

Abstract
O2-sensing in the carotid body occurs in neuroectoderm-derived type I glomus cells where hypoxia elicits a complex chemotransduction cascade involving membrane depolarization, Ca2+ entry and the release of excitatory neurotransmitters. Efforts to understand the exquisite O2-sensitivity of these cells currently focus on the coupling between local PO2 and the open-closed state of K+-channels. Amongst multiple competing hypotheses is the notion that K+-channel activity is mediated by a phagocytic-like multisubunit enzyme, NADPH oxidase, which produces reactive oxygen species (ROS) in proportion to the prevailing PO2. In O2-sensitive cells of lung neuroepithelial bodies (NEB), multiple studies confirm that ROS levels decrease in hypoxia, and that EM and K+-channel activity are indeed controlled by ROS produced by NADPH oxidase. However, recent studies in our laboratories suggest that ROS generated by a non-phagocyte isoform of the oxidase are important contributors to chemotransduction, but that their role in type I cells differs fundamentally from the mechanism utilized by NEB chemoreceptors. Data indicate that in response to hypoxia, NADPH oxidase activity is increased in type I cells, and further, that increased ROS levels generated in response to low-O2 facilitate cell repolarization via specific subsets of K+-channels.
1. Introduction
The survival of multicellular aerobic organisms is dependent on a continuous supply of molecular oxygen. O2 is consumed in enzymatically catalyzed electron transfer reactions which occur in cell membranes, at multiple sites throughout the cell, and within particular organelles, such as mitochondria. Much of the O2 used in these reactions is completely reduced via the transfer of 4 electrons to form 2 molecules of H2O. However, important amounts of O2 can also be partially reduced, by single electron reduction to form the highly reactive metabolite, superoxide anion (O2−). O2− is converted to hydrogen peroxide (H2O2) by an isozyme of superoxide dismutase (SOD) either in the cytosolic compartment (ZnCuSOD), or in mitochondria (MnSOD). In the presence of transition metal ions, O2− can also undergo conversion to the highly reactive hydroxyl radical (OH−)(Thannickal et al., 2000). These partially reduced metabolites of O2 have been designated “reactive oxygen species” (ROS) because they are variously more reactive than molecular O2. High levels of ROS have traditionally been regarded as toxic (oxidative stress), and their numerous potentially damaging effects collectively constitute the basis for one of the most popular theories of aging. However, recent investigations have also demonstrated that low concentrations of ROS, produced locally and acting on nearby effectors, are important signaling molecules(Thannickal et al., 2000; Sauer et al., 2001; Griendling et al., 2000; Lambeth, 2002; Finkel, 2000). Thus, an emerging body of data indicate that specific extracellular stimuli including cytokines, neurotransmitters and hypoxia initiate signaling cascades involving the production of ROS from mitochondria and non-phagocyte forms of NADPH oxidase(Chandel et al., 2000b; Thannickal et al., 2000). Like cyclic nucleotides (cAMP, cGMP), inositol phosphates (IPN) and nitric oxide (NO), ROS affect specific targets within local cellular compartments(Pani et al., 2001), frequently involving select cysteine and methionine residues which are highly labile for oxidation/reduction(Thannickal et al., 2000; Xu et al., 2002). Changes in the redox state at these sites has been demonstrated to critically alter important effector molecules such as ion channels (K+-channels(Hoshi and Heinemann, 2001; Tang et al., 2001)), protein kinases(Thannickal et al., 2000) and transcription factors (HIF-1α, NF-κB, AP-1(Thannickal et al., 2000; Görlach et al., 2001; Zhu et al., 2002; Chandel et al., 2000b)).
1.1 ROS formation in mitochondria
Mitochondrial cytochrome oxidase catalyzes the 4-electron reduction of O2 in the final step of an elaborate electron transport chain which captures metabolic energy in the terminal phosphate bond of ATP. In addition, 1–3% of the O2 consumed by mitochondria is partially reduced to O2− by enzyme-complex I, the ubisemiquinone site of complex III, and other electron transfer proteins(Chandel et al., 2000b; Thannickal et al., 2000). Extended periods of hypoxia (1–2 hr) may depress the Vmax of cytochrome oxidase, and produce a shift in the redox state within mitochondria (i.e., increased levels of NADH), thereby promoting the formation of O2−(Chandel et al., 2000b). Although its negative charge prevents this anion from crossing mitochondrial membranes, it is rapidly converted by MnSOD to highly membrane permeant H2O2(Sauer et al., 2001), which appears to mediate apoptotic signaling and cell proliferation(Irani, 2000). Mitochondria have also been implicated as cellular O2 sensors in erythropoietin (Epo)-producing Hep3B cells and pulmonary artery smooth muscle cells. Mitochondrial ROS production evoked by hypoxia has been correlated with modulation of K+-channels and vasoconstriction (hypoxic pulmonary vasoconstriction, HPV(Michelakis et al., 2002; Chandel and Schumacker, 2000b; Waypa et al., 2001)). However, the mechanism by which mitochondrial ROS are coupled to contractile elements has not been elucidated, and the role of cell redox in HPV is a point of considerable controversy(Sylvester, 2001).
1.2 ROS formation by NADPH oxidase
For many years it has been known that large quantities of ROS are produced by phagocyte cells as part of an extracellular killing mechanism activated in response to invading micro-organisms. ROS-generating phagocyte NADPH oxidase is a complex enzyme comprised of two trans-membrane and four cytosolic subunits (Figure 1). The large 91 kD glycoprotein (gp91phox ; phox: phagocyte oxidase) and a 22 kD protein (p22phox) form a membrane bound cytochrome b558(Sauer et al., 2001; Diebold et al., 2001; Babior, 1999). Immunologic stimulation initiates a protein kinase C (PKC)-dependent process in which the cytosolic subunits p67phox, p40phox, p47phox and a small GTPase (Rac-1 or Rac-2), unite at the membrane to form the active enzyme. An electron is then transferred from NADPH (produced in the hexose monophosphate shunt pathway [HMS] of glucose metabolism) to O2, thus forming O2− plus NADP+ and H+(Babior, 1999; Diebold et al., 2001). Despite advances in demonstrating the importance of ROS in cell signaling, the role of the phagocyte oxidase is perplexing because it is inactive in resting cells, and produces O2− primarily on the extracellular side of the plasma membrane(Diebold et al., 2001), whose location is thus entirely inconsistent with the action of an intracellular messenger. However, recent studies of non-phagocyte cells have shown that NADPH oxidase may also be constitutively active, and produce ROS intracellularly at low levels (Brar et al., 2002b; Brar et al., 2002a; Li et al., 2002; Szöcs et al., 2002; Görlach et al., 2001). For example, vascular endothelial cells contain an NADPH oxidase in which cytochrome b558 and the cytosolic subunits are preassembled on cytoskeletal elements in the perinuclear region, where they are engaged in constitutive production of intracellular ROS(Li et al., 2002). Moreover, recent studies in the lung have documented the involvement of ROS generated by NADPH oxidase in chemosensory cells of neuroepithelial bodies (NEB) which respond to hypoxia by inhibition of a voltage-dependent K+-current. Furthermore, K+-channels in these cells are activated by low concentrations of H2O2, and hypoxia-evoked depression of channel activity is occluded in the presence of oxidase inhibitors, suggesting that ROS production is a key factor which couples local PO2 to cell activity(Wang et al., 1996). Importantly, the depolarizing response is absent in null-mutant mice lacking gp91phox (Wang et al., 1996; O'Kelly et al., 2000; Fu et al., 2000a)
Figure 1.
