

Abstract
Purpose
Most transgenic animal models of retinal degeneration caused by rhodopsin mutations express the rhodopsin transgene on a wild-type (WT) genetic background. Previous studies have demonstrated that one mechanism of retinal degeneration is rhodopsin overexpression. To study the effect of C-terminal truncation of rhodopsin without the confounding factors of overexpression, several lines of transgenic mice were generated that expressed a C-terminal rhodopsin mutation on rhodopsin-knockout backgrounds.
Methods
Two lines of transgenic mice, expressing different levels of C-terminal truncated rhodopsin (S334ter) were mated with heterozygous rhodopsin-knockout (rho+/−) mice to express S334ter rhodopsin on a background with reduced endogenous rhodopsin expression. S334ter mice were mated to homozygous knockout (rho−/−) mice to examine the effect of S334ter rhodopsin on a null rhodopsin background. S334ter rhodopsin expression was estimated by Western blot. Retinal function was assessed by ERG and retinal degeneration by histopathology and morphometry. C-terminal rhodopsin sorting and trafficking was examined by fluorescence immunocytochemistry with detection by electron microscope.
Results
Expression of S334ter truncated rhodopsin at low levels in the presence of decreased total rhodopsin in rods (S334ter, rho+/−) increased the rate of rod cell death in comparison to rho+/− littermates. In addition, S334ter rhodopsin prolonged the recovery time of the rod ERG to a light flash and diminished the a-wave amplitudes in comparison to their (rho+/−) littermates. Photoreceptors of S334ter mice on a homozygous rhodopsin-knockout background (S334ter+, rho−/−) had a fraction of mutant rhodopsin localized to the ciliary membranes.
Conclusions
Expression of S334ter rhodopsin without overexpression of total opsin in the rod photoreceptor decreased rod cell contribution to the ERG and compromised rod cell survival in adult mice. The increased cell death may be a consequence of C-terminal truncated rhodopsin mislocalization in membranes of the inner segment. Another possible pathologic mechanism is prolonged activation of phototransduction from the presence of mutant rhodopsin in the outer segment lacking the normal C-terminal binding sites for shutoff by arrestin and phosphorylation. These results suggest that rhodopsin lacking a C-terminal trafficking signal can be transported to the rod outer segment without cotransporting with full-length rhodopsin.
Rhodopsin mutations are common, with more than 100 distinct rhodopsin mutations described in patients who have autosomal dominant retinitis pigmentosa (ADRP).1 Symptoms of RP include loss of sensitivity to dim light, abnormal visual function, and characteristic bone spicule deposits of pigment in the retina. Affected individuals progressively lose visual field and visual acuity, and photoreceptor cell death can ultimately lead to blindness. Although mutations in several photoreceptor-specific and some nonspecific genes cause RP, mutations in rhodopsin are the most prevalent class identified to date, resulting in 30% to 40% of all ADRP.2 A fraction of these rhodopsin mutations alter the C-terminal tail of the protein, such as the point mutations P347L, P347S, P347R, and V345M.3–5 In addition, two frame-shift mutations (fs 341del and fs 341-343del) are predicted to add additional residues to the C terminus,6 whereas Q344ter results in a C-terminal truncation, and an intron splice mutation (N88) is thought to remove the entire C-terminal tail of rhodopsin.7,8 Previously, transgenic mice were generated with a C-terminal truncation S334ter to remove all the putative sites of rhodopsin shutoff by arrestin and phosphorylation, to study the effect on phototransduction.9 With the subsequent identification of several mutations within this region in patients with RP, these mice have become increasingly useful in the study of RP pathogenesis, as well as intracellular sorting of rhodopsin in photoreceptors. Other transgenic mouse models, Q334ter and P347S, have been created with mutations in the C-terminal cytoplasmic domain.10,11 In these animals, extracellular mislocalization of rhodopsin P347S, and intracellular delocalization of Q344ter to inner segment and synaptic terminal membranes strongly suggest a role for the C-terminal tail in opsin sorting and transport.
Rhodopsin trafficking has been further investigated in vitro and in vivo with other systems. Evidence supporting the role of the C-terminal residues for proper sorting into post-Golgi vesicles has been obtained with a retinal cell-free system.12 The final five amino acids of the C-terminal (QVAPA in human) were found to be critical for this sorting process. In other studies, the dynein light chain Tctex-1 has been shown to interact directly with the C-terminal tail of rhodopsin and has been hypothesized to mediate transport from the Golgi to the apical inner segment membrane.13 Transgenic Xenopus have also been used to examine the distribution of rhodopsin tail–GFP fusion proteins, uncovering transport signals in the last 44 amino acids that are sufficient for outer segment localization, and specifically, the last eight amino acids directed a nonpolarized protein exclusively to the outer segments.14 In addition to delocalizing, a fraction of GFP-tail fusion proteins lacking these eight C-terminal amino acids was present within the outer segment, despite removal of its transport signal. The authors suggest this may occur through cotransport with full-length rhodopsin. This mechanism has been independently suggested by another group based on observations that outer segments failed to form in photoreceptors expressing truncated rhodopsin on a rhodopsin-knockout background.15 However, this hypothesis has yet to be studied in detail and warrants further examination, to understand the transport mechanism of truncated rhodopsin in rod photoreceptors.
Aberrant transport and mislocalization of truncated rhodopsin may have a detrimental effect on photoreceptors’ health and survival. Similar to transgenic Q344ter rhodopsin mice, transgenic animals expressing S334ter rhodopsin lacking the terminal 15 amino acids mislocalize a fraction of truncated opsin to ectopic photoreceptor membranes and undergo a progressive retinal degeneration.8,15,16 Various studies, however, have documented that overexpression of rhodopsin itself causes photoreceptor cell death and may induce photoreceptor cell loss in transgenic animals expressing truncated rhodopsin on a wild-type (WT) genetic background.11,17,18 Therefore, it is still unknown whether truncated rhodopsin is harmful to photoreceptors in these transgenic animals. With the creation of rhodopsin-knockout mice,19,20 it is now possible to lower the concentration of endogenous rhodopsin in vivo by genetically removing one or both rhodopsin allele(s), and this technique has been used in several studies.15,21,22 In one, Concepcion et al.15 placed S334ter transgenic mice on a +/− endogenous rhodopsin genetic background, and did not observe greater photoreceptor degeneration than in control mice. However, outer nuclear layer (ONL) thickness was observed only up to P50, and long-term effects on photoreceptors were not assessed.
In the current report, we expanded on the work of Concepcion et al.15 by completing a long-term time-course study to determine whether truncated rhodopsin induces photoreceptor degeneration. We also performed detailed immunoelectron microscopy after the subcellular trafficking of S334ter rhodopsin in the absence of endogenous rhodopsin to determine whether the truncated form reaches the outer segment independent of WT rhodopsin. Truncated rhodopsin was found to affect rod function and increased the rate of photoreceptor degeneration. In addition, our data indicate that a fraction of truncated rhodopsin reaches the outer segment without requiring cotransport along with normal rhodopsin.
Material and Methods
Generation of Transgenic Animals
All animals were treated in accordance with both the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research and procedures approved by the University of California Animal Care and Use Committee.
