

Abstract
Recent findings including computerized live imaging suggest that polyploidy cells transiently emerging after severe genotoxic stress (and named ‘endopolyploid cells’) may have a role in tumour regrowth after anti-cancer treatment. Until now, mostly the factors enabling metaphase were studied in them. Here we investigate the mitotic activities and the role of Aurora B, in view of potential de-polyploidisation of these cells, because Aurora B- kinase is responsible for coordination and completion of mitosis. We observed that endopolyploid giant cells are formed in irradiated p53 tumours in several ways: (1) by division/fusion of daughter cells creating early multi-nucleated cells; (2) by asynchronous division/fusion of sub-nuclei of these multinucleated cells; (3) by a series of polyploidising mitoses reverting replicative interphase from aborted metaphase and forming giant cells with a single nucleus; (4) by micronucleation of arrested metaphases enclosing genome fragments; or (5) by incomplete division in the multipolar mitoses forming late multi-nucleated giant cells. We also observed that these activities are able to release para-diploid cells, although they do so infrequently. Although after a substantial delay, apoptosis typically occurs in these cells, we also found that roughly 2% of endopolyploid cells evade apoptosis and senescence arrest and continue mitotic activities. In this article we describe that catalytically active aurora B-kinase is expressed in the nuclei of many interphase endopolyploid cells, as well as being present at the centromeres, mitotic spindle and cleavage furrow during their mitotic efforts. The totally micronucleated giant cells (containing subgenomic fragments in multiple micronuclei) represented the only minor fraction, which failed to undergo mitosis and Aurora B was absent from it. These observations suggest that most endopolyploid tumour cells are not reproductively inert and that aurora B may contribute to the establishment of resistant tumours post-irradiation.
Keywords: mitotic catastrophe, tumours, polyploidy, aurora B-kinase
Background
The application of genotoxic insults including irradiation is an established method of treatment of malignant tumours. Tumours lacking functional p53 are defective in many cell cycle checkpoints and often respond to genotoxic stress by undergoing mitotic catastrophe (MC). Although MC is defined as ‘cell death occurring during or shortly after a failed mitosis” (Kroemer et al., 2005; Galuzzi et al., 2007), p53- deficient tumours undergoing MC are resistant to genotoxic treatments. As a result of mitotic failure cells alternatively reset interphase becoming tetraploid (Casteda et al., 2004a; 2004b). Therefore, MC has been also defined as mitotic events that produce tetraploid progeny cells in the first post-damage generation (Andreassen et al., 2001). p53 mutant tumour cells that have incurred genotoxic stress and became tetraploid can continue endoreplication and achieve DNA content up to 8C–64C (Illidge et al., 2000; Chu et al., 2004). Association of genotoxic resistance with the induced endopolyploidy was found in rodent and human tumours (Baroja et al., 1998; Come et al., 1999). Our earlier observations revealed that transient endopolyploid p53- Burkitt lymphoma cells were able to facilitate DNA repair and release para-diploid mitotic progeny post-irradiation (Illidge et al., 2000; Erenpreisa et al., 2000; Ivanov et al., 2003). From these observations we hypothesised that transient endopolyploid cells, which are capable for de-polyploidisation, may actually constitute an alternative survival pathway (Erenpreisa and Cragg, 2001; 2007). Similarly Baroja et al. (1996, 1998) reported that high ploidy rodent tumour cells are capable of proliferating, despite certain peculiarities in their cell cycle. Using computerized live imaging we (Ianzini and Mackey, 2002) have demonstrated that a small proportion of endopolyploid cells formed in vitro post-mitotic catastrophe can successfully undergo polyploidy reduction and form viable clones. Prieur-Carrillo and colleagues (2003) found that about 2% of human bladder carcinoma giant cells formed after irradiation release potentially clonogenic 2N progeny. Stewenius and colleagues (2005) revealed that events of mitotic catastrophe in colorectal cancer are compatible with survival and underlined the role of anaphase bridged mitoses in clonogenic growth. Furthermore the striking live-imaging studies of Chu with co-authors (2004) on CDKN1A-deficient cells (CDKN1A is upregulated by the tumour suppressor p53 controlling G1/S checkpoint) have clearly shown the viability of the endopolyploid cells produced by multiple mitotic catastrophe events. The authors concluded that the occurrence of MC is not directly responsible for individual cell deaths. Similar observations were provided and reviewed by Rajaraman et al. (2006).
These intriguing reports underscore the importance to study further the division potential of endopolyploid cells in p53-deficient tumours. Although the presence of high-ploidy cells in malignant tumours has long been documented (Baroja et al., 1998), the biological significance of these cells is not well understood and there is still much controversy over their proliferative potential. However, if as a result of genotoxic treatment genetically unstable giant cells can give rise even to a few selected clones, these might be genetically changed promoting resistant regrowth and further tumour progression. Therefore detailed study of the mechanisms of the reproductive/apoptotic behaviour of giant cells is very important.
Here we have investigated the reproductive activities of endopolyploid cells post-irradiation in p53 defective human cell-lines through the involvement of aurora-B kinase, the essential regulator of mitosis (Carmena and Earnshaw, 2003; Vagnarelli and Earnshaw, 2004). Aurora B belongs to the group of mitosis regulators called ‘chromosome passengers’. Within this group, Aurora B-kinase provides for fidelity and procession of mitosis by coordinating chromosome alignment onto metaphase spindle with anaphase and cytotomy (Ditchfield et al., 2003). The easily recognizable immunocytochemical markers of its presence are the attachment of aurora B to centromeres in metaphase plate, to microtubules of the central mitotic spindle during anaphase B and participation in the formation of the midbody in ana-telophase. The midbody is marked by the two bands of aurora B and two lateral bands of tubulin. In immunofluorescent staining for the two proteins, these two-coloured bands and a central split in the mature midbody (the place of the centriolin ring) assigns the whole structure its unique appearance. While the main events of mitosis occur within 1 h, the midbody, which is responsible for cytotomy completion, is persisting in the cytoplasmic bridge between daughter cells for two or more hours longer (Gromley et al., 2005). Thus the midbody represents a characteristic marker of the processed mitosis. Our data reveal that catalytically active aurora B-kinase is intimately associated with the formation, divisions, and extended survival of endopolyploid cells resulting from MC, in p53 function deficient tumour cell lines.
Methods
Cell Lines
The Namalwa Burkitt’s lymphoma cells (ATCC) were grown as suspension cultures in RPMI 1640 medium, 10% Foetal Calf Serum (Gibco or Sigma) at 37°C in a 5% CO2 humidified incubator. HeLa S3 cells (ATCC) were grown either in suspension culture or as adherent clone 3. Suspension HeLa culture was grown under constant rotation in Joklik’s MEM media containing 10% heat-inactivated calf serum (Hyclone) and antibiotics. Suspension cultures were maintained in log phase of growth for at least 24 h prior to irradiation. Namalwa cells were further cultivated by replenishing culture medium every 48–72 h, and HeLa S3, every 24 h.
HeLa adherent clone 3 cells were grown as monolayer in F-10 medium (Hyclone) containing 10% heat-inactivated fetal calf serum (Sigma or Hyclone) and antibiotics (100 U/ml penicillin and 100 mkg/ml streptomycin) in a 37°C incubator supplied with 5% CO2, either on 13 mm polylysine-coated coverglasses in 24x wells, for immunocytochemistry and DNA cytometry or in a T-25 tissue culture flasks, for live-imaging.
