med Biol. 2003;510:337–41. doi: 10.1007/978-1-4615-0205-0_55. [DOI] [PubMed] [Google Scholar]
- 64.Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O'Rourke J, Mole DR, Mukherji M, Metzen E, Wilson MI, Dhanda A, Tian YM, Masson N, Hamilton DL, Jaakkola P, Barstead R, Hodgkin J, Maxwell PH, Pugh CW, Schofield CJ, Ratcliffe PJ. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;107:43–54. doi: 10.1016/s0092-8674(01)00507-4. [DOI] [PubMed] [Google Scholar]
- 65.Stroka DM, Burkhardt T, Desbaillets I, Wenger RH, Neil DA, Bauer C, Gassmann M, Candinas D. HIF-1 is expressed in normoxic tissue and displays an organ-specific regulation under systemic hypoxia. Faseb J. 2001;15:2445–53. doi: 10.1096/fj.01-0125com. [DOI] [PubMed] [Google Scholar]
- 66.Wiesener MS, Maxwell PH. HIF and oxygen sensing; as important to life as the air we breathe? Ann Med. 2003;35:183–90. doi: 10.1080/0785389031000458233. [DOI] [PubMed] [Google Scholar]
- 67.Leonard MO, Cottell DC, Godson C, Brady HR, Taylor CT. The role of HIF-1 alpha in transcriptional regulation of the proximal tubular epithelial cell response to hypoxia. J Biol Chem. 2003;278:40296–304. doi: 10.1074/jbc.M302560200. [DOI] [PubMed] [Google Scholar]
- 68.Manalo DJ, Rowan A, Lavoie T, Natarajan L, Kelly BD, Ye SQ, Garcia JG, Semenza GL. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood. 2005;105:659–69. doi: 10.1182/blood-2004-07-2958. [DOI] [PubMed] [Google Scholar]
- 69.Josko J, Mazurek M. Transcription factors having impact on vascular endothelial growth factor (VEGF) gene expression in angiogenesis. Med Sci Monit. 2004;10:RA89–98. [PubMed] [Google Scholar]
- 70.Marti HH. Erythropoietin and the hypoxic brain. J Exp Biol. 2004;207:3233–42. doi: 10.1242/jeb.01049. [DOI] [PubMed] [Google Scholar]
- 71.Nagy Z, Simon L, Bori Z. Regulatory mechanisms in focal cerebral ischemia. New possibilities in neuroprotective therapy. Ideggyogy Sz. 2002;55:73–85. [PubMed] [Google Scholar]
- 72.Scortegagna M, Ding K, Zhang Q, Oktay Y, Bennett MJ, Bennett M, Shelton JM, Richardson JA, Moe O, Garcia JA. HIF-2alpha regulates murine hematopoietic development in an erythropoietin-dependent manner. Blood. 2005;105:3133–40. doi: 10.1182/blood-2004-05-1695. [DOI] [PubMed] [Google Scholar]
- 73.Zelko IN, Folz RJ. Extracellular superoxide dismutase functions as a major repressor of hypoxia-induced erythropoietin gene expression. Endocrinology. 2005;146:332–40. doi: 10.1210/en.2004-1007. [DOI] [PubMed] [Google Scholar]
- 74.Dawson LA, Djali S, Gonzales C, Vinegra MA, Zaleska MM. Characterization of transient focal ischemia-induced increases in extracellular glutamate and aspartate in spontaneously hypertensive rats. Brain Res Bull. 2000;53:767–76. doi: 10.1016/s0361-9230(00)00363-4. [DOI] [PubMed] [Google Scholar]
- 75.Liu J, Narasimhan P, Yu F, Chan PH. Neuroprotection by hypoxic preconditioning involves oxidative stress-mediated expression of hypoxia-inducible factor and erythropoietin. Stroke. 2005;36:1264–9. doi: 10.1161/01.STR.0000166180.91042.02. [DOI] [PubMed] [Google Scholar]