A low-expressing S334ter rhodopsin transgenic mouse line, CTC,9,15 was crossed with rhodopsin-knockout mice20 to generate S334ter+, rho+/− progeny. F1 generation siblings were mated to produce littermates carrying the transgene on various rhodopsin-knockout backgrounds (rho+/−, rho−/−). Control littermate mice lacking the transgene were also created from crosses. Presence of the transgene was detected by PCR amplification using primers Rho2 (5′ TGGGAGATGACGACGCCTAA 3′) and Rho3 (5′ TGAGGGAGGGGTACAGATCC 3′). The rhodopsin-knockout allele was detected with a separate amplification of the neomycin cassette using primers Neo4 (5′CGGGAGCGGCGATACCGTAAAGC 3′) and Neo7 (5′ GAAGCGGGAAGGGACTGGCTGCTA 3′). Matings were repeated with CTA9, a high-expressing S334ter mouse line. All mouse lines created were reared in cyclic light.
Rhodopsin Western
Retinas from P20 animals were individually isolated and homogenated by sonication in a chilled buffer (5 mM Tris-acetate buffer with 65 mM NaCl, 2 mM MgCl2) containing protease inhibitors (Sigma-Aldrich, St. Louis, MO). Equivalent amounts of protein were incubated with Laemmli loading dye/SDS with β-mercaptoethanol (2%) overnight at 4°C and separated electrophoretically on a 12% Tris gel (Bio-Rad, Hercules, CA) at 65 mV for ~2.5 hours. Transfer of proteins to a polyvinylidene difluoride (PVDF)–charged membrane was performed with a miniprotean gel transfer apparatus (Bio-Rad). Overnight transfer was run at 25 mV and 100 amps at 4°C. Membranes were blocked with 5% nonfat dried milk in phosphate-buffered saline (PBS) with Tween-20. Primary rhodopsin antibody, Rho4D2, was diluted 1:100 in 4 mL blocking buffer and used to probe the membrane. After they were washed, the membranes were incubated with horseradish peroxidase (HRP)–conjugated anti-mouse secondary antibodies (Sigma-Aldrich) at 1:1000 dilution. Chemiluminescence detection of labeling was performed with ECL reagents (NEN Life Science Products, Boston, MA) in dark conditions. X-ray film (Kodak, Rochester, NY) was exposed for 2 to 30 minutes until the bands were detectable. Densitometric measurements of Western blot analysis were completed with NIH Image software (available by ftp at zippy.nimh.nih.gov/or at http://rsb.info.nih.gov/nih-image; developed by Wayne Rasband, National Institutes of Health, Bethesda, MD). Two animals were used per genotype.
Electroretinography
For electroretinography, the animals were dark adapted overnight before testing. They were anesthetized with 100 μg ketamine and 16 μg xylazine per gram body weight in dim red light conditions. Eyes were dilated for 1 minute with phenylephrine hydrochloride 2.5% (Wilson, Mustang, OK) and atropine sulfate 1% (Alcon, Fort Worth, TX). Proparacaine 0.5% (Bausch & Lomb, Tampa, FL) was used as a local anesthetic. Before flash stimulus, the animals were further kept in the dark for 20 minutes for consistent, full pupil dilation. Body temperature was kept at 38°C by placement on a heating pad, and ERG recordings were measured with silver chloride contact electrodes placed on the cornea with a layer of clear 2% hydroxypropyl methyl-cellulose (Gonisol; Wilson). Silver reference and ground electrodes were placed subcutaneously in both cheeks and tail, respectively. A 0.5 log cd/m2 scotopic flash was presented by a mounted Ganzfeld field burst stimulator controlled by a commercial system (Espion; Diagnosys, Littleton, MA). For experiments using a short interstimulus interval (ISI), an initial 0.5-log cd/m2 flash was followed 10 seconds later by a second flash. For analysis, the a-wave was measured as the distance from the baseline to trough of the negative curve, whereas the b-wave was measured from the trough to peak of the positive curve. Only the measurements from the right eye were used to ensure reproducibility of light stimuli. The mean ± SD was calculated from measurements of seven animals per genotype for animals at postnatal day (P)30 to P35 and P100 to P110 and four animals per genotype at P210 to P220.
Histology and Morphometry
Eyes were isolated from P35 to P40, P110 to P120, and P220 to P230 littermate and age-matched animals after cardiac perfusion with 2.5% formaldehyde/2% glutaraldehyde/PBS and immersion in fixative for 2 to 10 days. Eyes were cauterized to mark the poles and bisected hemispherically along the superior/inferior pole. The samples were osmicated, washed, and dehydrated in a graded series of ethanol baths before they were embedded in Epon-Araldite resin. Polymerization occurred at 65°C and 1-μm sections were cut on a microtome (UltraCut; Leica, Deerfield, IL) and stained with 1% toluidine blue. Images were taken with a cooled charge-coupled device (CCD) camera mounted on a microscope (Axiocam; Carl Zeiss Meditec, Inc., Thornwood, NY). ONL thickness of the superior and inferior hemispheres was measured every 100 μm along 20 to 26 equally spaced intervals from optic nerve head to ora serrata, by using a camera lucida drawing tube and software (Axiovision; Zeiss) interfaced with a digitizing tablet (Wacom, Inc., Vancouver, WA). The mean ± SD was calculated from measurements of six to seven animals per genotype at P35 to P40 and P110 to P120 and four animals per genotype at P220 to P230.
Immunohistochemistry
Eyes of P20 to P25 mice were enucleated, pierced through the cornea, and immersed in 4% formaldehyde-PBS for 5 to 10 minutes. Cornea and lens were removed and eyecups were fixed for 1 hour, followed by PBS washes and cryoprotection in 15% and 30% sucrose. Eyecups were embedded in OCT medium (Sakura Fine-Tek USA, Torrance, CA) and frozen at −80°C. Seven-micrometer-thick frozen sections were cut, placed on glass slides, and blocked with 2% BSA, 0.1% FBS, and 1% Triton X-100, probed with affinity-purified anti-rom-1 Rom1 C-1 (acidic; 1:500) antibody, washed with PBS, and incubated with Cy3 sheep anti-rabbit secondary antibody (1:1000; Sigma-Aldrich). The sections were incubated with Hoechst 33342 nuclear dye (Invitrogen-Molecular Probes, Inc., Eugene, OR) at 1:10,000 dilution for 1 minute before addition of antifade medium (Prolong Antifade; Invitrogen-Molecular Probes) to reduce bleaching. For rhodopsin detection, Rho4D2 (1:10) primary monoclonal antibody and Cy2 secondary antibody (1:100; Jackson ImmunoResearch Laboratories, Inc., West Grove, PA) were used. Labeling was performed on two mice per genotype. Images were then captured by microscope (Axiophot; Carl Zeiss Meditec, Inc.).
Immunoelectron Microscopy
The eyes were processed for immunoelectron microscopy with a low-temperature embedding unit (Bal-Tec LTE 020; Furstentum, Liechtenstein). Retinas were dissected from the eyecup and lightly fixed in 4% formaldehyde/0.5% glutaraldehyde for 2 hours. The retinas were washed in PBS and dehydrated in a graded series of ethanol washes at progressively lower temperatures, to preserve antigenicity. Samples were embedded (Lowicryl K11M medium; Polysciences, Inc., Warrington, PA) and polymerized under UV at −20°C. Seventy- to 90-nm sections were cut with an ultramicrotome (Leica) and placed on formvar-coated nickel grids. The sections were probed with the primary rhodopsin antibodies Rho4D2 or Rho1D4 at 1:150 dilution. Five-or 10-nm gold-conjugated secondary antibodies were used at 1:40 dilution (Ted Pella, Redding, CA). The grids were postfixed in 0.5% glutaraldehyde, rinsed with water, and counterstained with 2% uranyl acetate and 0.5% lead citrate. The images were captured on film with an electron microscope at 80 kV (JEOL USA., Peabody, MA). Measurements of S334ter rhodopsin density in photoreceptor plasma membranes were made by counting gold particles per micrometer of external membrane. The mean ± SD was calculated from measurements of three CTCKO photoreceptors at P9 and P30.
