

Abstract
The glutathione S-transferase P1 (GSTP1) is involved in multiple cellular functions, including phase II metabolism, stress response, signaling, and apoptosis. The mechanisms underlying the significantly high GSTP1 expression in many human tumors are, however, currently not well understood. We report here that the GSTP1 gene is a heretofore unrecognized downstream transcriptional target of the tumor suppressor p53. We identified a p53-binding motif comprising two consecutive half-sites located in intron 4 of the GSTP1 gene and is highly homologous to consensus p53-binding motifs in other p53-responsive genes. Using a combination of electrophoretic mobility shift assay and DNase I footprinting analyses, we showed that wild-type p53 protein binds to the GSTP1 p53 motif and luciferase reporter assays showed the motif to be transcriptionally functional in human tumor cells. In a temperature-sensitive p53-mutant cells, levels of both p21/WAF1 and GSTP1 gene transcripts increased time dependently when cells were switched from the inactive mutant state to the wild-type p53 state. Small interfering RNA — mediated reduction of p53 expression resulted in a specific decrease in GSTP1 expression and in tumor cells with mutated p53; adenovirally mediated expression of wild-type p53 increased GSTP1 expression significantly. In a panel of early-passage brain tumor cultures from patients, high levels of GSTP1 transcripts and protein were associated with wild-type p53 and, conversely, low GSTP1 levels with mutant p53. p53 expression knockdown by small interfering RNA increased cisplatin sensitivity. The ability of wild-type p53 to transcriptionally activate the human GSTP1 gene defines a novel mechanism of protecting the genome and, potentially, of tumor drug resistance.
Introduction
The human glutathione S-transferase P1 (GSTP1) protein plays several critical roles in both normal and neoplastic cells. Notably, it functions in cellular phase II metabolism by catalyzing the conjugation of the tripeptide glutathione with a wide variety of electrophilic compounds, including genotoxins, carcinogens, and chemotherapeutic agents (1, 2). GSTP1 also regulates cellular signaling through binding to a variety of important signaling proteins, including c-Jun NH2-terminal kinase, ASK1, and TRAF2, thereby regulating their downstream signaling functions (3, 4). In most normal tissues, GSTP1 expression is very low but increases significantly in tumors arising from these tissues, including brain tumors, leukemias, lymphomas, sarcomas, and many carcinomas (5-7). The high GSTP1 protein expression and its nuclear localization have been shown to correlate not only with drug resistance but also with increased tumor grade and poor patient survival (6) and to enhance survival of tumor cells under hypoxic condition.
The p53 tumor suppressor, similar to the GSTP1 gene, is involved in multiple important cellular and molecular events, including cellular proliferation and differentiation, DNA damage/repair, and apoptosis (8, 9). Mutations in the p53 gene are among the most frequent somatic genetic changes observed in human cancers, and consequently, p53 mutational status has been extensively studied for its correlation with tumor grade, patient survival, and treatment outcome in many cancers (10). The results of these studies have, however, been inconsistent with some showing a positive correlation of p53 mutations with tumor growth, progression, and drug resistance (11-14), whereas others showed the mutant p53 status to be deleterious to survival and to favor drug sensitivity (15-17). The findings indicate a high level of complexity in the role of p53 in human cancer and suggest that multiple upstream and downstream pathways of p53 may interact to mediate its effects in this disease.
Despite the important role that GSTP1 plays in oncogenesis (18) and therapeutic outcome (19) in many human tumors, currently, the molecular mechanisms and pathways regulating its expression in tumors are not well understood. Because GSTP1 and the transcription factor p53 are both independently involved in regulating cell growth, cell death, and survival and in cellular protection against genotoxins, we examined, in this study, the possibility that a cross-talk between these two genes could contribute to the regulation of GSTP1 expression in tumor cells. Using both primary tumors and cell lines of human malignant gliomas, we discovered that an intronic p53 consensus binding site present in the GSTP1 gene was transcriptionally active and responsive to wild-type but not mutant p53. In glioma cells, the GSTP1 gene expression was significantly affected by the p53 mutational status and p53-mediated GSTP1 gene activation may contribute to cisplatin resistance.
Results
Identification of an Intronic p53-Binding Motif in the Human GSTP1 Gene
We conducted a search for putative p53-binding sites in the human GSTP1 gene using a web-based search engine3 and identified a putative p53-binding motif located in intron 4 of the GSTP1 gene and spanning nucleotides +983 and +1,002. The motif comprised two half-sites with no spacer nucleotide between them (Fig. 1A) and sequence wise was highly homologous to the consensus p53-binding sites in several wellknown p53 target genes, such as p21/WAF1, MDM2, GADD45, and ribosomal gene cluster (RGC; Fig. 1A; refs. 20-24). The absence of a nucleotide spacer between the two half-sites is consistent with the 0- to 13-nucleotide spacer requirement in functional p53 motifs and similar to that of the p53 motifs found in p53-responsive genes, such as MDM2, GADD45, and RGC.
FIGURE 1.