- 76.Mackensen GB, Patel M, Sheng H, Calvi CL, Batinic-Haberle I, Day BJ, Liang LP, Fridovich I, Crapo JD, Pearlstein RD, Warner DS. Neuroprotection from delayed postischemic administration of a metalloporphyrin catalytic antioxidant. J Neurosci. 2001;21:4582–92. doi: 10.1523/JNEUROSCI.21-13-04582.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Baranova O, Miranda LF, Pichiule P, Dragatsis I, Johnson RS, Chavez JC. Neuron-specific inactivation of the hypoxia inducible factor 1 alpha increases brain injury in a mouse model of transient focal cerebral ischemia. J Neurosci. 2007;27:6320–32. doi: 10.1523/JNEUROSCI.0449-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bergeron M, Gidday JM, Yu AY, Semenza GL, Ferriero DM, Sharp FR. Role of hypoxia-inducible factor-1 in hypoxia-induced ischemic tolerance in neonatal rat brain. Ann Neurol. 2000;48:285–96. [PubMed] [Google Scholar]
- 79.Mu D, Jiang X, Sheldon RA, Fox CK, Hamrick SE, Vexler ZS, Ferriero DM. Regulation of hypoxia-inducible factor 1alpha and induction of vascular endothelial growth factor in a rat neonatal stroke model. Neurobiol Dis. 2003;14:524–34. doi: 10.1016/j.nbd.2003.08.020. [DOI] [PubMed] [Google Scholar]
- 80.Sharp FR, Bergeron M, Bernaudin M. Hypoxia-inducible factor in brain. Adv Exp Med Biol. 2001;502:273–91. doi: 10.1007/978-1-4757-3401-0_18. [DOI] [PubMed] [Google Scholar]
- 81.Ralph GS, Parham S, Lee SR, Beard GL, Craigon MH, Ward N, White JR, Barber RD, Rayner W, Kingsman SM, Mundy CR, Mazarakis ND, Krige D. Identification of potential stroke targets by lentiviral vector mediated overexpression of HIF-1 alpha and HIF-2 alpha in a primary neuronal model of hypoxia. J Cereb Blood Flow Metab. 2004;24:245–58. doi: 10.1097/01.WCB.0000110532.48786.46. [DOI] [PubMed] [Google Scholar]
- 82.Ratan RR, Siddiq A, Aminova L, Lange PS, Langley B, Ayoub I, Gensert J, Chavez J. Translation of ischemic preconditioning to the patient: prolyl hydroxylase inhibition and hypoxia inducible factor-1 as novel targets for stroke therapy. Stroke. 2004;35:2687–9. doi: 10.1161/01.STR.0000143216.85349.9e. [DOI] [PubMed] [Google Scholar]
- 83.Chrivia JC, Kwok RP, Lamb N, Hagiwara M, Montminy MR, Goodman RH. Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature. 1993;365:855–9. doi: 10.1038/365855a0. [DOI] [PubMed] [Google Scholar]
- 84.Kwok RP, Lundblad JR, Chrivia JC, Richards JP, Bachinger HP, Brennan RG, Roberts SG, Green MR, Goodman RH. Nuclear protein CBP is a coactivator for the transcription factor CREB. Nature. 1994;370:223–6. doi: 10.1038/370223a0. [DOI] [PubMed] [Google Scholar]
- 85.Beitner-Johnson D, Millhorn DE. Hypoxia induces phosphorylation of the cyclic AMP response element-binding protein by a novel signaling mechanism. J Biol Chem. 1998;273:19834–9. doi: 10.1074/jbc.273.31.19834. [DOI] [PubMed] [Google Scholar]
