

Abstract
Reactive oxygen species (ROS) are reactive derivatives of O2 metabolism, including superoxide anion, hydrogen peroxide, hydroxyl radical and nitric oxide. All types of vascular cells produce ROS, primarily via cell membrane-associated NAD(P)H oxidase. Cardiovascular diseases, such as hypertension, are associated with increased ROS formation (oxidative stress). Oxidative excess in the vasculature reduces levels of the vasodilator nitric oxide, causes tissue injury, promotes protein oxidation and DNA damage, and induces proinflammatory responses. ROS are also important intracellular signalling molecules that regulate vascular function by modulating vascular cell contraction/dilation, migration, growth/apoptosis, and extracellular matrix protein turnover, which contribute to vascular remodelling. Interventions to decrease ROS bioavailability regress remodelling and reduce blood pressure in experimental hypertension. Such strategies may have therapeutic potential in cardiovascular diseases.
Keywords: Endothelium, Free radicals, Inflammation, Redox signaling, Smooth muscle cells, Vessels
Abstract
Les espèces oxygénées radicalaires (EOR) sont des dérivés radicalaires du métabolisme de l’oxygène, y compris l’anion de superoxyde, le peroxyde d’hydrogène, l’hydroxyle et le monoxyde d’azote. Tous les types de cellules vasculaires produisent des EOR, surtout par l’oxydase NAD(P)H associée aux membranes cellulaires. Les maladies cardiovasculaires, comme l’hypertension, sont reliées à une formation accrue d’EOR (stress oxydatif). L’excédent oxydatif dans le système vasculaire réduit les taux de monoxyde d’azote vasodilatateur, provoque des lésions tissulaires, favorise l’oxydation protéique et les dommages à l’ADN et induit des réponses pro-inflammatoires. Les EOR sont également d’importantes molécules de signalisation intracellulaire, qui régularisent la fonction vasculaire en modulant la contraction et la dilatation, la migration la croissance et l’apoptose des cellules vasculaires, ainsi que le renouvellement des porines extracellulaires, qui contribuent au remodelage vasculaire. Les interventions en vue de réduire la biodisponibilité de l’EOR font régresser le remodelage et réduisent la tension artérielle dans les cas d’hypertension expérimentale. Ces stratégies peuvent avoir un potentiel thérapeutique en présence de maladies cardiovasculaires.
Reactive oxygen species (ROS) are ubiquitous reactive derivatives of O2 metabolism found in all biological systems. ROS were traditionally regarded as byproducts of aerobic metabolism. However, ROS have recently been recognized to act as signalling molecules in vascular cells and to play a role in cellular events associated with vascular remodelling in cardiovascular diseases (1). ROS function as important intracellular and intercellular second messengers to modulate many downstream signalling molecules, such as protein tyrosine phosphatases (PTPs), protein tyrosine kinases, transcription factors, mitogen-activated protein kinases (MAPKs) and ion channels. Induction of these signalling cascades leads to vascular smooth muscle cell (VSMC) growth and migration, expression of proinflammatory mediators and modification of the extracellular matrix (ECM). In addition, ROS increase intracellular free Ca2+ concentrations, a major determinant of vascular reactivity. ROS influence signalling molecules by altering the intracellular reduction-oxidation (redox) state and by oxidative modification of proteins. In physiological conditions, these events are highly regulated and play an important role in maintaining vascular function and integrity. Under pathological conditions, dysregulation of ROS, due to enhanced production and/or reduced antioxidant potential, contributes to increased bioavailability of free radicals and consequently to oxidative stress, which results in vascular dysfunction and remodelling through oxidative damage.
Among ROS, attention has focused on the highly reactive free radical superoxide anion (•O2−) and the more stable hydrogen peroxide (H2O2). ROS are formed as intermediates in redox processes leading from O2 to H2O. The univalent reduction of O2, in the presence of a free electron, yields •O2−, which is cell membrane-impermeable (Figure 1). In physiological conditions, the favoured reaction of •O2− is dismutation by superoxide dismutase (SOD) yielding H2O2, which is scavenged by catalase and glutathione peroxide to produce H2O. In the vasculature, all types of cells, including endothelial cells (ECs), VSMCs and fibroblasts, produce •O2− and H2O2 to varying degrees (2). When generated in excess, •O2− reacts with nitric oxide (NO) to produce peroxynitrite, a potentially deleterious oxidant, leading to decreased NO bioavailability. H2O2 is lipid-soluble and crosses cell membranes. A mismatch between ROS formation and the defense ability of antioxidants results in increased bioavailability of ROS, leading to a state of oxidative stress (3).