Identification and functional characterization of the GSTP1 p53-binding sites. A. Comparison of the p53-binding motif in intron 4 of the GSTP1 gene with consensus p53-binding site (24) and those in known p53-responsive genes. Spacer nucleotides between two p53 half-sites are indicated. Nucleotides that do not match the consensus p53-binding motifs are in lower case and underlined. B. Binding of wild-type p53 protein to the GSTP1 p53-binding motif. Electrophoretic mobility shift assay was done with oligonucleotides corresponding to the GSTP1 p53-binding motif and protein extracts from Sk-Ala cells containing wild-type p53. a, lane 1, 32P-labeled GSTP1 p53 motif with no protein extract; lane 2, 32P-labeled probe and p53-containing protein extracts; lane 3, competition with 50-fold excess of unlabeled GSTP1 p53 motif probe; lanes 4 to 6, competition experiments with 50-, 100-, and 200-fold excess of unlabeled (cold) consensus binding sites (24). b, supershift of p53 protein bound to GSTP1-binding motif. The p53 monoclonal antibody DO-1 was added to protein extract from Sk-Ala cells preincubated with the 32P-end—labeled probe. Arrow, supershifted band. C. DNase I footprinting analysis of wild-type p53 protein binding to the GSTP1 p53 motif. Wild-type p53 protein extracts from Sk-Ala cells were used in these studies. Lane 1, Maxam-Gilbert sequencing of the nucleotide sequence (A+G) of the footprint probe; lane 2, sequence of the GSTP1 p53 motif without protein extract; lanes 3 and 4, contained 200 and 400 μg of p53-containing protein from Sk-Ala cells, respectively. Brackets, the sequence of the protected region corresponds to the GSTP1 p53-binding motif.
Wild-Type p53 Binds to the GSTP1 p53 Motif
The results of the analysis of the binding affinity and specificity of wild-type p53 protein to the GSTP1 p53 motif, done by electrophoretic mobility shift assay, are summarized in Fig. 1B and show significant binding of proteins from wild-type p53-expressing Sk-Ala cells (lane 2) to the 32P-end— labeled GSTP1 p53-binding motif (a). The binding was outcompeted by cold GSTP1 p53 motif (lane 3) and progressively competed out by increasing amounts of cold consensus p53-binding site (lanes 4-6), indicating specificity of the binding. Fig. 1B (b) shows a supershift (arrow) of the p53 protein/p53-binding motif complex on addition of an anti— p53-specific antibody.
The results of the DNase I footprinting analysis (Fig. 1C) show the Maxam-Gilbert sequencing (adenine and guanine) of the footprint probe (lane 1) and the standard footprint pattern of the DNase I—digested GSTP1 intron 4 probe (lane 2). Fig. 1C (lanes 3 and 4) shows increased protection of the GSTP1 p53-binding sites in the presence of increasing amounts of protein from the wild-type p53-expressing Sk-Ala cells. The results of the gel shift/supershift and DNase footprinting analysis, taken together, show the specificity of the binding of wild-type p53 protein to the GSTP1 p53-binding sites.
Wild-Type p53 Transactivates Luciferase Gene Expression via the GSTP1 p53 Motif
The structures of the four vectors used in these studies are shown in Fig. 2. The results summarized in Table 1 show that untransfected and pFR-Luc—transfected tumor cells express low levels of luciferase at both temperatures 32°C and 38°C. In contrast, luciferase expression increased significantly in pRGC-p53BS-Luc—transfected cells maintained at 32°C for 24 and 48 h relative to cells at 38°C. In cells transfected with pGSTP1-p53BS(3X)-Luc and pGSTP1-p53BS(4X)-Luc containing three and four GSTP1 p53 motifs, there was a marked increase in luciferase expression, consistent with wild-type p53-mediated transactivation of the GSTP1 gene via the GSTP1 p53-binding sites.
FIGURE 2.

Wild-type p53 activates luciferase reporters driven by the GSTP1 p53-binding motif. Structure of GSTP1 p53-binding motif-containing vectors.
TABLE 1.
Functional p53 Time Dependently Activates Promoters Containing GSTP1 p53 Motif
| Vectors | 38°C |
32°C |
||
|---|---|---|---|---|
| 24 h | 48 h | 24 h | 48 h | |
| None | 1.86 ± 0.44 | 1.53 ± 0.14 | 1.48 ± 2.57 | 0.24 ± 0.20 |
| pFR-Luc | 0.14 ± 0.18 | 0.87 ± 0.66 | 0.14 ± 0.00 | 3.89 ± 2.15 |
| pRGC-p53BS-Luc | 2.00 ± 0.10 | 0.91 ± 0.91 | 3,755.05 ± 112.61 | 6,095.77 ± 190.09 |
| pGSTP1-p53BS(3X)-Luc | 2.11 ± 0.43 | 1.81 ± 0.47 | 393.72 ± 27.36 | 1,734.02 ± 83.67 |
| pGSTP1-p53BS(4X)-Luc | 0.47 ± 0.81 | 1.26 ± 1.09 | 2,592.93 ± 245.43 | 8,846.25 ± 69.83 |
NOTE: Sk-Ala cells were transfected with various constructs and maintained at 38°C or 32°C for 24 and 48 h before luciferase assays. Relative luciferase activity was computed as described earlier and SDs from three independent experiments.