- 86.Taylor CT, Furuta GT, Synnestvedt K, Colgan SP. Phosphorylation-dependent targeting of cAMP response element binding protein to the ubiquitin/proteasome pathway in hypoxia. Proc Natl Acad Sci U S A. 2000;97:12091–6. doi: 10.1073/pnas.220211797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Comerford KM, Leonard MO, Karhausen J, Carey R, Colgan SP, Taylor CT. Small ubiquitin-related modifier-1 modification mediates resolution of CREB-dependent responses to hypoxia. Proc Natl Acad Sci U S A. 2003;100:986–91. doi: 10.1073/pnas.0337412100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zaman K, Ryu H, Hall D, O'Donovan K, Lin KI, Miller MP, Marquis JC, Baraban JM, Semenza GL, Ratan RR. Protection from oxidative stress-induced apoptosis in cortical neuronal cultures by iron chelators is associated with enhanced DNA binding of hypoxia-inducible factor-1 and ATF-1/CREB and increased expression of glycolytic enzymes, p21 (waf1/cip1), and erythropoietin. J Neurosci. 1999;19:9821–30. doi: 10.1523/JNEUROSCI.19-22-09821.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Deak M, Clifton AD, Lucocq LM, Alessi DR. Mitogen- and stress-activated protein kinase-1 (MSK1) is directly activated by MAPK and SAPK2/p38, and may mediate activation of CREB. Embo J. 1998;17:4426–41. doi: 10.1093/emboj/17.15.4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tan Y, Rouse J, Zhang A, Cariati S, Cohen P, Comb MJ. FGF and stress regulate CREB and ATF-1 via a pathway involving p38 MAP kinase and MAPKAP kinase-2. Embo J. 1996;15:4629–42. [PMC free article] [PubMed] [Google Scholar]
- 91.Wiggin GR, Soloaga A, Foster JM, Murray-Tait V, Cohen P, Arthur JS. MSK1 and MSK2 are required for the mitogen- and stress-induced phosphorylation of CREB and ATF1 in fibroblasts. Mol Cell Biol. 2002;22:2871–81. doi: 10.1128/MCB.22.8.2871-2881.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Widmann C, Gibson S, Jarpe MB, Johnson GL. Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human. Physiol Rev. 1999;79:143–80. doi: 10.1152/physrev.1999.79.1.143. [DOI] [PubMed] [Google Scholar]
- 93.Bu X, Huang P, Qi Z, Zhang N, Han S, Fang L, Li J. Cell type-specific activation of p38 MAPK in the brain regions of hypoxic preconditioned mice. Neurochem Int. 2007 doi: 10.1016/j.neuint.2007.04.028. [DOI] [PubMed] [Google Scholar]
- 94.Gao Y, Gao G, Long C, Han S, Zu P, Fang L, Li J. Enhanced phosphorylation of cyclic AMP response element binding protein in the brain of mice following repetitive hypoxic exposure. Biochem Biophys Res Commun. 2006;340:661–7. doi: 10.1016/j.bbrc.2005.12.064. [DOI] [PubMed] [Google Scholar]
- 95.Long AA, Chapman NR, Innes B, Europe-Finner GN, Robson SC. Expression and interaction of the transcriptional coregulators, CBP/p300, in the human myometrium during pregnancy and labor. J Soc Gynecol Investig. 2005;12:92–7. doi: 10.1016/j.jsgi.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 96.Whitmarsh AJ, Davis RJ. Transcription factor AP-1 regulation by mitogen-activated protein kinase signal transduction pathways. J Mol Med. 1996;74:589–607. doi: 10.1007/s001090050063. [DOI] [PubMed] [Google Scholar]
- 97.Cummins EP, Berra E, Comerford KM, Ginouves A, Fitzgerald KT, Seeballuck F, Godson C, Nielsen JE, Moynagh P, Pouyssegur J, Taylor CT. Prolyl hydroxylase-1 negatively regulates IkappaB kinase-beta, giving insight into hypoxia-induced NFkappaB activity. Proc Natl Acad Sci U S A. 2006;103:18154–9. doi: 10.1073/pnas.0602235103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Singh H, Sen R, Baltimore D, Sharp PA. A nuclear factor that binds to a conserved sequence motif in transcriptional control elements of immunoglobulin genes. Nature. 1986;319:154–8. doi: 10.1038/319154a0. [DOI] [PubMed] [Google Scholar]