Figure 1.
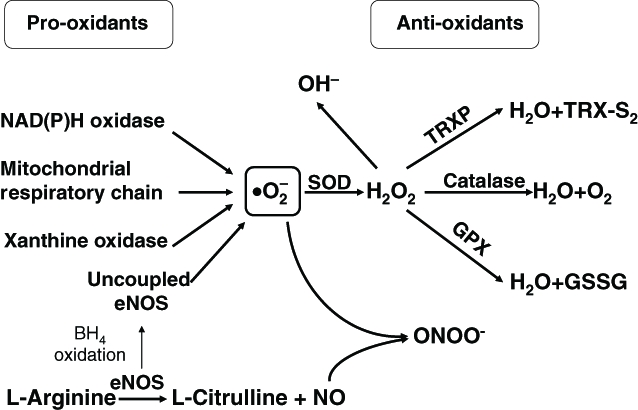
Possible role of reactive oxygen species in vascular remodelling and implications in the pathogenesis of hypertension. Angiotensin II-stimulated NAD(P)H oxidase in vascular cells results in the univalent reduction of O2 in the presence of a free electron to yield superoxide anion (•O2−), which in turn is dismutated to hydrogen peroxide (H2O2). In the presence of nitric oxide (NO), peroxynitrite (ONOO−) can be formed. Increased intracellular levels of reactive oxygen species contribute to vascular inflammation, growth, altered contraction and dilation (vascular tone), and endothelial dysfunction. These events in turn lead to vascular remodelling, arterial narrowing, increased peripheral resistance and consequently to increased blood pressure. BH4 Tetrahydrobiopterin; eNOS Endothelial nitric oxide synthase; GPX Glutathione peroxidase; GSSG Oxidized glutathione; •OH− Hydroxyl radical; SOD Superoxide dismutase; TRXP Thioredoxin peroxidase; TRX-S2 Thioredoxin disulphide
Through their cell-damaging effects, ROS have been implicated to play a role in vascular injury associated with cardiovascular diseases, such as hypertension, atherosclerosis, restenosis and diabetic vascular complications (1–3). In hypertension, small arteries undergo structural remodelling due, in large part, to increased cell growth, cell migration, ECM deposition and inflammation (4). All of these processes are influenced to varying degrees by ROS (2) (Figure 1).
GENERATION OF ROS IN THE VASCULATURE
Potential sources of vascular •O2− generation include NAD(P)H oxidase, uncoupled nitric oxide synthase (NOS), xanthine oxidase and mitochondria. Among them, NAD(P)H oxidase appears to be the major source in vascular cells. Vascular NAD(P)H oxidases that are low-output, slow-release enzymes differ from phagocytic NAD(P)H oxidases in their structural and biochemical characteristics (5) (Table 1). Phagocytic NAD(P)H oxidases comprise a plasma membrane-spanning cytochrome b558 composed of a large catalytic subunit, gp91phox (nox2), and a small subunit, p22phox, together with cytosolic regulatory subunits p47phox, p67phox, p40phox and the small GTPase rac (6). Phagocytic NAD(P)H oxidase uses intracellular NADPH and transfers electrons across the membrane to extracellular O2 (6). However, ROS generation in vascular cells appear to be intracellular. gp91phox (nox2), p22phox, p47phox and p67phox have been identified in ECs, adventitial fibroblasts (5) and VSMCs from small human resistance arteries (7). Studies on VSMCs from aortas and other large arteries demonstrate that p22phox, p47phox and rac are expressed, whereas nox2 and p67phox are either absent or present in very low concentrations. Instead, the nox2 homologues, nox1 and nox4, appear to be the major catalytic subunits in these cells (8).
TABLE 1.