Statistical Analysis
The two-tailed unpaired t-test was used for statistical analysis. P < 0.05 was considered significant.
Results
Identification of Mutant and Normal Mice on Multiple Rhodopsin-Knockout Backgrounds
The following genotypes were generated: WT (rho+/+), WTKO (rho+/−), CTCWTKO (S334ter+, rho+/−), CTCKO (S334ter+ rho−/−), and KO (rho−/−). The animals were genotyped and phenotyped by using PCR analysis in addition to functional and/or histologic screening. Mice carrying the rhodopsin transgene were identified by PCR with primers specific for the mutant rhodopsin (Fig. 1A). The presence of the targeted rhodopsin-knockout cassette was identified with a separate set of gene-specific primers for neomycin. Homozygous rhodopsin-knockout mice (rho−/−) were phenotypically identified by ERG analysis and verified histologically, as rho−/− mice have an absent a- and small b-wave response due to loss of long outer segment structures.19,23,24 CTCKO mice also fail to produce full outer segments,15 and were screened from CTCWTKO animals by using ERG and histologic analysis. WTKO mice missing one rhodopsin allele have shorter outer segments due to decreased opsin expression,19–21 and this morphologic difference distinguished WT and WTKO animals.
Figure 1.
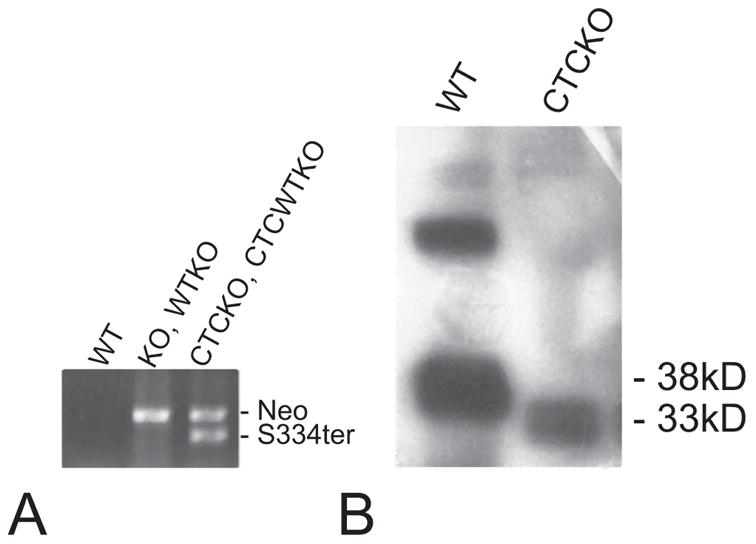
Western blot and transgene PCR analysis of WT and mutant mice. (A) PCR analysis of genomic DNA with primers specific for the Neomycin (Neo) KO cassette and the S334ter transgene confirmed their presence or absence. KO (rho−/−) and WTKO (rho+/−) mice are positive for the neomycin cassette. CTCKO (S334ter+, rho+/−) and CTCWTKO (S334ter+, rho−/−) animals are positive for both Neo and S334ter PCR products. WT (rho+/+) animals are negative for either product. (B) Anti-rhodopsin antibody Rho4D2 was used to probe immunoblots prepared from P20 WT and CTCKO (S334ter+, rho−/−) retinas. S334ter rhodopsin migrated as a smaller fragment due to the removal of 15 amino acids from its C terminus.
Mutant Rhodopsin Expression
Western blot analysis was performed to compare the levels of mutant rhodopsin relative to normal rhodopsin by measuring protein concentrations in retinas of CTCKO (S334ter+, rho−/−) and WT (rho+/+) mice. The shorter, truncated S334ter rhodopsin had a molecular mass of ~33 kDa because of the 15-amino-acid deletion from the C terminus and migrated farther than the 38-kDa full-length WT rhodopsin. The amount of truncated opsin was found to be ~10% of WT levels (Fig 1B), consistent with other reports.9,15
Transport of Truncated Rhodopsin in Mutant Photoreceptors
To determine the distribution of rhodopsin and other outer segment proteins within rod photoreceptors, frozen retinal sections of S334ter+, rho+/− mice were labeled with the anti-rhodopsin antibody, Rho4D2, and anti-rom-1, Rom C-1 acidic. Rho4D2 antibody binds to the N terminus of rhodopsin and thus recognizes both endogenous and mutant forms. Full-length rhodopsin was localized to the outer segments of rho+/− photoreceptors, and was weakly detected in the ONL (Fig. 2A). Rhodopsin was detected in various membrane domains within the CTCWTKO (S334ter+, rho+/−) photoreceptors including the outer segment, inner segment, perinuclear region, and synaptic terminal (Fig. 2B). Immunohistochemistry performed on CTCKO (S334ter+, rho−/−) mice photoreceptors lacking endogenous rhodopsin also showed prominent mislocalization of truncated rhodopsin within the inner segment, cell body, and synaptic terminal membranes (Fig. 2C). Although long outer segments do not form in CTCKO animals, membranes distal to the inner segment were labeled with rhodopsin antibody (Fig. 2C). The disc rim protein, Rom-1, was localized to outer segments in both WTKO and CTCWTKO mice, indicating that the presence of truncated rhodopsin does not alter the localization of Rom-1 (data not shown). In CTCKO photoreceptors, Rom-1 trafficked to the distal connecting cilium, was incorporated into ciliary membranes, and was virtually absent from inner segments and the cell body (data not shown). This observation is similar to Rom-1 localization in rhodopsin-knockout mice,25 and suggests that the mechanisms governing outer segment protein transport are not severely altered in CTCKO mice, despite the absence of large, organized outer segments.
Figure 2.