GSTP1 Gene Expression Is Induced by Wild-Type p53 in Tumor Cells
The results of the Northern blot analysis (Fig. 3A) show a significant increase in GSTP1 gene transcripts in Sk-Ala cells maintained at 32°C (wild-type p53) compared with those at 38°C (mutant p53). On switching from the mutant (38°C) to the wild-type (32°C) p53 state, the levels of GSTP1 gene transcripts in the cells increased time dependently to 4.3-, 12-, 18-, and 29-fold of controls after 6, 12, 24, and 48 h, respectively. In contrast, no change in GSTP1 gene transcript levels was observed in the parental SKLMS-1 cells on switching from 38°C to 32°C. Transcripts of p21/WAF1, a p53 responsive gene, also increased significantly on switching of Sk-Ala cells from the mutant p53 state at 38°C to the wild-type p53 state at 32°C for 48 h (Fig. 3B). On switching back to 38°C for 72 h, p21/WAF1 transcripts were undetectable. The increase in GSTP1 and p21/WAF1 gene transcripts at 32°C and decline to the basal level at 38°C indicate that both genes are under the control of the functional wild-type p53. The Western blot analysis (Fig. 3C) also shows higher levels of GSTP1 protein in Sk-Ala cells expressing wild-type p53 (32°C) than those with the mutant p53 (38°C). Consistent with these observations, tumor cells infected with the adenoviral vector Ad/CMV-p53 expressed higher levels of GSTP1 protein than the controls (Fig. 3D). Small interfering RNA (siRNA)-mediated down-regulation of wild-type p53 in U87MG cells was associated with a reduction of GSTP1 protein expression (Fig. 3E) and a decrease of luciferase expression in cells carrying the pGSTP1-p53BS(3X)-Luc construct compared with cells transduced with nonspecific control siRNA (Fig. 3F). Taken together, these results show that wild-type p53, but not mutant p53, transcriptionally activates the human GSTP1 gene.
FIGURE 3.

GSTP1 gene expression is induced by wild-type p53 in tumor cells. Sk-Ala cells stably express wild-type p53 at 38°C and inactive mutant at 32°C. SKLMS-1 expresses inactive p53 at both temperatures. A. GSTP1 gene transcript was examined by Northern blotting in both cell lines maintained at 38°C for 48 h and then switched to 32°C for 6, 12, 24, and 48 h. The membranes were blotted for GSTP1, p53, p21/WAF1, and the loading control GAPDH. Band signals were quantified densitometrically and used to compute GSTP1 to GAPDH ratios. B. GSTP1 gene is transcriptionally activated in cells with wild-type. Sk-Ala cells were maintained at 38°C (mutant p53; lane 1), switched to 32°C (wild-type p53) for 48 h (lane 2), returned to 38°C (mutant p53) for 72 h (lane 3), harvested, and subjected to Northern blot analyses for transcripts of the GSTP1, p21/WAF1, and GAPDH genes. C. p53 protein with the wild-type conformation/function increases GSTP1 protein expression. Sk-Ala cells were maintained at 38°C and switched to 32°C for 48 h. Equal loading is indicated by Coomassie blue staining. D. Forced expression of the wild-type p53 increased GSTP1 expression. MGR1 cells were infected with the adenovirus carrying wild-type p53, Ad/CMV-p53, and the control virus Ad/CMV. After 48 h, infected cells were harvested and subjected to Western blotting to determine expression of GSTP1 and p53. Equal loading is indicated by Coomassie blue staining. E and F. p53 knockdown suppresses GSTP1 expression. In E, U87MG cells were transfected with nonspecific control siRNA and p53 siRNA for 48 h and lysed and total protein was extracted. Western blot analyses were then done to determine levels of p53, GSTP1, and β-actin. In F, U87MG cells were transfected with pGSTP1-p53BS(3X)-Luc/pRL-CMV and control siRNA or p53 siRNA and luciferase activity was determined 48 h after transfection, as described earlier. Columns, mean of three independent experiments; bars, SD. Student’s t test was subsequently carried.
Relationship between p53 Mutational Status and GSTP1 Expression in Brain Tumor Cells
The relationship between p53 and GSTP1 was examined in seven human gliomas in early-passage culture and the results are shown in Table 2. DNA sequencing showed three of the cell lines (MGR3, UW28, and UW281) to harbor the wild-type p53 gene, whereas UW78 cells had a G→A silent mutation at codon 36. MGR2 contained a G→A transition at codon 273, resulting in a substitution of arginine to histidine. MGR1 and UW228 both had C→A transversions at codon 155, changing the encoded amino acid from threonine to asparagine. GSTP1 gene transcript levels were determined by densitometry of the Northern blots and normalized against glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The results (Fig. 4A) showed that wild-type p53-containing cell lines contained 2- to 5-fold higher levels of GSTP1 gene transcripts compared with those with mutant p53. GSTP1 protein expression, determined by Western blotting (Fig. 4B), also showed GSTP1 to be consistently elevated in tumors with the wild-type p53 compared with those with the mutant forms.
TABLE 2.
Mutational Status of p53 Gene in Brain Tumor Cells
| Brain Tumor Cell Line | p53 Status | Mutated Exon | Mutated Codon | Nucleotide Changes | Amino Acid Substitution |
|---|---|---|---|---|---|
| MGR3 | Wild-type | - | - | - | - |
| UW28 | Wild-type | - | - | - | - |
| UW281 | Wild-type | - | - | - | - |
| UW78 | Wild-type | 4 | 36 | CCG→CCA | None |
| MGR2 | Mutant | 8 | 273 | CGT→CAT | Arg→His |
| MGR1 | Mutant | 5 | 155 | ACC→AAC | Thr→Asn |
| UW228 | Mutant | 5 | 155 | ACC→AAC | Thr→Asn |
FIGURE 4.
