

Abstract
Dopaminergic neurons in the substantia nigra (SN) selectively die in Parkinson’s disease (PD), but it is unclear how and why this occurs. Recent findings implicate prostaglandin E2 (PGE2) and two of its four receptors, namely EP1 and EP2, as mediators of degenerative and protective events in situations of acute and chronic neuronal death. EP1 activation can exacerbate excitotoxic damage in stroke models and our recent study showed that EP1 activation may explain the selective sensitivity of dopaminergic neurons to oxidative stress. Conversely, EP2 activation may be neuroprotective, although toxic effects have also been demonstrated. Here we investigated if and how EP2 activation might alter the survival of dopaminergic neurons following selective low-level oxidative injury evoked by the neurotoxin 6-hydroxydopamine (6-OHDA) in primary neuronal cultures prepared from embryonic rat midbrain. We found that cultured dopaminergic neurons displayed EP2 receptors. Butaprost, a selective EP2 agonist, significantly reduced 6-OHDA neurotoxicity. EP2 receptors are coupled to stimulatory G-proteins (Gs), which activate adenylate cyclase, increasing cAMP synthesis, which then activates protein kinase A (PKA). Both dibutyryl cAMP and forskolin reduced dopaminergic cell loss after 6-OHDA exposure. Conversely, KT5720 and H-89, two structurally distinct high-affinity PKA inhibitors, abolished the protective effect of butaprost, implicating cAMP-dependent PKA activity in the neuroprotection by EP2 activation. Finally, we show that melanized dopaminergic neurons in the human SN express EP2. This pathway warrants consideration as a neuroprotective strategy for PD.
Keywords: Parkinson’s disease, inflammation, neuroprotection, PGE2, 6-OHDA, cAMP, PKA, butaprost
INTRODUCTION
Neurodegenerative diseases often target specific neuronal groups in the CNS. In Parkinson’s disease (PD), midbrain dopaminergic neurons from the substantia nigra pars compacta (SNc) are selectively vulnerable [18]. We successfully mimicked this selective loss in cultures of embryonic rat midbrain neurons with 6-hydroxydopamine (6-OHDA), imposing low oxidative stress [8]. Inflammation can contribute to or accelerate neuronal death through the induction of cyclooxygenases (COX’s), neurotoxic cytokines, and production of reactive oxygen species [17]. We previously showed that COX-2, the COX isoform induced in the periphery by inflammation, was required for selective loss of dopaminergic neurons exposed to 6-OHDA [6].
Prostaglandin E2 (PGE2), a major product of COX activity, acts on four receptors, EP1–4 [12]. We previously identified EP1 and EP2 receptors on dopaminergic neurons in rat SNc using immunohistochemistry and reported that PGE2, by activating EP1, accounted for selective toxicity in culture [7]. In contrast, several studies suggest that EP2 activation may be neuroprotective in situations that produce neuronal death such as ischemia [22, 26], excitoxicity [4], and oxidative stress [13], although EP2-mediated neurotoxicity was also reported [20, 33, 35]. Here, we examined the effects of EP2 activation on dopaminergic cell death in our in vitro selective neurotoxicity model. We demonstrate for the first time that dopaminergic neurons in both human SNc and primary cultures of embryonic rat midbrain possess EP2 receptors. Importantly, EP2 agonists protected these neurons against 6-OHDA-mediated oxidative stress in culture. Neuroprotection was also conferred by cAMP analogs and was blocked by PKA inhibitors, introducing the EP2 receptor signal transduction pathway as a possible strategy for neuroprotection in PD.
MATERIALS AND METHODS
Cultures from 14–15 day embryonic rat midbrain were established as previously described [6–8] in accordance with NIH and Institutional guidelines. Cells were plated onto polyornithine-coated 24-well plates at 1.3 × 105 cells per well, and maintained at 100% humidity at 37°C in an atmosphere of 6–8% CO2/92–94% air throughout. After 36–40 hours, medium was replaced with defined medium (DM) [6–8]. Approximately 5% of the cells were dopaminergic neurons.
Treatments were performed on day-in-vitro 7 (DIV7). Medium was replaced with fresh DM containing drugs (Sigma, unless otherwise noted) in vehicle or vehicle alone (final vehicle concentrations noted in parentheses) for 30 minutes before and during exposure to 6-OHDA overnight. 6-OHDA stock solutions were prepared fresh in ice-cold 0.001N HCl (1 × 10−6 N). Butaprost (Cayman Chemical, Ann Arbor, MI) was dissolved in methyl acetate (0.2%). Dibutyryl cAMP was dissolved in water (1%). Forskolin and protein kinase A inhibitors H-89 and KT5720 were dissolved in ethanol (0.5–0.01%). Vehicles had no effect on cell survival ([6] and data not shown). Cultures were fixed for immunocytochemistry (ICC) after treatment.