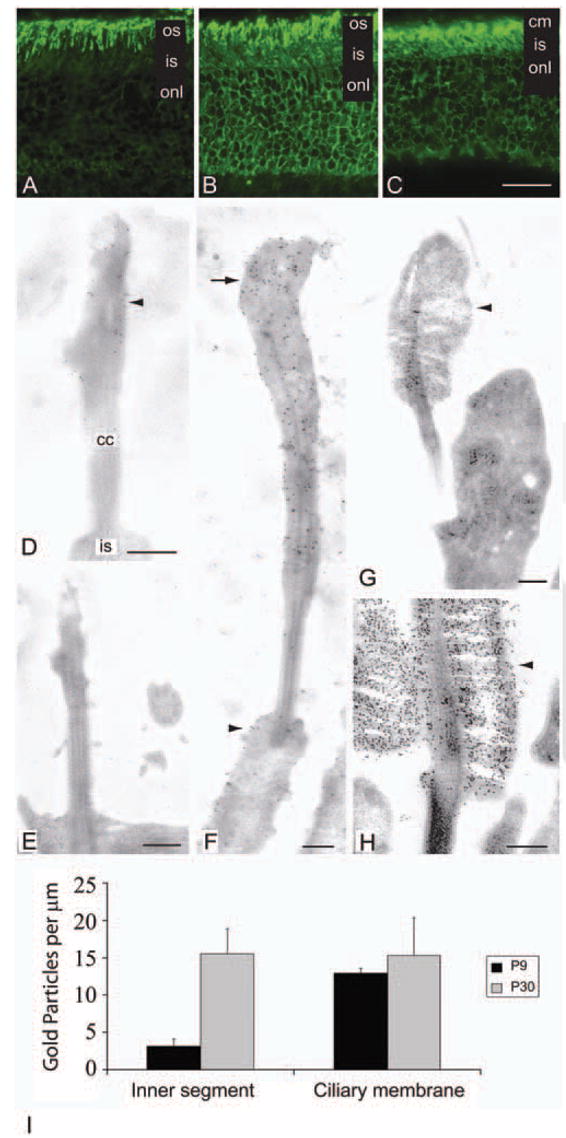
Rhodopsin distribution in WT and mutant photoreceptors. (A–C) Immunohistochemical labeling with anti-rhodopsin antibody, Rho4D2 on frozen sections of P23 WTKO, CTCWTKO, and CTCKO mice. Rho4D2 antibody recognizes the N terminus of rhodopsin and thus labels both WT and mutant forms. (A) In WTKO (rho+/−) photoreceptors, Rho4D2 predominantly immunolabeled outer segments. (B) Membranes of the outer segments, inner segments, nuclei, and synaptic terminal were labeled in the CTCWTKO (S334ter+, rho+/−) mice. (C) In CTCKO photoreceptors expressing only S334ter rhodopsin, the mutant opsin was detected in the ciliary, inner segment, perinuclear, and synaptic terminal membranes. (D–H) Electron micrographs of WT and mutant photoreceptors immunolabeled with Rho4D2. (D) In P9 CTCKO photoreceptors, a low concentration of truncated rhodopsin (arrowhead) and membranes were detected in the distal connecting cilium. (E) An aged-matched rhodopsin KO photoreceptor showing no rhodopsin labeling. (F) Larger rudimentary ciliary membranes lacking organized discs were observed at P30, with higher levels of truncated S334ter rhodopsin in these distal ciliary membranes (arrow). A portion of truncated rhodopsin also mislocalized (arrowhead) to inner segment membranes. (G) Control P9 WTKO (rho+/−) photoreceptors formed small outer segments (arrowhead) heavily labeled for rhodopsin. (H) Rhodopsin localized to the rod outer segments discs (arrowhead) in P30 WTKO mice. (I) Immunogold S334ter rhodopsin labeling density in CTCKO (S334ter+, rho−/−) mice photoreceptors. Ciliary and inner segment plasma membrane densities were measured at P9 and P30. Results are expressed as the mean ± SD of three photoreceptors per age. os, outer segment; is, inner segment; onl, outer nuclear layer; cm, ciliary membrane; cc, connecting cilium. Scale bars: (A–C) 20 μm; (D–H) 250 nm.
Immunoelectron microscopy of ultrathin sections was performed to determine the subcellular localization of rhodopsin in CTCKO mice and to investigate whether truncated rhodopsin transports to the ciliary membranes in photoreceptors lacking endogenous rhodopsin. Truncated rhodopsin was detected within the distal connecting cilium tip where morphogenesis of the outer segment normally initiates (Fig. 2D), and in the inner segment of P9 developing photoreceptors (data not shown). At this age, very little membrane was observed at the tip of the connecting cilium, resembling the morphology of rhodopsin-knockout photoreceptors. As anticipated, rhodopsin was not detected in KO (rho−/−) photoreceptors (Fig. 2E). In P30 CTCKO mice, further incorporation of truncated rhodopsin and membranes at the connecting cilium tip created short, thin ciliary membranes labeled with Rho4D2 antibody (Fig. 2F). P9 WTKO mice generally produced small, rudimentary outer segments heavily labeled with rhodopsin antibodies, whereas at P30, outer segments with rhodopsin-laden discs were observed (Figs. 2G, 2H).
To determine the distribution levels of truncated rhodopsin within the ciliary and inner segment membranes, counts of gold particles labeling S334ter rhodopsin were made in these two membrane domains. In developing P9 CTCKO mice, S334ter rhodopsin increased approximately fourfold in the external ciliary plasma membrane compared with inner segment membranes (Fig. 2I). At P30, the density of S334ter rhodopsin was similar between the overlying ciliary and inner segment membranes. Although moderate interphotoreceptor variability of truncated rhodopsin was observed at this age, intraphotoreceptor differences in concentration were minimal between the two membrane domains.
Photoreceptor Development and Transport of Truncated Rhodopsin in Mice Overexpressing Mutant Opsin
The absence of outer segment formation in CTCKO (S334ter+, rho−/−) animals is probably a consequence of the severely reduced levels of truncated rhodopsin. To determine whether an increase in the concentration of truncated rhodopsin would induce outer segment formation, we placed a transgenic mouse line overexpressing S334ter on a homozygous rhodopsin-knockout background, removing endogenous opsin. Retinas of CTA9KO (S334ter+++, rho−/−) animals degenerated rapidly and therefore, we examined developing photoreceptors for outer segment formation and rhodopsin localization at P8, when degeneration was moderate. P8 CTA9KO photoreceptors failed to form normal-sized outer segments and at best, recruited small amounts of membrane to the distal connecting cilium (Fig. 3A). This result suggests that a relatively small fraction of truncated opsin can reach the outer segment on its own. In comparison with P9 CTCKO mice (Fig. 2D), larger amounts of membranes were recruited to the distal connecting cilium of CTA9KO mice, indicating a correlation between the level of truncated rhodopsin expression and the amount of rhodopsin transported to the ciliary membranes. As a control, we restored one copy of WT opsin, to determine whether the lack of outer segment formation was a secondary effect of degeneration induced by opsin overexpression. In CTA9WTKO (S334ter+++, rho+/−) littermate animals, we observed partial rescue in a small fraction of photoreceptors, creating rudimentary outer segments that were in general smaller than aged-matched WT photoreceptors (Figs. 3B–D). This signifies that small outer segments still have the potential to form in these rapidly degenerating retinas. Photoreceptors lacking outer segment material were also observed in both CTA9KO and CTA9WTKO retinas.
Figure 3.