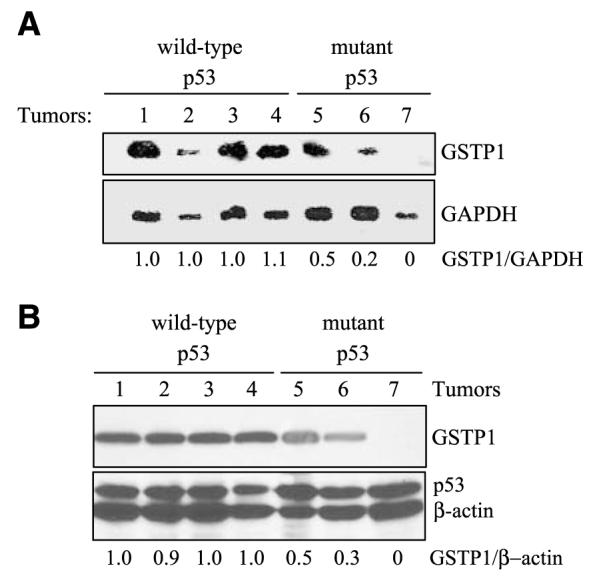
Relationship between p53 mutational status and GSTP1 expression in brain tumor cells. Seven early-passage cultures of human gliomas were used in these studies. A. Higher GSTP1 transcript levels in brain tumor cells with wild-type p53. GSTP1 gene transcript levels were examined by Northern blotting and relative GSTP1 expression (GSTP1/ GAPDH) was determined using MGR3 as standard. B. Increased GSTP1 protein expression in brain tumor cells correlates with the presence of wild-type p53. Levels of GSTP1 and p53 proteins were determined by Western blotting. Relative GSTP1 levels are expressed as GSTP1/β-actin with MGR3 as expression standard.
Furthermore, we hypothesized that p53-mediated GSTP1 gene activation may play a role in the resistance of glioblastomas to cisplatin given the ability of GSTP1 to inactivate cisplatin. If our hypothesis is correct, p53 expression knockdown by siRNA is expected to lead to reduced GSTP1 protein levels and increased cisplatin sensitivity. We used human glioblastoma U87MG cells in these studies as they express the wild-type p53 (25) and are highly resistant to cisplatin with an IC50 of 134 μmol/L. U87MG cells were ideal for these studies as they can be transfected with high efficiency, which was critical for sufficient p53 knockdown by siRNA. Moreover, U87MG cells transfected with nonspecific siRNA were highly resistant to cisplatin, whereas those with the p53 siRNA showed significantly increased sensitivity (P = 0.027; Table 3). Collectively, these data indicate that wild-type p53 expression correlates with high GSTP1 gene activity in brain tumors and that high levels of wild-type p53 contribute to cisplatin resistance of glioblastoma cells, in part, due to p53-mediated GSTP1 transcriptional up-regulation.
TABLE 3.
Reduced Expression of Both p53 and GSTP1 Rendered Resistant Glioblastoma Cells to Cisplatin Treatments
| Control | CDDP | |
|---|---|---|
| Nonspecific control siRNA | 100% | 72.5 ± 6.6% |
| p53 siRNA | 100% | 45.8 ± 4.5% |
NOTE: U87MG cells in 96-well culture plates were transfected with the nonspecific control siRNA or p53 siRNA, and 24 h after transfection, cells were treated with and without 200 μmol/L cisplatin for 24 h and harvested and cell viability was determined using the CellTiter Blue Assay kit. All data represent means and SDs from three independent experiments. Student’s t test was conducted to determine whether a significant difference existed in the survival rate between the nonspecific control siRNA — transfected and p53 siRNA — transfected cells. P value was subsequently determined to be 0.027.
Discussion
The tumor suppressor p53 is a transcriptional factor, the biological effects of which are mediated largely through the downstream target genes that it transactivates. This study reports that the human GSTP1 gene is a heretofore unrecognized downstream target of p53 and is transcriptionally activated by p53 through binding of p53 to a p53-binding motif present in the GSTP1 gene, located in intron 4 of the GSTP1 gene at nucleotides +983 to +1,002 relative to the transcription start site. The motif 5′-GGGCAGGCATGGGCAAGCCT-3′ is homologous in sequence to the p53 motifs in other p53 responsive genes, including p21/WAF1, MDM2, GADD45, and RGC (20-24), and comprises two half-sites. One of the half-sites is a perfect match to the consensus p53 sequence 5′-PuPuPuC(A/T)(T/A)GPyPyPy-3′ (24), whereas the other half-site contains two mismatches from the consensus motif. No spacer nucleotides separate the p53 half-sites in the GSTP1 gene. Such a spacerless p53-binding site has also been previously observed in MDM2 (21), GADD45 (22), and RGC (23) genes. Molecular and functional analysis showed that the wild-type p53 protein binds to the p53-binding site in the human GSTP1 gene and activates its transcription.
The p53 tumor suppressor has been extensively investigated for its role in oncogenesis and tumor progression and in therapeutic outcome in many human cancers (10). Based on the high frequency of p53 mutations in human tumors, efforts have been made to treat tumors by restoring the wild-type p53 function (26-28). Despite the initial promise, clinical trials of wild-type p53 gene therapy, alone or in combination with chemotherapy, have shown little clinical efficacy (29, 30). Simultaneously, the results of studies of p53 mutational status as a correlate of patient response to therapy have also yielded mixed results. Thus, in gliomas, the presence of wild-type p53 results in higher resistance to cisplatin than when the gene is mutated and/or functionally inactive and that abrogation of the wild-type p53 (e.g., via antisense oligonucleotides) sensitizes tumor cells to cisplatin (31). Similar observations of wild-type p53 conferring drug resistance or decreased drug sensitivity have also been made in other cancer types, such as breast, bladder, and ovarian carcinomas (15-17). In contrast, several other reports showed that p53 mutations are associated with drug resistance and that restoration of p53 function reverses such resistance (11-14).