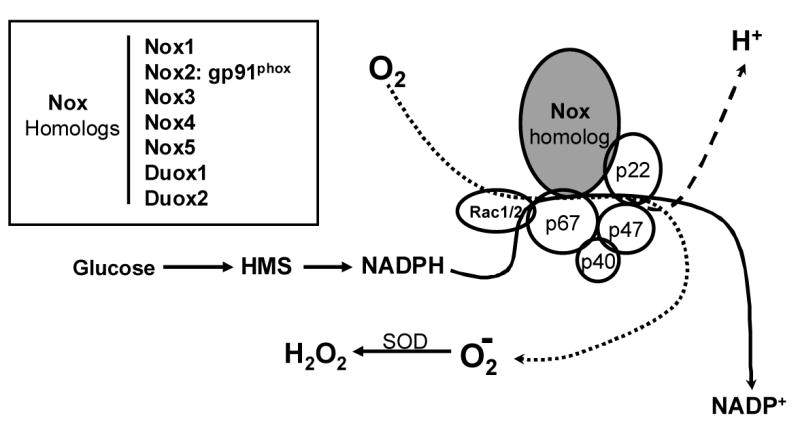
Composition and function of NADPH oxidase (Nox). A heme-containing Nox-homolog plus p22phox form a cytochrome b558 which in the phagocytic enzyme is embedded in plasma membrane. Cytosolic components [p67phox, p47phox, p40phox, plus a GTPase (Rac 1 or 2)] transmigrate in a protein kinase-dependent process to form the functional enzyme. The cofactor, NADPH, is formed in the hexose monophosphate shunt (HMS) pathway of glucose metabolism. Superoxide anion, O2−, is converted to hydrogen peroxide (H2O2) by superoxide dismutase (SOD). Inset indicates various superoxide-generating Nox homologs (Nox 1-5) which may be associated with membranes, organelles or cytoskeletal components. Duox1 and Duox2 function as hydrogen ion transporters.
Homologs of gp91phox (Figure 1) have been cloned and sequenced from a variety of cell types, suggesting that there are multiple forms of the enzyme which are adapted to function in specific signaling pathways (Cheng et al., 2001; Lambeth, 2002). Some of these alternative forms of NADPH oxidase (Nox1–5, plus Duox1and Duox2) have likewise been proposed as O2-sensors which produce ROS in proportion to local levels of PO2 (Acker, 1994; Porwol et al., 2001). ROS generated by Nox homologs may also mediate cell proliferation and expression of transcription factors in multiple cell types (e.g., airway smooth muscle, melanoma cells, vascular endothelial cells(Brar et al., 2002b; Brar et al., 2002a; Irani, 2000)) in response to a variety of physical (e.g., shear stress) and chemical (e.g., cytokines, neurotransmitters) stimuli(Griendling et al., 2000; Irani, 2000). Chemosensory type I cells in the carotid body, like NEB cells in the lung, are O2-sensors which respond to acute hypoxia (AH) by depolarization, Ca2+ entry, the release neuroactive agents, and activation of sensory nerves. ROS have been proposed as intracellular mediators of chemotransduction events in type I cells, coupling moment-to-moment cellular activity to the prevailing PO2 (Acker, 1994), but efforts to determine the precise mode(s) of involvement of Nox in ROS production have resulted in conflicting views(Obeso et al., 1999).
2. Oxygen Sensing in the Carotid Body and the Potential Involvement of ROS
Exposure of the carotid body to AH elicits increased neural activity in the carotid sinus nerve (CSN), and reflex cardio-pulmonary adjustments which mitigate the adverse effects of hypoxia. Increased carotid body activity occurs at low to moderate arterial PO2 levels, in contrast to the severe hypoxia required to elicit metabolic and functional adjustments in non-O2 sensing tissues(Fidone et al., 1997; Fidone et al., 1995; Gonzalez et al., 1994). Neuroectodermal type I cells are responsible for this exquisite sensitivity, and contemporary efforts to understand their unique hypoxic chemotransduction mechanisms have focused primarily on the relationship between PO2 and the activity of K+-channels(Gonzalez et al., 1994; Peers et al., 1995; Lopez-Lopez et al., 1989; Lopez-Barneo, 1994; Lopez-Barneo et al., 2001). In the rat, studies of type I cells have demonstrated that hypoxia inhibits two types of K+-channel: 1) a large-conductance, voltage- and calcium-dependent channel (maxiK(Peers, 1990; Wyatt et al., 1995)), and 2) a voltage-independent TASK-type channel which mediates a background leak current(Buckler, 1997; Buckler et al., 2000; Buckler et al., 2006). Available data indicate that TASK-like channels play a major role in setting the Em, and they strongly suggest that closure of these channels initiates cell depolarization (Donnelly, 1997; Buckler, 1999). The properties of maxiK, however, are not consistent with the initiation of cell activity evoked by hypoxia because their current-voltage characteristics suggest that they do not open until the membrane potential is more positive than -20 mV. Because of these properties, and the fact that specific maxiK blockers fail to activate type I cells, some authors conclude that maxiK channels modulate cell activity and facilitate repolarization of the membrane(Prabhakar, 2000; Buckler, 1999; Donnelly, 1997). In this view, hypoxic inhibition of the maxiK current would contribute to cell activity by delaying recovery of the cell to the resting state.
There is general agreement that the K+-channels which are also expressed in non-O2 sensing tissues are not inherently sensitive to molecular oxygen and must somehow be coupled to responsive cellular components modulated by the prevailing PO2 in type I cells(Patel et al., 2001). Foremost amongst the differing points of view regarding O2-sensing are that either an O2-binding heme-protein in the plasma membrane(Lopez-Lopez et al., 1992), or specialized O2-sensitive cytoplasmic components (i.e., mitochondria or Nox(Acker, 1994; Wilson et al., 1994; Buckler et al., 2006)), modulate the activity of K+-channels. Importantly, a recent review by Prabhakar pointed out that these different mechanisms are not necessarily mutually exclusive, and that chemoreceptor cells may integrate signals from multiple O2-sensors, which collectively determine K+-channel activity(Prabhakar, 2000). This concept is particularly attractive because it also embodies the notion that hypoxia targets multiple cellular components, and that the response depends on both the severity and duration of the stimulus.
3. Diverse Views of Nox Function in Carotid Body
The Nox hypothesis of O2-sensing (figure 2) in type I cells suggests that a phagocyte Nox2 or other isoform couples PO2 to K+-channel activity by generating ROS in proportion to available O2(Acker, 1994). Accordingly, H2O2 derived from O2− by the action of SOD would facilitate K+-channel activity either by direct action on channel protein or via a shift in the cell redox balance. Indeed, application of low concentrations of H2O2 to the in vitro rat carotid body/CSN preparation has been shown to depress chemoreceptor nerve activity(Acker et al., 1992), and a non-specific inhibitor of the oxidase, diphenyleneiodonium (DPI), alters nerve activity evoked by hypoxia(Cross et al., 1990). Furthermore, certain subunits common to the phagocyte and non-phagocyte forms of the enzyme, including p22phox, gp91phox (or a closely related Nox isoform), p47phox, and p67phox, have been localized to type I cells by immunocytochemical staining(Kummer et al., 1995). The spectral absorption studies of Acker and his colleagues have enhanced the Nox hypothesis by showing that hypoxia reduces an isoform of cytochrome b558 (Lahiri et al., 1999). Also, a specific Nox inhibitor, 4-(2-aminoethyl)-benzenesulfonyl fluoride (AEBSF), was shown to first excite carotid sinus nerve activity, and then block the chemorecptor response to hypoxia (Lahiri et al., 1999).
Figure 2.
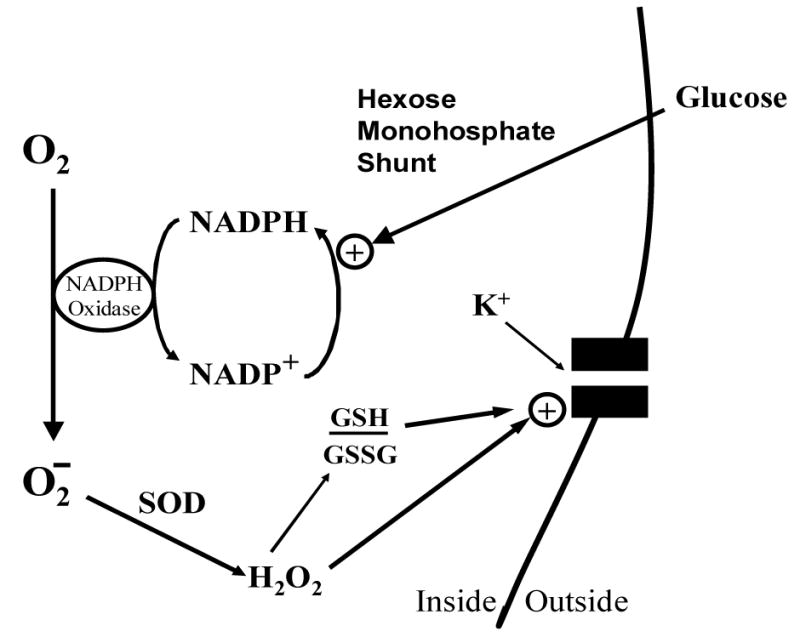
Nox function in O2-sensitive chemoreceptor cells. Diagram depicts activation of K+-channel by H2O2, and/or shift in ratio of reduced (GSH) versus oxidized (GSSG) glutathione (i.e., cellular redox potential). The rate of superoxide generation may depend on the concentrations of molecular O2, and/or NADPH cofactor produced by glucose flux through the hexose monophosphate shunt. SOD: superoxide dismutase.
