

Abstract
Inflammatory bowel disease is a chronic inflammatory condition of the intestinal mucosa whose etiology is unclear but is likely to be multifactorial. We have shown previously that an increased amount of hyaluronan (HA) is present both in the inflamed mucosa of inflammatory bowel disease patients and in isolated human cells after polyI:C treatment. The signal transducer and activator of transcription (STAT)1 protein plays an important role in many signaling pathways that are associated with inflammation. We therefore investigated the role of STAT1 in adhesive interactions that occur between leukocytes and polyI:C-induced mucosal smooth muscle cells (M-SMCs). Activation of STAT1 was observed after the polyI:C treatment of M-SMCs. Specific phosphorylation of tyrosine and serine residues of STAT1 was observed in polyI:C-treated, but not untreated, M-SMC cultures. To evaluate further the role of STAT1, a corresponding STAT-1-null mouse was used. PolyI:C-induced, HA-mediated leukocyte adhesion to colon SMCs from STAT1-null mice was significantly decreased compared with that from wild-type control mice. In vivo, using the dextran sulfate sodium-induced model of colon inflammation, both tissue damage and HA deposition were attenuated in STAT1-null mice compared with that in wild-type control mice. Additionally, the inter-α-trypsin inhibitor (IαI), a proteoglycan essential for facilitating leukocyte binding to the HA matrix, was reduced in STAT1-null mice. Together, these results demonstrate that STAT1 plays an important role in HA-mediated inflammatory processes.
The pathogenesis of chronic inflammatory diseases such as inflammatory bowel disease (IBD) is not well understood. The origin of IBD is unknown and likely to be multifactorial. Environmental factors, including viruses or microbial infections, alter the normal immune balances in the intestine of genetically susceptible people that may lead to Crohn’s disease and ulcerative colitis. Pathological changes in IBD include destruction of normal tissue architecture, increase of mononuclear leukocyte influx into the intestinal mucosa, and hyperplasia of the mucosal smooth muscle cells (M-SMCs).
Alterations in cytokine levels are thought to have critical roles in the pathogenesis of IBD. The mucosal concentrations of several cytokines, including tumor necrosis factor-α, interleukin (IL)-12, IL-6, IL-23, IL-17, and interferon (IFN)-γ are increased in IBD1,2,3,4,5,6,7,8,9 and contribute to tissue damage and chronic disease. Janus kinases (Jak)-signal transducer and activator of transcription (STAT)-dependent pathways are major signaling pathways for biological functions of IFN-γ.10,11,12,13 However, STAT-independent pathways are also known.14,15 Jaks and STATs are present in the cytoplasm of most cell types. In general, cytokines and growth factors that signal through Jak-STAT-dependent pathways form specific Jaks and STAT-factor complexes with their respective receptors. Subsequently, Jaks are activated through auto- and trans-phosphorylation before phosphorylating the receptor at a specific residue near the C-terminus that creates a docking site on the receptor for STAT factors. Complex-bound STAT is then similarly phosphorylated by Jaks and dissociates from the receptor complex as a dimer. The liberated, phosphorylated STAT dimer then translocates to the nucleus and binds with the gamma-activating sequence present in many cytokine and growth factor-inducible gene promoters that stimulates the transcription and mediates the biological responses of these gene products.10,11,12,13,16 Acetylation of STATs also occurs after cytokine stimulation that promotes stable dimer formation and subsequent transcriptional stimulation of target genes.17,18
Hyaluronan (HA) is a glycosaminoglycan composed of glucuronic acid and N-acetyl-d-glucosamine. Our laboratory has established that virus or viral mimic double-stranded RNA (poly I:C) treatment increases cell surface HA deposition and formation of long cable-like structures of HA that are important for leukocyte attachment.19 In IBD patients, the association of viruses such as cytomegalovirus, respiratory syncytial virus, herpesvirus, mumps, and measles has been reported.20,21,22,23 We also demonstrated that increased HA staining was observed in inflamed tissue sections from patients with ulcerative colitis or Crohn’s disease compared to noninflamed and less inflamed areas from the same patient.19 In addition, we showed by immunohistochemical staining that inter-α-trypsin inhibitor (IαI), a HA-binding protein, is localized in these HA cable structures.24 Recent reports have demonstrated the activation of STAT1 and also STAT3 in IBD patients.25,26,27,28 Constitutive activation of STAT3 in IBD has also been reported.29 In this study, we demonstrate that STAT1 has a role in the attachment of U937 cells to poly I:C-treated M-SMCs and STAT1 is required for dextran sulfate sodium (DSS)-induced intestinal damage in mice.
Materials and Methods
Materials
Cell culture media and additives were obtained from our institute core facility. Fetal bovine serum was from Bio- Whitaker, Walkersville, MD, cycloheximide, poly I:C, and α-actin antibody were from Sigma, St. Louis, MO. STAT1 and STAT3 antibodies were from Cell Signaling Technology, Beverly, MA, and poly dI:dC, protease inhibitors, EGCG, and AG490 were from Calbiochem, La Jolla, CA.
Cell Culture
The techniques of cell isolation, culture, and adhesion assays of M-SMCs (obtained from either human colon or mouse specimens) were described in our earlier publication.19 Smooth muscle cell (SMC) cultures were derived from the large bowel of STAT-1 knockout and control wild-type mice by explant culture. Briefly, after isolation, M-SMCs were cultured in Dulbecco’s modified Eagle’s medium/F12 medium supplemented with 10% fetal bovine serum, 100 U/ml of penicillin, 100 μg/ml of streptomycin, and 0.25 μg/ml of fungizone in a humidified cell culture incubator at 37°C with 5% CO2 unless otherwise indicated. All experiments were done within passage 3 of cells in culture. Approximately 80% confluent cell cultures were either untreated or treated with 50 μg/ml of poly I:C in fresh medium unless otherwise indicated. Experiments were performed at least in triplicate and representative data from one such experiment are shown.