Dopaminergic neurons were identified by ICC with a mouse monoclonal antibody to tyrosine hydroxylase (anti-TH; 1 µg/ml; Chemicon, Temecula, CA) and visualized with the anti-mouse peroxidase ABC kit using 3,3’-diaminobenzidine (DAB) as the chromagen (both from Vector Labs, Burlingame, CA) [6, 7]. Dopaminergic neurons were quantified by counting TH immunoreactive (TH+) cells (a stained cell body with at least one process) in perpendicular strips spanning the diameter of the culture well using a Nikon inverted microscope (100x magnification) under bright-field illumination..
To localize EP2 receptors in vitro, fixed cultures underwent double-label ICC with a polyclonal rabbit anti-EP2 antibody (Cayman) and mouse anti-TH (above). After fixation and blocking, cultures were incubated with anti-EP2 (0.25 µg/ml) or non-immune rabbit IgG (0.25 µg/ml; Cayman) and anti-TH (1 µg/ml) in ICC buffer (0.2% Triton X-100, 1% bovine serum albumin in 0.01 M phosphate buffered saline, pH 7.4) overnight at 4°C. EP2 was visualized using peroxidase-conjugated goat anti-rabbit antibody (Vector) and DAB with nickel enhancement. Dopaminergic neurons were visualized with biotinylated anti-mouse antibody (Vector) and streptavidin-Alexa Fluor 488 (Invitrogen, Carlsbad, CA).
Adult autopsy brain specimens, a kind gift from the Section on Neuropathology/Clinical Brain Disorders Branch, National Institute of Mental Health, NIH, US Department of Health and Human Services, Bethesda, MD, were obtained through the Medical Examiner’s office of the District of Columbia under IRB-approved protocol 90-M-0142 with informed consent from next-of-kin. Tissue was examined and processed under anonymous conditions as previously described, without any traceable identifier [2]. Fresh-frozen 14 µm serial coronal sections through the midbrain containing the substantia nigra were cut and stored at −80°C. For staining, sections were thawed, air-dried for 30 minutes and fixed with 4% paraformaldehyde (30 minutes) at room temperature (RT). Endogenous peroxidase activity was quenched with 3% H2O2 in ICC buffer with 10% normal goat serum (NGS) for 30 minutes at RT. Sections were blocked for 90 minutes at RT with 10% NGS in ICC buffer and incubated overnight at 4°C, with anti-EP2 receptor antibody or non-immune rabbit IgG (both 0.25 µg/ml, Cayman) in 1% NGS/ICC buffer. EP2 receptors were visualized using the Vectastain peroxidase ABC kit and DAB/nickel (Vector). Dopaminergic neurons were identified by melanin content.
Statistical analysis was performed using GraphPad software (San Diego, CA). Data from 2–5 independent experiments (treatment groups containing quadruplicate cultures) are presented as mean percent survival relative to untreated controls. Error bars represent the standard errors of the mean (SEM). Significant differences between groups were identified by analysis of variance (ANOVA), followed by post hoc analysis with Dunnett’s test for multiple comparisons, or Student’s t-test for single comparisons. Significance was set at p<0.05.
RESULTS/DISCUSSION
Previously we showed that EP2 receptors were present on dopaminergic neurons in the SNc of adult rat [7]. Now we asked whether dopaminergic neurons and/or other cells displayed EP2 receptors in cultures of the embryonic rat midbrain. EP2 and TH double-label ICC was performed on DIV7 cultures. EP2-immunoreactive (EP2+) cells were visualized by HRP-DAB and dopaminergic neurons were identified by TH-immunoreactivity (TH+) and visualized with a green fluorophore. It should be noted that only dopaminergic neurons contain TH in these cultures. Results demonstrate that EP2+ cells are present in midbrain cultures (Fig. 1A; black arrows), and that some EP2+ cells are dopaminergic neurons (Fig.1B; white arrows). Co-localization is apparent in a merged composite of bright field and fluorescent signals (Fig. 1C; black arrows). Note that not all TH+ cells are EP2+. Due to the absence of (light-quenching) DAB, a brightly fluorescent TH+ neuron devoid of EP2 immunoreactivity is prominent in the merged micrograph (Fig. 1C; dashed black arrow). In addition, not all EP2+ cells are dopaminergic neurons, although the spheroid morphology of EP2-stained cell bodies suggests that they are neurons (Fig. 1C; white arrowhead). Antibody specificity for EP2 was verified by substituting primary antibody with non-immune rabbit IgG and processing sister cultures in parallel (Fig. 1D–F). Immunostaining was absent in these cultures (compare Fig. 1D with Fig. 1A), and in the images showing a DAB-negative TH+ neuron (Fig 1E, F; arrows). Surrounding cells were also devoid of DAB staining (Fig. 1D, F).