Opposing these findings are multiple observations which question the validity of the NADPH oxidase hypothesis of chemotransduction. First, studies in the rat have shown that the heme-binding molecule, carbon monoxide (CO), activates K+-channels, and blocks the inhibitory effects of hypoxia, suggesting that a heme-protein physically associated with channel protein mediates the effects of hypoxia on channel activity(Lopez-Lopez et al., 1992). Second, although early studies demonstrated that maxiK and TASK-like channels fail to respond to hypoxia following excision of membrane patches from type I cells (Wyatt et al., 1995; Buckler, 1999), more recent studies have obtained opposite results indicating that cytosolic factors (i.e., subunits of Nox) are not required for low-O2 inhibition of the K+-current(Riesco-Fagundo et al., 2001). Third, studies by Gonzalez and his colleagues showed that inhibitors of Nox (neopterin and arsine oxide) did not promote catecholamime (CA) release from the normoxic carotid body, nor block hypoxia-evoked CA release, indicating that O2-sensing in type I cells is not dependent on Nox function(Obeso et al., 1999). Fourth, although physiological hypoxia increases NAD(P)H levels in type I cell mitochondria (Duchen et al., 1992), it does not change the primary factor controlling the cytosolic redox state, namely, the reduced-to-oxidized glutathione (GSH/GSSH) ratio; moreover, pharmacological manipulation of the redox environment is not correlated with chemoreceptor excitation(Gonzalez et al., 2004; Gonzalez et al., 2002). Finally, experiments using genetically modified mice lacking the gp91phox subunit (targeted gene deletion: knockout, KO) demonstrated normal inhibition of IK by hypoxia in type I cells, as well as normal hypoxia-evoked Ca2+-responses and CSN activity(He et al., 2002; Roy et al., 2000). On the other hand, mice lacking p47phox displayed an enhanced electrophysiological chemoreceptor response to hypoxia (Sanders et al., 2002). Thus, inhibiting Nox function via p47phox gene KO resulted in an outcome (enhanced CSN response) which is diametrically opposed to findings obtained with the Nox inhibitor AEBSF (absence of CSN response), albeit in different species. On the other hand, the finding that chemosensitivity is elevated in the absence of the cytosolic subunit, p47phox, but remains unaltered following KO of gp91phox indicates that type I cells express a non-phagocyte isoform of Nox.
4. Chemoreceptor Function in Normal and Nox-Gene-Deleted Cells
These controversial issues led us to focus further studies on whether the enhanced chemoreceptor discharge following p47phox gene-deletion reflected an underlying negative modulation of type I cells by Nox-generated ROS. In patch-clamp experiments greater than 70% of the voltage-dependent current in mouse type I cells was sensitive to iberiotoxin, consistent with the presence of large conductance maxiK channels(He et al., 2005). Moreover, we found that application of H2O2 decreased hypoxic inhibition of voltage-dependent K+-currents in normal mouse type I cells (figure 3), a result that agreed with previous studies of rat NEB cells and their analog, the H146cell-line(O'Kelly et al., 2000). In accord with Nox-mediated negative modulation, hypoxia-evoked depression of the maxiK-like current was greater in the p47phox KO model, and the Nox inhibitor, AEBSF, enhanced hypoxia-induced current depression in normal, but not KO type I cells (fig. 3). Studies of Ca2+-levels in type I cells further showed that basal and hypoxia-evoked [Ca2+]I was higher in Nox gene-deleted cells, and that AEBSF enhanced Ca2+-responses in normal, but not KO type I cells(He et al., 2005)(figure 4). In new experiments we have found that AEBSF enhances hypoxia-evoked CSN activity in the rat carotid body (figure 5), a result which conflicts with earlier studies (Lahiri et al., 1999). Collectively, these recent findings strongly suggest that ROS generated by Nox enhance maxiK-like currents, and promote cell repolarization. The data further confirm that hypoxia-induced depression of the voltage-sensitive K+-current occurs independently of Nox function.
Figure 3.
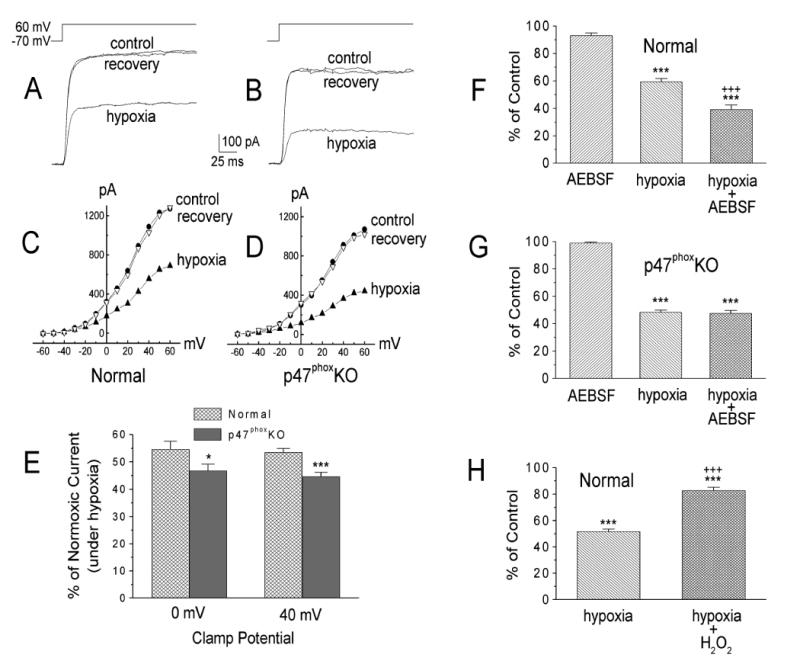
A and C: sample records (A) and current-voltage curves (C) obtained in a normal type I cell in normoxia, hypoxia, and again in normoxia (recovery). B and D: sample records (B) and current-voltage curves (D) obtained in a cell from a p47phoxKO mouse. E: mean hypoxic depression of the normoxic voltage-dependent K+ current (IK) at membrane potentials (Em) of 0 and +40 mV obtained in 31 normal and 32 p47phox KO cells. *P < 0.05; ***P< 0.001 vs. normal cells. F and G: effects of AEBSF (3 mM) in normal (n =10; F) and p47phox KO type I cells (n=12; G). ***P< 0.001 vs. AEBSF. +++, P< 0.001 vs. hypoxia. H: H2O2 (100μM) decreased hypoxic inhibition of IK in normal cells. Values are means± SE; n =12 cells. ***P< 0.001 vs. normoxia (100%). +++P < 0.001 vs. hypoxia. From (He et al., 2005) with permission.
Figure 4.