Electrophoretic Mobility Shift Assays
Electrophoretic mobility shift assays were done with a synthetic double-stranded hSIE oligonucleotide DNA (labeled by kinase reaction using γ-32P-ATP) as a probe, using an equal amount of protein from whole cell extracts according to standard procedures. Equal amounts of extracts were incubated in 20-μl reaction volumes in binding buffer [20 mmol/L HEPES (pH 7.9), 5% glycerol, 50 mmol/L NaCl, 1 mmol/L MgCl2, 1 mmol/L dithiothreitol, and 0.5 mmol/L ethylenediaminetetraacetic acid] containing 2 μg of poly (dI-dC), and 0.2 ng of labeled probe. After 20 minutes of incubation at 22°C, the reaction mixture was resolved in a 5% nondenaturing polyacrylamide gel in TBE buffer (45 mmol/L Tris, 45 mmol/L boric acid, and 1 mmol/L ethylenediaminetetraacetic acid) at 180 V for 2 hours. The gel was dried and visualized by autoradiography.30,31 Supershift analyses were done by preincubating extracts with the specific antibody for 10 minutes at 22°C in the reaction mixture before the addition of the probe.
Immunoblot and Binding Assay
Western blots were done with equal amounts of protein for each lane, electrophoresed in 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and transferred to polyvinylidene difluoride-type transfer membrane at 4°C by a standard procedure.31 Both phosphorylated STAT1 and actin antibodies were used according to the manufacturers’ protocols. Binding of leukocytes to SMCs was measured with radiolabeled U937 cells according to our previously published procedure.19,24 Cells binding values were presented as the mean data from at least triplicate wells ± SE.
Animal Experiments and Fluorescence Histochemistry for Confocal Microscopy
All animal experiments were performed following guidelines and approvals from the institutional animal care and use committee. Age-matched, 8- to 10-week-old, 129S6/SvEv strain of STAT-1-null and wild-type mice (obtained from Taconic Farms, Germantown, NY) were treated with or without 5% DSS, average molecular mass 10,000 Da, in the drinking water. Mice were sacrificed after 4 or 7 days of treatment and their colons were removed and processed for immunohistochemical staining from paraffin sections. The sections were examined by Leica (Wetzlar, Germany) TCS-SP laser-scanning confocal microscopy.24 Detection of HA and heavy chains of IαI were performed using biotinylated HA binding protein (Seikagaku, Tokyo, Japan) and an IαI antibody, respectively.19,24 We used secondary fluorescein isothiocyanate-conjugated streptavidin for HA detection and secondary Texas Red-conjugated antibody for IαI detection, and also 4,6-diamidino-2-phenylindole for nuclei staining.
Laser Microdissection
Colon tissue was obtained directly after surgical resection from the Cleveland Clinic Surgery Department according to institutional review board guidelines with their approval. The colon tissues from noninflamed and inflamed areas were embedded in OCT medium and stored at −80°C until further processing for experiments.32 A small portion of tissue was also obtained for homogenization in ice-cold buffer containing a cocktail of protease inhibitors. Tissue debris was then removed by low-speed centrifugation. Protein concentrations were determined and an equal amount of protein was loaded in each lane for gel electrophoresis. Sections of muscularis mucosae layers were selected and laser cuts were done similarly from inflamed and noninflamed areas. The cells in the selected regions were automatically acquired in a protein extraction buffer (Tris/HCl buffer containing all protease inhibitors). Extracts with an equal volume were loaded in SDS-PAGE gels for Western blots.31
Results
STAT1 Activation in Poly I:C-Treated M-SMCs
We previously demonstrated that STAT1 is the most important factor among the cytoplasmic components of the Jak-STAT pathway for induction of IFN-inducible gene, p56, by poly I:C using mutant cells lacking individual components of the Jak-STAT pathway.33 Many reports have demonstrated the activation of STAT1 by cytokines and growth factors.10,11,12,13,16 Because STAT1 is important for poly I:C-stimulated IFN-inducible gene induction,33 we investigated whether poly I:C activates STAT1 in primary human colon M-SMCs. Cultures of human M-SMCs were treated with or without poly I:C for 0, 2, and 4 hours, and STAT-DNA binding activity was measured by gel-shift assay. Activation of STAT is observed by 2 hours and the level of induction is greater at 4 hours after poly I:C treatment (Figure 1a). Control experiments using an excess amount of the same and different cold oligoneucleotides were performed and confirmed the specificity of DNA binding activity (data not shown). IL-6 primarily activates STAT3, but in many cells IL-6 also activates STAT1.34,35 We determined whether STAT is induced by IL-6 in M-SMCs by treating primary M-SMCs with 10 ng/ml of IL-6 for 30 minutes. The results indicate that the DNA binding STAT protein is present in IL-6-treated extracts (Figure 1a). To characterize whether the poly I:C-induced DNA-binding protein band is STAT1 or STAT3, we used antibody supershift