Figure 1. EP2 receptors are present on cultured midbrain dopaminergic neurons and protect dopaminergic neurons against 6-OHDA toxicity.
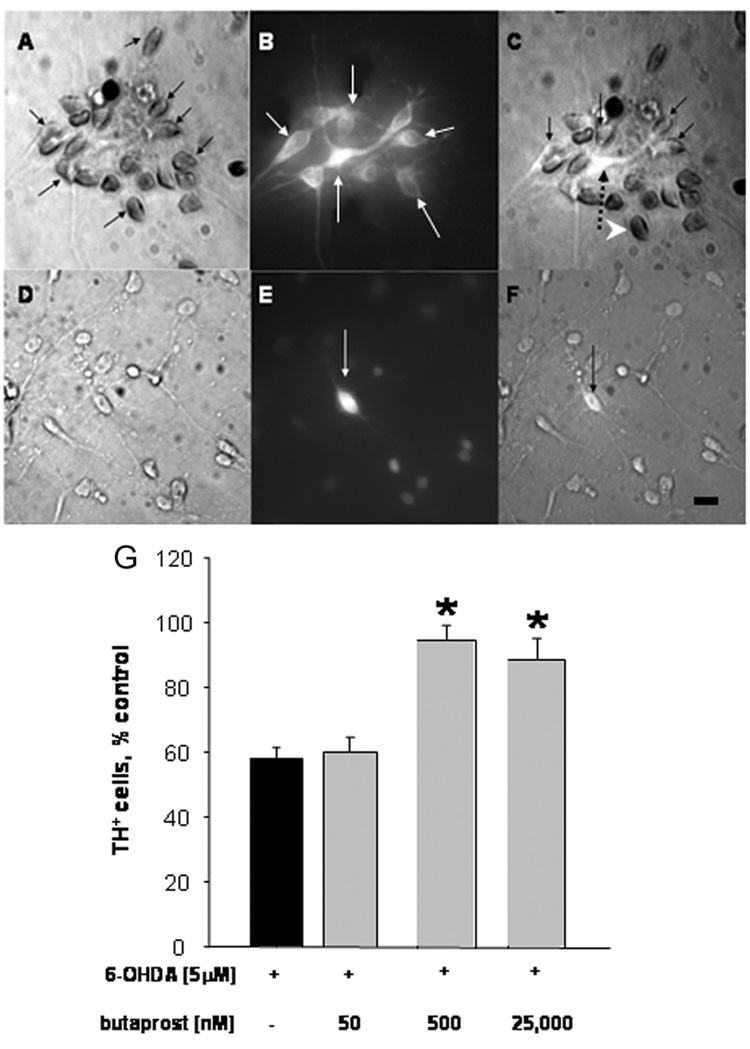
Cultures were established and processed at DIV7 for double-label ICC with anti-TH to identify dopaminergic neurons by fluorescence (B, C, E, and F), and anti-EP2 (A, C), or non-immune rabbit IgG (D, F) by peroxidase/DAB. Composite images are depicted in (C) and (F), respectively. (A): EP2+ cells (black arrows), (B): TH+ cells (white arrows); (C): TH+/EP2+ cells (black arrows), TH+/EP2− cell (dotted arrow), TH−/EP2+ cell (white arrowhead). Incubation with non-immune rabbit IgG and anti-TH only visualizes fluorescent TH+ cells (E; white arrow and F; black arrow). Scale bar = 25 µm. (G): Cultures were pre-treated for 30 minutes on DIV7 with 0 (black bar), 500 and 25,000 nM butaprost (grey bars) followed by overnight exposure to 5 µM 6-OHDA. After fixation, cultures were processed for TH ICC to identify surviving dopaminergic neurons. Bars represent percent survival of TH+ cells (+ SEM; error bars). Mean of controls: 47 ± 5, n = 22. *p < 0.001 compared to 6-OHDA alone, Dunnett’s test.