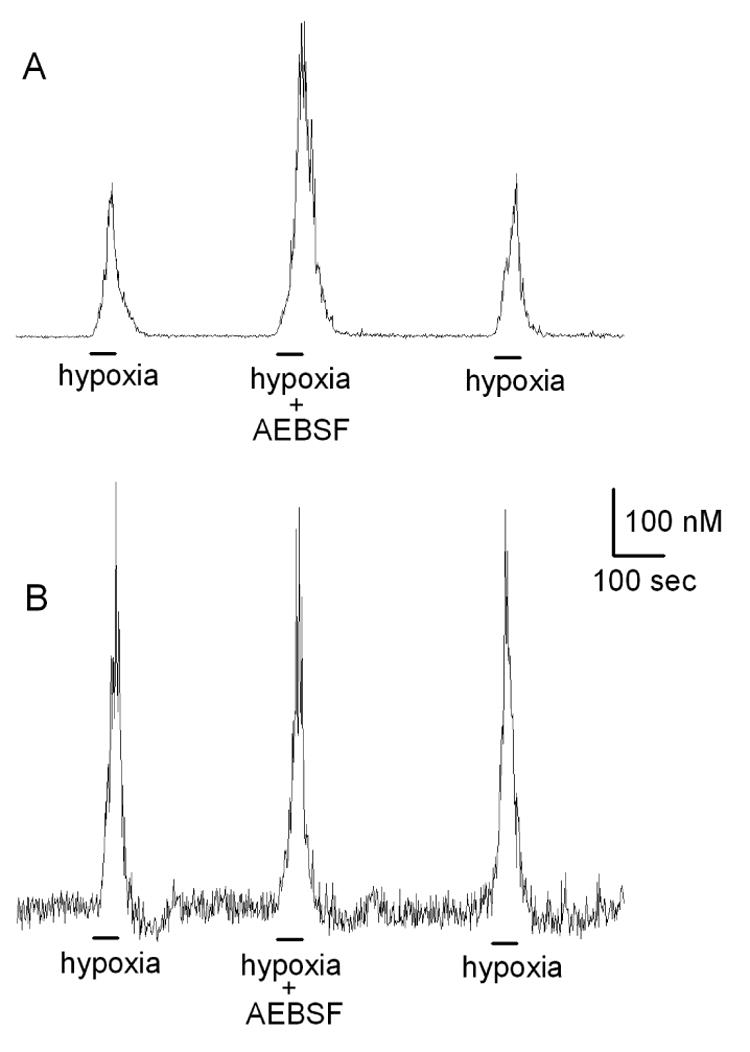
Hypoxia-evoked Ca2+-responses in Fura-2-loaded mouse type I cells. A: Response in normal cell is potentiated in presence of a specific Nox inhibitor, 4-(2-Aminoethyl)-benzenesulfonyl fluoride (AEBSF; 3 mM). B: In a cell from a p47phox KO mouse, hypoxia evokes a larger response; however, AEBSF has no effect, consistent with the absence of functioning Nox.
Figure 5.
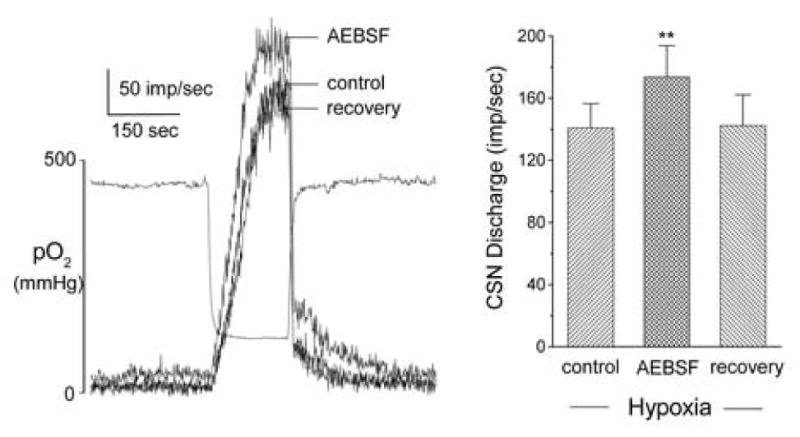
Effect of specific Nox inhibitor, AEBSF (3 mM) on rat carotid sinus nerve (CSN) activity evoked by acute hypoxia. In vitro superfused preparation. Left panel shows 3 superimposed traces of integrated nerve activity. Separate record indicates bath PO2. Left panel summarizes data from 4 preparations, and demonstrates significant (p<0.01) enhancement of hypoxia-evoked response in presence of AEBSF.
A hypothetical scheme has emerged in which ROS levels increase in hypoxia, in a mechanism which opposes the excitatory effects of low-O2. Testing this idea required direct evaluation of the effects of hypoxia on ROS generation in type I cells. Experimental conditions for measuring ROS were established using mouse polymorphonuclear (PMN) cells loaded with the dye, dihydroethidium (DHE), which is oxidized by O2− to fluorescent ethidium. Normal mouse PMN displayed a rapid increase in fluorescence when stimulated with a cocktail consisting of the chemotactic peptide Nformyl-Met-Leu-Phe (fMLP; 1 μM), arachidonic acid (AA; 10 μM), and cytochalasin B (CB; 5 μg/ml). This response was blocked by the Nox inhibitor diphenyleneiodonium (DPI, 10 μM)(He et al., 2005)(figure 6). The mitochondrial uncoupler, azide (5 μM), also elicited a substantial increase in cell fluorescence. PMN from p47phox KO mice did not respond to the cocktail, but did fluoresce like normal cells when treated with azide. The metabolic uncoupler also elicited a large increase in fluorescence in normal and p47phox KO type I cells, but when stimulated by hypoxia, only normal type I cells generated an AEBSF-sensitive response, indicating increased production of O2− originating from Nox (He et al., 2005)(figure 7). We have since reported similar results in rat carotid body lending further support to the hypothesis that low levels of O2 activate a unique form of Nox in type I cells(He et al., 2006). Because hypoxia has been shown to lower ROS levels in the H146 cell-line, in accord with a primary role for Nox in O2-sensing(O'Kelly et al., 2000), the data strongly indicate that hypoxia triggers opposite effects with respect to Nox activation in carotid body versus airway chemoreceptors.
Figure 6.
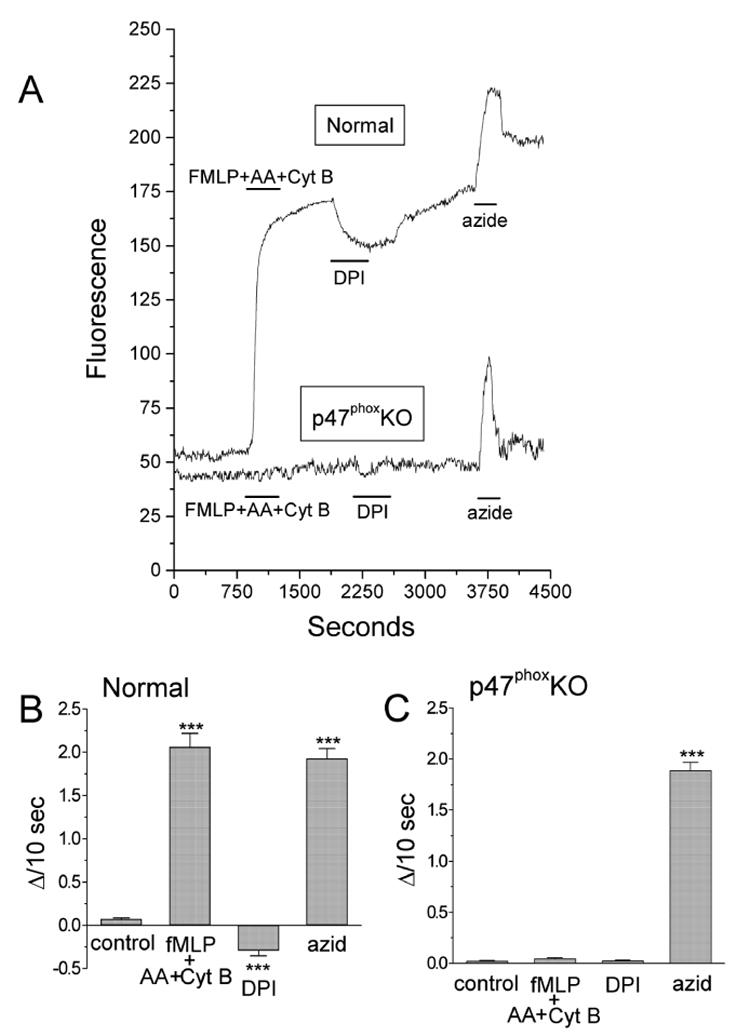
A: a cocktail of neutrophil-stimulating agents [N-formyl-Met-Leu-Phe (fMLP peptide; 1 μM), arachidonic acid (AA; 10 μM), and cytochalasin B (CytB; 5 μg/ml)] produced a sharp increase in dihydroethidium (DHE) fluorescence in a single attached polymorphonuclear neutrophil (PMN) from a normal mouse and no response in a p47phox knockout (KO) PMN. Diphenyleneiodonium (DPI; 10 μM) decreased the fluorescence signal in the normal mouse and had no effect on the p47phox KO PMN. Azide (5 μM) caused a similar increase in fluorescence in the normal and p47phox KO PMN. B and C: summaries of DHE fluorescence for 98 normal (B) and 185 p47phox KO PMN (C). Data are expressed as slopes of fluorescence intensity change (ΔI/10 s) during the initial 100 s of the experimental conditions designated in A. ***P < 0.001 vs. drug-free conditions. From (He et al., 2005) with permission.