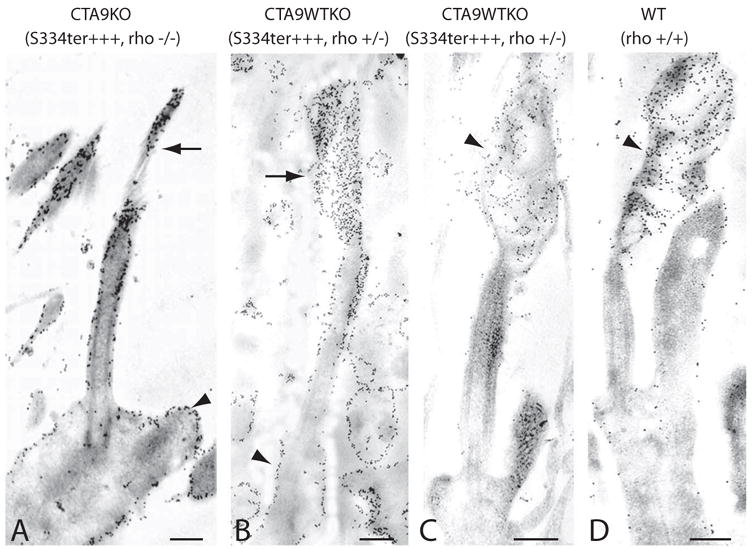
Overexpression of truncated rhodopsin in CTA9KO and CTA9WTKO photoreceptors. Retinal sections were labeled with anti-rhodopsin antibody Rho4D2 (N-terminal) or Rho1D4 (C-terminal). (A) Only a very small concentration of membranes with truncated rhodopsin (arrow) accumulated at the connecting cilium tip in P8 CTA9KO (S334ter+++, rho−/−) photoreceptors labeled with Rho4D2. Connecting cilium and inner segment membranes (arrowhead) were densely labeled in this mouse line, which overexpressed truncated rhodopsin on a homozygous rhodopsin-knockout background. (B, C) Restoring one allele of endogenous full-length rhodopsin induced formation of small rudimentary outer segments (arrow) in a fraction of P8 CTA9WTKO photoreceptors. (B) The rhodopsin 4D2 antibody heavily labeled photoreceptor membranes (arrowhead) except for the proximal connecting cilium. (C) Anti-rhodopsin antibody, Rho1D4, recognizes residues in the C terminus of rhodopsin and specifically labeled full-length rhodopsin (arrowhead) in our animals. Endogenous, full-length rhodopsin localized to the outer segment and rarely delocalized to inner segment membranes. (D) In P8 WT photoreceptors, rhodopsin distributed predominantly to the outer segment (arrowhead), although inner segment labeling was detected in a portion of photoreceptors. Scale bars, 250 nm.
Labeling of CTA9KO photoreceptors with anti-rhodopsin antibody, Rho4D2, revealed a high concentration of truncated rhodopsin within cell membranes including the distal tip of the connecting cilium and inner segment membranes (Fig. 3A). Anti-rhodopsin antibody also showed saturated labeling of inner segment membranes in CTA9WTKO photoreceptors (Fig. 3B). In contrast, labeling of CTA9WTKO with the C-terminal rhodopsin antibody (Rho1D4), detected little, if any, full-length rhodopsin in the inner segment membrane, whereas the outer segment membranes remained more heavily labeled (Fig. 3C). This finding emphasizes that normal trafficking of WT rhodopsin is not severely disrupted by truncated S334ter opsin and indicates that delocalized rhodopsin is mainly composed of truncated rhodopsin.
ERG Measurements of Rod Function in S334ter Photoreceptors
In addition to the removal of transport signals, S334ter truncated rhodopsin also lacks C-terminal residues for termination of its activity by rhodopsin kinase phosphorylation and arrestin binding. Chen et al.9 have reported that the presence of S334ter truncated rhodopsin in rods expressing full-length rhodopsin results in a prolonged response to light. Therefore, to verify the expression of truncated opsin in photoreceptors of our animals, we conducted dark-adapted ERG recordings on CTCWTKO (S334ter+, rho+/−) mice at P30 to P35 with a 10-second interstimulus interval and similarly observed a decrease in a- and b-wave amplitudes after a second flash (data not shown). When flashes were paired with a 10-minute inter-stimulus interval, a decrease was not observed in either mouse line. These experiments indicated that these animals were indeed expressing truncated rhodopsin and that recovery of bleached truncated rhodopsin after a bright flash occurs within 10 minutes. Mice expressing only truncated rhodopsin did not elicit an a-wave response and displayed only a small b-wave that remained unaltered after stimulation with a second flash (data not shown). Similarly, homozygous rhodopsin-knockout mice (rho−/−) generated only small b-wave responses, consistent with other reports.19,23,24 This finding suggest that the truncated opsins found in the ciliary membrane or delocalized in the inner segment of CTCKO photoreceptors do not contribute to the photoresponse.
In P30 to P35 animals, a slight decrease was observed in the a-wave of CTCWTKO mice compared with that of WTKO littermate control animals. We performed a time-course study using single full-flash ERGs to examine whether this difference was significant and to determine whether the truncated rhodopsin effects overall retinal function. In young (P30–P35) animals, the a-wave amplitudes of CTCWTKO animals were lower than those in control WTKO animals (Fig. 4A). This decrease was a small, but significant difference (P < 0.01 two-tail unpaired t-test). The a-waves of older CTCWTKO mice diminished progressively with age, as P100 to P110 and P210 to P220 animals expressing the mutant rhodopsin had reduced a-waves compared with WTKO (rho+/−) mice (P < 0.0005, P < 0.01; Fig. 4A). Conversely, b-wave amplitudes were maintained in P30 to P35 and P100 to P110 animals, and were only slightly, but significantly lower in older (P210–P220) animals (P < 0.05; Fig. 4B).
Figure 4.
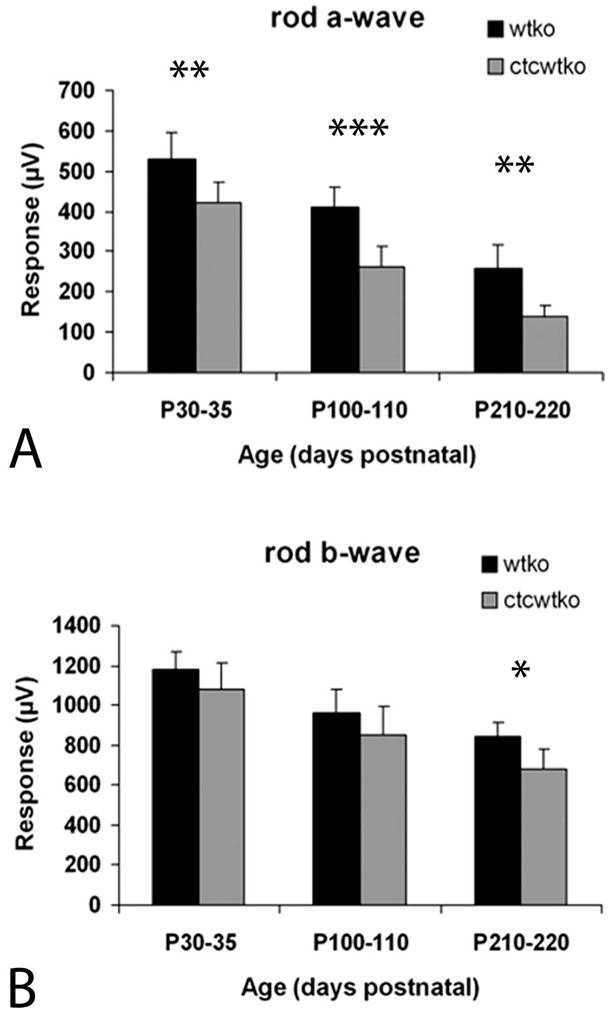
Scotopic electroretinogram recordings in WT and transgenic mice. (A, B) ERG responses after a single bright flash. (A) ERG response from CTCWTKO and WTKO animals showed a significant reduction of scotopic a-wave amplitude in CTCWTKO mice relative to WTKO mice at all ages measured. Over time, mean a-wave amplitudes decreased in both mouse lines. (B) The b-waves were significantly lower in CTCWTKO mice only at the oldest age tested (P210–P220). *P < 0.05; **P < 0.01; ***P < 0.0005 (two-tailed, unpaired t-test).