Having verified that a number of dopaminergic neurons possess EP2 receptors, we tested whether EP2 stimulation would influence the survival of dopaminergic neurons in response to low-level oxidative stress. Five micromolar concentrations of 6-OHDA will selectively and reproducibly kill 40–60% of the dopaminergic neurons in these cultures without affecting the survival of other neurons [8]. We chose to use butaprost, a well-characterized and highly preferential EP2 agonist with a reported affinity for EP2 of 91 nM [1]. Butaprost failed to cause EP1-mediated neurotoxicity at concentrations up to 25 µM, in sharp contrast to EP1 agonists and PGE2 itself [7]. Butaprost has a reported affinity for EP3 of 1.6 µM [1], but these receptors were only found in the midbrain on non-dopaminergic cells outside the SNc [7]. Cultures were incubated with 50 nM, 500 nM and 25 µM butaprost 30 minutes prior to and throughout a 24-hour challenge with 5 µM 6-OHDA (Fig. 1G). As expected, 5 µM 6-OHDA decreased the survival of dopaminergic neurons by 40% (p<0.001). Concentrations of butaprost of 500 nM and 25 µM significantly attenuated 6-OHDA toxicity (p< 0.001); numbers of surviving dopaminergic neurons were similar to those of controls (94 ± 4.9% and 89 ± 6.6%, respectively). A concentration of butaprost (50 nM) below its Ki for EP2 failed to protect, indicating that protection was due to receptor activation. Thus, stimulation of the EP2 receptor is neuroprotective for dopaminergic neurons exposed to 5 µM 6-OHDA, consistent with a report showing that EP2 receptor activation protected cultured cortical neurons against oxidative stress [13]. While EP2 presence on dopaminergic neurons suggests that neuroprotection is likely direct, it is also possible that these effects are indirect and mediated by non-dopaminergic cells that also bear these receptors.
EP2 receptors are coupled to Gs proteins, initiating a signal transduction cascade that begins with increased production of cAMP [30]. Increased cAMP levels activate a protein kinase (PKA) that initiates downstream signaling events that can change protein structure, activity, and gene expression [25, 34]. Others have shown that the cAMP/PKA pathway mediated estrogen-stimulated outgrowth of neuritic processes of dopaminergic neurons in culture [3] and similarly, dopamine-induced process outgrowth of striatal neurons [31]. Importantly, cAMP improves survival of dopaminergic neurons by itself [28], when challenged with 1-methyl-4-phenyl-pyridinium ion [16, 19], and PC12cells in response to 6-OHDA [15].
If the neuroprotective effect of EP2 activation is mediated through cAMP-dependent PKA, then it should be mimicked by increasing intracellular cAMP. Cultures were incubated with 1 mM dibutyryl cAMP, a membrane-permeable cAMP analogue, or 10 nM forskolin, an activator of adenylate cyclase [32], for 30 minutes prior to and throughout overnight incubation with 5 µM 6-OHDA. Both dibutyryl cAMP (Fig. 2A) and forskolin (Fig. 2B) significantly improved the survival of dopaminergic neurons