Figure 7.
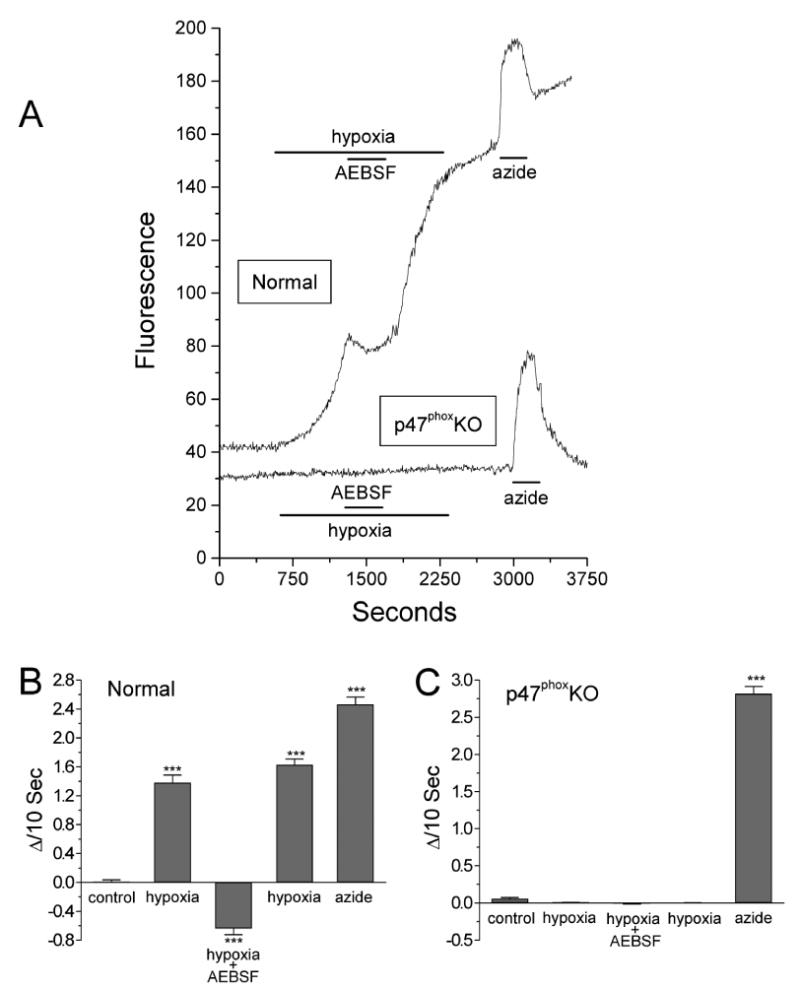
A: effect of hypoxia on DHE fluorescence in carotid body type I cells isolated from normal and p47phox KO mice. In normoxia, fluorescence gradually increases and is markedly elevated by hypoxia in the normal but not in the p47phox KO cell. 4-(2-Aminoethyl)-benzenesulfonyl fluoride (AEBSF; 3 mM) depresses hypoxic increase of reactive oxygen species (ROS) production in the normal type I cell. Azide (5μM) increased the fluorescence in the normal and p47phox KO cells. B and C: summaries of DHE fluorescence for 32 normal (B) and 44 p47phox KO type I cells (C) harvested from 4 normal and 4 KO animals. Slopes (ΔI/10 s) were quantified as described in Fig. 6. ***P<0.001 vs. control, normoxic conditions. From (He et al., 2005) with permission.
The finding that ROS generation increases as O2 levels decrease may appear as overtly counterintuitive to fundamental concepts of enzyme-substrate interactions. However, the affinity of the type I cell Nox isoform for O2 is unknown, and the reported mean Km for O2 of phagocyte Nox is PO2 =12.7 Torr (Cross et al., 1986), about half of PO2 used to stimulate ROS production(He et al., 2005). Thus in physiological hypoxia, O2 availability may not limit Nox activity. In fact, the phagocyte Nox is adapted to function in naturally hypoxic conditions, including wounds. On the other hand, a potentially relevant factor for Nox activation is the cellular concentration of enzyme cofactor, NADPH. In a previous study we demonstrated that hypoxia increases ouabain-sensitive glucose uptake in carotid body type I cells(Obeso et al., 1993). This finding is consonant with the hypothesis that hypoxia-evoked cell depolarization activates Na+/K+-ATPase, thereby triggering metabolic flux of glucose through the HMS. The resulting increase of cofactor could thus be responsible for activation of Nox after the initial hypoxia-induced depolarization. In preliminary experiments, we have found support for this scheme by showing that inhibitors of the HMS (epiandrosterone, 6-aminonicotinomide) block hypoxia-induced ROS production in rat type I cells, and enhance hypoxia-evoked CSN activity.
5. Summary: Adaptation of Nox Isoforms in Airway versus Arterial Chemoreceptor Cells
Nox isoforms are present in airway (NEB) and arterial (carotid body) chemoreceptos where they subserve diverse signaling functions(Kummer et al., 1995; Youngson et al., 1997). In NEB, Nox appears to be a primary O2-sensor which produces ROS in proportion to local PO2. K+-currents are activated by H2O2, and pharmacological inhibition of Nox partially blocks low-O2 depression of currents(O'Kelly et al., 2000; O'Kelly et al., 2001; Fu et al., 2000b). Gene-deletion of the phagocyte gp91phox subunit (Nox2), inhibits responses to low-O2(Fu et al., 2000a). Finally, detailed studies of the NEB analog cell-line (H146) suggest that Nox-mediated O2-sensing may be only one of multiple mechanisms detecting O2 in airway chemoreceptor cells(O'Kelly et al., 2001).
In arterial chemoreceptor type I cells, the available data indicate that primary O2-sensing occurs via a membrane-delimited (but also see Buckler, 1999; Wyatt et al., 1995) detector which responds to a decrease in PO2 by closure of K+-channels(Riesco-Fagundo et al., 2001). Cell depolarization activates voltage-dependent Ca2+-channels and exocytotic release of neurotransmitters. Unlike NEB cells, functional Nox is not necessary for the full expression of O2-sensing in type I cells. Nox gene-deletion (p47phox) in fact enhances chemoreceptor sensitivity, consistent with a modulatory role for ROS. This conclusion is supported by studies of voltage-sensitive K+-currents, cell Ca2+-responses, and measurement of ROS production in type I cells. Data from the normal versus p47phox KO mice, and rats, uniformly indicate that hypoxia activates Nox-dependent ROS production, and that ROS increase voltage-dependent K+-currents, thereby promoting cell repolarization. Importantly, the primary O2-sensing machinery in type I cells appears to be insensitive to the cytosolic redox state. Moreover, hypoxia does not alter the cytosolic GSH/GSSH redox potential in carotid body, suggesting that changes in the redox status are confined to microdomains closely associated with target molecules(Gonzalez et al., 2002; Gonzalez et al., 2004). Consistent with this conclusion is a recent report showing that co-expression of TASK-1 K+-channels and Nox4 in HEK293 cells results in association in the plasma membrane(Lee et al., 2006).