experiments using STAT1 and STAT3 antibodies. Extracts from poly I:C-treated (4 hours) and IL-6-treated (30 minutes) cells were incubated with STAT1 or STAT3 antibody. STAT1 antibody supershifted both bands almost completely whereas STAT3 antibody did not shift either band in extracts of poly I:C-treated cells. IL-6 activates both STAT1 and STAT3 as shown by band supershifting with both antibodies (Figure 1b) and primarily serves as a positive control for determining supershift of both STAT1 and STAT3 in this experiment. STAT1 activation was also observed in M-SMCs after IFN-γ treatment (data not shown). STAT1 was not activated at 30 minutes or 1 hour after poly I:C treatment and activation started to decline after 4 hours and was completely gone after 8 hours (data not shown). Maximum STAT1 activation was observed at 4 hours, although activation of STAT1 was also detected at the 2-hour time point (similar as Figure 1a). We investigated the requirement of ongoing protein synthesis for STAT1 activation by poly I:C. M-SMCs were treated with and without poly I:C in the presence or absence of cycloheximide. Poly I:C induces the STAT1 band in electrophoretic mobility shift assay, and induction of the STAT1 band is reduced in poly I:C plus cycloheximide treatment (Figure 1c). Partial activation of STAT1 in the presence of cycloheximide indicates that STAT1 is likely induced by poly I:C, and other secreted factors that might be induced by poly I:C treatment could contribute to full STAT1 activation. To further understand the role of secreted factors induced by poly I:C in STAT1 activation, we treated human M-SMCs with poly I:C for 4 hours and then collected the medium for subsequent treatment of untreated human M-SMCs. These cellular extracts were also subjected to a similar gel-shift assay to see whether STAT1 is activated by any secreted factors that are initially produced because of poly I:C treatment. Figure 1d shows no STAT1-activated band by treatment with supernatant obtained from poly I:C-treated cells. The results of STAT1 activation by poly I:C in the presence of cycloheximide and also the lack of STAT1 activation by medium obtained from poly I:C-stimulated cells indicate that STAT1 activation in M-SMCs by poly I:C is partially mediated by poly I:C and not by other secondary secreted factors induced by poly I:C.
Figure 1.

STAT1 activation by poly I:C treatment in M-SMCs. Electrophoretic mobility shift assays using labeled hSIE as a probe with equal amounts of whole cell extracts (15 μg) prepared from cells in the presence of 2% fetal bovine serum as indicated in figures. a: Activation of STAT protein in 2- or 4-hour poly I:C-treated and 30-minute IL-6-treated extracts. b: Supershift analyses with 4-hour poly I:C-treated extract or with 30-minute IL-6-treated extract were performed after pre-incubation with STAT1- or STAT3-specific antibody. c: Cells treated with and without poly I:C and cycloheximide 30 minutes before the poly I:C treatment. d: Cultured M-SMCs were treated for 4 hours with medium obtained from 4-hour poly I:C-treated M-SMCs. Several exposures were taken from the autoradiogram and one of the exposures was used for this figure.
Tyrosine and Serine Phosphorylation of STAT1 by Poly I:C Treatment of Human M-SMCs
Because STAT1 is activated in human M-SMCs by poly I:C, we sought to determine whether STAT1 is phosphorylated at 701-tyrosine in primary human M-SMCs after poly I:C treatment. Because poly I:C activation of STAT1 was observed in the previous experiment in M-SMCs starting at 2 hours and achieving a maximum at 4 hours, we prepared cell extracts from M-SMCs treated with or without poly I:C for 2 hours and 4 hours for immunoblot analysis. Extracts were subjected to SDS-PAGE followed by immunoblot and phosphotyrosine analysis of STAT1 protein using a specific STAT1-phosphotyrosine antibody. Figure 2a shows the 701-tyrosine-phosphorylated STAT1 band in extracts prepared from cells treated with poly I:C for 2 hours and a more intense band at 4 hours. However, phosphorylated bands are not observed in extracts of cells not treated with poly I:C (designated medium 2 hours and medium 4 hours in Figure 2a). This result indicates that STAT1 is tyrosine-phosphorylated after poly I:C treatment in human M-SMCs. We also determined whether poly I:C treatment causes phosphorylation of the serine residue at the 727-amino acid position of STAT1, which is required for further transcriptional activation. Human M-SMCs were similarly treated with and without poly I:C for 2 hours and 4 hours and cell extracts were examined by SDS-PAGE followed by Western blots with a 727-serine-specific antibody. The results indicate that STAT1 serine phosphorylation is barely visible after 2 hours of poly I:C treatment but is easily visible at 4 hours after poly I:C treatment (Figure 2b). The same blots were also used for α-actin expression using a specific antibody to verify equal sample loading in each lane. Therefore, both critical 701-tyrosine and 727-serine residues of STAT1 are phosphorylated in M-SMCs after poly I:C treatment.
Figure 2.
