

Abstract
Over the last couple of decades of neuro-immunological research, the p40 family of cytokines has emerged out as one of the most intriguing areas of interest because of multi-faceted roles of these cytokine in immune-modulation and inflammation. The IL-12, the most widely studied cytokine of this family, is well-characterized for its Th1-favoring activity, and therefore plays a key role in the pathophysiology of Th1-mediated autoimmune diseases like multiple sclerosis (MS). On the other hand, the IL-23, another member of the p40 family with shared p40 subunit, drives polarization of Th17, a subset of T cell suspected to have a key role in the pathophysiology of MS and experimental allergic encephalomyelitis (EAE), poses a challenge to our current understanding of Th1/Th2 hypotheses in autoimmune diseases. However, the more puzzling issues, the researchers are currently confronted with, are the biological roles of other two members of this family, the p40 monomer and the p402, the homodimer. Predominance of the mRNA level of p40 over p35 in the central nervous system of EAE and MS suggests a possible involvement of p40 in the pathogenesis of MS. However, the distinctive biological role of monomeric and dimeric form of p40 is not clearly understood yet. Initially, it was thought that p402 does not have any biological activity and only involved in antagonizing bioactive IL-12 but according to recent evidences, both p402 and p40 appear to have a proinflammatory role, therefore might be a crucial molecule in the pathogenesis of MS. The current review focuses on biological function of p40 family of cytokines with particular emphasis on MS.
Keywords: Cytokines, Autoimmune diseases, Multiple sclerosis
The p40 family of cytokines has four members which are IL-12 or p70, the p40 monomer (p40), the p40 homodimer (p402), and the IL-23 (Figure 1). The two of the members IL-12 and IL-23 are heterodimers. The IL-12 is a heterodimer of p40 and p35 subunits whereas IL-23 consists of p40 and p19 subunits (Figure 1). The p35 and p19 subunits are always secreted in association with p40. However, the p40 subunit apart from constituting heterodimers, are also secreted independently forming either monomer or homodimer. A key feature of the p40 family is the presence of a shared subunit p40 in all the members of this family. This distinctive characteristics makes it little difficult to study the functional importance of the p40 family of cytokines.
Figure 1.
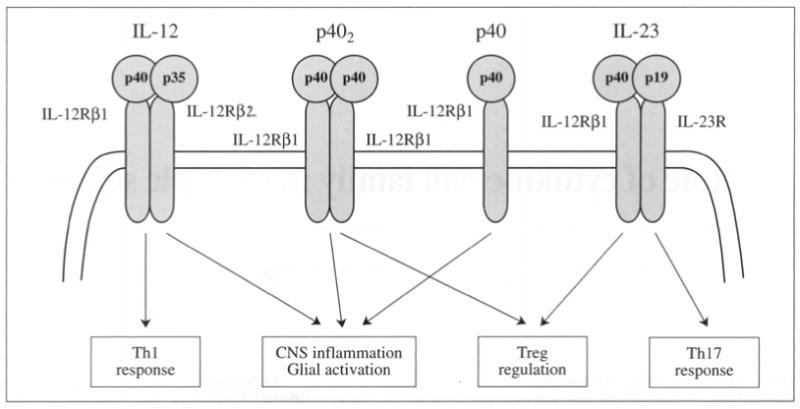
Modulation of immune responses by p40 family of cytokines.
Although in general, the p40 family members are very critical for immunoregulation and inflammation, precise functional roles of all these cytokines are not fully explored yet. One of the convenient approaches to functionally characterize a protein is gene-knockout study. However, because the p40 subunit is shared by all four members, the gene knockout of p40 knocks down all four cytokines of this family and therefore, correct interpretation of result becomes extremely difficult. Although, the IL-12 and IL-23 have at least one different subunit, the p40 and p402 are composed of same subunit differing only by its monomeric and dimeric conformations (Figure 1). Therefore it is extremely difficult to characterize the functional aspects of p40 and p402 unless specific monoclonal antibodies against them are developed. Because multiple sclerosis (MS), the most common human autoimmune demyelinating disease, presents a T-helper-1 (Th1)-mediated immune dysregulation, the p40 family of cytokines is strongly implicated in the pathogenesis of MS.1-3 The present review will illustrate the role of p40 family of cytokines in MS with particular stress on the molecular mechanisms involved in immune-regulation and central nervous system (CNS) inflammation.