Differences between phagocytic and vascular NAD(P)H oxidase
| Characteristic | Phagocytic oxidase | Vascular oxidase |
|---|---|---|
| Name | NAD(P)H oxidase | nox enzymes |
| Activity | Basal state inactive | Constitutively active |
| Primary function | Host:defense reactions | Signal transduction |
| Mode of activity | Inducible | Inducible |
| Nox isoform | gp91phox (nox2) | gp91phox/nox1/nox4/nox5 |
| Pattern of •O2− release | Burst-like | Slow and sustained |
| Concentration released | High | Low (1% to10% of phagocytic) |
| Site of •O2− release | Extracellular | Intracellular |
| Substrate | NAD(P)H | NAD(P)H/NADH |
| Small guanine nucleotide-binding protein | rac2 | rac1 |
On cell stimulation, p47phox becomes phosphorylated and the cytosolic subunits form a complex, which then migrates to the membrane, where it associates with cytochrome b558 to assemble the active oxidase, which transfers electrons from the substrate to O2, leading to •O2− generation (6). In vascular cells, nox4 expression is abundant and may play an important role in constitutive •O2− production in nonproliferating cells. On stimulation, nox1 is upregulated and may be important in vascular pathology (9). Recently, nox organizer 1 (NOXO1) and nox activator 1, homologues of p47phox and p67phox, respectively, have been cloned from colon epithelial cells (10). Similar to p47phox, NOXO1 binds to p22phox, which is required for nox1-dependent activity. NOXO1 is prelocalized at membranes together with nox1 and p22phox in unstimulated cells (10). However, the functional significance of these homologues in the vasculature remains unclear.
Endothelial nitric oxide synthase (eNOS) is a calcium-dependent flavoenzyme that generates NO in a process involving oxidation of the amino acid L-arginine by the reduction of molecular O2. All three NOSs, eNOS, neuronal NOS and inducible NOS can also generate •O2− in conditions of substrate (arginine) or cofactor (tetrahydrobiopterin [BH4]) deficiency (11). These findings have led to the concept of ‘NOS uncoupling’, where the activity of the enzymes for NO production is decreased in association with an increase in NOS-dependent •O2− formation. BH4 itself is highly susceptible to oxidative degradation, and the initial oxidative loss of BH4 in response to increased ROS production by NAD(P)H oxidases has been shown to amplify oxidative stress through the resulting loss of NO production and increased NOS-dependent •O2− generation (8,11). In spontaneously hypertensive rats (SHR), in spite of the increased expression and activity of NOS, •O2− is elevated and NO production is reduced. In deoxycorti- costerone acetate-salt hypertensive mice, BH4, and NO are improved or restored by treatment with BH4, eNOS gene deletion, apocynin, or p47phox gene deletion, suggesting a role for NAD(P)H oxidase in NOS uncoupling (8).
Electron leakage from the mitochondrial electron transport chain constitutively produces •O2−, usually rapidly degraded by manganese SOD. However, under some pathological conditions, such as hypoxia/reoxygenation, mitochondria may be a significant source of •O2− in a ceramide-dependent fashion (12,13). In deoxycorticosterone acetate-induced hypertension, a model of endothelin (ET)-dependent hypertension, although NAD(P)H oxidase and xanthine oxidase activities are increased, only mitochondrial generation of ROS was normalized by ETA receptor antagonist, indicating that mitochondria may play a role in ET-1-driven oxidative stress (14).
Xanthine oxidase requires reduction of molecular O2 to catalyze oxidation of hypoxanthine to xanthine and xanthine to urate, thereby generating •O2−. The possible contribution of xanthine oxidase to ROS elevation in hypertension has been assessed using specific inhibitors. Such treatments normalize ROS formation in microvessels from rats fed a high-salt diet, and increase endothelial-dependent relaxation in arteries from SHR and rats overexpressing renin and angiotensinogen genes, suggesting that xanthine oxidase is another potential source of elevated ROS generation in hypertension that could impair vascular function and structure (8).
ROS IN HUMAN AND EXPERIMENTAL HYPERTENSION
Clinical studies demonstrated increased ROS production in patients with essential hypertension, renovascular hypertension, malignant hypertension and preeclampsia (15). In general, these findings are based on increased levels of plasma thiobarbituric acid reactive substances and 8-epi-isoprostanes, biomarkers of lipid peroxidation and oxidative stress. Studies in cultured VSMCs derived from resistance arteries of hypertensive patients revealed enhanced formation of ROS (16). Patients with essential hypertension have decreased levels of antioxidant glutathione, and activity of SOD is reduced (17).