in 6-OHDA-treated cultures (p < 0.04 and p < 0.01, respectively). However, neither was able to completely prevent 6-OHDA neurotoxicity. That is, survival of TH+ cells was improved by dibutyryl cAMP from 53 ± 4.4% to 72 ± 8.3%, and by forskolin from 43 ± 4.1% to 67 ± 7.1 %, or reductions in toxicity of about 60%. Incomplete protection could be explained by differences in the kinetics of cAMP elevation between these compounds and physiologically localized EP2/Gs-mediated induction. Higher concentrations of these agents can also be neurotoxic (unpublished observations).
Figure 2. EP-2 receptor-mediated neuroprotection requires the cAMP/PKA pathway.
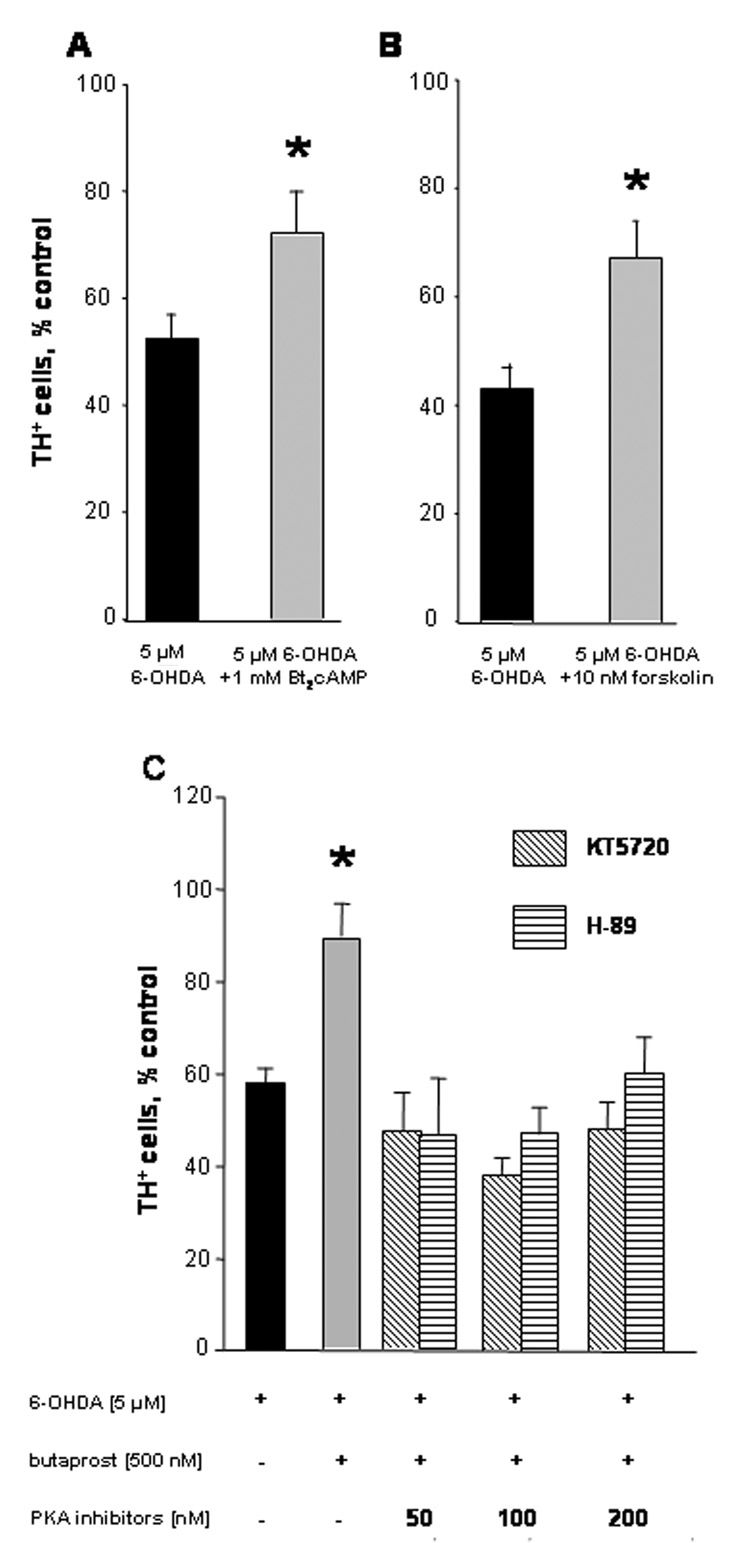
(A): DIV 7 cultures were pre-treated for 30 minutes with 1 mM dibutyryl cAMP (Bt2cAMP) or 10 nM forskolin and throughout overnight exposure to 5 µM 6-OHDA. TH ICC was performed to identify dopaminergic neurons. Bars represent percent survival of TH+ cells (+ SEM; error bars). Mean of controls: 64 ± 2.3, n = 20. *p < 0.05 compared to 6-OHDA alone, Student’s t-test. (B): DIV 7 cultures were pre-treated with 0, 50, 100, and 200 nM KT5720 or H-89 for 30 minutes before and throughout overnight exposure to 5 µM 6-OHDA. TH ICC was performed to identify dopaminergic neurons. Bars represent percent survival of TH+ cells (+ SEM; error bars). Mean of controls: 59 ± 5, n = 40. Black bar: 6-OHDA alone; gray bar: 6-OHDA + butaprost; bars with diagonal stripes: 6-OHDA + butaprost + KT5720; bars with horizontal stripes: 6-OHDA + butaprost + H-89. *p < 0.001 compared to 6-OHDA alone, Dunnett’s test.
The transcription factor CREB, a target of PKA activity, has a confirmed role in promoting neuronal survival [24]. However, cross talk between the cAMP/PKA system and other signal transduction pathways must also be considered. For example, elevated levels of cAMP stimulate the mitogen-activated protein (MAP) kinase cascade in PC12 cells [14]. To test whether PKA activity was required for neuroprotection we used two selective and structurally distinct PKA inhibitors, H-89 (Ki= 48 nM; [10]) and KT5720 (Ki= 60 nM; [21]). Neither inhibitor alone altered neuronal survival in 6-OHDA treated or control cultures during 24-hour exposure (data not shown). Next, neuronal cultures were incubated with 500 nM butaprost for 30 minutes prior to and throughout exposure to 6-OHDA in the presence or absence of the PKA inhibitors (Fig. 2C). As before, 6-OHDA killed about 40% of dopaminergic neurons (black bar; p<0.001) and butaprost protected these neurons; survival was similar to untreated control cultures (90 ± 7.2 %; grey bar). However, this protection was attenuated by the presence of 50, 100 and 200 nM concentrations of KT5720 (diagonally striped bars) and H-89 (horizontally striped bars). Essentially, the survival of dopaminergic neurons was reduced to that of cultures exposed to 6-OHDA alone. Thus, the protection of dopaminergic neurons against oxidative stress by selective activation of Gs-coupled EP2 receptors apparently required PKA activity.