The interleukin-12
An overview of structure and function
The interleukin-12 (IL-12 or IL-12p70) is a heterodimeric cytokine consisting of p40 and p35 subunits covalently linked by two disulfide linkages.4 The IL-12p35 subunit has four α-helix structures typical to many other cytokines and shows significant structural homology to IL-6 and G-CSF.5 On the other hand, IL-12p40 subunit with no homology to other cytokines belongs to hemoprotein receptor family of proteins and shows strong structural resemblance with extracellular domain of IL-6 receptor-α subunit and ciliary neurotrophic factor receptor.5
The IL-12 receptor complex (IL-12R) is also made up of two subunits IL-12Rβ1 and IL-12Rβ2 and both of these belong to gp130 subgroup of cytokine receptor superfamily. The IL-12R is primarily expressed on activated T cells and NK cells.4 It appears that IL-12p40 recognizes IL-12Rβ1 which is associated with Tyk2 while the IL-12p35 recognizes IL-12Rβ2 associated with Jak2.6, 7 However, there are significant differences of binding affinity of receptor subunits between human and mouse despite significant structural homology. The mouse IL-12Rβ1 mediates both low and high affinity binding in contrast to human IL-12Rβ1 which provides very high affinity binding with IL-12. On the other hand both mouse and human IL-12Rβ2 mediate low affinity IL-12 binding but for mouse IL-12Rβ2 it is too low to quantify.4 Therefore, it appears that IL-12Rβ1 provides most of the binding energy for high affinity IL-12 binding whereas IL-12Rβ2 primarily acts as a signal transducing subunit. However, biological activity of IL-12p40 monomer and dimer that has been studied recently, suggests that IL-12Rβ1 may also be involved in signal transduction mechanism. Activation of IL-12 receptor complex results in phosphorylation, dimerization and nuclear translocation of several STAT transcription factors which will be discussed in subsequent sections of this review.
The IL-12 is primarily produced by professional antigen presenting cells (APCs) such as monocytes, macrophages and dendritic cells, upon interaction with CD4+ T cells and exerts its immunoregulatory effects on T cells and natural killer cells (NK) cells.4 Therefore, it plays very crucial role in both adaptive and innate immunity. The innate recognition of pathogen-associated molecular patterns by different Toll-like receptors activates APCs to induce IL-12. Involvement of MHC-II and CD40 during T cell-APC interaction is very critical for IL-12 production. The most pronounced function of IL-12 is to promote differentiation of cells from naïve T cells as well as to generate memory Th1 cells in response to antigenic stimulation. It has been a potent inducer of interferon-γ (IFNγ), the prototypic Th1 cytokine therefore implying its central role in cell-mediated immune responses predominantly against intracellular microbial pathogens. Initially during the first encounter with antigen the IL-12 induces differentiation of naive T cells into a population of Th1 cells that secrete IFNγ. It also acts as a co-stimulus for excessive IFNγ production by differentiated Th1 cells on their subsequent encounter to specific antigens. Finally, it stimulates proliferation of IFNγ producing memory Th1 cells from resting memory cells during interaction with specific antigens to which they were previously exposed. The IFNγ via induction of inducible nitric oxide synthases (iNOS) produces reactive nitrogen intermediates in infected macrophages thereby increases their antimicrobial activity. The IFNγ also induces cytotoxic activity of NK cells and CD8+ T cells. In addition, recombinant IL-12 has been found to have significant therapeutic efficacy in mouse tumor model, mouse models of infectious disease and airway inflammation.4