- 99.O'Neill LA, Kaltschmidt C. NF-kappa B: a crucial transcription factor for glial and neuronal cell function. Trends Neurosci. 1997;20:252–8. doi: 10.1016/s0166-2236(96)01035-1. [DOI] [PubMed] [Google Scholar]
- 100.Kaltschmidt B, Baeuerle PA, Kaltschmidt C. Potential involvement of the transcription factor NF-kappa B in neurological disorders. Mol Aspects Med. 1993;14:171–90. doi: 10.1016/0098-2997(93)90004-w. [DOI] [PubMed] [Google Scholar]
- 101.Kaltschmidt C, Kaltschmidt B, Baeuerle PA. Brain synapses contain inducible forms of the transcription factor NF-kappa B. Mech Dev. 1993;43:135–47. doi: 10.1016/0925-4773(93)90031-r. [DOI] [PubMed] [Google Scholar]
- 102.Kaltschmidt C, Kaltschmidt B, Lannes-Vieira J, Kreutzberg GW, Wekerle H, Baeuerle PA, Gehrmann J. Transcription factor NF-kappa B is activated in microglia during experimental autoimmune encephalomyelitis. J Neuroimmunol. 1994;55:99–106. doi: 10.1016/0165-5728(94)90151-1. [DOI] [PubMed] [Google Scholar]
- 103.Kaltschmidt C, Kaltschmidt B, Neumann H, Wekerle H, Baeuerle PA. Constitutive NF-kappa B activity in neurons. Mol Cell Biol. 1994;14:3981–92. doi: 10.1128/mcb.14.6.3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Fridmacher V, Kaltschmidt B, Goudeau B, Ndiaye D, Rossi FM, Pfeiffer J, Kaltschmidt C, Israel A, Memet S. Forebrain-specific neuronal inhibition of nuclear factor-kappaB activity leads to loss of neuroprotection. J Neurosci. 2003;23:9403–8. doi: 10.1523/JNEUROSCI.23-28-09403.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Collart MA, Baeuerle P, Vassalli P. Regulation of tumor necrosis factor alpha transcription in macrophages: involvement of four kappa B-like motifs and of constitutive and inducible forms of NF-kappa B. Mol Cell Biol. 1990;10:1498–506. doi: 10.1128/mcb.10.4.1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sarmiere PD, Freeman RS. Analysis of the NF-kappa B and PI 3-kinase/Akt survival pathways in nerve growth factor-dependent neurons. Mol Cell Neurosci. 2001;18:320–31. doi: 10.1006/mcne.2001.1021. [DOI] [PubMed] [Google Scholar]
- 107.Middleton G, Hamanoue M, Enokido Y, Wyatt S, Pennica D, Jaffray E, Hay RT, Davies AM. Cytokine-induced nuclear factor kappa B activation promotes the survival of developing neurons. J Cell Biol. 2000;148:325–32. doi: 10.1083/jcb.148.2.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Taglialatela G, Robinson R, Perez-Polo JR. Inhibition of nuclear factor kappa B (NFkappaB) activity induces nerve growth factor-resistant apoptosis in PC12 cells. J Neurosci Res. 1997;47:155–62. [PubMed] [Google Scholar]
- 109.Briggs MR, Kadonaga JT, Bell SP, Tjian R. Purification and biochemical characterization of the promoter-specific transcription factor, Sp1. Science. 1986;234:47–52. doi: 10.1126/science.3529394. [DOI] [PubMed] [Google Scholar]
- 110.Lania L, Majello B, De Luca P. Transcriptional regulation by the Sp family proteins. Int J Biochem Cell Biol. 1997;29:1313–23. doi: 10.1016/s1357-2725(97)00094-0. [DOI] [PubMed] [Google Scholar]