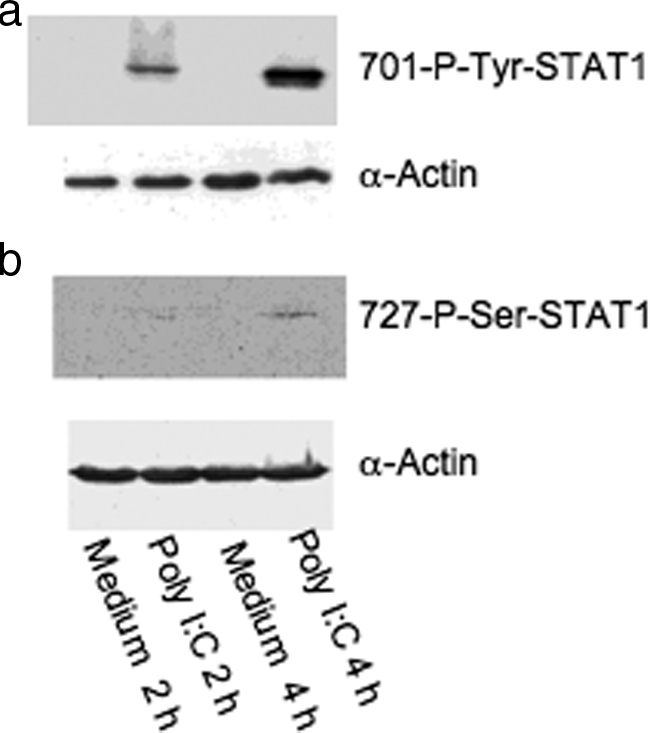
Phosphorylation of STAT1 by Western blots. M-SMCs were serum-starved for 18 hours and then treated for 2 or 4 hours with poly I:C in 2% serum-containing medium as indicated and equal protein (30 μg) amount of cellular extracts were used for Western blot analyses using phosphor-specific antibodies 701-tyrosine (a) and 727-serine (b) residues of STAT1 protein. The same blot was then reused with α-actin antibody to compare equal loading of each sample.
Inhibition of Poly I:C-Stimulated Leukocyte Adhesion by Inhibitors
We previously reported that cellular stress of M-SMCs induced by treatment with poly I:C stimulated HA-mediated leukocyte attachment.19,36 Therefore, we investigated whether STAT1 has any role in poly I:C-stimulated HA-mediated leukocyte adhesion. To test this, we treated M-SMCs with poly I:C in the presence and absence of STAT1 inhibitor (EGCG) or Jak inhibitor (AG490). In agreement with our earlier publication,19 Figure 3 shows that poly I:C treatment induces cell adhesion much more than the untreated control. The induction of HA-mediated U937 cell adhesion by poly I:C was inhibited ∼60% in the presence of EGCG or AG490. We did not observe toxic effects under these conditions on M-SMCs by treatment either with EGCG or AG490 as determined by trypan blue dye exclusion (data not shown). To verify that the induced adhesion is HA-mediated, cells were treated with hyaluronidase after attachment as described previously.19,24 These results demonstrated abrogation of adhesion indicating that poly I:C-stimulated, HA-mediated cell recruitment is at least partially STAT1-dependent.
Figure 3.

Reduced U937 cell attachment to poly I:C-stimulated M-SMCs with Jak or STAT inhibitors. M-SMCs were untreated or treated with poly I:C for 18 hours in the presence or absence of 100 μmol/L of AG490 or EGCG as indicated. Labeled U937 cells were used for the binding assay as described in Materials and Methods section.
Attenuated Leukocyte Adhesion to Poly I:C-Stimulated Murine STAT1-Null SMCs
Figure 3 indicates that STAT1 has a role in poly I:C-stimulated, HA-mediated adhesion in vitro. To further investigate the role of STAT1 in poly I:C-induced, HA-mediated leukocyte adhesion, we isolated SMCs from the colon of STAT1-null and wild-type mice and treated them with and without poly I:C and assayed them for their ability to bind U937 cells. Parallel cultures were treated similarly for HA staining using confocal microscopy. Our results demonstrate that poly I:C treatment of SMCs induces U937 cell attachment in wild-type cells whereas the attachment is substantially reduced in STAT1-null cells. In untreated cultures, the U937 cell attachment to STAT1-null cells is slightly higher compared to wild-type cells (Figure 4a). The U937 cells binding to poly I:C-stimulated SMCs was approximately eightfold higher in poly I:C-treated wild-type cells, and only approximately twofold higher in STAT1-null cells compared to respective unstimulated control cells (Figure 4a). As previously reported in isolated primary human M-SMCs,19 hyaluronidase treatment of poly I:C-stimulated SMCs demonstrates that the induced attachment of U937 cells in both cells derived from wild-type and null mice are HA-mediated. These results indicate that STAT1 is important for the poly I:C-stimulated attachment of leukocytes in isolated mouse primary SMCs. Results of Figure 4b show no quantitative differences in the overall HA staining after poly I:C treatment of STAT1-null and wild-type SMCs. The HA staining in green showing less cable-like structure in STAT1-null cells and also binds less U937 cells compared to wild-type SMCs. The role of STAT1 in the assembly of HA cable formation and is only correlative at this point. Similar to our previously published report in human M-SMCs,19 wild-type cells also form cable-like HA structures (green) after poly I:C treatment (Figure 4b).
Figure 4.

Poly I:C-stimulated HA-mediated cell attachment is less in STAT1-null SMCs compared to wild-type cells. a: Isolated SMCs from STAT1 wild-type (+/+) and null (−/−) mice were treated with or without poly I:C for 18 hours and a quantitative assay was performed using labeled U937 cells for attachment as described in the Materials and Methods. b: Parallel cultures on coverslips were treated similarly, methanol-fixed, and stained for HA (green) and nuclei (blue, 4,6-diamidino-2-phenylindole) as described in the Materials and Methods section.