IL-12 in EAE and MS
Clinical and pathophysiological evidences
Although IL-12 is critically important for normal host defense mechanism against intracellular pathogens which is mediated by optimal Th1 response, the excessive IL-12 secretion is associated with many adverse consequences. The overproduction of IL-12 plays an instrumental role in pathogenesis of several organ-specific autoimmune disease models such as experimental colitis, collagen-induced arthritis, insulin-dependent diabetes and experimental autoimmune encephalomyelitis (EAE).8, 9 Because MS is a Th1-mediated autoimmune disease and EAE the animal model of MS that clinically and histopathologically mimics MS, the Th1 biasness by uncontrolled IL-12 activity may have severe outcome in the pathogenesis of MS and EAE. There are several evidences that show involvement of IL-12 in the pathogenesis of the disease. In adoptive transfer model of EAE, treatment of T cells with rIL-12 during in-vitro priming resulted in early onset as well as increased the severity of the disease.4 This study demonstrates that IL-12 increases the encephalogenicity of myelin-specific T cells. Similarly, treatment of rIL-12 in adoptively transferred mice worsened EAE.4 Furthermore, attenuation of disease progression and severity in adoptively transferred EAE by neutralization of IL-12 strongly suggests a role of endogenous IL-12 in disease pathogenesis of EAE.4 Parallel to EAE, there are also significant evidences that support the role of IL-12 in MS pathogenesis. Increased IL-12 production was observed when APCs were stimulated by T cells isolated from MS patients compared to T cells of normal subjects.4 Moreover, elevated level of IL-12 was found from serum of secondary progressive MS patients but not from control subjects.4 These evidences clearly suggest a deleterious role IL-12 in pathogenesis of MS.
How does IL-12 interplay in the pathology of MS and EAE? A close look into insights for possible mechanisms
The key steps of MS and EAE pathogenesis are activation and proliferation of autoreactive T cells and infiltration of these T cells associated with other mononuclear cells into CNS followed by CNS inflammation, glial activation and demyelination. In order to explore drug intervention in the treatment of MS it is essential to understand the molecular mechanisms underlying the pathogenesis of the disease process. However, as there are several factors that come into play in a concerted but complicated manner leading to pathogenesis of MS, it is very difficult to identify any particular pathway or mechanism to be exclusively responsible for disease generation. Rather, it seems more logical to understand the key molecular events and measure their relative contribution that is integrated in the final outcome.
The IL-12, the Th1 driving cytokine, has multi-faceted mode of action in pathophysiology of MS. In one hand, it drives the differentiation of myelin-reactive Th1 cells and may facilitate their entry into the CNS through the leaky blood-brain-barrier.10 On the other; two other pronounced effects of IL-12 in the pathophysiology of MS are augmentation of CNS inflammation and glial activation. IL-12 may execute these effects via production of IFNγ and NO.
Induction of IFNγ by IL-12
Although the primary Th1 cytokine IFNγ is necessary for host defense, the uncontrolled production of IFNγ as a result of homeostasis shift to Th1 is likely to be associated with deleterious effect in autoimmune diseases like MS. Once myelin-specific Th1 cells reach CNS parenchyma, it re-encounters antigens presented by macrophages and microglia which triggers IL-12 production that in turn strongly induces the production of IFNγ by Th1 cells and thereby stimulates proinflammatory milieu in the CNS parenchyma.