To determine the cells in S-phase, BrdU was added to concentration 10 mkg/ml in the cell culture for 60 min prior to cell fixation on slides with methanol. DNA denaturation was performed by 2N HCl, 37° C for 20 min. After washes in PBS, the primary and secondary antibodies were applied (Table 1). In some experiments, proteasome inhibitors (Sigma) Mg-132 (5 mkg/ml), inhibitor of calpain (25 mkg/ml), and lactocystin (10 mkg/ml) were added for 2hs prior to cell harvest. Autophagic vacuoles were detected by monodansylcadaverin (MDC) and by Sa-b-galactosidase. For MDC (Sigma) staining the cultures were incubated with 0.05mM MDC at 37°C for 60 min followed by fixation in 4% paraformaldehyde, washing twice in PBS. The slides were mounted into Perm Mount and immediately scored in the DAPI channel. For Sa-b-galactosidase detection the instruction of the Sigma kit (Code CS0030) was followed (staining was extended overnight).
Table 1.
Antibodies: source and usage
| Primary antibodies against: | Secondary antibodies |
|---|---|
| Aurora-B-kinase polyclonal in a rabbit(Abcam, ab2254), 1:300 | goat a-rabbit- IgG-.Alexa Fluor 480 or 594(Invitrogen) 1:400 |
| a-tubulin mouse monoclonal (Sigma, B-512) 1:2,000 | goat a-mouse IgG-.Alexa Fluor 480 or 594(Invitrogen) 1:400 |
| b-tubulin mouse monoclonal (Neomarkers; clone DM1B) 1:100 | Goat a-mouse IgG-Alexa Fluor 480(Invitrogen) 1:400 |
| Cyclin B mouse monoclonal (SC-245; Santa Cruz Biotech) 1:50 | goat a-mouse IgG-.Alexa Fluor 480 or 594(Invitrogen) 1:400 |
| Human-anti-centromere/kinetochore(Antibodies Inc; 15-234), 1:100 | sheep a-h-IgG-FITC (Dr. MS Cragg) 1:50 |
| Lamin B, goat polyclonal (Santa Cruz Biotech),1:50 | Donkey a-goat IgG Alexa Fluor 594(Invitrogen) 1:400 |
| Active caspase-3 rabbit polyclonal (Promega, G7481) 1:50 | goat a-rabbit- IgG-.Alexa Fluor 594(Invitrogen) 1:400 |
| phospho-H3ser10rabbit polyclonal (Santa Cruz Biotech.) 1:300 | goat a-rabbit- IgG-.Alexa Fluor 480 or 594(Invitrogen) 1:400 |
| Bromodeoxyuridine (BdU) mouse monoclonal(Invitrogen) 1:200 | Goat a-mouse IgG-Alexa Fluor 480(Invitrogen) 1:400 |
Irradiation
Irradiation for live- imaging at the University of Iowa was delivered at room temperature using a PANTAK Bipolar 2HF320 irradiator (200 kV, 0.35 Cu/1.5 Al filter). Dosimetry was performed using a Victoreen electroscope; the dose rate was 1.27 Gy min−1. In vitro cell culture irradiation was applied using a Gulmay D3 225 X-ray source at a dose rate of 0.77 Gy/min. In all experiments, a single dose of 10 Gy was delivered.
LSDCAS Imaging Analysis
Large Scale Digital Cell Analysis System (LSDCAS) a live cell imaging and analysis system that includes quantitative cell analysis software, amenable to determine the kinetics of various cellular mechanisms on a cell-by-cell basis (Ianzini and Mackey, 2002; Davis et al., 2007). For live-imaging, HeLa adherent clone 3 cells were maintained for irradiation by the scheme described above. 48 h prior to irradiation 1×105 cells were plated as a cell suspension and placed into the CO2 incubator. After irradiation the cells were returned to the incubator for 2 h; at which time an equal volume of fresh, complete and warmed up media were added and the flask was positioned onto the LSDCAS stage for imaging acquisition for 72h. One hundred random fields were manually chosen. Division-related events were categorised as follows: normal, normal followed by cleavage regression (sister cells fusion), and un-related cell fusion (non-sister cell fusion). The number of cells alive at the end of the experiment was also counted.
Immunofluorescent (IF) staining
Suspension cells were pelleted, re-suspended in FCS and cytospun onto polylysine-coated slides; cultures on cover-glasses were rinsed before fixation in PBS and FCS. Cells were allowed to semi-dry for 1 min (when tubulins were detected, drying was not allowed) and were fixed at −20°C in methanol followed by short 5–10 rinses in cold acetone or in methanol/acetone (1:1) at −20°C. 1-min rinse in 70% methanol and several in PBS followed, afterwards blocking in 1% bovine-serum albumin in PBS (for some stains adding 0.01–0.05% tween-20) was performed for 15 min, application of the primary antibody and further manipulations were performed by usual schemes. A list of antibodies is presented on Table 1. Post-staining was performed with Propidium Iodide (5 mkg/ml), DAPI (1 mkg/ml) or 1 mkg/ml of amino actinomycin (7-AAD). Cells were finally embedded in Prolong Gold (Invitrogen).
DNA image cytometry
For DNA in situ cytometry, cells were fixed in ethanol/acetone (1:1, v:v) for 30 min at room temperature and then stained with by modified Feulgen reaction using Toluidine blue. Images were obtained using a Leitz Ergolux L03–10 microscope equipped with a calibrated Sony DXC 390P colour video camera. DNA content was measured as the integral optical density in the green channel or in the red channel with interference filter 289 nm using Image Pro Plus 4.1 software (Media Cybernetics; REO 2001, Riga, Latvia). The stoichiometry of DNA staining was verified using the values obtained for metaphases compared to anaphases and telophases (ratio 2.0); arbitrary diploid (2C) DNA values were averaged from measuring 50 anaphases in non-treated tumour cells. The device error was estimated as to be 0.5%. The variation coefficient for DNA content was also assessed in normal human lymphocytes where it was determined as being in the range 2–5%, while for HeLa mitotic cells it reached 20%.
Fluorescent in situ hybridization (FISH)
HeLa cells were harvested, treated with 75 mM KCl at RT for 10 min and fixed in five changes of methanol/glacial acetic acid (3:1), this suspension was dropped onto slides and allowed to dry. Satellite probes for chromosomes 10 and X were used in the kits provided by Molecular Cytogenetics (QBIOgene) by the applied instruction. These chromosomes were chosen as containing three normal copies and not participating in clonal markers (Macville et al., 1999). The number of labels per individual nuclei was counted. The results were grouped as normal (three labels) and abnormal (all other counts) and compaired in pairs by Fisher’s Exact Test (http://www.matforsk.no/ola/fisher.htm).
Fluorescent, bright field, and confocal microscopy
Leica confocal laser microscope DM 600 and Leitz Ergolux L03–10 microscope were used.