DSS-Induced Colon Damage Is Attenuated in STAT1-Null Mice
To investigate the role of STAT1 to inflammation in vivo, we used an established mouse model of colitis by adding 5% DSS in drinking water. We compared STAT1-null mice to wild-type animals throughout the 1-week course of colitis development. In this model pathological changes are known to develop first in the distal colon likely because of the bacterial burden on this tissue. Distal colon sections from wild-type mice show distinct pathological changes throughout time of DSS treatment including submucosal thickening and mucosal architecture destruction. The HA deposition (green staining) also changes during the course of colitis induction as previously reported.37 In untreated animals, the HA is regularly distributed among the crypts in the mucosa, but after 4 days of DSS treatment epithelial associated HA is diminished and large submucosal deposits are seen (Figure 5a). By 7 days the largest deposition of HA is at the damaged mucosal surface. Although the HA loss from the epithelium in STAT1-null animals is equivalent to the wild type, the intestinal damage and HA redistribution in the colon of null mice were greatly reduced at 4 and 7 days (Figure 5a). These observations indicate that stimulated HA deposition induced by DSS treatment is partially STAT1-dependent. We have previously reported increased HA-associated heavy chain IαI deposition in intestinal tissues of IBD patients and have shown in vitro that IαI components are necessary for leukocyte attachment to the HA matrix.24 Therefore, we assayed the deposition of heavy chain IαI in the colon tissue of wild-type and STAT1-null animals during the development of colitis. The results demonstrate that heavy chain IαI staining of distal colon sections is also greatly reduced in STAT1-null mice compared to wild-type mice (Figure 5b).
Figure 5.

Reduced HA deposition and less inflammatory changes in STAT1−/− mice after DSS treatment. a: Confocal image demonstrates HA polymers (green) in mouse colon sections after DSS treatment in STAT1−/−-null (top) and STAT1+/+ wild-type animals (bottom). The null mice section demonstrates minimal staining on day 4 and moderate staining on day 7 that is localized to the submucosa (arrow), whereas the wild-type mice have moderate staining of the submucosa (arrow) on day 4 and heavy staining of the lamina propria (asterisks) on day 7; cells are identified by their nuclei (blue). b: HA (green) and IαI (red) staining in colon designated as wild-type STAT1 (WT) and STAT1-null mice (KO) sections after a 4- or 7-day treatment with DSS.
Phosphorylated STAT1 Is Expressed in Inflamed Human Colon
Because STAT1 is tyrosine phosphorylated at the 701 amino acid residue (Figure 2), we investigated whether activated STAT1 is present in inflamed human tissue. To do this, we used inflamed and noninflamed portions from human colon tissues and prepared whole cell extracts for detection of tyrosine-phosphorylated STAT1 by immunoblot using phospho-tyrosine-specific STAT1 antibody. Figure 6a shows that the phosphorylated STAT1 band is prominent in inflamed tissue extracts, but only a faint band is observed in extracts from a noninflamed area from the same sample. The total STAT1 level was comparable in both inflamed and noninflamed samples (Figure 6a). To investigate whether phosphorylated STAT1 is present in smooth muscle cells in situ, we made extracts from laser-microdissected muscle cells derived from inflamed and noninflamed regions from the same colon sample used in Figure 6a. Figure 6b shows that the tyrosine-phosphorylated STAT1 band is present only in the SMCs from inflamed sections; the phosphorylated STAT1 band is undetectable in SMCs from noninflamed microdissected sections. These results demonstrate that 701-tyrosine-phosphorylated STAT1 is present in inflamed human colon tissue, specifically in muscle cells. Figure 6c demonstrates an example of microdissected cells obtained from the muscularis mucosa shown by asterisks.
Figure 6.

Tyrosine phosphorylation of STAT1 in inflamed whole tissue extracts and specifically in muscularis mucosa. Western blot analyses using equal amounts of protein from whole cell extracts (50 μg) (a) and equal volume of laser-dissected muscularis mucosa extracts (b) from both noninflamed and inflamed area from same patient sample are shown with 701-phospho-specific tyrosine antibody. c: Microscopic picture demonstrated by asterisks that the microdissected cells derived from the muscularis mucosa.
Discussion
Emerging evidence suggests that STATs can mediate cellular stress in response to various stimuli.38 We have previously shown that viral stress of human SMCs in vitro, induced either by viruses or by a viral intermediate (poly I:C), results in the production of unique cable-like HA structures that facilitate leukocyte binding in cultured cells. Similar HA structures are present in vivo in the intestine of IBD patients.19
Our previous study suggested that STAT1 is the most important component of the Jak-STAT pathway for the induction of IFN-stimulated gene by poly I:C.33 In this report we demonstrated that cellular stress induced by poly I:C in human primary M-SMCs activates STAT1. Because the presence of soluble factors in poly I:C-treated culture medium is unable to activate STAT1, this STAT1 activation is most likely attributable to direct effects of poly I:C, although we could not rule out the possibility of the contribution of other factors independently or in combination with poly I:C to activate STAT1. The phosphorylations of 701-tyrosine and 727-serine STAT1 residues are necessary for maximum transcriptional activation of STAT1-dependent genes.10,11,39 We provide evidence that both critical residues of STAT1 are phosphorylated on poly I:C treatment.
We identified a role for activated STAT1 in poly I:C-induced leukocyte adhesion by showing abrogation of adhesion in cells treated with inhibitors of the Jak-STAT pathway. In contrast, M-SMCs treated with IFN-α/β, IFN-γ, or tumor necrosis factor-α alone, or in combination, did not attach more leukocytes compared to untreated cells in a HA-dependent manner (data not shown). STAT1-null cells exhibit greatly attenuated leukocyte adhesiveness after poly I:C stimulation of intestinal SMCs compared with wild-type cells. This provides further compelling evidence that STAT1 has an important role in this unique HA-mediated cell-cell interaction.