Effect of Truncated Rhodopsin on Photoreceptor Degeneration
To determine whether truncated rhodopsin affects photoreceptor morphology and survival, we evaluated the histology of CTCWTKO (S334ter+, rho+/−) retinas. At every age examined (P35–P40, 110–P120, and 220–P230), the outer segments of WTKO and CTCWTKO photoreceptors were shorter than those in WT eyes (WT; Fig. 5). Furthermore, at P110 to P120, the outer segments of S334ter+, rho+/− mice appeared slightly shorter than those in rho+/−, and minor outer segment disorganization was evident in a fraction of the CTCWTKO animals (Figs. 5E, 5F). At P220 to P230, mild outer segment disorganization of WTKO photoreceptors was also observed, although the outer segments of CTCWTKO animals appeared overall shorter and more disorganized at this age (Figs. 5H, 5I). The ONL thickness of WTKO and CTCWTKO retinas were close to normal thickness at P35 to P40 (Figs. 5B, 5C). Older P110 to P120 WTKO animals had a thinner ONL relative to WT animals with the loss of one to two rows of photoreceptors, and CTCWTKO retinas expressing truncated opsin had further reduction of the number of photoreceptor (Figs. 5E, 5F). Thinning of the ONL continued in older P220 to P230 animals for all genotypes (Figs. 5H, 5I).
Figure 5.
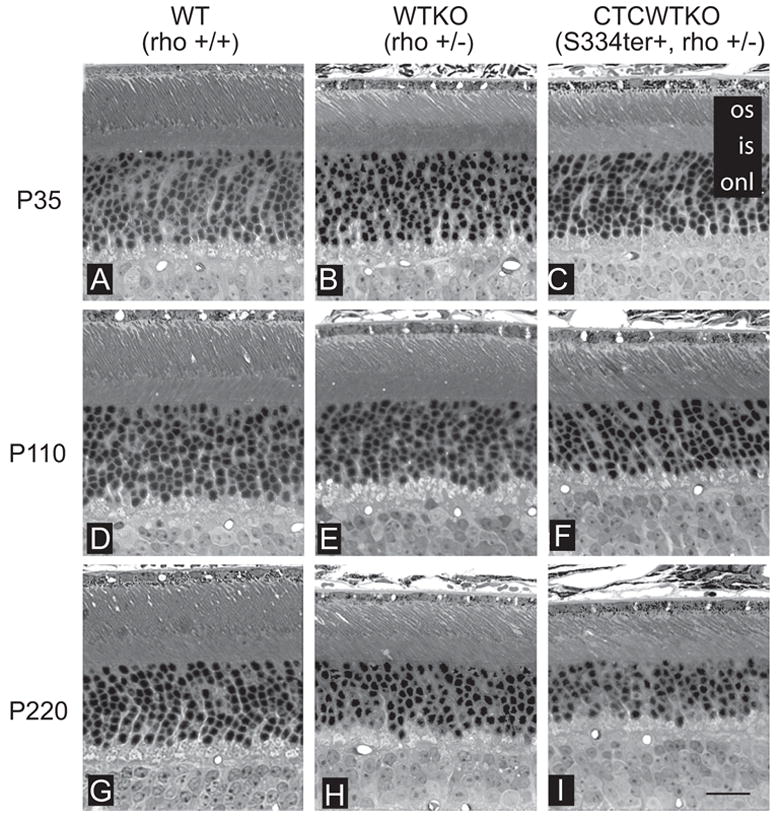
Histologic examination of WT and transgenic retinas. Light micrographs from the inferior retina were taken at different ages. (A–C) P35 retinas of WT, WTKO, and CTCWTKO mice had similar ONL thicknesses (10–12 rows). Outer segments were slightly shorter in WTKO and CTCWTKO animals compared with WT controls. (D–F) At P110, WTKO animals had a slightly reduced ONL thickness relative to WT animals, whereas thicknesses in CTCWTKO animals were further reduced one to two rows. Outer segments of CTCWTKO mice were occasionally shorter and slightly more disorganized than those in WTKO at this age. (G–I) By P220, each genotype examined had lost one to three more rows of photoreceptors. The ONL of CTCWTKO mice was thinner, and the outer segments were generally shorter and more disorganized than in WTKO mice. Abbreviations are as in Figure 2. Scale bar, 20 μm.
Photoreceptor degeneration was quantified more precisely by measuring the ONL thickness of WTKO and CTCWTKO mice at P35 to P40, P110 to P120, and P220 to P230. ONL thickness has been shown to be a comparative measure of the number of photoreceptors.26 Measurements were taken in 100-μm intervals from the optic nerve head to the ora serrata in both the superior and inferior halves of the eye. Figure 6A shows a spider graph plotted from P220 littermate animals. The ONL thickness was largest in WT animals, followed by WTKO animals, and was the thinnest in CTCWTKO animals. Averages of ONL thickness were plotted in WTKO and CTCWTKO animals against age, to measure the loss of photoreceptors over time (Fig. 6B). In P35 to P40 animals, the mean ONL thickness was comparable. By P110 to P120, photoreceptors had degenerated in both mouse lines, although the loss occurred at a faster rate in animals expressing truncated rhodopsin. The average ONL thickness of mice expressing the mutant opsin on a heterozygous rhodopsin-knockout background was reduced 9% compared with control WTKO (rho+/−) littermate and age-matched animals. This was a small, but a significant decrease in the number of photoreceptors as measured by mean ONL thickness (P < 0.005). In P220 to P230 animals, the average number of photoreceptors of CTCWTKO mice was significantly reduced 10% compared with that in control mice (P < 0.0001).
Figure 6.

Truncated rhodopsin accelerates photoreceptor degeneration in adult mice. (A) Spider graphs of ONL thickness measurements taken in 100-μm increments from the optic nerve head (ONH) to the ora serrata (ORS) in both the inferior and superior retina indicate in the P220 littermate animals that overall thickness was greatest in WT mice, followed by WTKO, and was thinnest in CTCWTKO animals. (B) Average ONL thickness plotted against age in WTKO and CTCWTKO mice. The ONL of S334ter+, rho+/− mice was significantly thinner in older animals (P110–P120 and P220–P230). Note that the x-axis is not to scale. *P < 0.005; **P < 0.001 (significant by two-tailed, unpaired t-test).
Discussion
Our data indicate that expression of truncated rhodopsin compromises rod cell survival. In previous reports, transgenic animals with a truncated rhodopsin mutation on a WT rhodopsin background (rho+/+) also lost photoreceptors over time, although degeneration may have been a consequence of rhodopsin overexpression. By placing S334ter transgenic mice on a heterozygous rhodopsin-knockout background, we removed the variable of overexpression to show that truncated rhodopsin itself negatively affects both photoreceptor function and health. The pathologic occurrences observed in our animals were signaled by a slow, progressive decline in the number of photoreceptors that persisted in P230 animals, the latest time point measured in our study.