Increased levels/activity of vascular NAD(P)H oxidase has been implicated as the primary source of excess •O2− in essential hypertension (5). Activation of the renin-angiotensin system has been proposed as a major stimulator of NAD(P)H oxidase activation and ROS production in human hypertension (5). Because of this interaction between renin-angiotensin II and •O2−-generating systems, it is not surprising that some of the therapeutic blood pressure-lowering actions of renin-angiotensin converting enzyme inhibitors and renin-angiotensin II type I receptor blockers may be mediated by inhibiting NAD(P)H oxidase activity and reducing ROS production.
Polymorphisms in the p22phox gene have been suggested to play a role in altered NAD(P)H oxidase-generated •O2− on in human cardiovascular disease (18). In particular, the –930A/G polymorphism in the p22phox promoter may be a novel genetic marker associated with hypertension (18). A single nucleotide polymorphism in the p22phox gene has been linked to altered arterial compliance (18). However, to confirm that these polymorphisms are indeed markers for hypertension, studies in large populations are necessary.
Although clinical studies provide compelling evidence that oxidative stress is important in the pathophysiology of hypertension, not all human hypertension is redox-dependent. In never-treated, mild-to-moderate hypertension, lipid peroxidation is not increased (19). In some studies, renin-angiotensin II type I receptor blockade did not improve endothelial function and •O2− production was unaltered in hypertensive subjects (20). Furthermore, many large clinical trials on antioxidants failed to demonstrate beneficial therapeutic effects on blood pressure and cardiovascular outcomes (21–3). Reasons for these discrepancies probably relate to the heterogeneous nature of hypertension and to the complexities of redox biology in the cardiovascular system.
Oxidative stress has been convincingly demonstrated in various models of genetic and experimental hypertension. SHR and stroke-prone SHR exhibit increased NAD(P)H oxidase-driven •O2− generation in mesenteric and aortic arteries (8,15). Increased activation of vascular NAD(P)H oxidase, xanthine oxidase and uncoupling of eNOS have been implicated in enhanced •O2− generation in experimental hypertension, such as renin-angiotensin II-induced hypertension, Dahl salt-sensitive hypertension and lead-induced hypertension. Inhibition of ROS generation with apocynin and scavenging of free radicals with antioxidants or SOD mimetics decreases blood pressure and prevents development of hypertension in most experimental models (2).
ROLE OF ROS IN VASCULAR REMODELLING
ROS regulate vascular function by modulating cell growth, apoptosis/anoikis, migration, inflammation, secretion and ECM protein production. During vascular damage in hypertension, when oxidative stress is increased, redox-sensitive growth processes may lead to accelerated proliferation and hypertrophy, further contributing to vascular injury and remodelling (Figure 1). Rao and Berk (24) showed that VSMC DNA synthesis and cell number increased by increasing intracellular ROS. Agonists, such as renin-angiotensin II (5), platelet-derived growth factor (PDGF) (25) and thrombin (26), stimulate VSMC proliferation and migration by induction of intracellular ROS generation, whereas application of p22phox antisense, catalase or diphenyleneiodinium (DPI), which inhibit NAD(P)H oxidase activity, reduced effects by agonists. However, a recent study (27) showed that moderate concentrations of exogenous H2O2 (0.1 mM) cause cell cycle arrest, and high concentrations induce apoptosis in vascular VSMCs, or multiphase cell cycle arrest in fibroblasts. The differential responses of vascular cells to ROS may be related to different species generated to varying concentrations, and to compartmentalization of ROS. Differential signalling by ROS, due to concentration or time dependency, may also contribute to variable responses (28).
Redox-sensitive inflammatory processes, including expression of proinflammatory molecules, such as monocyte chemoattractant protein 1 and interleukin (IL)-6, expression of adhesion molecules, including vascular cell adhesion molecule-1 and intracellular adhesion molecule-1, further contribute to vascular remodelling in hypertension (2). In cultured ECs and VSMCs, renin-angiotensin II upregulates expression of vascular cell adhesion molecule-1, intracellular adhesion molecule-1, E-selectin, and proinflammatory cytokines/chemokines, IL-6 and IL-8. These effects are mediated via redox-sensitive processes (29).
ROS also influence vascular remodelling by increasing deposition of ECM proteins, such as collagen, and by modulating matrix metalloproteinases (MMPs), which degrade collagen and other ECM proteins. In cultured ECs, lysophosphatidylcholine, a major component of oxidized low-density lipoprotein, induced generation of ROS and secretion of MMP-2 (30). Inhibition of NAD(P)H oxidase attenuated the effects of lysophosphatidylcholine (30).