- 111.Suske G. The Sp-family of transcription factors. Gene. 1999;238:291–300. doi: 10.1016/s0378-1119(99)00357-1. [DOI] [PubMed] [Google Scholar]
- 112.Yang CR, Wilson-Van Patten C, Planchon SM, Wuerzberger-Davis SM, Davis TW, Cuthill S, Miyamoto S, Boothman DA. Coordinate modulation of Sp1, NF-kappa B, and p53 in confluent human malignant melanoma cells after ionizing radiation. Faseb J. 2000;14:379–90. doi: 10.1096/fasebj.14.2.379. [DOI] [PubMed] [Google Scholar]
- 113.Dunah AW, Jeong H, Griffin A, Kim YM, Standaert DG, Hersch SM, Mouradian MM, Young AB, Tanese N, Krainc D. Sp1 and TAFII130 transcriptional activity disrupted in early Huntington's disease. Science. 2002;296:2238–43. doi: 10.1126/science.1072613. [DOI] [PubMed] [Google Scholar]
- 114.Li SH, Cheng AL, Zhou H, Lam S, Rao M, Li H, Li XJ. Interaction of Huntington disease protein with transcriptional activator Sp1. Mol Cell Biol. 2002;22:1277–87. doi: 10.1128/mcb.22.5.1277-1287.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Li F, Altieri DC. Transcriptional analysis of human survivin gene expression. Biochem J. 1999;344(Pt 2):305–11. doi: 10.1042/0264-6021:3440305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tanaka T, Kurabayashi M, Aihara Y, Ohyama Y, Nagai R. Inducible expression of manganese superoxide dismutase by phorbol 12-myristate 13-acetate is mediated by Sp1 in endothelial cells. Arterioscler Thromb Vasc Biol. 2000;20:392–401. doi: 10.1161/01.atv.20.2.392. [DOI] [PubMed] [Google Scholar]
- 117.Chou CF, Peng HW, Wang CY, Yang YT, Han SH. An Sp1 binding site involves the transcription of the Fas ligand gene induced by PMA and ionomycin in Jurkat cells. J Biomed Sci. 2000;7:136–43. doi: 10.1007/BF02256620. [DOI] [PubMed] [Google Scholar]
- 118.Xiao S, Matsui K, Fine A, Zhu B, Marshak-Rothstein A, Widom RL, Ju ST. FasL promoter activation by IL-2 through SP1 and NFAT but not Egr-2 and Egr-3. Eur J Immunol. 1999;29:3456–65. doi: 10.1002/(SICI)1521-4141(199911)29:11<3456::AID-IMMU3456>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 119.Liu YW, Arakawa T, Yamamoto S, Chang WC. Transcriptional activation of human 12-lipoxygenase gene promoter is mediated through Sp1 consensus sites in A431 cells. Biochem J. 1997;324(Pt 1):133–40. doi: 10.1042/bj3240133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lin KI, DiDonato JA, Hoffmann A, Hardwick JM, Ratan RR. Suppression of steady-state, but not stimulus-induced NF-kappaB activity inhibits alphavirus-induced apoptosis. J Cell Biol. 1998;141:1479–87. doi: 10.1083/jcb.141.7.1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bouwman P, Philipsen S. Regulation of the activity of Sp1-related transcription factors. Mol Cell Endocrinol. 2002;195:27–38. doi: 10.1016/s0303-7207(02)00221-6. [DOI] [PubMed] [Google Scholar]
- 122.Ryu H, Lee J, Zaman K, Kubilis J, Ferrante RJ, Ross BD, Neve R, Ratan RR. Sp1 and Sp3 are oxidative stress-inducible, antideath transcription factors in cortical neurons. J Neurosci. 2003;23:3597–606. doi: 10.1523/JNEUROSCI.23-09-03597.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lee TH, Chang HC, Chuang LY, Hung WC. Involvement of PKA and Sp1 in the induction of p27 (Kip1) by tamoxifen. Biochem Pharmacol. 2003;66:371–7. doi: 10.1016/s0006-2952(03)00258-2. [DOI] [PubMed] [Google Scholar]