Although several transcriptional mechanisms, such as those mediated by Sp1 (32), AP1 (33), retinoic acid response element (34), and PKA/CREB1 (35), have been shown to regulate GSTP1 gene expression, the best-characterized regulating mechanism of GSTP1 expression in tumors is that mediated by the differential methylation of the GSTP1 promoter (36, 37). Our results in this study indicate that the cross-talk between the p53 tumor suppressor and the GSTP1 gene is a novel, heretofore, unrecognized mechanism by which GSTP1 gene expression can be regulated in tumor cells. This interaction between the p53 and GSTP1 genes could shed light on the complex role that these two genes and their encoded proteins play in protecting the cellular genome against genotoxic compounds and in regulating cellular response to DNA damage, oxidative stress, and other stimuli (1, 3, 4) and as a determinant of cancer risk and outcome of cancer therapy. Our findings that the GSTP1 gene contains a p53-binding site that is functionally active and can be activated by wild-type p53 are thus highly significant. The results with the cells from seven gliomas suggest that tumors with wild-type p53 express high levels of GSTP1 and, thus, are likely to be more resistant to alkylating agents. This is supported by a large body of evidence indicating that high levels of GSTP1 expression are associated with increased resistance to chemotherapy in many cancers (5, 38-40).
Although the mechanisms underlying the association of wild-type p53 with drug resistance is likely to be complex and to include effects on the cell cycle and altered apoptotic response (41), our data suggest that, in GSTP1-expressing tumors, the ability of p53 to increase GSTP1 expression will be an important part of this mechanism of drug resistance and stress response.
Together, our findings define a novel mechanism by which wild-type p53 regulates GSTP1 expression and protects the genome from alkylating and free radical—generating compounds. In tumors, the transcriptional activation of the GSTP1 gene by p53 will lead to increased survival and increased drug resistance, whereas in normal cells this could lead to protection against genotoxins. Further studies are necessary to define the effect and significance of these findings in these and other important cellular GSTP1-mediated processes and in outcome of cancer therapy.
Materials and Methods
Cell Lines and Cell Culture
The SKLMS-1 is a human leiomyosarcoma cell line with a codon 245 mutation (glycine to serine) in the p53 protein and is routinely maintained in DMEM with 10% FCS and antibiotics. Sk-Ala cells are stable transfectants generated from SKLMS-1 cells that harbor a temperature-sensitive p53 with a V143A mutation. At 38°C, Sk-Ala cells express a p53 gene in the mutant conformation/function but, at 32°C, assume the wild-type p53 conformation/function. The human anaplastic astrocytoma (MGR1), the human glioblastoma (MGR2, MGR3, UW28, UW281, and UW78), and the human medulloblastoma (UW228) cell lines were all early-passage cultures established from primary specimens in the laboratory of Dr. Francis Ali-Osman (5). U87MG human glioblastoma cells were obtained from the American Type Culture Collection and express the wild-type p53 (25). All cells were maintained in DMEM supplemented with 10% FCS.
Reagents and Chemicals
p53 siRNA and nonspecific control siRNA were purchased form Millipore/Upstate. p53 and control viruses, Ad/CMV-p53 and Ad/CMV, respectively, were obtained from the Institutional Vector Core at The University of Texas M. D. Anderson Cancer Center. Unless otherwise stated, all biochemicals and chemicals were purchased from Sigma.
Identification of p53-Binding Motifs in the GSTP1 Gene
A web-based search engine3 was used to search the human GSTP1 gene for sequences matching the p53 consensus motif 5′-PuPuPuC(A/T)(T/A)GPyPyPy-3′. The target sequence analyzed covered nucleotides —2,000 to +2,975 and spanned the entire GSTP1 gene, including all seven exons and six introns (34).
Electrophoretic Mobility Shift Assay
As part of its functional characterization, we examined the ability of wild-type p53 protein to bind to the GSTP1 putative p53-binding motif by electrophoretic mobility shift assay, as previously described (35). Oligonucleotides corresponding to the GSTP1 p53-binding motif and control consensus p53-binding sites (24), namely, 5′-CCAGGGCAGGCATGGGCAAGCCTCTG-3′ and 5′-TAGGCATGTCTAGGCATGTCTA-3′, respectively, were 32P labeled and used as binding probes. For competition experiments, the p53-containing protein extracts were preincubated with 50-fold excess of unlabeled GSTP1 p53 motif (Fig. 1B, lane 3) or with 50-, 100-, and 200-fold excess (Fig. 1B, lanes 4-6) of unlabeled consensus p53 motif probe for 15 min before the addition of the 32P-labeled probes. For the gel supershift assays, 1 μg of an anti-p53 monoclonal antibody (DO-1; Santa Cruz Biotechnology) was added to the p53-containing protein extracts that had been preincubated with 32P-labeled GSTP1 p53 motif for 30 min at room temperature.
DNase I Footprinting Assays
DNase I footprinting was done as previously described (35) to determine the direct binding of the p53 protein to p53-binding motif. The GSTP1 intron 4 probe containing the putative p53-binding sites was generated by PCR using the primers 5′-GTGAGTCTTGAACCTCCAAGT-3′ (forward) and 5′-CTGGGGTTGGGTGGAGGGGAG-3′ (reverse) and genomic DNA from the MGR3 glioblastoma cell line as template. An aliquot of the end-labeled probe was subjected to Maxam-Gilbert DNA sequencing (42).