Results
Irradiation causes anaphase bridging and bi-nucleation
Irradiation (10Gy dose) induces a G2 arrest that persists for 8–10 h in HeLa cells. Cells then enter mitosis displaying elevated mitotic indices of 10–11% at 24 h and 15%, at 48 hours. About 25% of metaphases in HeLa become arrested and many of them restitute interphase as micronucleated (many small nuclear vesicles enclosing separate chromosomes or their groups) or mono-nucleated polyploidy cells. However, the majority of irradiated HeLa cells proceed through anaphase. About 70% of all HeLa cells in the first mitosis at 19–24 hrs and 96% of cells in the second mitosis at 42– 48 hrs display anaphase bridges due to dicentric chromosomes (checked by CREST-immunoserum staining for centromere proteins), as compared to 2–5% in controls. As a result, 40% of cells become bi-nucleated on day 1 post-irradiation. Live-imaging confirmed that mitotic cleavage furrow regression between nuclei-bridged daughter cells was responsible for the initiation of bi-nuclearity in these cells, as 82.2% of daughter cells fused (n=107), of which 96.7% were still alive at the end of the three-day filming session (Figure 1 and Table 2). The nuclei of the bridged post-mitotic daughter cells often display irregular contours and/or contain in addition a few micronuclei, however segregation as judged by DNA image cytometry was mostly equal (Fig. 2a–c, e–f). Also FISH studies using pericentromeric probes for chromosomes X and 10, which do not participate in HeLa clonal markers, revealed that a small proportion of nuclei (5%) aneusomic by these chromosomes was present in the control population, which increased up to 15% after the first cell cycles in irradiated samples (Table 3). A second smaller wave of bridged anaphases appeared 3 days post- irradiation, with increased bi-nucleation following 24 h later.
Fig. 1.

A series of images of irradiated HeLa cells in LINDCAS system starting live-imaging 12 h post-irradiation – four cells (A,B,C, D) divide remaining connected by nuclear bridges and then each daughter pair regresses cleavage furrow forming a bi-nucleated cell.
Table 2.
Results of computed live imaging.
| Analyzed cells that divide normally in 72 h | 16.6% (107/645 |
|---|---|
| Analyzed cells that divide normally and then fuse | 82.2% (88/107) |
| Cells that divide -> fuse and live to the end of the movie | 96.7% (85/88) |
| non-sister cell fusions in 72 h | 3% (20/645) |
Fig. 2.

Characteristic patterns of the DNA cytometry images of HeLa cells: e–c,e,f – DNA- bridged karyoplasts arising by bi-polar mitoses in the first three days post-irradiation; h–j – late multinuclear cells on days 6–9 with circularly arranged subnuclei. ‘2c’ DNA content is an arbitrary value which correspond average of 50 anaphase halfs in the control not irradiated cells.
Table 3.
Statistical results of studies of aneusomy by FISH reaction for pericentromeric probes of chromosomes X and 10 in HeLa cell nuclei, in control and 48 h after irradiation.
| Chr#X | Chr#10 | |||
|---|---|---|---|---|
| Samples | Control | 48 h | Control | 48h |
| Number of normal nuclei (3 labels) | 577 | 450 | 637 | 667 |
| Number of abnormal nuclei | 28 | 69 | 35 | 101 |
| % abnormal(aneusomic) nuclei | 4.85 | 15 | 5.5 | 15 |
| Fischer Exact Test, p-value | p<0.001 | p<0.001 | ||
Irradiated Namalwa cells arrest in G2 for 24 hs, start aberrant mitoses from d.2, which are most arrested in metaphase showing increased mitotic index on dd.3–4 (Ivanov et al., 2003). Contrary to HeLa which mostly pass anaphase and form bi- and multinucleated giant cells (MNGC), the later contain three or more nuclei, the majority of Namalwa cells restitute the arrested metaphases as mono-nucleated giant cells (MONGC).
Aurora-B localises normally in cells during the first division events post-irradiation
During mitosis in untreated tumour cells, the Aurora B-kinase localises sequentially to the centromeres, spindle midzone and equatorial cortex, and the mid-body (Figure 3a and c). Despite the presence of bridged chromosomes in irradiated cells well seen on Fig. 2a,b, aurora-B kinase localised normally and the microtubules forming the mitotic spindle, the anaphase midzone, and the midbody, were organised correctly (Figure 3b and c). However, it was noted that some of these cells showed additional diffuse staining in the cytoplasm (Figure 3b).
Fig. 3.

Aur B/a-tubulin/DNA cellular localisation in the first mitotic activities of HeLa cells: (a) non-treated control: in metaphase Aur B is equatorially attached to centrosomes (arrow), a mid-body connecting post-telophasic sister cells is arrowheaded (; (b–d) after irradiation: (b) in metaphase; (c) a proper mid-body in late anaphase; d) formation of a bi-nuclear cell enclosing mid-body in its cytoplasm (arrow) – a witness of post-anaphasic or post-mitotic re-union of sister nuclei/cells. For. 3a,b,d - Aurora B green, alfa-tubulin red, DAPI – blue; for 3c – Aurora B is red, alfa-tubulin green, DAPI – blue. Bars=20 μm
Polyploidy doubles with a periodicity of one generation time
After the second round of mitosis (day 2) the frequency of bi-nucleated cells (4C) reduced in HeLa cells from 40% to 20%. However, the incidence of MNGC increased from 10% to about 70%, with most multinucleated cells containing 8 chromosome sets. Polyploidy increased each day post-damage. By day 4, HeLa cells typically displayed DNA contents of 8C, 16C, and 32C corresponding to completion of four polyploidising cycles-phases over the 4 day period (Figure 4). The proportion of polyploid cells reached 50–60%. Consistent with our previously published results, the same polyploidy limit (32C, occasionally 64C) was exhibited in irradiated Namalwa cells which also increased in ploidy by a daily doubling (Erenpreisa et al., 2000).
Fig. 4.

A histogram of the DNA content in giant HeLa cells on d.4 after irradiation (n=110). It is seen that 8C and 16C cells are in the cell cycle, while 24C and 32C represent apparently the end-point of polyploidisation.
Asynchronous mitosis occurs in multinucleated cells
At 2–4 days post-irradiation, approximately 30% of HeLa cells were paired with long DNA bridges (Fig. 5 a–g) surrounded by the nuclear envelope and thin layer of cytoplasm as judged by the staining for lamin B and tubulin (Fig. 5f). Frequently the two daughter cells comprising the DNA-bridged pair exhibited a loss in synchrony during their second or third division, such that one daughter nucleus/cell was in mitosis, while the other was in interphase (Fig. 2c, e, f). About 5% of MNGC exhibited asynchronous division of only one of sub-nucleus (Fig. 5a, d–e). The same phenomenon, although at a smaller scale and at later terms was found in Namalwa (Fig. 5h). These divisions had normal or nearly normal aurora-B positive mid-bodies, suggesting that elements of mitosis were retained during this event (Fig. 5a–c, g–h). Similar quantities of DNA appeared to be segregating by this asynchronous division, as judged by voxel intensity of the DAPI signal (see Figs. 5d and e). These secondary divisions were also often DNA-and nuclear envelope bridged (Fig. 5 d and f). These apparently symmetric “daughter nuclei” on the either side of the bridge suggest that early bi-and multi-nucleated cells have mostly originated from successful previous mitosis followed by fusion of daughter nuclei or cells. At a smaller scale, this process continued later, where both asynchronous bi-polar mitosis and cytoplasmic mid-bodies were found in multi-nucleated cells (Fig. 5 h–i). Synchronous divisions of two sub-nuclei in bi-nucleated cells were observed but only rarely (Fig. 2d). Totally micronucleated cells, which represented a minority, were aurora B-negative (6a, arrow); they never entered mitosis and often died (by necrosis). However, some of them, as seen from inclusion of BrdU and increasing size, grew indefinitely solely by endoreduplication (data not shown).