6. Perspectives and Future Directions
The Nox model of O2-sensing as originally formulated by Acker and his colleagues(Acker et al., 1989; Cross et al., 1990) stimulated numerous studies which elucidated novel mechanisms involving ROS in cellular and systemic responses to hypoxia(Acker, 2005). Compelling evidence for Nox-mediated O2-sensing has been obtained in NEB, and ROS have been implicated in regulating ion channel activity and gene expression in multiple cells and tissues(Acker, 2005). In the carotid body available evidence suggests that Nox fills a unique supporting role as a modulator of type I cell activity. However, several issues remain which require further investigation for complete understanding of Nox involvement in arterial chemoreception. Of paramount importance is the identification of the Nox isoform expressed in type I cells, and detailed examination of the role of NADPH cofactor in regulating ROS production. To this end, it will be important to examine the relationship between glucose uptake and cofactor production in the HMS. It is encouraging that recent studies of pulmonary versus coronary vascular smooth muscle have shown that inherently different levels of ROS production by Nox4 are correlated with cellular concentrations of NADPH(Gupte et al., 2005).
Although our data strongly support a role for ROS in the enhancement of maxiK currents in type I cells, virtually nothing is known about the influence of ROS on TASK-like channel activity in carotid body. This is particularly important in view of the fact that these O2-sensitive channels appear to control type I cell EM via a background leak current(Buckler, 1999; Buckler et al., 2000). Importantly, a study of TASK-1 channels co-expressed in HEK293 cells along with Nox4 showed that background K+-current was correlated with Nox activity(Lee et al., 2006). Thus, hypoxia-initiated ROS production may promote type I cell recovery via both maxiK and TASK-like channels. Although not fully understood, recent findings indicate that hypoxia inhibits TASK-1 and TASK-3 in type I cells via changes in mitochondrial energy metabolism, perhaps involving cytosolic concentrations of ATP(Buckler et al., 2006).
A final important consideration is the role of Nox in adaptation of the carotid body during chronic exposure to hypoxia. It has been repeatedly demonstrated that chronic hypoxia (CH) elicits remarkable structural changes and chemosensory hypersensitivity in mammalian carotid body(Dinger et al., 2003). Moreover, systemic hypoxia is known to increase levels of ROS in tissues, consistent with increased O2− production in mitochondria (Chandel et al., 2000a)and/or via enhanced Nox activity. The finding that type I cell activity in acute hypoxia is routinely modulated by ROS suggests the possibility that Nox acts as an important regulatory mechanism which could limit CH-induced hyperexcitability. In contrast, a different paradigm of prolonged exposure, namely, chronic intermittent hypoxia (CIH), has been shown to elicit chemoreceptor hypersensitivity involving cellular mechanisms which differ fundamentally from CH(Peng et al., 2004). Interestingly, Prabhakar and his colleagues have demonstrated that ROS scavengers prevent the CIH-induced increase in chemosensitivity(Peng et al., 2003). Future studies may determine that diverse mechanisms involving Nox are critical for resetting chemosensitivity in carotid body following CH versus CIH.
Acknowledgments
Supported by USPHS Grants NS 12636, NS 07938, NL-50153, and Spanish DGICYT (BFU2004-06394), FISS (PI042462) and JCyL (VA011C05).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Acker H. Mechanisms and meaning of cellular oxygen sensing in the organism. Respir Physiol. 1994;95(1):1–10. doi: 10.1016/0034-5687(94)90043-4. [DOI] [PubMed] [Google Scholar]
- Acker H. The oxygen sensing signal cascade under the influence of reactive oxygen species. Philos Trans R Soc Lond B Biol Sci. 2005;360:2201–2210. doi: 10.1098/rstb.2005.1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acker H, Bolling B, Delpiano MA, Dafau E, Gorlach A, Holtermann G. The meaning of H2O2 generation in carotid body cells for PO2 chemoreception. J Auton Nerv Syst. 1992;41(1–2):41–51. doi: 10.1016/0165-1838(92)90125-z. [DOI] [PubMed] [Google Scholar]
- Acker H, Dufau E, Huber J, Sylvester D. Indications to an NADPH oxidase as a possible pO2 sensor in the rat carotid body. FEB. 1989;256(12):75–78. doi: 10.1016/0014-5793(89)81721-1. [DOI] [PubMed] [Google Scholar]
- Babior BM. NADPH oxidase: An update. Blood. 1999;93(5):1464–1476. [PubMed] [Google Scholar]
- Brar SS, Kennedy TP, Sturrock AB, Huecksteadt TP, Quinn MT, Murphy TM, Chitano P, Hoidal JR. NADPH oxidase promotes NF-κB activation and proliferation in human airway smooth muscle. Am J Physiol Lung Cell Molec Physiol. 2002a;282:L782–L795. doi: 10.1152/ajplung.00206.2001. [DOI] [PubMed] [Google Scholar]
- Brar SS, Kennedy TP, Sturrock AB, Huecksteadt TP, Quinn MT, Whorton AR, Hoidal JR. An NAD(P)H oxidase regulates growth and transcription in melanoma cells. Am J Physiol Cell Physiol. 2002b;282:C1212–C1224. doi: 10.1152/ajpcell.00496.2001. [DOI] [PubMed] [Google Scholar]
- Buckler KJ. A novel oxygen-sensitive potassium current in rat carotid body type I cells. J Physiol. 1997;498(3):649–66. doi: 10.1113/jphysiol.1997.sp021890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckler KJ. Background leak K+-currents and oxygen sensing in carotid body type 1 cells. Resp Physiol. 1999;115:179–187. doi: 10.1016/s0034-5687(99)00015-8. [DOI] [PubMed] [Google Scholar]
- Buckler KJ, Williams BA, Honore E. An oxygen-, acid- and anaesthetic-sensitive TASK-like background potassium channel in rat arterial chemoreceptor cells. J Physiol. 2000;525(1):135–142. doi: 10.1111/j.1469-7793.2000.00135.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckler KJ, Williams BA, Orozco RV, Wyatt CN. The role of TASK-like K+ channels in oxygen sensing in the carotid body. Novartis Found Symp. 2006;272:73–85. [PubMed] [Google Scholar]
- Chandel NS, Schumacker PT. Cellular oxygen sensing by mitochondria: old questions, new insight. J Appl Physiol. 2000b;88:1880–1889. doi: 10.1152/jappl.2000.88.5.1880. [DOI] [PubMed] [Google Scholar]
- Chandel NS, Schumacker PT. Cellular oxygen sensing by mitochondria: old questions, new insight. J Appl Physiol. 2000a;88:1880–1889. doi: 10.1152/jappl.2000.88.5.1880. [DOI] [PubMed] [Google Scholar]
- Cheng G, Cao Z, Xu X, Van Meir EG, Lambeth JD. Homologs of gp91phox: cloning and tissue expression of Nox3, Nox4 and Nox5. Gene. 2001;269:131–140. doi: 10.1016/s0378-1119(01)00449-8. [DOI] [PubMed] [Google Scholar]
- Cross AR, Henderson L, Jones OTG, Delpiano MA, Hentschel J, Acker H. Involvement of an NAD(P)H oxidase as a pO2 sensor protein in the rat carotid body. Biochem J. 1990;272:743–747. doi: 10.1042/bj2720743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross AR, Jones OTG. The effect of the inhibitor diphenylene iodonium on the superoxide-generating system of neutrophils. Biochem J. 1986;237:111–116. doi: 10.1042/bj2370111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diebold BA, Bokoch GM. Molecular basis for Rac2 regulation of phagocyte NADPH oxidase. Nature Immunol. 2001;2(3):211–215. doi: 10.1038/85259. [DOI] [PubMed] [Google Scholar]