Plasmid Constructs, Transfection, and Luciferase Reporter Assay
To study the functionality of the GSTP1 p53-binding motifs further, we examined its ability to activate gene expression p53 dependently using a luciferase reporter vector, pFR-Luc (Stratagene), containing the firefly luciferase cDNA. DNA fragments containing three and four tandem repeats of the GSTP1 p53-binding motif, 5′-AGGCAGGCATGGGCAAGCCT-3′, were synthesized by PCR and cloned into the vector at a site 36 nucleotides upstream of the luciferase start codon. The resulting constructs, each containing a TATA box, control promoter, and the firefly luciferase cDNA, were designated pGSTP1-p53BS(3X)-Luc and pGSTP1-p53BS(4X)-Luc, respectively. The pRGC-p53BS-Luc plasmid (Stratagene) is consisted of 15 tandem repeats of the RGC p53-binding site, 5′-TGCCTGGACTTGCCTGG-3′ (23). A Renilla luciferase expression vector, pRL-CMV (Promega), was used as a transfection efficiency control. Transfections were done using MBS Mammalian Transfection kit (Stratagene), Lipofectamine LTX (Invitrogen), and FuGENE HD (Roche). The cells were then lysed and luciferase activity was measured in the supernatant using the Dual-Luciferase Assay kit (Promega). Relative luciferase activity was compared following normalization of the firefly luciferase against that of the Renilla luciferase.
Gene Expression Analyses by Northern Blotting
Tumor cells treated as per experimental protocol were harvested and total RNA was extracted using the acid guanidinium thiocyanate-phenol-chloroform (34, 43). Northern blotting was done using GSTP1 and p53 cDNA generated by PCR as we had described previously (34, 44). The human p21/WAF1 cDNA probe was generated by PCR using primers with sequences 5′-TAAGAGGAGGCGCCATGTCAGAAC-3′ (forward) and 5′-TCCTGTGGGCGGATTAGGGCTTCCT-3′ (reverse). GAPDH cDNA probe was purchased from Clontech Laboratories. Following autoradiography, the bands were quantitated by densitometry and target gene transcripts were normalized against GAPDH.
Western Blot Analysis
Tumor cells in exponential growth were treated as per experimental protocol and lysed by sonication, and particle-free supernatants were prepared by centrifugation at 15,000 rpm for 20 min. The protein extracts were subjected to Western blot analysis, as previously described (45), using the following antibodies: anti-GSTP1 rabbit polyclonal antibody (Stressgen), anti-p53 mouse monoclonal antibody (DO-1), and anti-β-actin mouse monoclonal antibody (Sigma).
Analysis of p53 Mutational Status in Brain Tumor Cell Lines
Total genomic DNA was isolated from the eight brain tumor cell lines, MGR1, MGR2, MGR3, UW28, UW281, UW78, and UW228, using the phenol-chloroform-isoamyl alcohol procedure. The extracted DNA was used to amplify each of exons 2 to 8 of the p53 gene. Each PCR product was cloned into the pCR2.1 using the TA cloning kit (Invitrogen). Positive clones were subjected to DNA sequencing, as we previously described (46).
Statistical Analysis
Statistical analysis was done using the Student’s t test. Statisca (StatSoft) and Microsoft Excel were used to analyze the data. Statistical significance was set at P < 0.05.
Acknowledgments
Grant support: NIH grants RO1CA112519 and P50CA108786-01 (F. Ali-Osman) and 1K01-CA118423-01 (H-W. Lo) and National Cancer Institute Career Development Awards from Duke Brain Cancer Specialized Program of Research Excellence and Duke Comprehensive Cancer Center (H-W. Lo).
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Hayes JD, Flanagan JU, Jowsey IR. Glutathione transferases. Annu Rev Pharmacol Toxicol. 2005;45:51–88. doi: 10.1146/annurev.pharmtox.45.120403.095857. [DOI] [PubMed] [Google Scholar]
- 2.Ishikawa T, Ali-Osman F. Glutathione-associated cis-diamminedichloroplatinum(II) metabolism and ATP-dependent efflux from leukemia cells. Molecular characterization of glutathione-platinum complex and its biological significance. J Biol Chem. 1993;268:20116–25. [PubMed] [Google Scholar]
- 3.Adler V, Yin Z, Fuchs SY, et al. Regulation of JNK signaling by GSTp. EMBO J. 1999;18:1321–34. doi: 10.1093/emboj/18.5.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu Y, Fan Y, Xue B, et al. Human glutathione S-transferase P1-1 interacts with TRAF2 and regulates TRAF2-ASK1 signals. Oncogene. 2006;25:5787–800. doi: 10.1038/sj.onc.1209576. [DOI] [PubMed] [Google Scholar]
- 5.Ali-Osman F, Stein DE, Renwick A. Glutathione content and glutathione-S-transferase expression in 1,3-bis(2-chloroethyl)-1-nitrosourea-resistant human malignant astrocytoma cell lines. Cancer Res. 1990;50:6976–80. [PubMed] [Google Scholar]