a) Mechanism of IFN-γ-production
As mentioned earlier high affinity binding of IL-12 to its receptor complex is executed by recognition of p40 subunit by IL-12Rβ1 and p35 subunit by IL-12Rβ2. The IL-12Rβ1 is associated with janus kinase (Jak) family member Tyk2 whereas the IL-12Rβ2 is associated with Jak2.6, 7 Although activation of IL-12 receptor complex activates several signal transducers and activators of transcriptions (STAT) transcription factors, the most striking biological responses of IL-12 is mediated by STAT4. The STAT4 has been found to be critical transcription factor for induction of IFNγ by IL-12. The binding of IL-12 to IL-12R results in ligand induced autophosphorylation and transphosphorylation of receptor-associated Jaks and the Jaks in turn phosphorylate tyrosine residues located in the cytoplasmic domain of the receptor subunits. The phosphorylated IL-12Rβ2 subunit serves as a docking site for STAT4 binding. There is a marked difference between STAT4 binding to human and mouse IL-12Rβ2 subunits. In human, only tyrosin 800 (Y800) of IL-12Rβ2 subunit is responsible for STAT4 binding whereas in mouse Y757, Y804 and Y811 that corresponds to Y800 of mouse, provide the binding sites for STAT4.11-13 Upon receptor activation STAT4 is phosphorylated at its conserved tyrosine residue and then undergoes dimerization through intermolecular association between SH2 domain of one STAT4 and a conserved phosphotyrosine residue of another leading to classical activation of STAT4 molecule.14 The phosphorylated STAT4 dimer then translocates into nucleus, binds to IFNγ promoter and induces transcriptional activation IFNγ.
Apart from JAK/STAT signaling, activation of p38MAPK pathway by IL-12 also plays a crucial role in induction of IFNγ. The IL-12 signaling activates MKK3/6, the upstream activator of p38MAPK which in turn activates p38MAPK via phosphorylation. The p38MAPK has been demonstrated to phosphorylate serine 721 (S721) in STAT4 and S721 phosphorylation of STAT4 has been shown to be essential for enhanced IFNγ production induced by IL-12.15 The MKK3-deficient mice show decreased production of IFNγ clearly suggesting an essential role p38MAPK in maximal production of IFNγ.16 Therefore, it is likely that STAT4 activation alone may be sufficient for optimal production of IFNγ which is necessary for clearance of intracellular pathogens, but p38MAPK mediated S721 phosphorylation of STAT4 may be responsible for increased production of IFNγ that triggers proinflammatory events resulting in CNS inflammation and gliosis during pathogenesis of MS. However further experimental studies are required to establish this hypotheses. The IL-12-mediated IFNγ induction may also involve activation of PI3K/Akt and NF-κB, but evidences supporting these facts are controversial and poorly understood.
b) Role of IFN-γ in demyelination
Experimental evidences suggest that IFNγ plays a deleterious role in MS and its level in CSF correlates with disease severity.11 Interaction of IFNγ with oligodendrocytes is strongly implicated in the pathogenesis of EAE and MS. In-vitro studies show that pretreatment of oligodendrocytes with IFNγ results in severe upregulation of chemokines like CXCL10, MCP-1, MCP-1〈, and RANTES.11 These chemokines play an active role in enhancement of parenchymal infiltration, CNS inflammation and demyelination. However, the most pronounced effect of IFNγ is exerted on myelinating oligodendrocytes which undergo both apoptotic and necrotic death in response to IFNγ.12 It has been demonstrated that IFNγ mediates its deleterious effects on via activation of STAT-1 signaling and inhibition of STAT-1 signaling prevents IFNγ-responsiveness to oligodendrocytes and many other cell types. On the other hand, IFNγ may also have protective effect on disease pathogenesis such as preventing oligodendrocytes from oxidative stress.12 Moreover, IFNγ activates endoplasmic reticulum stress response in oligodendrocytes which in turn activates pancreatic endoplasmic reticulum kinase (PERK). Activation of PERK in mature oligodendrocytes attenuates severity of EAE and in PERK-deficient mice this beneficial effect of IFNγ is abrogated.12 Therefore, it is like that optimal level of IFNγ is beneficial while its overproduction is severely deleterious to the disease pathogenesis of MS.