- 124.Sanchez-Elsner T, Ramirez JR, Sanz-Rodriguez F, Varela E, Bernabeu C, Botella LM. A cross-talk between hypoxia and TGF-beta orchestrates erythropoietin gene regulation through SP1 and Smads. J Mol Biol. 2004;336:9–24. doi: 10.1016/j.jmb.2003.12.023. [DOI] [PubMed] [Google Scholar]
- 125.Xu Q, Ji YS, Schmedtje JF., Jr. Sp1 increases expression of cyclooxygenase-2 in hypoxic vascular endothelium. Implications for the mechanisms of aortic aneurysm and heart failure. J Biol Chem. 2000;275:24583–9. doi: 10.1074/jbc.M003894200. [DOI] [PubMed] [Google Scholar]
- 126.Chatterjee S, Zaman K, Ryu H, Conforto A, Ratan RR. Sequence-selective DNA binding drugs mithramycin A and chromomycin A3 are potent inhibitors of neuronal apoptosis induced by oxidative stress and DNA damage in cortical neurons. Ann Neurol. 2001;49:345–54. [PubMed] [Google Scholar]
- 127.Ratan RR, Murphy TH, Baraban JM. Oxidative stress induces apoptosis in embryonic cortical neurons. J Neurochem. 1994;62:376–9. doi: 10.1046/j.1471-4159.1994.62010376.x. [DOI] [PubMed] [Google Scholar]
- 128.Ratan RR, Murphy TH, Baraban JM. Macromolecular synthesis inhibitors prevent oxidative stress-induced apoptosis in embryonic cortical neurons by shunting cysteine from protein synthesis to glutathione. J Neurosci. 1994;14:4385–92. doi: 10.1523/JNEUROSCI.14-07-04385.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Berra E, Pages G, Pouyssegur J. MAP kinases and hypoxia in the control of VEGF expression. Cancer Metastasis Rev. 2000;19:139–45. doi: 10.1023/a:1026506011458. [DOI] [PubMed] [Google Scholar]
- 130.Milanini J, Vinals F, Pouyssegur J, Pages G. p42/p44 MAP kinase module plays a key role in the transcriptional regulation of the vascular endothelial growth factor gene in fibroblasts. J Biol Chem. 1998;273:18165–72. doi: 10.1074/jbc.273.29.18165. [DOI] [PubMed] [Google Scholar]
- 131.Hoffmann A, Gloe T, Pohl U. Hypoxia-induced upregulation of eNOS gene expression is redox-sensitive: a comparison between hypoxia and inhibitors of cell metabolism. J Cell Physiol. 2001;188:33–44. doi: 10.1002/jcp.1092. [DOI] [PubMed] [Google Scholar]
- 132.Ameyar M, Wisniewska M, Weitzman JB. A role for AP-1 in apoptosis: the case for and against. Biochimie. 2003;85:747–52. doi: 10.1016/j.biochi.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 133.Eferl R, Wagner EF. AP-1: a double-edged sword in tumorigenesis. Nat Rev Cancer. 2003;3:859–68. doi: 10.1038/nrc1209. [DOI] [PubMed] [Google Scholar]
- 134.Ema M, Hirota K, Mimura J, Abe H, Yodoi J, Sogawa K, Poellinger L, Fujii-Kuriyama Y. Molecular mechanisms of transcription activation by HLF and HIF1alpha in response to hypoxia: their stabilization and redox signal-induced interaction with CBP/p300. Embo J. 1999;18:1905–14. doi: 10.1093/emboj/18.7.1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ema M, Taya S, Yokotani N, Sogawa K, Matsuda Y, Fujii-Kuriyama Y. A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1alpha regulates the VEGF expression and is potentially involved in lung and vascular development. Proc Natl Acad Sci U S A. 1997;94:4273–8. doi: 10.1073/pnas.94.9.4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Jochum W, Passegue E, Wagner EF. AP-1 in mouse development and tumorigenesis. Oncogene. 2001;20:2401–12. doi: 10.1038/sj.onc.1204389. [DOI] [PubMed] [Google Scholar]