SIGNALLING PATHWAYS OF ROS IN REGULATING VASCULAR REMODELLING
ROS influence vascular function by modulating various redox-sensitive signalling proteins, such as MAPKs (31). MAPKs, including extracellular signal-regulated kinase (ERK)-1/2, ERK5, p38 MAPK and c-jun n-terminal kinase (JNK), are serine/threonine kinases involved in a number of intracellular signalling pathways that regulate vascular cell growth, apoptosis, contraction, migration and inflammation. H2O2 also influences cell growth by inhibiting nuclear protein transport through ERK1/2-dependent pathways (32). Potential mechanisms for ROS-mediated activation of MAPKs may be via oxidant-induced inactivation of PTPs (33). In VSMC, renin-angiotensin II stimulates ERK1/2 and p38 MAPK activation, whereas transfection of VSMCs with nox1 antisense significantly inhibits p38 MAPK phosphorylation (9). In ECs, increased intracellular ROS activated JNK activity, whereas antioxidant n-acetyl cysteine or diphenyleneiodonium inhibited the rise in JNK activity.
Receptor and nonreceptor tyrosine kinases are also influenced by ROS. Exogenous H2O2 induces tyrosine phosphorylation and activation of PDGF receptor and epidermal growth factor receptor (EGFR), probably due to redox-mediated inhibition of dephosphorylation of PDGF receptor and EGFR by inactivation of membrane-associated PTPs (34). Under pathological conditions associated with oxidative stress, such as hypertension, ROS may directly activate cell surface receptors, thereby amplifying the process of •O2− generation (2,3,5). The cell survival kinase protein kinase B (Akt), which is a nonreceptor tyrosine kinase, is another redox-sensitive kinase. Both exogenous H2O2 and renin-angiotensin II stimulate Akt activation in VSMC (35). Importantly, renin-angiotensin II-induced Akt phosphorylation was inhibited by DPI, overexpression of catalase, and nox1 anti-sense (9), indicating a role for NAD(P)H oxidase in agonist-induced Akt activation.
The proinflammatory transcription factor, nuclear factor kappa B (NFκB) is regulated in a redox-dependent manner. NFκB regulates the expression of a number of genes involved in immune and inflammatory responses. In cultured ECs, H2O2 increases the nuclear translocation of NFκB and binding to target DNA (36), whereas n-acetyl cysteine inhibits activation of NFκB by tumour necrosis factor-beta by inhibiting inhibitor kappa B phosphorylation and degradation (37). Both SOD and catalase prevented activation of NFκB by inhibiting renin-angiotensin II-induced inhibitor kappa B degradation (38). These observations indicate that ROS are important for NFκB activation.
In addition to influencing cellular processes associated with growth and inflammation, ROS modulate intracellular free Ca2+ concentration, a major determinant of vascular contraction and dilation. Both •O2− and H2O2 increase intracellular free Ca2+ concentration in VSMCs and ECs (39). These processes involve mobilization from reticular stores and activation of Ca2+ channels (40,41). These redox-regulated Ca2+ processes may be more important in stress responses than in receptor-mediated signalling by growth factors or cytokines, and may play a role in altered vascular contractility in hypertension.
CONCLUSIONS
ROS are produced in the vasculature in a highly regulated manner. •O2− and H2O2 have important signalling properties, mainly through oxidative modification of proteins and activation of transcription factors that maintain vascular, cardiac and renal function and structure. In hypertension, dysregulation of enzymes such as NAD(P)H oxidase, NOS, xanthine oxidase, mitochondrial enzymes or SOD that generate •O2−, H2O2 and hydroxyl radical or reduced scavenging by antioxidants, results in increased formation of ROS, which has damaging actions on the vasculature. ROS in hypertension contribute to vascular injury by promoting vascular cell growth, ECM protein deposition, activation of MMPs, inflammation, endothelial dysfunction and increased vascular tone. In experimental hypertension oxidative stress is increased and antioxidant levels and activity are decreased. Clinical data suggest that hypertensive patients, especially those with severe hypertension, salt-sensitive hypertension and renovascular hypertension, exhibit oxidative excess. Although inconclusive at present, therapeutic modalities to alter ROS bioavailability by decreasing production and/or by increasing radical scavenging, may regress vascular remodelling, prevent further vascular injury and reduce blood pressure and associated target organ damage in hypertensive patients. Whether it would be preferable, from a clinical viewpoint, to reduce ROS generation rather than increase scavenging of free radicals is unclear at present. However, the potential of SOD mimetics, NAD(P)H oxidase inhibitors and gene transfer strategies to enhance NO and antioxidant bioactivity may provide additional and improved therapeutic options in the management of hypertension and other cardiovascular diseases.