Having shown that EP2 receptors are indeed present on, and, if activated, protective for dopaminergic neurons of the rat midbrain, we determined whether our observations had relevance to the human CNS. Such validation is often omitted, even though substantial interspecies variations have been documented. For example, differences in EP1–EP4 receptor mRNA expression in neuronal cortical cultures depend on the species from which they were prepared even within the same genus (rat versus mouse) [5, 36]. Therefore, we probed the SNc of two autopsy cases for the presence of EP2 receptors on dopaminergic neurons. Both cases, a 31 year-old African-American female and a 66 year-old Caucasian female, had undergone neuropathological examination and displayed no noticeable pathology indicative of any neurodegenerative disease. Post-mortem intervals were recorded as 26.5 and 29.5 hours, respectively. Midbrain sections were processed for EP2 immunohistochemistry and dopaminergic neurons were identified by the presence of neuromelanin (brown), a feature that has been employed in human neuroanatomical studies [11, 23].
Results from the first case (cause of death; arteriosclerotic cardiovascular disease) are illustrated (Fig. 3), and reflect findings from both cases. Immunoreactivity for EP2 receptors (blue-black) was detected on a number of melanin-containing (brown) dopaminergic neurons (Fig. 3A; arrows), as well as on a few non-melanized cells (e.g. see arrowhead). Because not all of the dopaminergic neurons of the human SNc contain melanin, it is possible that these neurons are also dopaminergic. The specificity of EP2 labeling was confirmed by the absence of labeling when EP2 antibodies were replaced by an identical concentration of non-immune rabbit IgG. In this case, only melanin-positive cells were visible (Fig. 3B). The presence of EP2 receptors on a majority of dopaminergic neurons in the human SNc raises the possibility that EP2 receptor activation may protect these neurons as well.
Figure 3. EP2 receptors are present on dopaminergic neurons in the human substantia nigra.

Adjacent 14 µm coronal sections of adult human midbrain were processed for immunohistochemistry with anti-EP2 (A) or non-immune rabbit IgG (B). EP2 immunoreactivity is demonstrated in melanized dopaminergic neurons (arrows) and non-melanized cells (arrowhead) in a representative section. No staining is apparent in the control section. Scale bar = 25 µm.
While non-steroidal anti-inflammatory drugs that inhibit COX activity were the first to indicate that suppression of inflammation could have a neuroprotective effect [27], COX-2 inhibition can have deleterious cardiovascular effects [29] and might adversely affect memory consolidation [9], necessitating new therapeutic approaches. Direct activation of EP2 by its natural ligand PGE2 or broadly active EP receptor agonists would be unlikely to provide effective neuroprotection because cross-activation of other EP receptors that lead to neuronal death, as we have shown for EP1, may override the neuroprotective potential of EP2 receptor activation [7]. This could explain why butaprost failed to protect dopaminergic neurons challenged directly with 2.5 nM PGE2 (data not shown). Nevertheless, it is clear that PGE2 and at least two of its receptors are critically positioned to play decisive roles in the survival of dopaminergic neurons and that EP2-selective agonists deserve further exploration as therapeutic candidates for PD.
ACKNOWLEDGEMENTS
Dr. Cynthia Shannon Weickert, currently, University of New South Wales and Dr. Joel E. Kleinman, Intramural Research Program, National Institute of Mental Health, NIH, Bethesda, MD for generously providing human autopsy tissue. Study support, in part, by the Goldman Charitable Trust (DC). EC was a fellow of the NIH Experimental Neuropathology Training Grant NS07098.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Abramovitz M, Adam M, Boie Y, Carriere M, Denis D, Godbout C, Lamontagne S, Rochette C, Sawyer N, Tremblay NM, Belley M, Gallant M, Dufresne C, Gareau Y, Ruel R, Juteau H, Labelle M, Ouimet N, Metters KM. The utilization of recombinant prostanoid receptors to determine the affinities and selectivities of prostaglandins and related analogs. Biochim Biophys Acta. 2000;1483:285–293. doi: 10.1016/s1388-1981(99)00164-x. [DOI] [PubMed] [Google Scholar]