Fig. 5.

Asymmetric bi-polar divisions of sub-nuclei in multinucleated HeLa and Namalwa cells after irradiation: (a–f) day 2; (g–i) days 4–7. (a,d,f) lamin B(red)/a-tubilin (green)/DAPI; (b,c,) Aur B (red)/a-tubulin (green)/DAPI; (g–i) Aur B (green)/a-tubulin (red)/DAPI; (e) DAPI only, fluorescence intensity measured. The presence of midbodies (arrowed) indicates to the mitotic character of these divisions. On (a) telophase chromatin figures are outlined by lamin B; (b)- after mitotic division of a presumably tetraploid HeLa cell, one of the daughter cell nuclei (asterics) has further divided a-mitotically; (c) - a multilobulated HeLa giant cell is joined with a smaller one by a tubulin bridge with remnant midbody; (d–e) is illustrating an equal division of one of the nuclei in a bi-nuclear cell; (g) - a low ploidy HeLa cell is still connected to the high ploidy multinuclear cell by a tubulin bridge with a mid-body. This well shaped mid-body displays a clear central split – the place of the centriolin ring, necessary for the completion of cytokinesis; (h) a sample was collected after 2h-treatment with lactocystin, proteasome inhibitor. On ‘h’ two subsequent asymmetric reduction bi-polar divisions of a giant Namalwa cell are seen displaying two persisting midbodies. These divisions result in a small ana-telophase. Persistence of the two subsequent midbodies and small final mitotic figures suggest that the second division rapidly followed the first, most probably without intermitting S-phase. (i) shows a midbody within a multinucleated cell – a probable result of the failed cytotomy and reverse of asymmetric mitotic division in a HeLa cell. Bars=20 μm.
In summary, the majority of early multinucleated He la cells (days 1– 3) resulted from a series of bi-polar mitosis/sister fusion events, while approximately 10-fold smaller amount, by non-sister fusions of stressed cells, and a minor population, by micronucleation of the failed mitosis. For irradiated Namalwa, the majority of emerged endopolyploid cells (around 70%) were mononucleated.
Aurora B-kinase is present in MONC and MN GC nuclei but is targeted for degradation
In control cells, those undergoing mitosis stain positively for aurora-B. Aurora-B is also present in the nuclei of G2-cells and rare giant cells, albeit far less abundant than in mitotic cells (Fig. 3a). About 60% of giant cells on days 3–4 post-irradiation display enhanced nuclear positivity for aurora B. However, aurora-B-negative MONG and MNGC cells without signs of degradation are also present in the population (Fig. 6 b). On day 5 the positive giant cells comprise about 40%, on day 7 – only 10%. MONGC sometimes have a very strong karyoplasmic reaction for aurora B (Fig. 6a). Expression of aurora-B in the interphase nuclei of giant cells was much more prevalent after treatment with the proteasome inhibitors, lactocystin, MG132 or inhibitor of calpain (2h), suggesting that aurora-B is normally targeted for proteasome-mediated degradation when present in the nucleus (see Fig. 6d and e). We also noted that aurora-B was absent from annexin-V positive apoptotic cells (Fig. 6f), but again its expression could be rescued in them by the application of proteasome inhibitors (not shown).
Fig. 6.

Distribution of Aurora-B kinase in and among irradiated cells: (a) strongly positive MONGC, however always negative micronucleated HeLa cells (arrow); (b) a proportion of cells with Aurora-B-positive nuclei in Namalwa; (c) A positive MNGC of HeLa (green for Aurora, counterstained with red for a-tubulin); (d) thin dotted arrays of aur-B (green) in the chromatin of the nucleus restituting from the arrested polyploid metaphase: fixed 2 hs after application of Mg-132; (e) a rare finding: clear aurora-B dots over minimally condensed preprophasic chromosomes red – for Aur-B, green – a false colour for overexposed DAPI; (f) annexin-positive dying cells (red ) are negative for aurora-B kinase (Namalwa cells). Bars=20 μm.
As aurora-B is generally considered a mitotic kinase, and its activity is stimulated primarily by its mitotic partner protein, inner centromere protein INCENP (Adams et al., 2001), we studied the co-localisation of Aurora B with centromere proteins as revealed by CREST immunoserum(see Fig. 7a–c). In metaphase plates of control cells aurora B colocalises with individual centromeres (Fig. 7a). After irradiation, the majority of aurora B-rich nuclei of giant cells contain one large central nucleolus and clustered centromeres. Aurora B positive nuclear foci in giant interphase cells rarely fully colocalise with individual centromeres (Fig. 7b) but rather are found as larger patches in centromere clusters, mostly in the perinucleolar heterochromatin, in chromocentres, and, in some cells, at the nuclear envelope (Fig. 7c–d). To test whether the aurora-B present in giant interphase nuclei was active, we used the immunoprobe for phospho-H3ser10, a specific substrate of aurora-B kinase. Some giant cell nuclei contained speckles of phosphorylated histone H3, mostly around nucleoli (Fig. 7e). However, giant aberrant metaphases were extensively positive for phosphorylated histone H3, indicating a high level of aurora-B kinase activity (Fig. 7f).
Fig. 7.
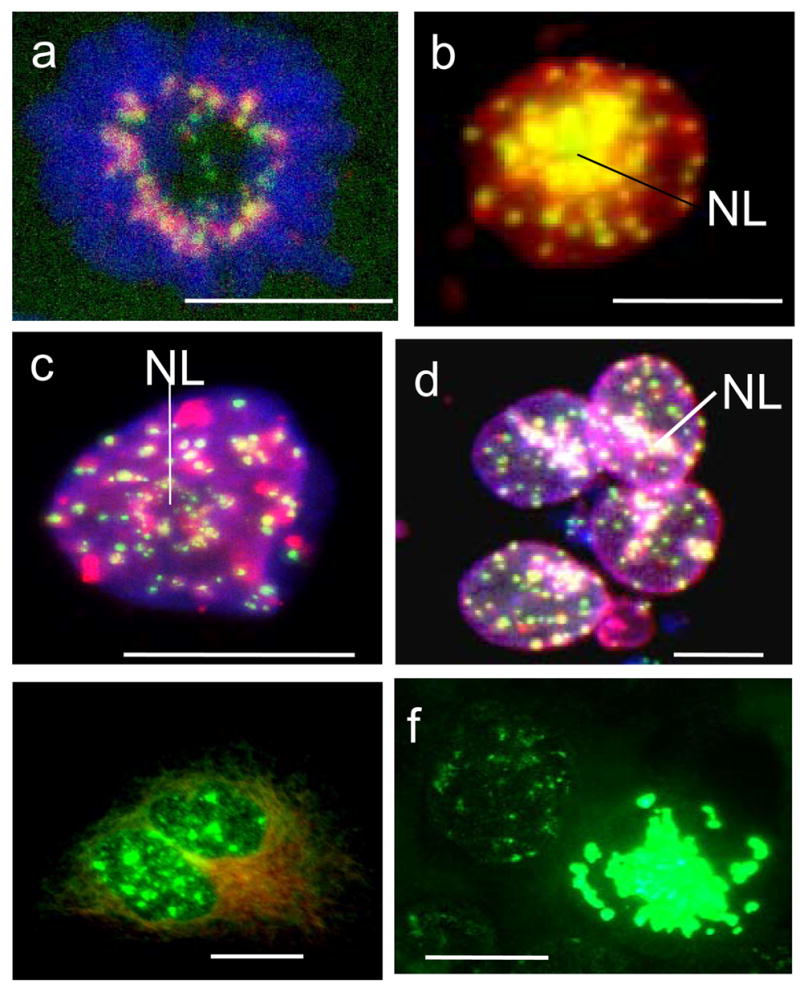
Specific activities of aurora-B kinase in polyploid tumour cells after irradiation: (a) in control, foci of aurora-B (Alexa, red) colocalise with kinetochore proteins (FITC, green) in the metaphase plate (DAPI); (b–f) after irradiation: (b) nearly full colocalisation of aurora-B and kinetochores, especially heavy around central nucleolus (NL) in the interphase cell, presumably G2; DAPI channel is not overlaid; (c) large patches of aurora-B at centromere clusters and chains in a late giant cell; (d) heavy precipitation of aurora-B onto the centromeres around nucleoli (NL), tendency of colocalisation at the nuclear envelope, however absence of colocalisation in most centromeres inside the nuclei; (e,f) IF for substrate of aurora-B phosphorylation phospho-H3ser10(green) in interphase and mitotic giant cells shows catalytical activity of kinase. This activity is very high in the numerous polyploid aberrant mitoses arrested at this stage (on d.4 post-irradiation) in metaphase, one of them is seen on ‘f’. (a–d) sequential scanning by confocal microscope in three colour channels. Bars=20 μm.