REFERENCES
- 1.Griendling KK, Sorescu D, Lassegue B, Ushio-Fukai M. Modulation of protein kinase activity and gene expression by reactive oxygen species and their role in vascular physiology and pathophysiology. Arterioscler Thromb Vasc Biol. 2000;20:2175–83. doi: 10.1161/01.atv.20.10.2175. [DOI] [PubMed] [Google Scholar]
- 2.Touyz RM, Schiffrin EL. Reactive oxygen species in vascular biology: Implications in hypertension. Histochem Cell Biol. 2004;122:339–52. doi: 10.1007/s00418-004-0696-7. [DOI] [PubMed] [Google Scholar]
- 3.Landmesser U, Harrison DG. Oxidative stress and vascular damage in hypertension. Coron Artery Dis. 2001;12:455–61. doi: 10.1097/00019501-200109000-00004. [DOI] [PubMed] [Google Scholar]
- 4.Intengan HD, Schiffrin EL. Vascular remodeling in hypertension: Roles of apoptosis, inflammation and fibrosis. Hypertension. 2001;38:581–7. doi: 10.1161/hy09t1.096249. [DOI] [PubMed] [Google Scholar]
- 5.Lassegue B, Clempus RE. Vascular NAD(P)H oxidases: Specific features, expression, and regulation. Am J Physiol Regul Integr Comp Physiol. 2003;285:R277–97. doi: 10.1152/ajpregu.00758.2002. [DOI] [PubMed] [Google Scholar]
- 6.Babior BM, Lambeth JD, Nauseef W. The neutrophil NADPH oxidase. Arch Biochem Biophys. 2002;397:342–4. doi: 10.1006/abbi.2001.2642. [DOI] [PubMed] [Google Scholar]
- 7.Touyz RM, Chen X, Tabet F, et al. Expression of a functionally active gp91phox-containing neutrophil-type NAD(P)H oxidase in smooth muscle cells from human resistance arteries: Regulation by angiotensin II. Circ Res. 2002;90:1205–13. doi: 10.1161/01.res.0000020404.01971.2f. [DOI] [PubMed] [Google Scholar]
- 8.Lassegue B, Griendling KK. Reactive oxygen species in hypertension; An update. Am J Hypertens. 2004;17:852–60. doi: 10.1016/j.amjhyper.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 9.Lassegue B, Sorescu D, Szocs K, et al. Novel gp91(phox) homologues in vascular smooth muscle cells: Nox1 mediates angiotensin II-induced superoxide formation and redox-sensitive signaling pathways. Circ Res. 2001;88:888–94. doi: 10.1161/hh0901.090299. [DOI] [PubMed] [Google Scholar]
- 10.Banfi B, Clark RA, Steger K, Krause KH. Two novel proteins activate superoxide generation by the NADPH oxidase Nox1. J Biol Chem. 2003;278:3510–3. doi: 10.1074/jbc.C200613200. [DOI] [PubMed] [Google Scholar]
- 11.Landmesser U, Dikalov S, Price SR, et al. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest. 2003;111:1201–9. doi: 10.1172/JCI14172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sato H, Sato M, Kanai H, et al. Mitochondrial reactive oxygen species and c-Src play a critical role in hypoxic response in vascular smooth muscle cells. Cardiovasc Res. 2005;67:714–22. doi: 10.1016/j.cardiores.2005.04.017. [DOI] [PubMed] [Google Scholar]
- 13.Therade-Matharan S, Laemmel E, Carpentier S, et al. Reactive oxygen species production by mitochondria in endothelial cells exposed to reoxygenation after hypoxia and glucose depletion is mediated by ceramide. Am J Physiol Regul Integr Comp Physiol. 2005;289:R1756–62. doi: 10.1152/ajpregu.00480.2004. [DOI] [PubMed] [Google Scholar]
- 14.Callera GE, Tostes RC, Yogi A, Montezano AC, Touyz RM. Endothelin-1-induced oxidative stress in DOCA-salt hypertension involves NADPH-oxidase-independent mechanisms. Clin Sci (Lond) 2006;110:243–53. doi: 10.1042/CS20050307. [DOI] [PubMed] [Google Scholar]