- 137.Mechta-Grigoriou F, Gerald D, Yaniv M. The mammalian Jun proteins: redundancy and specificity. Oncogene. 2001;20:2378–89. doi: 10.1038/sj.onc.1204381. [DOI] [PubMed] [Google Scholar]
- 138.Shaulian E, Karin M. AP-1 in cell proliferation and survival. Oncogene. 2001;20:2390–400. doi: 10.1038/sj.onc.1204383. [DOI] [PubMed] [Google Scholar]
- 139.Millhorn DE, Raymond R, Conforti L, Zhu W, Beitner-Johnson D, Filisko T, Genter MB, Kobayashi S, Peng M. Regulation of gene expression for tyrosine hydroxylase in oxygen sensitive cells by hypoxia. Kidney Int. 1997;51:527–35. doi: 10.1038/ki.1997.73. [DOI] [PubMed] [Google Scholar]
- 140.Salnikow K, Kluz T, Costa M, Piquemal D, Demidenko ZN, Xie K, Blagosklonny MV. The regulation of hypoxic genes by calcium involves c-Jun/AP-1, which cooperates with hypoxia-inducible factor 1 in response to hypoxia. Mol Cell Biol. 2002;22:1734–41. doi: 10.1128/MCB.22.6.1734-1741.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Semenza GL. Hydroxylation of HIF-1: oxygen sensing at the molecular level. Physiology (Bethesda) 2004;19:176–82. doi: 10.1152/physiol.00001.2004. [DOI] [PubMed] [Google Scholar]
- 142.Yamashita K, Discher DJ, Hu J, Bishopric NH, Webster KA. Molecular regulation of the endothelin-1 gene by hypoxia. Contributions of hypoxia-inducible factor-1, activator protein-1, GATA-2, AND p300/CBP. J Biol Chem. 2001;276:12645–53. doi: 10.1074/jbc.M011344200. [DOI] [PubMed] [Google Scholar]
- 143.Meyer T, Starr DB, Carlstedt-Duke J. The rat glucocorticoid receptor mutant K461A differentiates between two different mechanisms of transrepression. J Biol Chem. 1997;272:21090–5. doi: 10.1074/jbc.272.34.21090. [DOI] [PubMed] [Google Scholar]
- 144.Fantozzi I, Zhang S, Platoshyn O, Remillard CV, Cowling RT, Yuan JX. Hypoxia increases AP-1 binding activity by enhancing capacitative Ca2+ entry in human pulmonary artery endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2003;285:L1233–45. doi: 10.1152/ajplung.00445.2002. [DOI] [PubMed] [Google Scholar]
- 145.Premkumar DR, Adhikary G, Overholt JL, Simonson MS, Cherniack NS, Prabhakar NR. Intracellular pathways linking hypoxia to activation of c-fos and AP-1. Adv Exp Med Biol. 2000;475:101–9. doi: 10.1007/0-306-46825-5_10. [DOI] [PubMed] [Google Scholar]
- 146.Minet E, Michel G, Mottet D, Piret JP, Barbieux A, Raes M, Michiels C. c-JUN gene induction and AP-1 activity is regulated by a JNK-dependent pathway in hypoxic HepG2 cells. Exp Cell Res. 2001;265:114–24. doi: 10.1006/excr.2001.5180. [DOI] [PubMed] [Google Scholar]
- 147.Gerald D, Berra E, Frapart YM, Chan DA, Giaccia AJ, Mansuy D, Pouyssegur J, Yaniv M, Mechta-Grigoriou F. JunD reduces tumor angiogenesis by protecting cells from oxidative stress. Cell. 2004;118:781–94. doi: 10.1016/j.cell.2004.08.025. [DOI] [PubMed] [Google Scholar]