Multipolar mitoses and formation of late multinucleated cells
A small proportion of HeLa cells undergo tri-polar mitoses in the first days post-irradiation, which show Aurora B-kinase-positive mid-body with three spindle twigs (Fig. 8a). These cells often complete division into three daughters (not shown). However, the majority of multipolar mitoses in giant cells which they attempt before day 5 become arrested in metaphase showing clumped, poorly condensed, often partially polytenised chromosomes (Fig. 8b). In metaphase arrested cells Aurora-B-kinase was often found both on the chromosomes and in cytoplasm. Massive apoptosis which was observed around days 5–6, closely follows this period in both cell-lines as determined by cell morphology and caspase 3 activity (not shown), involving around 30% of giant HeLa cells and up to 80–90% of giant Namalwa cells as reported previously (Illidge et al., 2000; Ivanov et al., 2003). In survivors, occasionally from day 5–6 and then more frequently from day 10 post-irradiation, MNGC and MOGC underwent true endomitosis (as initially defined by Geitler (1937)). In these cells individual chromosomes were condensed but the nuclear envelope and nucleoli remained intact. Despite the absence of nuclear envelope break down, aurora-B strongly accumulated at these endomitotic chromosomes (Fig. 8c). At the same late period, in a number of multipolar mitoses aurora-B was found concentrated on metaphase centromeres of well condensed chromosomes (Fig. 8d) and on the central spindles of multipolar (usually tri-polar) anaphase cells (Fig. 8e). So, after a series of mitoses aborted in metaphase at the earlier post-irradiation period, these cells attained the capacity to pass a spindle checkpoint and were found segregating their DNA and attempting to cleave cytoplasm into a number of discrete progeny. In most cases, full cytokinesis did not occur and mid-bodies did not form. However, in rarer cells, daughter sub-cells were released tearing (disjoining?) the chromosome bridges as previously shown (Erenpreisa et al., 2005). The non-segregated anaphases reverted in a circular or semi-circular arrangement of sub-nuclei at the periphery of giant cytoplasm (Fig. 8f). DNA image cytometry revealed circularly arranged post-mitotic subnuclei (Fig. 2h–j), ranging from 1C to 4C (Fig. 2h) and often in odd numbers. However, about 60% of such subnuclei were para-2C. At later times (1–2 weeks post-irradiation), when regrowth of para-diploid mitotic fraction was renewed, post-mitotic giant cells underwent replicative senescence as revealed by positive staining for monodansylcadaverine and Sa-β-galactosidase, absence of BrdU inclusion and by aurora B-negativity (not shown). However about 10% of late giant cells were aurora B positive (Fig. 8f) and also labelled for BrdU and cyclin B (not shown) indicating that they remained in an active cell cycle during at least 2 week observation. This data demonstrate that active aurora-B may contribute to the long-lasting reproductive capacity of endopolyploid cells.
Fig. 8.
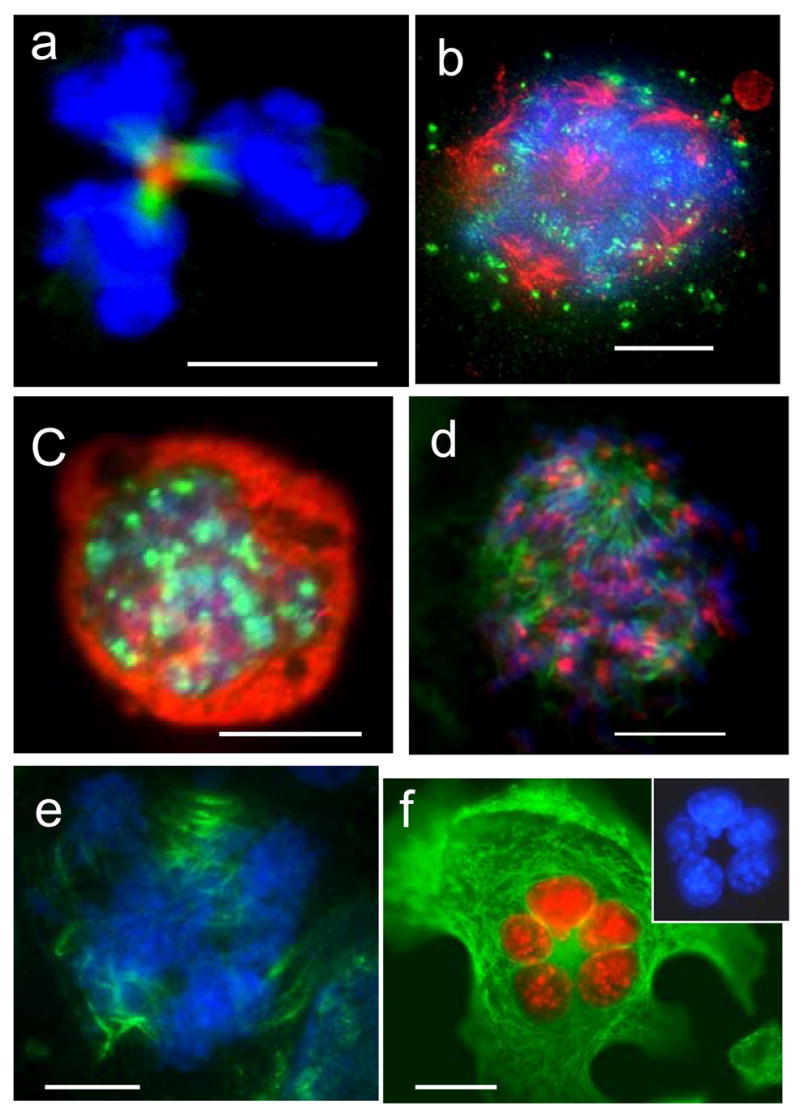
Multipolar mitoses in irradiated samples: (a) clear central mid-body, aurora B (red) and a-tubulin (green) in a tri-twigged spindle, however telophase nuclei (DAPI) possess very irregular contours; (b) arrested multipolar metaphase: alfa-tubulin revealing poles is red, aurora B (green) is poorly attached to chromosomes (blue), which are not sufficiently condensed and seem polytenised; (c) true endomitosis with condensed chromosomes (DAPI) and richly bound to them aurora-B (green), a-tubulin is red; (d,e) the cells were fixed 2hs after application of the calpain inhibitor: (d) a tri-polar metaphase, chromosomes are well condensed and aurora B (red) is richly attached to them; a-tubulin of the poles is green; (e) three spindles are seen decorated in their central part with the tandemly arranged aurora-B-positive grains (green; tubulins were not stained), while DAPI reveals several anaphase figures; (f) radial arrangement of the equal-sized aurora-B-imunopositive (red) nuclei in the large adherent HeLa cell with active microtubular skeleton (green). On insert – the nuclei in DAPI channel. (a) 2days; (b) 5 days; (c–e) 6 days; (f) 8 days post-irradiation. Bars=20 μm.