Accelerated degeneration of the CTCWTKO (S334ter+, rho+/−) mice may be due to a toxic build-up of mislocalized, truncated rhodopsin in various ectopic membrane locations throughout the rod cell. The presence of truncated opsin may impair synaptic transmission or other cellular processes and eventually cause cell death. The metabolic burden placed on the cell due to the constant degradation of a large concentration of truncated rhodopsin may also lead to degeneration. Studies of dissociated salamander cultures have suggested that the presence of delocalized opsin in the inner segment membranes triggers apoptosis through a G-protein cascade on reception of light.27 However, a recent report by Tam et al.28 showed mislocalized truncated rhodopsin modified to be incapable of activating transducin could not prevent photoreceptor loss. The authors hypothesized that large quantities of mislocalized opsin may decrease the availability of functional proteins in regions where truncated opsin is concentrated.
Prolonged photoreceptor responses may also cause or contribute to photoreceptor degeneration in animals expressing S334ter rhodopsin. In our animals, photoreceptors expressing both normal and S334ter mutant opsin displayed a slowed recovery to flash due to the presence of truncated rhodopsin lacking C-terminal tail phosphorylation sites for rapid deactivation. Abnormal rhodopsin deactivation may induce outer segment shortening and eventual photoreceptor death, similar to that in arrestin- and rhodopsin kinase–knockout mice. These mice have reduced outer segment length at an early age (P30, P42 respectively), followed by eventual photoreceptor death when reared in cyclic light—a consequence thought to occur through deleterious effects of prolonged activation.29,30 This effect may account for the degeneration observed in our cyclic-light–reared animals and lack of degeneration observed in dark-reared CTCWTKO mice, as the effect of prolonged stimulation was removed.15 However, ONL thickness in dark-reared animals was measured up to P50—an age that may have preceded the onset of degeneration; older animals should be assessed. Generation of a low-expressing Q344ter rhodopsin mouse line on a heterozygous rhodopsin-knockout background could also distinguish the effects of prolonged activation versus mislocalization on a-wave reduction and rod degeneration. The Q344ter truncated rhodopsin mislocalizes to ectopic locations similar to S344ter rhodopsin, but retains C-terminal phosphorylation sites for rhodopsin kinase and arrestin binding.11
Toxicity to photoreceptors by S334ter rhodopsin may cause or contribute to abnormal rod function observed in older CTCWTKO mice. Retinas of P110 to P120 or P220 to P230 CTWTKO animals producing decreased a-wave responses exhibited greater loss of photoreceptors and generally shorter and more disorganized outer segments than age-matched WTKO animals. Young CTCWTKO mice unexpectedly had lower a-waves relative to control WTKO animals, despite possessing close to normal the number of photoreceptors with no apparent ultrastructural outer segment defects. The reason for this decrease observed in young animals is currently unknown. Possibly, the mislocalization of S334ter rhodopsin to the lateral plasma membranes and synaptic terminal affect rod function by globally impacting the health of the cell, poisoning photoreceptors before degeneration. Alternatively, mild prolonged photoreceptor activation may reduce the number of channels in the outer segment or decrease the concentration of phototransduction proteins, thus reducing the amplification of the signal. However, the possibility that a short exposure to dim red light prolonged rhodopsin activity, partially light-adapting the retina, cannot be excluded, even though rods were allowed to recover for 20 minutes before ERG measurements were initiated.
As the presence of truncated rhodopsin in the outer segments causes functional abnormalities and localization of rhodopsin in the outer and/or inner segments induces increased photoreceptor cell death, another goal was to study the trafficking of truncated rhodopsin. We sought to determine whether S334ter truncated rhodopsin reaches the outer segment by cotransport with WT rhodopsin. To test this hypothesis, photoreceptors that expressed only truncated rhodopsin were created by genetically removing endogenous rhodopsin. Our results show that a portion of truncated opsin was detected in membranes protruding from the distal connecting cilium tip in both developing and mature CTCKO photoreceptors. This finding is strong evidence that in photoreceptors expressing both truncated and full-length forms, a fraction of truncated opsin can transport to the outer segment, independent of endogenous rhodopsin.
Our data support a mechanism in which truncated rhodopsin is nonspecifically targeted to various membrane domains. In P30 animals expressing only truncated rhodopsin, the density of S334ter rhodopsin in the ciliary membrane and inner segment were similar, suggesting nonspecific membrane localization. In addition, overexpression of truncated rhodopsin did not rescue photoreceptor outer segment formation when expressed on a homozygous rhodopsin-knockout background. Although there was an overall increase in truncated rhodopsin collecting at the tip of the connecting cilium compared with mice only expressing 10% levels, this increase is most likely due to higher concentration of truncated rhodopsin randomly transported to the distal connecting cilium. In accordance, the distribution between the ciliary and inner segment plasma membranes appeared similar in these overexpressing photoreceptors. The loss of the transport signal may affect rhodopsin trafficking by interfering with proper sorting, or the directed transport of post-Golgi carriers. Recently, the terminal 5 amino acids of rhodopsin was found to be involved in sorting into post-Golgi carriers through binding with ARF4,31 a small GT-Pase, and removal of this sorting–packaging signal disrupted regulated sorting.
In summary, our results have shown that a fraction of truncated opsin can reach the ciliary membranes independent of rhodopsin, and without cotransport with WT rhodopsin. Truncated opsin was also found to have a deleterious effect on rod photoreceptor physiology and viability, as mutant opsin impaired rod output to a bright flash and accelerated photoreceptor degeneration. Although the S334ter mutation has not been found to occur naturally in the human population, CTCWTKO animals are genetically similar to affected carriers with the loss of one rhodopsin allele. S334ter mice on a heterozygous rhodopsin-knockout background may be a useful model for testing potential therapies in addition to understanding the mechanism of functional loss and degeneration attributed to truncated rhodopsin.
Acknowledgments
The authors thank Jeannie Chen for the CTC mice, Melvin Simon for the CTA9 mice, Janis Lem for the rhodopsin KO mice, Robert Molday for the rhodopsin Rho4D2 antibody, Roderick McInnes for the Rom-1 C-1 antibody, Jason Lee for technical assistance, and others in the Flannery Laboratory for helpful comments on the manuscript.
Supported by National Eye Institute Grant EY013553 and The Foundation Fighting Blindness.