- 2.Akil M, Kolachana BS, Rothmond DA, Hyde TM, Weinberger DR, Kleinman JE. Catechol-O-methyltransferase genotype and dopamine regulation in the human brain. J Neurosci. 2003;23:2008–2013. doi: 10.1523/JNEUROSCI.23-06-02008.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beyer C, Karolczak M. Estrogenic stimulation of neurite growth in midbrain dopaminergic neurons depends on cAMP/protein kinase A signalling. J Neurosci Res. 2000;59:107–116. [PubMed] [Google Scholar]
- 4.Bilak M, Wu L, Wang Q, Haughey N, Conant K, St Hillaire C, Andreasson K. PGE2 receptors rescue motor neurons in a model of amyotrophic lateral sclerosis. Ann Neurol. 2004;56:240–248. doi: 10.1002/ana.20179. [DOI] [PubMed] [Google Scholar]
- 5.Carlson NG. Neuroprotection of cultured cortical neurons mediated by the cyclooxygenase-2 inhibitor APHS can be reversed by a prostanoid. J Neurosci Res. 2003;71:79–88. doi: 10.1002/jnr.10465. [DOI] [PubMed] [Google Scholar]
- 6.Carrasco E, Casper D, Werner P. Dopaminergic neurotoxicity by 6-OHDA and MPP+: differential requirement for neuronal cyclooxygenase activity. J Neurosci Res. 2005;81:121–131. doi: 10.1002/jnr.20541. [DOI] [PubMed] [Google Scholar]
- 7.Carrasco E, Casper D, Werner P. PGE(2) receptor EP1 renders dopaminergic neurons selectively vulnerable to low-level oxidative stress and direct PGE(2) neurotoxicity. J Neurosci Res. 2007;85:3109–3117. doi: 10.1002/jnr.21425. [DOI] [PubMed] [Google Scholar]
- 8.Carrasco E, Werner P. Selective destruction of dopaminergic neurons by low concentrations of 6-OHDA and MPP+: protection by acetylsalicylic acid aspirin. Parkinsonism Relat Disord. 2002;8:407–411. doi: 10.1016/s1353-8020(02)00022-6. [DOI] [PubMed] [Google Scholar]
- 9.Chen C, Bazan NG. Lipid signaling: sleep, synaptic plasticity, and neuroprotection. Prostaglandins Other Lipid Mediat. 2005;77:65–76. doi: 10.1016/j.prostaglandins.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 10.Chijiwa T, Mishima A, Hagiwara M, Sano M, Hayashi K, Inoue T, Naito K, Toshioka T, Hidaka H. Inhibition of forskolin-induced neurite outgrowth and protein phosphorylation by a newly synthesized selective inhibitor of cyclic AMP-dependent protein kinase, N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide (H-89), of PC12D pheochromocytoma cells. J Biol Chem. 1990;265:5267–5272. [PubMed] [Google Scholar]
- 11.Chu Y, Kompoliti K, Cochran EJ, Mufson EJ, Kordower JH. Age-related decreases in Nurr1 immunoreactivity in the human substantia nigra. J Comp Neurol. 2002;450:203–214. doi: 10.1002/cne.10261. [DOI] [PubMed] [Google Scholar]
- 12.Coleman RA, Smith WL, Narumiya S. International Union of Pharmacology classification of prostanoid receptors: properties, distribution, and structure of the receptors and their subtypes. Pharmacol Rev. 1994;46:205–229. [PubMed] [Google Scholar]
- 13.Echeverria V, Clerman A, Dore S. Stimulation of PGE receptors EP2 and EP4 protects cultured neurons against oxidative stress and cell death following beta-amyloid exposure. Eur J Neurosci. 2005;22:2199–2206. doi: 10.1111/j.1460-9568.2005.04427.x. [DOI] [PubMed] [Google Scholar]
- 14.Frodin M, Peraldi P, Van Obberghen E. Cyclic AMP activates the mitogen-activated protein kinase cascade in PC12 cells. J Biol Chem. 1994;269:6207–6214. [PubMed] [Google Scholar]
- 15.Fujita H, Ogino T, Kobuchi H, Fujiwara T, Yano H, Akiyama J, Utsumi K, Sasaki J. Cell-permeable cAMP analog suppresses 6-hydroxydopamine-induced apoptosis in PC12 cells through the activation of the Akt pathway. Brain Res. 2006;1113:10–23. doi: 10.1016/j.brainres.2006.06.079. [DOI] [PubMed] [Google Scholar]
- 16.Hartikka J, Staufenbiel M, Lubbert H. Cyclic AMP, but not basic FGF, increases the in vitro survival of mesencephalic dopaminergic neurons and protects them from MPP(+)-induced degeneration. J Neurosci Res. 1992;32:190–201. doi: 10.1002/jnr.490320208. [DOI] [PubMed] [Google Scholar]
- 17.Hirsch EC, Hunot S, Damier P, Faucheux B. Glial cells and inflammation in Parkinson's disease: a role in neurodegeneration? Ann Neurol. 1998;44:S115–S120. doi: 10.1002/ana.410440717. [DOI] [PubMed] [Google Scholar]
- 18.Hornykiewicz O. Dopamine (3-hydroxytyramine) and brain function. Pharmacol Rev. 1966;18:244–253. [PubMed] [Google Scholar]
- 19.Hulley P, Hartikka J, Lubbert H. Cyclic AMP promotes the survival of dopaminergic neurons in vitro and protects them from the toxic effects of MPP+ J Neural Transm Suppl. 1995;46:217–228. [PubMed] [Google Scholar]