- Dinger B, He L, Chen J, Stensaas L, Fidone S. Mechanisms of morphological and functional plasticity in the chronically hypoxic carotid body. In: Lahiri S, Semenza G, Prabhakar N, editors. Oxygen Sensing: Responses and Adaptation to Hypoxia. New York: Marcel Dekker; 2003. pp. 439–465. [Google Scholar]
- Donnelly DF. Are oxygen dependent K+ channels essential for carotid body chemo-transduction? Resp Physiol. 1997;110:211–218. doi: 10.1016/s0034-5687(97)00085-6. [DOI] [PubMed] [Google Scholar]
- Duchen MR, Biscoe TJ. Mitochondrial function in type I cells isolated from rabbit arterial chemoreceptors. J Physiol. 1992;450:13–31. doi: 10.1113/jphysiol.1992.sp019114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fidone S, Dinger BG, Gonzalez C. Mechanisms of carotid body chemoreception. In: Dempsey JA, Pack AI, editors. The Lung Biology in Health Disease, Vol X, The Regulation of Breathing. New York: Marcel Dekker, Inc; 1995. pp. 391–471. [Google Scholar]
- Fidone SJ, Gonzalez C, Almaraz L, Dinger B. Cellular mechanisms of peripheral chemoreceptor function. In: Crystal RG, West JB, et al., editors. The Lung: Scientific Foundations. Philadelphia: Lippincott-Raven Publishers; 1997. pp. 1725–1746. [Google Scholar]
- Finkel T. Redox-dependent signal transduction. FEBS Letters. 2000;476:52–54. doi: 10.1016/s0014-5793(00)01669-0. [DOI] [PubMed] [Google Scholar]
- Fu XW, Wang D, Nurse CA, Dinauer MC, Cutz E. NADPH oxidase is an O2 sensor in airway chemoreceptors: Evidence from K+ current modulation in wild-type and oxidase-deficient mice. PNAS. 2000a;97(8):4374–4379. doi: 10.1073/pnas.97.8.4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu XW, Wang D, Nurse CA, Dinauer MCCE. NADPH oxidase is an O2 sensor in airway chemoreceptors: Evidence from K+ current modulation in wild-type and oxidase-deficient mice. PNAS. 2000b;97(8):4374–4379. doi: 10.1073/pnas.97.8.4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez C, Almaraz L, Obeso A, Rigual R. Carotid body chemoreceptors: from natural stimuli to sensory discharges. Physiol Rev. 1994;74:829–898. doi: 10.1152/physrev.1994.74.4.829. [DOI] [PubMed] [Google Scholar]
- Gonzalez C, Sanz-Alfayate G, Agapito MT, Gomez-Nino A, Rocher A, Obeso A. Significance of ROS in oxygen sensing in cell systems with sensitivity to physiological hypoxia. Respir Physiol Neurobiol. 2002;132:17–41. doi: 10.1016/s1569-9048(02)00047-2. [DOI] [PubMed] [Google Scholar]
- Gonzalez C, Sanz-Alyayate G, Agapito MT, Obeso A. Effects of reducing agents on glutathione metabolism and the function of carotid body chemoreceptor cells. Biol Chem. 2004;385:265–274. doi: 10.1515/BC.2004.021. [DOI] [PubMed] [Google Scholar]
- Görlach A, Diebold I, Schini-Kerth VP, Berchner-Pfannschmidt U, Roth U, Brandes RP, Kietzmann T, Busse R. Thrombin activates the hypoxia-inducible factor-1 signaling pathway in vascular smooth muscle cells: Role of the p22phox-containing NADPH oxidase. Circ Res. 2001;89:47–54. doi: 10.1161/hh1301.092678. [DOI] [PubMed] [Google Scholar]
- Griendling KK, Sorescu D, Ushio-Fukai M. NAD(P)H oxidase: Role in cardiovascular biology and disease. Circ Res. 2000;86:494–501. doi: 10.1161/01.res.86.5.494. [DOI] [PubMed] [Google Scholar]
- Gupte SA, Kaminski PM, Floyd B, Agarwal R, Ali N, Ahmad M, Edwards J, Wolin MS. Cytosolic NADPH may regulate differences in basal Nox oxidase-derived superoxide generation in bovine coronary and pulmonary arteries. Am J Physiol Heart Circ Physiol. 2005;288:H13–H21. doi: 10.1152/ajpheart.00629.2004. [DOI] [PubMed] [Google Scholar]
- He L, Chen J, Dinger B, Sanders K, Sundar K, Hoidal J, Fidone S. Characteristics of carotid body chemosensitivity in NADPH oxidase-deficient mice. Am J Physiol Cell Physiol. 2002;282:C27–C33. doi: 10.1152/ajpcell.2002.282.1.C27. [DOI] [PubMed] [Google Scholar]
- He L, Dinger B, Gonzalez C, Obeso A, Fidone S. Function of NADPH oxidase and signaling by reactive oxygen species in rat carotid body type I cells. Adv Exp Med Biol. 2006;580:155–160. doi: 10.1007/0-387-31311-7_23. [DOI] [PubMed] [Google Scholar]
- He L, Dinger B, Sanders K, Hoidal J, Obeso A, Stensaas L, Fidone S, Gonzalez C. Effect of p47phox gene deletion on ROS production and oxygen sensing in mouse carotid body chemoreceptor cells. Am J Physiol Lung Cell Mol Physiol. 2005;289:L916–L924. doi: 10.1152/ajplung.00015.2005. [DOI] [PubMed] [Google Scholar]
- Hoshi T, Heinemann SH. Topical review: Regulation of cell function by methionine oxidation and reduction. J Physiol. 2001;531(1):1–11. doi: 10.1111/j.1469-7793.2001.0001j.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irani K. Oxidant signaling in vascular cell growth, death and survival: A review of the roles of reactive oxygen species in smooth muscle and endothelial cell mitogenic and apoptotic signaling. Circ Res. 2000;87:179–183. doi: 10.1161/01.res.87.3.179. [DOI] [PubMed] [Google Scholar]
- Kummer W, Acker H. Immunohistochemical demonstration of four subunits of neutrophil NAD(P)H oxidase in type I cells of carotid body. J Appl Physiol. 1995;78(5):1904–1909. doi: 10.1152/jappl.1995.78.5.1904. [DOI] [PubMed] [Google Scholar]
- Lahiri S, Ehleben W, Acker H. Chemoreceptor discharges and cytochrome redox changes of the rat carotid body: Role of heme ligands. Proc Natl Acad Sci USA. 1999;96:9427–9432. doi: 10.1073/pnas.96.16.9427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambeth JD. Nox/Duox family of nicotinamide adenine dinucleotide (phosphage) oxidases. Current Opinion in Hematology. 2002;9:11–17. doi: 10.1097/00062752-200201000-00003. [DOI] [PubMed] [Google Scholar]
- Lee YM, Kim BJ, Chun YS, So I, Choi H, Kim MS, Park JW. NOX4 as an oxygen sensor to regulate TASK-1 activity. Cell Signal. 2006;18:499–507. doi: 10.1016/j.cellsig.2005.05.025. [DOI] [PubMed] [Google Scholar]
- Li JM, Mullen AM, Yun S, Wientjes F, Brouns GY, Thrasher AJ, Shah AM. Essential role of the NADPH oxidase subunit p47phox in endothelial cell superoxide production in response to phorbol ester and tumor necrosis factor-α. Circ Res. 2002;90:143–150. doi: 10.1161/hh0202.103615. [DOI] [PubMed] [Google Scholar]
- Li JM, Shah AJ. Intracellular localization and preassembly of the NADPH oxidase complex in cultured endothelial cells. J Biol Chem. 2002;277(22):19952–19960. doi: 10.1074/jbc.M110073200. [DOI] [PubMed] [Google Scholar]
- Lopez-Barneo J. Oxygen-sensitive ion channels: how ubiquitous are they? TINS. 1994;17(4):133–135. doi: 10.1016/0166-2236(94)90084-1. [DOI] [PubMed] [Google Scholar]
- Lopez-Barneo J, Pardal R, Ortega-Saenz P. Cellular mechanism of oxygen sensing. Annu Rev Physiol. 2001;63:259–287. doi: 10.1146/annurev.physiol.63.1.259. [DOI] [PubMed] [Google Scholar]