The reproducible schedule of main reproductive activities of the two cell-lines after genotoxic insult is summarised in Table 3. Although with differences, the main timing of events is similar in them.
Discussion
In developmental systems, endopolyploidy is usually a terminal point of cell differentiation (Nagl, 1978). In Drosophila metamorphosis salivary glands, the giant polytenic cells cycle only between G1 and S-phase, with the down-regulated activity of mitotic cyclin kinase complex (Edgar and Orr-Weaver, 2001). However, this lack of mitotic activity is clearly not the case for most p53-deficient giant tumour cells formed by MC. Firstly, we observed that endopolyploid giant cells are formed in irradiated p53 tumours as a result of aberrant mitotic activities in several ways: (1) by division/fusion of daughter cells creating early MNGC; (2) by asynchronous division/fusion of sub-nuclei of these multinucleated cells; (3) by a series of polyploidising mitoses resetting replicative interphase from arrested metaphase and forming MOGC; (4) by micronucleation of arrested metaphases; or (5) by incomplete division in the multipolar mitoses forming late MNGC. We also observed that these activities are able to release para-diploid cells, although they do so infrequently. Some of those descendants are likely prone to start the next round of MC. All these events are displayed after genotoxic stress in a reproducible sequence extended for 1–2 weeks as summarized in Table 3. Roughly, they take the first period of aberrant mitoses, most bridged, the second (d.3–5) of increasing polyploidisation, mostly due to metaphase arrests of polyploidising mitoses, the third (d.5–6) – segregation by multipolar anaphases, and the fourth, from d.7, starts regrowth of paradiploid clones. It is evident that between day 5 and 6 is a border-line, when the cells shift from polyploidisation cycles to de-polyploidisation and it is also the start of apoptotic crisis for the cells which are evidently unable to do so. Therefore it is worth to note that preceding this trigger, endopolyploid Namalwa cells are undergoing on days 4–5 the delayed wave of recombination repair of DNA double-strand breaks which protects them from apoptosis (Ivanov et al., 2003). Roughly only 10–20% of giant cells evade apoptosis (Illidge et al., 2000; Ivanov et al., 2003) and as we see in this study, about 10% of those overcome further the senescence arrest and continue cyclic mitotic activities. So, the fraction of potential long-living survivors originating from endopolyploid cells may comprise in this tumour 1–2%. This estimation correspond the counts obtained by live-imaging analysis in other system (Prieur-Carrillo et al., 2003). The same reproductive activities involving giant cells were found by us in the untreated tumour cell cultures during prolonged cell cultivation (10–20 days), although occurring slowly and to a much smaller proportion and extent.
Secondly, we showed that four of the five aforementioned mechanisms producing endopolyploid cells were marked by the presence of aurora-B kinase. This chromosome passenger protein was localised at centromeres, the anaphase midzone, and the midbody. Moreover, it was present in the interphase nuclei of giant cells, where immunoprobing for phosphor-H3 revealed that it was catalytically active (supposedly in the G2-state). This activity was particularly high in true endomitotic and giant mitotic cells. During de-polyploidisation stage, aurora B shifted from centromeres to central spindles in cells starting multipolar anaphases and occasionally was found as a midbody in tripolar mitoses undergoing cytotomy. Similarly, association of overexpression of aurora B with increased proliferative potential of megakaryocytes has been shown in transgenic mice (Zhang et al., 2004) and in polyploidisation through aborted mitoses in vascular smooth vessel cells (Nagata et al., 2005), suggesting common mechanisms.
Thus in line with some previous reports (Chu et al., 2004, Baroja et al., 1998; Stewenius et al., 2005), we herein report that endopolyploid p53 defficient tumour cells are derived by active, yet aberrant mitotic events and that also de-polyploidisation can occur by mitotic mechanisms, although mostly modified. The only exception to this observation are entirely micronucleated cells which loose the ability to enter mitosis (and apoptosis as well) and which in our models represented a minor fraction. There is some evidence and arguments in literature that chromosome bridges in colon cancer do not prevent clonogenic growth, while multipolar mitoses likely may produce genomically less perspective cells (Stewenius et sl., 2003; Gisselson, 2005). However, the interference of recombination and true endomitosis found by us between these events may account for more crucial effects on the genomes than only their simple segregation and needs further studies on different models.
The literature data report that deregulation of aurora B kinase, both by overexpression or knockdown leads to polyploidy (Nguyen and Ravid, 2006). This paradox becomes more understandable in view of the present results showing the necessity of both mitosis and its failure or reverse for the formation of endopolyploid cells.
Overexpression of aurora-B kinase has been shown to be a characteristic of many tumours correlating with genetic instability, endopolyploidy and aggressive behaviour (Adams et al., 2001; Giet et al., 2005). These characteristics are particularly prevalent in tumours lacking functional p53 or its target (Hamada et al., 2003; Ditchfield et al., 2003). Deregulated aurora-kinase B is capable of transforming cells in vitro. Moreover, the in vivo anti-tumour activity of the inhibitors of aurora-kinases has been reported and the question of their use as anti-cancer targets is currently under investigation (Warner et al., 2003; Wilkinson et al., 2007; Tao et al., 2007).
Conclusions
We have shown that p53-deficient endopolyploid tumour cells are formed mostly as a result of abortive mitoses and that part of these cells retain a long-lasting reproductive potential and vitality supported by the activity of aurora B-kinase. This data suggests that endopolyploid cells and aurora B kinase may contribute to the formation of genotoxically resistant growth.
Table 4.