Induction of Glial iNOS by IL-12
The iNOS is a key proinflammatory gene whose activation results in substantial production of NO that mediates several deleterious effects in the pathogenesis of EAE and MS. NO is believed to be a primary proinflammatory molecule mediating key inflammatory responses in EAE and MS. Elevated mRNA expressions of iNOS has been reported in the brain of MS patients which correlates with the severity of CNS inflammation as decrease in iNOS expression is associated with reduced inflammation of CNS.17, 18 Demyelinated plaques of MS patients reveal that reduced inflammation corresponds to decreased expression of iNOS.17, 18 Because both increased IL-12 and upregulated iNOS expression correlates with disease severity of EAE an MS it can be speculated that IL-12 might have potential role in induction of iNOS during pathogenesis of the disease.
a) Indirect Induction of iNOS by IL-12 (via IFNγ)
The glial iNOS is primarily regulated at transcriptional level but its transcriptional regulation is highly complex. According to Taylor et al, a 7.2 kb segment of human iNOS promoter confers partial inducibility in response to cytokine whereas 8.3 kb or 16kb is required for full transcriptional activation simply suggesting the complex nature of transcriptional regulation of iNOS.19 The promoter of iNOS houses binding elements for some key transcription factors like NF-κB, C/EBP, STAT-1α and AP-1. However, the activation of STAT-1α is primarily responsible for IFNγ-mediated transcriptional activation of iNOS. The binding of IFNγ to its receptors (IFNγR) induces assembly of active receptor complex and subsequent phosphorylation of receptor-associated Jak2. The activated Jak2 leads to tyrosine phosphorylation of STAT-1α which in turn is released from receptor complex to form homodimer that subsequently translocates into the nucleus and binds to IFNγ activation site (GAS elements) contained in the iNOS promoter and thereby activates transcription of iNOS.20-22 This Jak-STAT signaling is considered as a key pathway for regulation glial iNOS by IFNγ. It appears that IFNγ-induced activation of Jak-STAT signaling has a major contribution to the induction of iNOS which may also be coupled with activation of other transcription factors such as NF-κB, C/EBP and AP-1 by several other proinflammatory stimuli generated in the CNS during pathogenesis of EAE and MS leading to exaggerated production of NO and subsequently neurodegeneration.
In addition to STAT-1α, IFNγ also activates interferon regulatory factor (IRF-1) as a secondary response. Till now, the interferon-stimulated responsive element (ISRE), the binding element for IRF-1 has been detected only in mouse and rat but not in human iNOS promoter.23 However, several related elements have been detected in human iNOS promoter. The binding of IRF-1 to ISRE bends the iNOS promoter in such a way as to facilitate its transcription, a unique property that has not been reported for any other transcription factors. However ablation of IRF-1 does not abolish glial expression of iNOS but diminishes it suggesting a supplemental role IRF-1 in IFNγ-induced activation of iNOS.24
b) Direct Induction of iNOS by IL-12
In addition to IFNγ-mediated induction of iNOS, the IL-12 by alone is capable of inducing iNOS and NO production (Figure 2). In earlier studies it was demonstrated that IL-12 dose-dependently induced production of NO as measured by nitrite production in BV-2 microglial cells and macrophages.3 The inhibition of nitrite production by arginase, an enzyme that degrades L-arginine, the substrate of NOS and L-NMA, a competitive inhibitor of NOS, strongly suggests that IL-12-induced NO production in BV-2 glial cells is dependent on NOS-mediated arginine metabolism. This was further confirmed by significant induction of iNOS at protein level by IL-12 in BV-2 microglial cells.3 Induction of iNOS by IL-12 may require activation of NF-κB, however, the detailed mechanism by which IL-12 directly induces iNOS is not understood yet.
Figure 2.