- Lopez-Lopez J, Gonzalez C, Urena J, Lopez-Barneo J. Low PO2 selectively inhibits K+ channel activity in chemoreceptor cells of the mammalian carotid body. J Gen Physiol. 1989;93:1001–1015. doi: 10.1085/jgp.93.5.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Lopez JR, Gonzalez C. Time course of K+ current inhibition by low oxygen in chemoreceptor cells of adult rabbit carotid body: effects of carbon monoxide. FEBS Lett. 1992;299:251–254. doi: 10.1016/0014-5793(92)80126-2. [DOI] [PubMed] [Google Scholar]
- Michelakis ED, Hampl V, Nsair A, Wu XC, Harry G, Haromy A, Gurtu R, Archer SL. Diversity in mitochondrial function explains differences in vascular oxygen sensing. Circ Res. 2002;90:1307–1315. doi: 10.1161/01.res.0000024689.07590.c2. [DOI] [PubMed] [Google Scholar]
- O'Kelly I, Lewis A, Peers C, Kemp PJ. O2 sensing by airway chemoreceptor-derived cells: Protein kinase C activation reveals functional evidence for involvement of NADPH oxidase. J Biol Chem. 2000;275(11):7684–7692. doi: 10.1074/jbc.275.11.7684. [DOI] [PubMed] [Google Scholar]
- O'Kelly I, Peers C, Kemp PJ. NADPH oxidase does not account fully for O2-sensing in model airway chemoreceptor cells. Biochem Biophys Res Comm. 2001;283:1131–1134. doi: 10.1006/bbrc.2001.4919. [DOI] [PubMed] [Google Scholar]
- Obeso A, Gomez-Nino A, Gonzalez C. NADPH oxidase inhibition does not interfere with low PO2 transduction in rat and rabbit CB chemoreceptor cells. Am J Physiol. 1999;276:C593–C601. doi: 10.1152/ajpcell.1999.276.3.C593. Cell Physiol 45. [DOI] [PubMed] [Google Scholar]
- Obeso A, Gonzalez C, Rigual R, Dinger B, Fidone S. Effect of low O2 on glucose uptake in rabbit carotid body. J Appl Physiol. 1993;74(5):2387–2393. doi: 10.1152/jappl.1993.74.5.2387. [DOI] [PubMed] [Google Scholar]
- Pani G, Bedogni B, Colavitti R, Anezvino R, Borrello S, Galeotti T. Cell compartmentalization in redox signaling. IUBMB Life. 2001;52:7–16. doi: 10.1080/15216540252774702. [DOI] [PubMed] [Google Scholar]
- Patel AJ, Honoré E. Molecular physiology of oxygen-sensitive potassium channels. Eur Respir J. 2001;18:221–227. doi: 10.1183/09031936.01.00204001. [DOI] [PubMed] [Google Scholar]
- Peers C. Hypoxic suppression of K+ currents in type I carotid body cells: Selective effect on the Ca2+-activated K+ current. Neurosci Lett. 1990;119:253–256. doi: 10.1016/0304-3940(90)90846-2. [DOI] [PubMed] [Google Scholar]
- Peers C, Buckler KJ. Transduction of chemostimuli by the type I carotid body cell. J Mem Biol. 1995;144:1–9. doi: 10.1007/BF00238411. [DOI] [PubMed] [Google Scholar]
- Peng YJ, Prabhakar NR. Reactive oxygen species in the plasticity of respiratory behavior elicited by chronic intermittent hypoxia. J Appl Physiol. 2003;94:2342–2349. doi: 10.1152/japplphysiol.00613.2002. [DOI] [PubMed] [Google Scholar]
- Peng YJ, Prabhakar NR. Effect of two paradigms of chronic intermittent hypoxia on carotid body sensory activity. J Appl Physiol. 2004;96:1236–1242. doi: 10.1152/japplphysiol.00820.2003. [DOI] [PubMed] [Google Scholar]
- Porwol T, Ehleben W, Brand V, Acker H. Tissue oxygen sensor function of NADPH oxidase isoforms, and an unusual cytochrome aa3; producing reactive oxygen species. Resp Physiol. 2001;128:331–348. doi: 10.1016/s0034-5687(01)00310-3. [DOI] [PubMed] [Google Scholar]
- Prabhakar NR. Oxygen sensing by the carotid body chemoreceptors. J Appl Physiol. 2000;88:2287–2295. doi: 10.1152/jappl.2000.88.6.2287. [DOI] [PubMed] [Google Scholar]
- Riesco-Fagundo AM, Pérez-García MT, Gonzalez C, López-López JR. O2 modulates large-conductance Ca2+-dependent K+ channels of rat chemoreceptor cells by a membrane-restricted and CO-sensitive mechanism. Circ Res. 2001;89:430–436. doi: 10.1161/hh1701.095632. [DOI] [PubMed] [Google Scholar]
- Roy A, Rozanov C, Mokashi A, Daudu P, Al-mehdi AB, Shams H, Lahiri S. Mice lacking in gp91 phox subunit of NAD(P)H oxidase showed glomus cell [Ca(2+)](i) and respiratory responses to hypoxia. Brain Res. 2000;872:188–193. doi: 10.1016/s0006-8993(00)02458-6. [DOI] [PubMed] [Google Scholar]
- Sanders KA, Sundar K, He L, Dinger B, Fidone S, Hoidal JR. Role of components of the phagocytic NADPH oxidase in oxygen sensing. J Appl Physiol. 2002;93(4):1357–1364. doi: 10.1152/japplphysiol.00564.2001. [DOI] [PubMed] [Google Scholar]
- Sauer H, Wartenberg M, Hescheler J. Reactive oxygen species as intracellular messengers during cell growth and differentiation. Cell Physiol Biochem. 2001;11:173–186. doi: 10.1159/000047804. [DOI] [PubMed] [Google Scholar]
- Sylvester JT. Hypoxic pulmonary vasoconstriction: A radical view. Circ Res. 2001;88:1228–1230. doi: 10.1161/hh1201.093167. [DOI] [PubMed] [Google Scholar]
- Szöcs K, Lassè B, Sorescu D, Hilenski LL, Valppu L, Couse TL, Wilcox JN, Quinn MT, Lambeth JD, Griendling KK. Upregulation of Nox-Based NAD(P)H oxidases in restenosis after carotid injury. Arterioscler Thromb Vasc Biol. 2002;22:21–27. doi: 10.1161/hq0102.102189. [DOI] [PubMed] [Google Scholar]
- Tang XD, Daggett H, Hanner M, Garcia ML, McManus OB, Brot N, Weissbach H, Heinemann SH, Hoshi T. Oxidative regulation of large conductance calcium-activated potassium channels. J Gen Physiol. 2001;117:253–273. doi: 10.1085/jgp.117.3.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thannickal VJ, Fanburg BL. Reactive oxygen species in cell signaling. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1005–L1028. doi: 10.1152/ajplung.2000.279.6.L1005. [DOI] [PubMed] [Google Scholar]
- Wang D, Youngson C, Wong V, Yeger H, Dinauer MC, Vega-Saenz De Miera E, Rudy B, Cutz E. NADPH-oxidase and a hydrogen peroxide-sensitive K+ channel may function as an oxygen sensor complex in airway chemoreceptors and small cell lung carcinoma cell lines. Proc Natl Acad Sci USA. 1996;93:13182–13187. doi: 10.1073/pnas.93.23.13182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waypa GB, Chandel NS, Schumacker PT. Model for hypoxic pulmonary vasoconstriction involving mitochondrial oxygen sensing. Circ Res. 2001;88:1259–1266. doi: 10.1161/hh1201.091960. [DOI] [PubMed] [Google Scholar]
- Wilson DF, Mokashi A, Chugh D, Vinogradov S, Osanai S, Lahiri S. The primary oxygen sensor of the cat carotid body is cytochrome α3 of the mitochondrial respiratory chain. FEBS Lett. 1994;351:370–374. doi: 10.1016/0014-5793(94)00887-6. [DOI] [PubMed] [Google Scholar]
- Wyatt CN, Peers C. Ca2+-activated K+ channels in isolated type I cells of the neonatal rat carotid body. J Physiol. 1995;483(3):559–565. doi: 10.1113/jphysiol.1995.sp020606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D, Rovira II, Finkel T. Oxidants painting the Cysteine Chapel: Redox regulation of PTPs. Developmental Cell. 2002;2:251–259. doi: 10.1016/s1534-5807(02)00132-6. [DOI] [PubMed] [Google Scholar]
- Youngson C, Nurse C, Yeger H, Curnutte JT, Vollmer C, Wong V, Cutz E. Immunocytochemical localization on O2-sensing protein (NADPH oxidase) in chemoreceptor cells. Microsc Res Tech. 1997;37:101–106. doi: 10.1002/(SICI)1097-0029(19970401)37:1<101::AID-JEMT10>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Zhu H, Jackson T, Bunn HF. Detecting and responding to hypoxia. Nephrol Dial Transplant. 2002;17 1:3–7. doi: 10.1093/ndt/17.suppl_1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]