Schedule of the tumour cell reproductive activities following 10 Gy insult
| Days after irradiation | HeLa | Namalwa |
|---|---|---|
| 1 | Bridged mitoses/reunion of sister nuclei/cells | Arrest in G2 |
| 2 | Bi-nucleation, bipolar mitoses of their subnuclei forming MNGC (about 70%); metaphase arrests of mono- nucleated cells | Aberrant mitoses, many of them are bridged. |
| 3 | Bi-polar mitoses of subnuclei in bi-and MNGC. A new, smaller wave of bi- nucleation. | Metaphase arrests, emergence of MONGC (about 80%) and MNGC (about 20%) |
| 4–5 | Metaphase arrests of most polyploidy mitoses, increase of polyploidy to 32 C; bi-polar mitoses of subnuclei in MNGC; A new smaller wave of multinucleation* | Metaphase arrests ofpolyploidy mitoses, incr ease of polyploidy to 32C; bi-polar mitoses of subnuclei in MNGC |
| 5–6 | Major cessation of further polyploidisation, apoptotic crisis, true endomitosis, bi- and multi-polar cell divisions processing anaphase in surviving cells | Major cessation of further polyploidisation, apoptotic crisis, true endomitosis, bi-and multi-polar cell divisions processing anaphase in surviving cells |
| 7–9 | Beginning of clonogenic regrowth of small para-diploid cells; cessation of proliferative activities and appearance of senescence markers in the majority of giant cells; continuation of mitotic attempts in the minor fraction of giant cells. | Beginning of clonogenic regrowth of small para-diploid cells; cessation of proliferative activities and appearance of senescence markers in the majority of giant cells; continuation of mitotic attempts in the minor fraction of giant cells. |
The second and possibly next waves of polyploidisation were noticed also in later terms
Acknowledgments
This work was supported by grant of the Latvian Scientific Council 05.1634.1, by the scientific exchange grant between USA and Latvia, by the grant of the Royal Society allowing exchange visits between Brighton, Manchester and Riga. In Iowa, this work was partly funded by the National Institutes of Health grant 1R33CA94801 and by the Whitaker Foundation Special Opportunity Award. Work in SPW and TMI laboratories are funded by Cancer Research-UK. We acknowledge the work of the undergraduate student of the Latvian University Evgenia Kuznetsova who assisted in interactive DNA cytometry, FISH staining, counts, and statistics funded by the Latvian Scientific Council. The advice of Prof. Dr. M. Hausmann (Heidelberg) for FISH staining technique is also acknowledged.
Footnotes
Author’ contributions: JE – designed experiments and carried them out cytologically and immunocytochemically in Riga, Iowa, Brighton, and Manchester, drafted the paper; SPW – participated in experimental design, in establishing of some immunocytochemical methods, in microscopy, in the article draft and editting; EAK maintained in Iowa the HeLa cell culture, helped in the gamma-irradiation, worked at the LSDCAS imaging acquisition, analyzed the LSDCAS images, carried out DNA cytometry; FI and MAM conceived the LSDCAS studies, participated in its design and coordination and drafted the part of the paper related to the LSDCAS studies. FI also performed the gamma-irradiation at UI; APA – contributed in the cytological analysis of HeLa irradiated and control samples; PJD designed and wrote the event analysis software for LSDCAS and helped in the analysis of the LSDCAS images; GP – carried out the irradiation experiments on HeLa in Riga and participated in the analysis of their results; AI – carried out with JE the experiments on Namalwa in Manchester and participated in analysis of the results; TMI – participated in experimental design, analysis of results, drafting and editing of the manuscript.
References
- Adams RR, Eckley DM, Vagnarelli P, Wheatley SP, Gerloff DL, Mackay AM, Svingen PA, Kaufmann SH, Earnshaw WC. Human INCENP colocalizes with the Aurora-B/AIRK2 kinase on chromosomes and is overexpressed in tumour cells. Chromosoma. 2001;110:65–74. doi: 10.1007/s004120100130. [DOI] [PubMed] [Google Scholar]
- Andreassen PR, Lacroix FB, Lohez OD, Margolis RL. Neither p21WAF1 nor 14–3–3 sigma prevents G2 progression to mitotic catastrophe in human colon carcinoma cells after DNA damage, but p21WAF1 induces stable G1 arrest in resulting tetraploid cells. Cancer Res. 2001;61:7660–68. [PubMed] [Google Scholar]
- Baroja A, de la Hoz C, Alvarez A, Ispizua A, Bilbao J, de Gandarias JM. Genesis and evolution of high-ploidy tumour cells evaluated by means of the proliferation markers p34(cdc2), cyclin B1, PCNA and 3[H]-thymidine. Cell Prolif. 1996;29:89–100. [PubMed] [Google Scholar]
- Baroja A, de la Hoz C, Alvarez A, Vielba R, Sarrat R, Aréchaga J, de Gandarias JM. Polyploidization and exit from cell cycle as mechanisms of cultured melanoma cell resistance to methotrexate. Life Sci. 1998;62:2275–82. doi: 10.1016/s0024-3205(98)00208-2. [DOI] [PubMed] [Google Scholar]
- Carmena M, Earnshaw WC. The cellular geography of aurora kinases. Nature Rev Mol Cell Biol. 2003;4:842–54. doi: 10.1038/nrm1245. [DOI] [PubMed] [Google Scholar]
- Castedo M, Perfettini JL, Roumier T, Andreau K, Medema R, Kroemer G. Cell death by mitotic catastrophe: a molecular definition. Oncogene. 2004a;23:2825–37. doi: 10.1038/sj.onc.1207528. [DOI] [PubMed] [Google Scholar]
- Castedo M, Perfettini J-L, Roumier T, Yakushijin K, Horne D, Medema R, Kroemer G. The cell cycle checkpoint kinase Chk2 is a negative regulator of mitotic catastrophe. Oncogene. 2004b;23:4353–61. doi: 10.1038/sj.onc.1207573. [DOI] [PubMed] [Google Scholar]
- Chu K, Teele N, Dewey MW, Albright N, Dewey WC. Computerized video time lapse study of cell cycle delay and arrest, mitotic catastrophe, apoptosis and clonogenic survival in irradiated14–3–3s and CDKN1A (p21) knockout cell lines. Rad Res. 2004;162:270–86. doi: 10.1667/rr3221. [DOI] [PubMed] [Google Scholar]
- Come MG, Skladanowski A, Larsen AK, Laurent G. Dual mechanism of daunorubicin-induced cell death in both sensitive and MDR-resistant HL-60 cells. Br J Cancer. 1999;79:1090–97. doi: 10.1038/sj.bjc.6690174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis PJ, Kosmacek EA, Sun Y, Ianzini F, Mackey MA. The large-scale digital cell analysis system: an open system for nonperturbing live cell imaging. J Microsc. 2007;228:296–308. doi: 10.1111/j.1365-2818.2007.01847.x. [DOI] [PubMed] [Google Scholar]
- Ditchfield C, Johnson VL, Tighe A, Ellston R, Haworth C, Johnson T, Mortlock A, Keen N, Taylor SS. Aurora B couples chromosome alignment with anaphase by targeting BubR1, Mad2, and Cenp-E to kinetochores. J Cell Biol. 2003;161:267–80. doi: 10.1083/jcb.200208091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar BA, Orr-Weaver TL. Endoreplication cell cycles: more for less. Cell. 2001;105:297–306. doi: 10.1016/s0092-8674(01)00334-8. [DOI] [PubMed] [Google Scholar]