The in vitro role of STAT1 in the production of adhesive HA by mouse colonic SMCs led us to determine whether the absence of the STAT1 gene could impact the severity of disease in the DSS-induced mouse colitis model. The specific mechanism through which DSS induces colitis is not fully characterized, but it is thought that DSS damages the intestinal epithelial layers thereby allowing microbes to induce inflammation starting in the distal colon where pathological changes are first seen. Evidence indicates that IFN-γ knockout mice are as sensitive to DSS-induced inflammation as the wild-type mice. Therefore, the presence or absence of IFN-γ has no significant effects in chemically induced colitis in mice.40,41,42 Additionally, lack of the IFN regulatory factor (IRF)-1 gene in mice increases susceptibility to DSS-induced inflammation, although these mice after DSS exposure produce significantly less or no IFN-γ in colon cultures.43 We compared STAT1-null and wild-type mice treated orally with DSS to induce colitis. Our results demonstrated that STAT1-null mice are less sensitive to DSS-induced colitis. We conclude that STAT1 exacerbates DSS-induced inflammation in wild-type mice. Therefore, we speculate that the relatively protective state of STAT1-null mice against DSS-induced colitis as compared to wild-type mice is likely not attributable to IFN-γ because other reports demonstrated that IFN-γ knockout mice or administration of the neutralizing anti-IFN-γ antibody in wild-type mice, or IFN-γ receptor-null mice, are equally susceptible in mouse colitis models.40,41,42,43 A contradictory report appeared recently however, that indicates IFN-γ-null mice are less susceptible to DSS-induced colitis.44
We showed greater deposition of HA in DSS-treated wild-type mouse colon. This result is consistent with our in vitro studies in which we observed increased HA deposition with a unique cable-like structure that binds more leukocytes.19 We also reported that IαI is critical to the formation of leukocyte adhesiveness in vitro, and associated with HA in IBD patients.24 In the DSS-treated STAT1-null mouse, we observed diminished IαI in the tissue compared to wild-type tissue. Reduced IαI deposition, in addition to reduced HA could be a reason why fewer leukocytes are recruited into the intestine of the STAT1-null mice during colitis.
Recent reports have indicated a link in STAT1 activation with IBD,25,26,27,28 and in this study, we have confirmed tyrosine phosphorylation of STAT1 in human IBD tissues. In addition, human muscularis mucosae samples obtained by microdissection also demonstrated tyrosine phosphorylation of STAT1 in inflamed section. This study suggests that STAT1 exacerbates DSS-induced inflammation in mice and that STAT1 phosphorylation is associated with the inflammation in human IBD.
Acknowledgments
We thank Drs. Ganes C. Sen and Alana Majors for critical reading of this manuscript; Drs. George Stark, Ian M. Kerr, and Claudio Fiocchi for helpful suggestions throughout this work; and Karl Krymowski for his help with tissue microdissection.
Footnotes
Address reprint requests to Sudip K. Bandyopadhyay, Lerner Research Institute, Cleveland Clinic, Department of Pathobiology, NC-2, 9500 Euclid Ave., Cleveland, OH 44195. E-mail: bandyos@ccf.org.
Supported in part by the National Institutes of Health (grant RO1 DK58867 to S.A.S.) and the Thomas C. and Sandra S. Sullivan family (endowed chair to S.A.S.).
References
- Beagley KW, Elson CO. Cells and cytokines in mucosal immunity and inflammation. Gastroenterol Clin North Am. 1992;21:347–366. [PubMed] [Google Scholar]
- Becker C, Dornhoff H, Neufert C, Fantini MC, Wirtz S, Huebner S, Nikolaev A, Lehr HA, Murphy AJ, Valenzuela DM, Yancopoulos GD, Galle PR, Karow M, Neurath MF. Cutting edge: IL-23 cross-regulates IL-12 production in T cell-dependent experimental colitis. J Immunol. 2006;177:2760–2764. doi: 10.4049/jimmunol.177.5.2760. [DOI] [PubMed] [Google Scholar]
- Podolsky DK. Inflammatory bowel disease. N Engl J Med. 2002;347:417–429. doi: 10.1056/NEJMra020831. [DOI] [PubMed] [Google Scholar]
- Fuss IJ, Neurath M, Boirivant M, Klein JS, de la Motte C, Strong SA, Fiocchi C, Strober W. Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn’s disease LP cells manifest increased secretion of IFN-gamma, whereas ulcerative colitis LP cells manifest increased secretion of IL-5. J Immunol. 1996;157:1261–1270. [PubMed] [Google Scholar]
- Fiocchi C, Fukushima K, Strong SA, Ina K. Pitfalls in cytokine analysis in inflammatory bowel disease. Aliment Pharmacol Ther. 1996;10(Suppl 2):61–70. doi: 10.1046/j.1365-2036.1996.22164022.x. [DOI] [PubMed] [Google Scholar]
- Mudter J, Neurath MF. IL-6 signaling in inflammatory bowel disease: pathophysiological role and clinical relevance. Inflamm Bowel Dis. 2007;13:1016–1023. doi: 10.1002/ibd.20148. [DOI] [PubMed] [Google Scholar]