Footnotes
Disclosure: E.S. Lee, None; J.G. Flannery, None
References
- 1.Weleber RG, Gregory-Evans K. Retinitis pigmentosa and allied disorders. In: Ryan SJ, editor. Retina. St Louis: Mosby; 2001. pp. 362–470. [Google Scholar]
- 2.The Retina Network (RetNet) Houston, TX: University of Texas-Houston Health Science Center; [Accessed June 10, 2005]. Available at http://www.sph.uth.tmc.edu/RetNet/disease/htm. [Google Scholar]
- 3.Sung CH, Davenport CM, Hennessey JC, et al. Rhodopsin mutations in autosomal dominant retinitis pigmentosa. Proc Natl Acad Sci USA. 1991;88:6481–6485. doi: 10.1073/pnas.88.15.6481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dryja TP, Hahn LB, Cowley GS, et al. Mutation spectrum of the rhodopsin gene among patients with autosomal dominant retinitis pigmentosa. Proc Natl Acad Sci USA. 1991;88:9370–9374. doi: 10.1073/pnas.88.20.9370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dryja TP, McGee TL, Hahn LB, et al. Mutations within the rhodopsin gene in patients with autosomal dominant retinitis pigmentosa. N Engl J Med. 1990;323:1302–1307. doi: 10.1056/NEJM199011083231903. [DOI] [PubMed] [Google Scholar]
- 6.Horn M, Humphries P, Kunisch M, et al. Deletions in exon 5 of the human rhodopsin gene causing a shift in the reading frame and autosomal dominant retinitis pigmentosa. Hum Genet. 1992;90:255–257. doi: 10.1007/BF00220073. [DOI] [PubMed] [Google Scholar]
- 7.Jacobson SG, Kemp CM, Cideciyan AV, et al. Phenotypes of stop codon and splice site rhodopsin mutations causing retinitis pigmentosa. Invest Ophthalmol Vis Sci. 1994;35:2521–2534. [PubMed] [Google Scholar]
- 8.Sung CH, Schneider BG, Agarwal N, et al. Functional heterogeneity of mutant rhodopsins responsible for autosomal dominant retinitis pigmentosa. Proc Natl Acad Sci USA. 1991;88:8840–8844. doi: 10.1073/pnas.88.19.8840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen J, Makino CL, Peachey NS, et al. Mechanisms of rhodopsin inactivation in vivo as revealed by a COOH-terminal truncation mutant. Science. 1995;267:374–377. doi: 10.1126/science.7824934. [DOI] [PubMed] [Google Scholar]
- 10.Li T, Snyder WK, Olsson JE, Dryja TP. Transgenic mice carrying the dominant rhodopsin mutation P347S: evidence for defective vectorial transport of rhodopsin to the outer segments. Proc Natl Acad Sci USA. 1996;93:14176–14181. doi: 10.1073/pnas.93.24.14176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sung CH, Makino C, Baylor D, Nathans J. A rhodopsin gene mutation responsible for autosomal dominant retinitis pigmentosa results in a protein that is defective in localization to the photoreceptor outer segment. J Neurosci. 1994;14:5818–5833. doi: 10.1523/JNEUROSCI.14-10-05818.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deretic D, Puleo-Scheppke B, Trippe C. Cytoplasmic domain of rhodopsin is essential for post-Golgi vesicle formation in a retinal cell-free system. J Biol Chem. 1996;271:2279–2286. doi: 10.1074/jbc.271.4.2279. [DOI] [PubMed] [Google Scholar]
- 13.Tai AW, Chuang JZ, Bode C, et al. Rhodopsin’s carboxy-terminal cytoplasmic tail acts as a membrane receptor for cytoplasmic dynein by binding to the dynein light chain Tctex-1. Cell. 1999;97:877–887. doi: 10.1016/s0092-8674(00)80800-4. [DOI] [PubMed] [Google Scholar]
- 14.Tam BM, Moritz OL, Hurd LB, Papermaster DS. Identification of an outer segment targeting signal in the COOH terminus of rhodopsin using transgenic Xenopus laevis. J Cell Biol. 2000;151:1369–1380. doi: 10.1083/jcb.151.7.1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Concepcion F, Mendez A, Chen J. The carboxyl-terminal domain is essential for rhodopsin transport in rod photoreceptors. Vision Res. 2002;42:417–426. doi: 10.1016/s0042-6989(01)00195-x. [DOI] [PubMed] [Google Scholar]
- 16.Green ES, Menz MD, LaVail MM, Flannery JG. Characterization of rhodopsin mis-sorting and constitutive activation in a transgenic rat model of retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2000;41:1546–1553. [PubMed] [Google Scholar]
- 17.Olsson JE, Gordon HW, Pawlyk BS, et al. Transgenic mice with a rhodopsin mutation (Pro23His): a mouse model of autosomal dominant retinitis pigmentosa. Neuron. 1992;9:815–830. doi: 10.1016/0896-6273(92)90236-7. [DOI] [PubMed] [Google Scholar]
- 18.Tan E, Wang Q, Quiamboa AB, et al. The relationship between opsin overexpression and photoreceptor degeneration. Invest Ophthalmol Vis Sci. 2001;42:589–600. [PubMed] [Google Scholar]
- 19.Humprhies MM, Rancout D, Farrar GJ, et al. Retinopathy induced in mice by targeted disruption of the rhodopsin gene. Nat Genet. 1997;2:216–219. doi: 10.1038/ng0297-216. [DOI] [PubMed] [Google Scholar]
- 20.Lem J, Krasnoperova NV, Calvert PD, et al. Morphological, physiological, and biochemical changes in rhodopsin knockout mice. Proc Natl Acad Sci USA. 1999;96:736–741. doi: 10.1073/pnas.96.2.736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frederick JM, Krasnoperova NV, Hoffmann K, et al. Mutant rhodopsin transgene expression on a null background. Invest Ophthalmol Vis Sci. 2001;42:826–833. [PubMed] [Google Scholar]
- 22.Naash MI, Wu TH, Chakraborty D, et al. Retinal abnormalities associated with the G90D mutation in opsin. J Comp Neurol. 2004;478:149–163. doi: 10.1002/cne.20283. [DOI] [PubMed] [Google Scholar]
- 23.Jaissle GB, May CA, Reinhard J, et al. Evaluation of the rhodopsin knockout mouse as a model of pure cone function. Invest Ophthalmol Vis Sci. 2001;42:506–513. [PubMed] [Google Scholar]
- 24.Toda K, Bush RA, Humphries P, Sieving PA. The electroretinogram of the rhodopsin knockout mouse. Vis Neurosci. 1999;16:391–398. doi: 10.1017/s0952523899162187. [DOI] [PubMed] [Google Scholar]
- 25.Lee ES, Burnside B, Flannery FG. Characterization of peripherin/rds and rom-1 in knockout and transgenic animals. Invest Ophthalmol Vis Sci. 2006;47:2150–2160. doi: 10.1167/iovs.05-0919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Michon JJ, Li ZL, Shioura N, et al. A comparative study of methods of photoreceptor morphometry. Invest Ophthalmol Vis Sci. 1991;32:280–284. [PubMed] [Google Scholar]
- 27.Alfinito PD, Townes-Anderson E. Activation of mislocalized opsin kills rod cells: a novel mechanism for rod cell death in retinal disease. Proc Natl Acad Sci USA. 2002;99:5655–5660. doi: 10.1073/pnas.072557799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tam BM, Xie G, Oprian DD, Moritz OL. Mislocalized rhodopsin does not require activation to cause retinal degeneration and neurite outgrowth in Xenopus laevis. J Neurosci. 2006;26:203–209. doi: 10.1523/JNEUROSCI.3849-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen CK, Burns ME, Spencer M, et al. Abnormal photoresponses and light-induced apoptosis in rods lacking rhodopsin kinase. Proc Natl Acad Sci USA. 1999;96:3718–3722. doi: 10.1073/pnas.96.7.3718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen J, Simon MI, Matthes MT, et al. Increased susceptibility to light damage in an arrestin knockout mouse model of Oguchi disease (stationary night blindness) Invest Ophthalmol Vis Sci. 1999;40:2978–2982. [PubMed] [Google Scholar]
- 31.Deretic D, Williams AH, Ransom N, et al. Rhodopsin C terminus, the site of mutations causing retina disease, regulates trafficking by binding to ADP-ribosylation factor 4 (ARF4) Proc Natl Acad Sci USA. 2005;102:3301–3306. doi: 10.1073/pnas.0500095102. [DOI] [PMC free article] [PubMed] [Google Scholar]