- 6.Ali-Osman F, Brunner JM, Kutluk TM, Hess K. Prognostic significance of glutathione S-transferase π expression and subcellular localization in human gliomas. Clin Cancer Res. 1997;3:2253–61. [PubMed] [Google Scholar]
- 7.Lopes JM, Bruland OS, Bjerkehagen B, et al. Synovial sarcoma: immunohistochemical expression of P-glycoprotein and glutathione S transferase-π and clinical drug resistance. Pathol Res Pract. 1997;193:21–36. doi: 10.1016/s0344-0338(97)80090-8. [DOI] [PubMed] [Google Scholar]
- 8.Lozano G. The oncogenic roles of p53 mutants in mouse models. Curr Opin Genet Dev. 2007;17:66–70. doi: 10.1016/j.gde.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 9.Donehower LA, Harvey M, Slagle BL, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–21. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 10.Soussi T. p53 alterations in human cancer: more questions than answers. Oncogene. 2007;26:2145–56. doi: 10.1038/sj.onc.1210280. [DOI] [PubMed] [Google Scholar]
- 11.Dorigo O, Turla ST, Lebedeva S, Gjerset RA. Sensitization of rat glioblastoma multiforme to cisplatin in vivo following restoration of wild-type p53 function. J Neurosurg. 1998;88:535–40. doi: 10.3171/jns.1998.88.3.0535. [DOI] [PubMed] [Google Scholar]
- 12.Ganjavi H, Gee M, Narendran A, Freedman MH, Malkin D. Adenovirus-mediated p53 gene therapy in pediatric soft-tissue sarcoma cell lines: sensitization to cisplatin and doxorubicin. Cancer Gene Ther. 2005;12:397–406. doi: 10.1038/sj.cgt.7700798. [DOI] [PubMed] [Google Scholar]
- 13.Fan S, el-Deiry WS, Bae I, et al. p53 gene mutations are associated with decreased sensitivity of human lymphoma cells to DNA damaging agents. Cancer Res. 1994;54:5824–30. [PubMed] [Google Scholar]
- 14.Kigawa J, Terakawa N. Adenovirus-mediated transfer of a p53 gene in ovarian cancer. Adv Exp Med Biol. 2000;465:207–14. doi: 10.1007/0-306-46817-4_19. [DOI] [PubMed] [Google Scholar]
- 15.Chang FL, Ling YF, Lai MD. Exogenous mutant p53 DNA enhanced cisplatin-induced apoptosis in TSGH-8301 human bladder cancer cells. Anticancer Res. 2000;20:329–36. [PubMed] [Google Scholar]
- 16.Fan S, Smith ML, Rivet DJ, II, et al. Disruption of p53 function sensitizes breast cancer MCF-7 cells to cisplatin and pentoxifylline. Cancer Res. 1995;55:1649–54. [PubMed] [Google Scholar]
- 17.Yazlovitskaya EM, DeHaan RD, Persons DL. Prolonged wild-type p53 protein accumulation and cisplatin resistance. Biochem Biophys Res Commun. 2001;283:732–7. doi: 10.1006/bbrc.2001.4849. [DOI] [PubMed] [Google Scholar]
- 18.Henderson CJ, Smith AG, Ure J, et al. Increased skin tumorigenesis in mice lacking π class glutathione S-transferases. Proc Natl Acad Sci U S A. 1998;95:5275–80. doi: 10.1073/pnas.95.9.5275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lo HW, Ali-Osman F. Genetic polymorphism and function of glutathione S-transferases in tumor drug resistance. Curr Opin Pharmacol. 2007;7:367–74. doi: 10.1016/j.coph.2007.06.009. [DOI] [PubMed] [Google Scholar]
- 20.el-Deiry WS, Tokino T, Waldman T, et al. Topological control of p21WAF1/ CIP1 expression in normal and neoplastic tissues. Cancer Res. 1995;55:2910–9. [PubMed] [Google Scholar]
- 21.Wu X, Bayle JH, Olson D, Levine AJ. The p53-mdm-2 autoregulatory feedback loop. Genes Dev. 1993;7:1126–32. doi: 10.1101/gad.7.7a.1126. [DOI] [PubMed] [Google Scholar]
- 22.Kastan MB, Zhan Q, el-Deiry WS, et al. A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell. 1992;71:587–97. doi: 10.1016/0092-8674(92)90593-2. [DOI] [PubMed] [Google Scholar]
- 23.Bargonetti J, Reynisdottir I, Friedman PN, Prives C. Site-specific binding of wild-type p53 to cellular DNA is inhibited by SV40 T antigen and mutant p53. Genes Dev. 1992;6:1886–98. doi: 10.1101/gad.6.10.1886. [DOI] [PubMed] [Google Scholar]
- 24.el-Deiry WS, Kern SE, Pietenpol JA, Kinzler KW, Vogelstein B. Definition of a consensus binding site for p53. Nat Genet. 1992;1:45–9. doi: 10.1038/ng0492-45. [DOI] [PubMed] [Google Scholar]
- 25.Xu GW, Nutt CL, Zlatescu MC, et al. Inactivation of p53 sensitizes U87MG glioma cells to 1,3-bis(2-chloroethyl)-1-nitrosourea. Cancer Res. 2001;61:4155–9. [PubMed] [Google Scholar]
- 26.Bykov VJ, Issaeva N, Shilov A, et al. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat Med. 2002;8:282–8. doi: 10.1038/nm0302-282. [DOI] [PubMed] [Google Scholar]
- 27.Snyder EL, Meade BR, Saenz CC, Dowdy SF. Treatment of terminal peritoneal carcinomatosis by a transducible p53-activating peptide. PLoS Biol. 2004;2:E36. doi: 10.1371/journal.pbio.0020036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roth JA. Adenovirus p53 gene therapy. Expert Opin Biol Ther. 2006;6:55–61. doi: 10.1517/14712598.6.1.55. [DOI] [PubMed] [Google Scholar]