- Erenpreisa JA, Cragg MS, Fringes B, Sharakhov I, Illidge TM. Release of mitotic descendants by giant cells from irradiated Burkitt’s lymphoma cell line. Cell Biol Int. 2000;24:635– 48. doi: 10.1006/cbir.2000.0558. [DOI] [PubMed] [Google Scholar]
- Erenpreisa J, Cragg MS. Mitotic death: A mechanism of survival? A review Cancer Cell Int. 2001;1:1. doi: 10.1186/1475-2867-1-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erenpreisa J, Cragg MS. Cancer: A matter of life-cycle? Cell Biol Int. 2007;31:1507–10. doi: 10.1016/j.cellbi.2007.08.013. [DOI] [PubMed] [Google Scholar]
- Erenpreisa J, Kalejs M, Ianzini F, Kosmacek EA, Mackey MA, Emzinsh D, Cragg MS, Ivanov A, Illidge TM. Segregation of genomes in polyploidy tumour cells following mitotic catastrophe. Cell Biol Int. 2005;29:1005–11. doi: 10.1016/j.cellbi.2005.10.008. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Maiuri MC, Vitale I, Zischka H, Castedo M, Zitvogel L, Kroemer G. Cell death modalities: classification and pathophysiological implications. Cell Death Differ. 2007;14:1237–43. doi: 10.1038/sj.cdd.4402148. [DOI] [PubMed] [Google Scholar]
- Geitler L. Die Analyse des Kernbaus und der Kernteilung der Wasserlaufer Gerris lateralis un Gerris lacustris (Hemiptera Heroptera) un die Somadifferenzierung. Z Zellforsch. 1937;26:641–72. [Google Scholar]
- Giet R, Petretti C, Prigent C. Aurora kinases, aneuploidy and cancer, a coincidence or a real link? TRENDS in Cell Biology. 2005;15:241–50. doi: 10.1016/j.tcb.2005.03.004. [DOI] [PubMed] [Google Scholar]
- Gisselson D. Mitotic instability in cancer : Is there method in the madness? Cell cycle. 2005;4:1007–10. doi: 10.4161/cc.4.8.1884. [DOI] [PubMed] [Google Scholar]
- Gromley A, Yeaman C, Rosa J, Redick S, Chen CT, Mirabelle S, Guha M, Sillibourne J, Doxsye SJ. Centriolin anchoring of exocyst and SNARE complexes at the midbody is required for secretory-vesicle-mediated abscission. Cell. 2005;123:75–87. doi: 10.1016/j.cell.2005.07.027. [DOI] [PubMed] [Google Scholar]
- Hamada M, Yakushijin Y, Ohtsuka M, Kakimoto M, Yasukawa M, Fujita S. Aurora2/BTAK/STK15 is involved in cell cycle checkpoint and cell survival of aggressive non-Hodgkin’s lymphoma. Br J Haematol. 2003;121:439–47. doi: 10.1046/j.1365-2141.2003.04311.x. [DOI] [PubMed] [Google Scholar]
- de la Hoz C, Baroja A. Proliferative behaviour of high-ploidy cells in two murine tumour lines. J Cell Sci. 1993;104:31–6. doi: 10.1242/jcs.104.1.31. [DOI] [PubMed] [Google Scholar]
- Ianzini F, Mackey MA. Development of the Large-Scale Digital Cell Analysis System. Radiation Protection Dosimetry. 2002;99:289–293. doi: 10.1093/oxfordjournals.rpd.a006787. [DOI] [PubMed] [Google Scholar]
- Illidge T, Cragg M, Fringe B, Olive P, Erenpreisa Je. Polyploid giant cells provide a survival mechanism for p53 mutant cells after DNA damage. Cell Biol Int. 2000;24:621–33. doi: 10.1006/cbir.2000.0557. [DOI] [PubMed] [Google Scholar]
- Ivanov A, Cragg MS, Erenpreisa J, Emzinsh D, Lukman H, Illidge TM. Endopolyploid cells produced after severe genotoxic damage have the potential to repair DNA double strand breaks. J Cell Sci. 2003;11:4095–106. doi: 10.1242/jcs.00740. [DOI] [PubMed] [Google Scholar]
- Kroemer G, El-Deiry WS, Golstein P, Peter ME, Vaux D, Vandenabeele P, Zhivotovsky B, Blagosklonny MV, Malorni W, Knight RA, Piacentini M, Nagata S, Melino G. Nomenclature Committee on Cell Death. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death. Cell Death Differ. 2005;(Suppl 2):1463–67. doi: 10.1038/sj.cdd.4401724. [DOI] [PubMed] [Google Scholar]
- Macville M, Schröck E, Padilla-Nash H, Keck C, Ghadimi BM, Zimonjic D, Popescu N, Ried T. Comprehensive and Definitive Molecular Cytogenetic characterization of HeLa cells by spectral karyotyping. Cancer Res. 1999;59:141–50. [PubMed] [Google Scholar]
- Nagata Y, Jones MR, Nguyen HG, McCrann DJ, St Hilaire C, Schreiber BM, Hashimoto A, Inagaki M, Earnshaw WC, Todokoro K, Ravid K. Vascular smooth muscle cell polyploidization involves changes in chromosome passenger proteins and an endomitotic cell cycle. Exp Cell Res. 2005;305:277–91. doi: 10.1016/j.yexcr.2004.12.028. [DOI] [PubMed] [Google Scholar]
- Nagl W. Endopolyploidy and polyteny in differentiation and evolution. Amsterdam: North-Holland/Elsevier Publ; 1978. [Google Scholar]
- Nguyen HG, Ravid K. Tetraploidy/aneuploidy and stem cells in cancer promotion: The role of chromosome passenger proteins. Review J Cell Physiol. 2006;208:12–22. doi: 10.1002/jcp.20565. [DOI] [PubMed] [Google Scholar]
- Prieur-Carrillo G, Chu K, Lindqvist J, Dewey WC. Computerized video time-lapse (CVTL) analysis of the fate of giant cells produced by X-Irradiating EJ30 human bladder carcinoma cells. Rad Res. 2003;159:705–12. doi: 10.1667/rr3009. [DOI] [PubMed] [Google Scholar]
- Rajaraman R, Guernsey DL, Rajaraman MM, Rajaraman SR. Stem cells, senescence, neosis and self-renewal in cancer. Cancer Cell Int. 2006;6:25. doi: 10.1186/1475-2867-6-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewenius Y, Gorunova L, Jonson T, Larsson N, Hoglund M, Mandahl N, Mertens F, Mitelman F, Gisselsson D. Structural and numerical chromosome changes in colon cancer develop through telomere-mediated anaphase bridges, not through mitotic multipolarity. Proc Natl Acad Sci USA. 2005;102:5541–46. doi: 10.1073/pnas.0408454102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagnarelli P, Earnshaw WC. Chromosomal passengers: the four dimensional regulation of mitotic events. Chromosoma. 2004;113:211–22. doi: 10.1007/s00412-004-0307-3. [DOI] [PubMed] [Google Scholar]
- Warner SL, Bearss DJ, Han H, Von Hoff DD. Targeting Aurora-2 kinase in cancer. Mol Cancer Ther. 2003;2:589–95. [PubMed] [Google Scholar]
- Wilkinson RW, Odedra R, Heaton SP, Wedge SR, Keen NJ, Crafter C, Foster JR, Brady MC, Bigley A, Byth KF, Barrass NC, Mundt KE, Foote KM, Heron NM, Jung FH, Mortlock AA, Boyle FT, Green S. AZD1152, a selective inhibitor of aurora B kinase, inhibits human tumour xenograft growth by inducing apoptosis. Clin Cancer Res. 2007;13:3682–88. doi: 10.1158/1078-0432.CCR-06-2979. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Nagata Y, Yu G, Nguyen HG, Jones MR, Toselli P, Jackson CW, Tatsuka M, Todokoro K, Ravid K. Aberrant quantity and localization of Aurora-B/AIM-1 and survivin during megakaryocyte polyploidization and the consequences of Aurora-B/AIM-1–deregulated expression. Blood. 2004;103:3717–26. doi: 10.1182/blood-2003-09-3365. [DOI] [PubMed] [Google Scholar]