- 15.Touyz RM. Reactive oxygen species, vascular oxidative stress, and redox signaling in hypertension: What is the clinical significance? Hypertension. 2004;44:248–52. doi: 10.1161/01.HYP.0000138070.47616.9d. [DOI] [PubMed] [Google Scholar]
- 16.Touyz RM, Schiffrin EL. Increased generation of superoxide by angiotensin II in smooth muscle cells from resistance arteries of hypertensive patients: Role of phospholipase D-dependent NAD(P)H oxidase-sensitive pathways. J Hypertens. 2001;19:1245–54. doi: 10.1097/00004872-200107000-00009. [DOI] [PubMed] [Google Scholar]
- 17.Kedziora-Kornatowska K, Czuczejko J, Pawluk H, et al. The markers of oxidative stress and activity of the antioxidant system in the blood of elderly patients with essential arterial hypertension. Cell Mol Biol Lett. 2004;9:635–41. [PubMed] [Google Scholar]
- 18.Zalba G, San Jose G, Moreno MU, Fortuno A, Diez J. NADPH oxidase-mediated oxidative stress: Genetic studies of the p22(phox) gene in hypertension. Antioxid Redox Signal. 2005;7:1327–36. doi: 10.1089/ars.2005.7.1327. [DOI] [PubMed] [Google Scholar]
- 19.Cracowski JL, Baguet JP, Ormezzano O, et al. Lipid peroxidation is not increased in patients with untreated mild-to-moderate hypertension. Hypertension. 2003;41:286–8. doi: 10.1161/01.hyp.0000050963.16405.e6. [DOI] [PubMed] [Google Scholar]
- 20.Ghiadoni L, Magagna A, Versari D, et al. Different effect of antihypertensive drugs on conduit artery endothelial function. Hypertension. 2003;41:1281–6. doi: 10.1161/01.HYP.0000070956.57418.22. [DOI] [PubMed] [Google Scholar]
- 21.Jialal I, Devaraj S. Antioxidants and atherosclerosis: Don’t throw out the baby with the bath water. Circulation. 2003;107:926–8. doi: 10.1161/01.cir.0000048966.26216.4c. [DOI] [PubMed] [Google Scholar]
- 22.Abrescia P, Golino P. Free radicals and antioxidants in cardiovascular diseases. Expert Rev Cardiovasc Ther. 2005;3:159–71. doi: 10.1586/14779072.3.1.159. [DOI] [PubMed] [Google Scholar]
- 23.Vivekananthan DP, Penn MS, Sapp SK, Hsu A, Topol EJ. Use of antioxidant vitamins for the prevention of cardiovascular disease: Meta-analysis of randomised trials. Lancet. 2003;361:2017–23. doi: 10.1016/S0140-6736(03)13637-9. [DOI] [PubMed] [Google Scholar]
- 24.Rao GN, Berk BC. Active oxygen species stimulate vascular smooth muscle cell growth and proto-oncogene expression. Circ Res. 1992;70:593–9. doi: 10.1161/01.res.70.3.593. [DOI] [PubMed] [Google Scholar]
- 25.Sundaresan M, Yu ZX, Ferrans VJ, Irani K, Finkel T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science. 1995;270:296–9. doi: 10.1126/science.270.5234.296. [DOI] [PubMed] [Google Scholar]
- 26.Patterson C, Ruef J, Madamanchi NR, et al. Stimulation of a vascular smooth muscle cell NAD(P)H oxidase by thrombin. Evidence that p47(phox) may participate in forming this oxidase in vitro and in vivo. J Biol Chem. 1999;274:19814–22. doi: 10.1074/jbc.274.28.19814. [DOI] [PubMed] [Google Scholar]
- 27.Deshpande NN, Sorescu D, Seshiah P, et al. Mechanism of hydrogen peroxide-induced cell cycle arrest in vascular smooth muscle. Antioxid Redox Signal. 2002;4:845–54. doi: 10.1089/152308602760599007. [DOI] [PubMed] [Google Scholar]