- 29.Schuler M, Herrmann R, De Greve JL, et al. Adenovirus-mediated wild-type p53 gene transfer in patients receiving chemotherapy for advanced non-small-cell lung cancer: results of a multicenter phase II study. J Clin Oncol. 2001;19:1750–8. doi: 10.1200/JCO.2001.19.6.1750. [DOI] [PubMed] [Google Scholar]
- 30.Fujiwara T, Tanaka N, Kanazawa S, et al. Multicenter phase I study of repeated intratumoral delivery of adenoviral p53 in patients with advanced non-small-cell lung cancer. J Clin Oncol. 2006;24:1689–99. doi: 10.1200/JCO.2005.03.4116. [DOI] [PubMed] [Google Scholar]
- 31.Datta K, Shah P, Srivastava T, et al. Sensitizing glioma cells to cisplatin by abrogating the p53 response with antisense oligonucleotides. Cancer Gene Ther. 2004;11:525–31. doi: 10.1038/sj.cgt.7700724. [DOI] [PubMed] [Google Scholar]
- 32.Moffat GJ, McLaren AW, Wolf CR. Sp1-mediated transcriptional activation of the human π class glutathione S-transferase promoter. J Biol Chem. 1996;271:1054–60. doi: 10.1074/jbc.271.2.1054. [DOI] [PubMed] [Google Scholar]
- 33.Xia C, Hu J, Ketterer B, Taylor JB. The organization of the human GSTP1-1 gene promoter and its response to retinoic acid and cellular redox status. Biochem J. 1996;313:155–61. doi: 10.1042/bj3130155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lo HW, Ali-Osman F. Genomic cloning of hGSTP1*C, an allelic human π class glutathione S-transferase gene variant and functional characterization of its retinoic acid response elements. J Biol Chem. 1997;272:32743–9. doi: 10.1074/jbc.272.52.32743. [DOI] [PubMed] [Google Scholar]
- 35.Lo HW, Ali-Osman F. Cyclic AMP mediated GSTP1 gene activation in tumor cells involves the interaction of activated CREB-1 with the GSTP1 CRE: a novel mechanism of cellular GSTP1 gene regulation. J Cell Biochem. 2002;87:103–16. doi: 10.1002/jcb.10275. [DOI] [PubMed] [Google Scholar]
- 36.Antoun G, Baylin SB, Ali-Osman F. DNA methyltransferase levels and altered CpG methylation in the total genome and in the GSTP1 gene in human glioma cells transfected with sense and antisense DNA methyltransferase cDNA. J Cell Biochem. 2000;77:372–81. [PubMed] [Google Scholar]
- 37.Lee WH, Isaacs WB, Bova GS, Nelson WG. CG island methylation changes near the GSTP1 gene in prostatic carcinoma cells detected using the polymerase chain reaction: a new prostate cancer biomarker. Cancer Epidemiol Biomarkers Prev. 1997;6:443–50. [PubMed] [Google Scholar]
- 38.Fruehauf JP, Brem H, Brem S, et al. In vitro drug response and molecular markers associated with drug resistance in malignant gliomas. Clin Cancer Res. 2006;12:4523–32. doi: 10.1158/1078-0432.CCR-05-1830. [DOI] [PubMed] [Google Scholar]
- 39.Yoshii Y, Saito A, Hyodo A, Tsurushima H, Sun L. Expression of enzymes and oncogene induced after radiotherapy and/or chemotherapy in patients with brain tumors. Hum Cell. 2001;14:95–103. [PubMed] [Google Scholar]
- 40.Lo HW, Ali-Osman F. Genetic polymorphism and function of glutathione S-transferases in tumor drug resistance. Curr Opin Pharmacol. 2007;4:367–74. doi: 10.1016/j.coph.2007.06.009. [DOI] [PubMed] [Google Scholar]
- 41.Andrews GA, Xi S, Pomerantz RG, et al. Mutation of p53 in head and neck squamous cell carcinoma correlates with Bcl-2 expression and increased susceptibility to cisplatin-induced apoptosis. Head Neck. 2004;26:870–7. doi: 10.1002/hed.20029. [DOI] [PubMed] [Google Scholar]
- 42.Sambrook J, Fritsch E, Maniatis T. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor Laboratory (NY): 1989. [Google Scholar]
- 43.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–9. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 44.Pollock R, Lang A, Ge T, et al. Wild-type p53 and a p53 temperature-sensitive mutant suppress human soft tissue sarcoma by enhancing cell cycle control. Clin Cancer Res. 1998;4:1985–94. [PubMed] [Google Scholar]
- 45.Lo H-W, Hsu S-C, Ali-Seyed M, et al. Nuclear interaction of EGFR and STAT3 in the activation of iNOS/NO pathway. Cancer Cell. 2005;7:575–89. doi: 10.1016/j.ccr.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 46.Ali-Osman F, Akande O, Antoun G, Mao JX, Buolamwini J. Molecular cloning, characterization, and expression in Escherichia coli of full-length cDNAs of three human glutathione S-transferase π gene variants. Evidence for differential catalytic activity of the encoded proteins. J Biol Chem. 1997;272:10004–12. doi: 10.1074/jbc.272.15.10004. [DOI] [PubMed] [Google Scholar]