- 20.Jin J, Shie FS, Liu J, Wang Y, Davis J, Schantz AM, Montine KS, Montine TJ, Zhang J. Prostaglandin E2 receptor subtype 2 (EP2) regulates microglial activation and associated neurotoxicity induced by aggregated alpha-synuclein. J Neuroinflammation. 2007;4:2. doi: 10.1186/1742-2094-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kase H, Iwahashi K, Nakanishi S, Matsuda Y, Yamada K, Takahashi M, Murakata C, Sato A, Kaneko M. K-252 compounds, novel and potent inhibitors of protein kinase C and cyclic nucleotide-dependent protein kinases. Biochem Biophys Res Commun. 1987;142:436–440. doi: 10.1016/0006-291x(87)90293-2. [DOI] [PubMed] [Google Scholar]
- 22.Liu D, Wu L, Breyer R, Mattson MP, Andreasson K. Neuroprotection by the PGE2 EP2 receptor in permanent focal cerebral ischemia. Ann Neurol. 2005;57:758–761. doi: 10.1002/ana.20461. [DOI] [PubMed] [Google Scholar]
- 23.Malagelada C, Ryu EJ, Biswas SC, Jackson-Lewis V, Greene LA. RTP801 is elevated in Parkinson brain substantia nigral neurons and mediates death in cellular models of Parkinson's disease by a mechanism involving mammalian target of rapamycin inactivation. J Neurosci. 2006;26:9996–10005. doi: 10.1523/JNEUROSCI.3292-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mantamadiotis T, Lemberger T, Bleckmann SC, Kern H, Kretz O, Martin Villalba A, Tronche F, Kellendonk C, Gau D, Kapfhammer J, Otto C, Schmid W, Schutz G. Disruption of CREB function in brain leads to neurodegeneration. Nat Genet. 2002;31:47–54. doi: 10.1038/ng882. [DOI] [PubMed] [Google Scholar]
- 25.Mayr B, Montminy M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol. 2001;2:599–609. doi: 10.1038/35085068. [DOI] [PubMed] [Google Scholar]
- 26.McCullough L, Wu L, Haughey N, Liang X, Hand T, Wang Q, Breyer RM, Andreasson K. Neuroprotective function of the PGE2 EP2 receptor in cerebral ischemia. J Neurosci. 2004;24:257–268. doi: 10.1523/JNEUROSCI.4485-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McGeer PL, Schulzer M, McGeer EG. Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer's disease: a review of 17 epidemiologic studies. Neurology. 1996;47:425–432. doi: 10.1212/wnl.47.2.425. [DOI] [PubMed] [Google Scholar]
- 28.Michel PP, Agid Y. Chronic activation of the cyclic AMP signaling pathway promotes development and long-term survival of mesencephalic dopaminergic neurons. J Neurochem. 1996;67:1633–1642. doi: 10.1046/j.1471-4159.1996.67041633.x. [DOI] [PubMed] [Google Scholar]
- 29.Mukherjee D, Nissen SE, Topol EJ. Risk of cardiovascular events associated with selective COX-2 inhibitors. Jama. 2001;286:954–959. doi: 10.1001/jama.286.8.954. [DOI] [PubMed] [Google Scholar]
- 30.Narumiya S, Sugimoto Y, Ushikubi F. Prostanoid receptors: structures, properties, and functions. Physiol Rev. 1999;79:1193–1226. doi: 10.1152/physrev.1999.79.4.1193. [DOI] [PubMed] [Google Scholar]
- 31.Schmidt U, Pilgrim C, Beyer C. Differentiative effects of dopamine on striatal neurons involve stimulation of the cAMP/PKA pathway. Mol Cell Neurosci. 1998;11:9–18. doi: 10.1006/mcne.1998.0668. [DOI] [PubMed] [Google Scholar]
- 32.Seamon KB, Padgett W, Daly JW. Forskolin: unique diterpene activator of adenylate cyclase in membranes and in intact cells. Proc Natl Acad Sci U S A. 1981;78:3363–3367. doi: 10.1073/pnas.78.6.3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shie FS, Montine KS, Breyer RM, Montine TJ. Microglial EP2 is critical to neurotoxicity from activated cerebral innate immunity. Glia. 2005;52:70–77. doi: 10.1002/glia.20220. [DOI] [PubMed] [Google Scholar]
- 34.Sihag RK, Nixon RA. Identification of Ser-55 as a major protein kinase A phosphorylation site on the 70-kDa subunit of neurofilaments. Early turnover during axonal transport. J Biol Chem. 1991;266:18861–18867. [PubMed] [Google Scholar]
- 35.Takadera T, Ohyashiki T. Prevention of rat cortical neurons from prostaglandin E2-induced apoptosis by glycogen synthase kinase-3 inhibitors. Neurosci Lett. 2006;400:105–109. doi: 10.1016/j.neulet.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 36.Takadera T, Yumoto H, Tozuka Y, Ohyashiki T. Prostaglandin E(2) induces caspase-dependent apoptosis in rat cortical cells. Neurosci Lett. 2002;317:61–64. doi: 10.1016/s0304-3940(01)02449-1. [DOI] [PubMed] [Google Scholar]