- 28.Tombes RM, Auer KL, Mikkelsen R, et al. The mitogen-activated protein (MAP) kinase cascade can either stimulate or inhibit DNA synthesis in primary cultures of rat hepatocytes depending upon whether its activation is acute/phasic or chronic. Biochem J. 1998;330:1451–60. doi: 10.1042/bj3301451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Touyz RM. Molecular and cellular mechanisms in vascular injury in hypertension: Role of angiotensin II. Curr Opin Nephrol Hypertens. 2005;14:125–31. doi: 10.1097/00041552-200503000-00007. [DOI] [PubMed] [Google Scholar]
- 30.Inoue N, Takeshita S, Gao D, et al. Lysophosphatidylcholine increases the secretion of matrix metalloproteinase 2 through the activation of NADH/NADPH oxidase in cultured aortic endothelial cells. Atherosclerosis. 2001;155:45–52. doi: 10.1016/s0021-9150(00)00530-x. [DOI] [PubMed] [Google Scholar]
- 31.Tabet F, Schiffrin EL, Touyz RM. Mitogen-activated protein kinase activation by hydrogen peroxide is mediated through tyrosine kinase-dependent, protein kinase C-independent pathways in vascular smooth muscle cells: Upregulation in spontaneously hypertensive rats. J Hypertens. 2005;23:2005–12. doi: 10.1097/01.hjh.0000185715.60788.1b. [DOI] [PubMed] [Google Scholar]
- 32.Czubryt MP, Austria JA, Pierce GN. Hydrogen peroxide inhibition of nuclear protein import is mediated by the mitogen-activated protein kinase, ERK2. J Cell Biol. 2000;148:7–16. doi: 10.1083/jcb.148.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee K, Esselman WJ. Inhibition of PTPS by H(2)O(2) regulates the activation of distinct MAPK pathways. Free Radic Biol Med. 2002;33:1121–32. doi: 10.1016/s0891-5849(02)01000-6. [DOI] [PubMed] [Google Scholar]
- 34.Yang S, Hardaway M, Sun G, Ries WL, Key LL., Jr Superoxide generation and tyrosine kinase. Biochem Cell Biol. 2000;78:11–7. [PubMed] [Google Scholar]
- 35.Ushio-Fukai M, Alexander RW, Akers M, et al. Reactive oxygen species mediate the activation of Akt/protein kinase B by angiotensin II in vascular smooth muscle cells. J Biol Chem. 1999;274:22699–704. doi: 10.1074/jbc.274.32.22699. [DOI] [PubMed] [Google Scholar]
- 36.Barchowsky A, Munro SR, Morana SJ, Vincenti MP, Treadwell M. Oxidant-sensitive and phosphorylation-dependent activation of NF-kappa B and AP-1 in endothelial cells. Am J Physiol. 1995;269:L829–36. doi: 10.1152/ajplung.1995.269.6.L829. [DOI] [PubMed] [Google Scholar]
- 37.Spiecker M, Darius H, Kaboth K, Hubner F, Liao JK. Differential regulation of endothelial cell adhesion molecule expression by nitric oxide donors and antioxidants. J Leukoc Biol. 1998;63:732–9. [PubMed] [Google Scholar]
- 38.Pueyo ME, Gonzalez W, Nicoletti A, Savoie F, Arnal JF, Michel JB. Angiotensin II stimulates endothelial vascular cell adhesion molecule-1 via nuclear factor-kappaB activation induced by intracellular oxidative stress. Arterioscler Thromb Vasc Biol. 2000;20:645–51. doi: 10.1161/01.atv.20.3.645. [DOI] [PubMed] [Google Scholar]
- 39.Lounsbury KM, Hu Q, Ziegelstein RC. Calcium signaling and oxidant stress in the vasculature. Free Radic Biol Med. 2000;28:1362–9. doi: 10.1016/s0891-5849(00)00222-7. [DOI] [PubMed] [Google Scholar]
- 40.Roveri A, Coassin M, Maiorino M, et al. Effect of hydrogen peroxide on calcium homeostasis in smooth muscle cells. Arch Biochem Biophys. 1992;297:265–70. doi: 10.1016/0003-9861(92)90671-i. [DOI] [PubMed] [Google Scholar]
- 41.Tabet F, Savoia C, Schiffrin EL, Touyz RM. Differential calcium regulation by hydrogen peroxide and superoxide in vascular smooth muscle cells from spontaneously hypertensive rats. J Cardiovasc Pharmacol. 2004;44:200–8. doi: 10.1097/00005344-200408000-00009. [DOI] [PubMed] [Google Scholar]