- Neurath MF. IL-23: a master regulator in Crohn disease. Nat Med. 2007;13:26–28. doi: 10.1038/nm0107-26. [DOI] [PubMed] [Google Scholar]
- Hue S, Ahern P, Buonocore S, Kullberg MC, Cua DJ, McKenzie BS, Powrie F, Maloy KJ. Interleukin-23 drives innate and T cell-mediated intestinal inflammation. J Exp Med. 2006;203:2473–2483. doi: 10.1084/jem.20061099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullberg MC, Jankovic D, Feng CG, Hue S, Gorelick PL, McKenzie BS, Cua DJ, Powrie F, Cheever AW, Maloy KJ, Sher A. IL-23 plays a key role in Helicobacter hepaticus-induced T cell-dependent colitis. J Exp Med. 2006;203:2485–2494. doi: 10.1084/jem.20061082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell JE, Jr, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264:1415–1421. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- Stark GR, Kerr IM, Williams BR, Silverman RH, Schreiber RD. How cells respond to interferons. Annu Rev Biochem. 1998;67:227–264. doi: 10.1146/annurev.biochem.67.1.227. [DOI] [PubMed] [Google Scholar]
- O'Shea JJ, Gadina M, Schreiber RD. Cytokine signaling in 2002: new surprises in the Jak/Stat pathway. Cell. 2002;109:S121–S131. doi: 10.1016/s0092-8674(02)00701-8. [DOI] [PubMed] [Google Scholar]
- Schindler C, Levy DE, Decker T. JAK-STAT signaling: from interferons to cytokines. J Biol Chem. 2007;282:20059–20063. doi: 10.1074/jbc.R700016200. [DOI] [PubMed] [Google Scholar]
- Ramana CV, Gil MP, Han Y, Ransohoff RM, Schreiber RD, Stark GR. Stat1-independent regulation of gene expression in response to IFN-gamma. Proc Natl Acad Sci USA. 2001;98:6674–6679. doi: 10.1073/pnas.111164198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramana CV, Gil MP, Schreiber RD, Stark GR. Stat1-dependent and -independent pathways in IFN-gamma-dependent signaling. Trends Immunol. 2002;23:96–101. doi: 10.1016/s1471-4906(01)02118-4. [DOI] [PubMed] [Google Scholar]
- Levy DE, Darnell JE., Jr Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3:651–662. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- Yuan ZL, Guan YJ, Chatterjee D, Chin YE. Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science. 2005;307:269–273. doi: 10.1126/science.1105166. [DOI] [PubMed] [Google Scholar]
- O'Shea JJ, Kanno Y, Chen X, Levy DE. Cell signaling. Stat acetylation—a key facet of cytokine signaling? Science. 2005;307:217–218. doi: 10.1126/science.1108164. [DOI] [PubMed] [Google Scholar]
- de la Motte CA, Hascall VC, Calabro A, Yen-Lieberman B, Strong SA. Mononuclear leukocytes preferentially bind via CD44 to hyaluronan on human intestinal mucosal smooth muscle cells after virus infection or treatment with poly(I.C). J Biol Chem. 1999;274:30747–30755. doi: 10.1074/jbc.274.43.30747. [DOI] [PubMed] [Google Scholar]
- Bernstein CN, Blanchard JF. Viruses and inflammatory bowel disease: is there evidence for a causal association? Inflamm Bowel Dis. 2000;6:34–39. doi: 10.1097/00054725-200002000-00005. [DOI] [PubMed] [Google Scholar]
- Rahbar A, Bostrom L, Lagerstedt U, Magnusson I, Soderberg-Naucler C, Sundqvist VA. Evidence of active cytomegalovirus infection and increased production of IL-6 in tissue specimens obtained from patients with inflammatory bowel diseases. Inflamm Bowel Dis. 2003;9:154–161. doi: 10.1097/00054725-200305000-00002. [DOI] [PubMed] [Google Scholar]
- Papadakis KA, Tung JK, Binder SW, Kam LY, Abreu MT, Targan SR, Vasiliauskas EA. Outcome of cytomegalovirus infections in patients with inflammatory bowel disease. Am J Gastroenterol. 2001;96:2137–2142. doi: 10.1111/j.1572-0241.2001.03949.x. [DOI] [PubMed] [Google Scholar]
- Wong NA, Herbst H, Herrmann K, Kirchner T, Krajewski AS, Moorghen M, Niedobitek F, Rooney N, Shepherd NA, Niedobitek G. Epstein-Barr virus infection in colorectal neoplasms associated with inflammatory bowel disease: detection of the virus in lymphomas but not in adenocarcinomas. J Pathol. 2003;201:312–318. doi: 10.1002/path.1442. [DOI] [PubMed] [Google Scholar]
- de la Motte CA, Hascall VC, Drazba J, Bandyopadhyay SK, Strong SA. Mononuclear leukocytes bind to specific hyaluronan structures on colon mucosal smooth muscle cells treated with polyinosinic acid:polycytidylic acid: inter-alpha-trypsin inhibitor is crucial to structure and function. Am J Pathol. 2003;163:121–133. doi: 10.1016/s0002-9440(10)63636-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber S, Rosenstiel P, Hampe J, Nikolaus S, Groessner B, Schottelius A, Kuhbacher T, Hamling J, Folsch UR, Seegert D. Activation of signal transducer and activator of transcription (STAT) 1 in human chronic inflammatory bowel disease. Gut. 2002;51:379–385. doi: 10.1136/gut.51.3.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plevy S. A STAT need for human immunologic studies to understand inflammatory bowel disease. Am J Gastroenterol. 2005;100:73–74. doi: 10.1111/j.1572-0241.2005.41382.x. [DOI] [PubMed] [Google Scholar]
- Pfitzner E, Kliem S, Baus D, Litterst CM. The role of STATs in inflammation and inflammatory diseases. Curr Pharm Des. 2004;10:2839–2850. doi: 10.2174/1381612043383638. [DOI] [PubMed] [Google Scholar]
- Mudter J, Weigmann B, Bartsch B, Kiesslich R, Strand D, Galle PR, Lehr HA, Schmidt J, Neurath MF. Activation pattern of signal transducers and activators of transcription (STAT) factors in inflammatory bowel diseases. Am J Gastroenterol. 2005;100:64–72. doi: 10.1111/j.1572-0241.2005.40615.x. [DOI] [PubMed] [Google Scholar]
- Lovato P, Brender C, Agnholt J, Kelsen J, Kaltoft K, Svejgaard A, Eriksen KW, Woetmann A, Odum N. Constitutive STAT3 activation in intestinal T cells from patients with Crohn’s disease. J Biol Chem. 2003;278:16777–16781. doi: 10.1074/jbc.M207999200. [DOI] [PubMed] [Google Scholar]
- Leaman DW, Salvekar A, Patel R, Sen GC, Stark GR. A mutant cell line defective in response to double-stranded RNA and in regulating basal expression of interferon-stimulated genes. Proc Natl Acad Sci USA. 1998;95:9442–9447. doi: 10.1073/pnas.95.16.9442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandyopadhyay SK, de la Motte CA, Williams BR. Induction of E-selectin expression by double-stranded RNA and TNF-alpha is attenuated in murine aortic endothelial cells derived from double-stranded RNA-activated kinase (PKR)-null mice. J Immunol. 2000;164:2077–2083. doi: 10.4049/jimmunol.164.4.2077. [DOI] [PubMed] [Google Scholar]
- Kristiansen G, Pilarsky C, Wissmann C, Kaiser S, Bruemmendorf T, Roepcke S, Dahl E, Hinzmann B, Specht T, Pervan J, Stephan C, Loening S, Dietel M, Rosenthal A. Expression profiling of microdissected matched prostate cancer samples reveals CD166/MEMD and CD24 as new prognostic markers for patient survival. J Pathol. 2005;205:359–376. doi: 10.1002/path.1676. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay SK, Leonard GT, Jr, Bandyopadhyay T, Stark GR, Sen GC. Transcriptional induction by double-stranded RNA is mediated by interferon-stimulated response elements without activation of interferon-stimulated gene factor 3. J Biol Chem. 1995;270:19624–19629. doi: 10.1074/jbc.270.33.19624. [DOI] [PubMed] [Google Scholar]
- Kerr IM, Costa-Pereira AP, Lillemeier BF, Strobl B. Of JAKs, STATs, blind watchmakers, jeeps and trains. FEBS Lett. 2003;546:1–5. doi: 10.1016/s0014-5793(03)00411-3. [DOI] [PubMed] [Google Scholar]
- Kishimoto T. Interleukin-6: from basic science to medicine—40 years in immunology. Annu Rev Immunol. 2005;23:1–21. doi: 10.1146/annurev.immunol.23.021704.115806. [DOI] [PubMed] [Google Scholar]
- Majors AK, Austin RC, de la Motte CA, Pyeritz RE, Hascall VC, Kessler SP, Sen G, Strong SA. Endoplasmic reticulum stress induces hyaluronan deposition and leukocyte adhesion. J Biol Chem. 2003;278:47223–47231. doi: 10.1074/jbc.M304871200. [DOI] [PubMed] [Google Scholar]
- Kessler S, Rho H, West G, Fiocchi C, Drazba J, de la Motte C. Hyaluronan (HA) deposition precedes and promotes leukocyte recruitment in intestinal inflammation. Clin Translational Sci. 2008;1:57–61. doi: 10.1111/j.1752-8062.2008.00025.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley AC, Thomas D, Best J, Jenkins A. The STATs in cell stress-type responses. Cell Commun Signal. 2004;2:8. doi: 10.1186/1478-811X-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker T, Kovarik P. Serine phosphorylation of STATs. Oncogene. 2000;19:2628–2637. doi: 10.1038/sj.onc.1203481. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Hanada T, Mitsuyama K, Yoshida T, Kamizono S, Hoshino T, Kubo M, Yamashita A, Okabe M, Takeda K, Akira S, Matsumoto S, Toyonaga A, Sata M, Yoshimura A. CIS3/SOCS3/SSI3 plays a negative regulatory role in STAT3 activation and intestinal inflammation. J Exp Med. 2001;193:471–481. doi: 10.1084/jem.193.4.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tozawa K, Hanai H, Sugimoto K, Baba S, Sugimura H, Aoshi T, Uchijima M, Nagata T, Koide Y. Evidence for the critical role of interleukin-12 but not interferon-gamma in the pathogenesis of experimental colitis in mice. J Gastroenterol Hepatol. 2003;18:578–587. doi: 10.1046/j.1440-1746.2003.03024.x. [DOI] [PubMed] [Google Scholar]
- Hans W, Scholmerich J, Gross V, Falk W. Interleukin-12 induced interferon-gamma increases inflammation in acute dextran sulfate sodium induced colitis in mice. Eur Cytokine Netw. 2000;11:67–74. [PubMed] [Google Scholar]
- Siegmund B, Sennello JA, Lehr HA, Senaldi G, Dinarello CA, Fantuzzi G. Frontline: interferon regulatory factor-1 as a protective gene in intestinal inflammation: role of TCR gamma delta T cells and interleukin-18-binding protein. Eur J Immunol. 2004;34:2356–2364. doi: 10.1002/eji.200425124. [DOI] [PubMed] [Google Scholar]
- Ito R, Shin-Ya M, Kishida T, Urano A, Takada R, Sakagami J, Imanishi J, Kita M, Ueda Y, Iwakura Y, Kataoka K, Okanoue T, Mazda O. Interferon-gamma is causatively involved in experimental inflammatory bowel disease in mice. Clin Exp Immunol. 2006;146:330–338. doi: 10.1111/j.1365-2249.2006.03214.x. [DOI] [PMC free article] [PubMed] [Google Scholar]