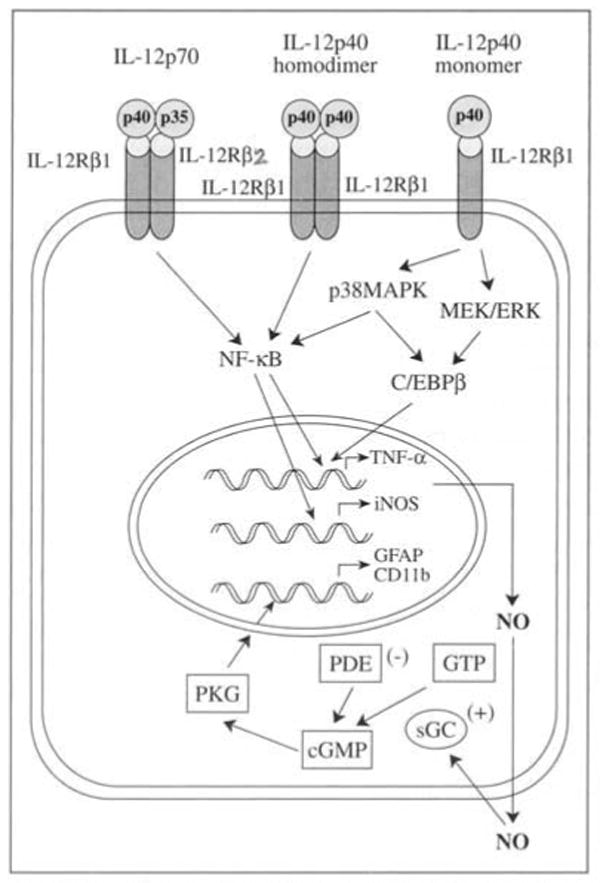
Glial activation by IL-12p70 and IL-12p40.
Although iNOS-derived NO production may be responsible for elimination of pathogens by macrophages during immune response, the augmentation of iNOS induction directly or indirectly by IL-12 can lead to excessive production of NO which plays devastating role in EAE and MS. The involvement of NO in the pathogenesis of EAE and MS will be discussed in subsequent section of this review.
Is IL-12 a key molecule in pathogenesis of EAE and MS? The other side of the coin
Despite IL-12 has been shown to be a potential player in the pathogenesis of MS, the critical role of IL-12 in the disease pathogenesis has been recently challenged by several studies. Most interestingly, the studies with knockout mice lacking IL-12p35, IL-12Rβ2, IFNγ or IFNγR show that instead of being resistant to EAE which was expected, these mice develop severe EAE.1, 25-27 These findings suggest that although uncontrolled IL-12 production is potentially hannful, the IL-12 and IFNγ are not essential for disease progression of EAE and moreover, the optimal production of IL-12 and IFNγ may be necessary for protection of EAE and MS.
Tarrant et al., have reported that IL-12 can promote apoptosis of antigen-specific T cells by upregulating pro-apoptotic members of Bcl-2 family of proteins such as Bax, Bad etc, via induction of IFNγ and iNOS.28 Thus IL-12 driven apoptosis of myelin antigen-specific T cells may be able to protect MS by preventing autoimmune attack. Furthermore IL-12 has also been shown to upregulate the expression of IL-2 receptor (CD25).29 The IL-12 induces binding of STAT4 to CD25 promoter which facilitates recruitment of cAMP response element-binding protein (CREB) associated with increased histone acetylation and decreased methylation of CD25 promoter resulting in transcriptional activation of CD25.29 Because CD4+CD25+T cells, known as regulatory T cells (Tregs), play a key role in suppression of activation of T cells, it may appear that IL-12 might play a significant role in the maintenance of normal Treg population which is very important for prevention of undesired activation of myelin-specific Th1 cells.
Taken together it is likely that endogenous level of IL-12 is the critical decider about its biological role. When IL-12 is produced at optimal level it is beneficial and it seems to be necessary for prevention of EAE or MS. However, exaggerated production of IL-12 is potentially harmful and plays a key role in CNS inflammation and glial activation during pathogenesis of EAE and MS.
The IL-12p40
Biological importance
As described earlier, the IL-12p40 subunit, in addition to forming heterodimer with p35 and pl9, is also secreted independently in the form of monomer and disulfide-linked homodimer.
Until recently, there was a prevailing thought that apart from antagonizing the activity of IL-12 by IL-12p40 homodimer (p402) that also binds to IL-12 receptor, the IL-12p40 do not have any biological function. However, there are several recent evidences that strongly suggest that p402 by alone may be able to carry out important biological functions. Firstly, there is a predominance of p40 mRNA level over p35 and subsequently its predominance in protein level is evidenced by secretion of 5 to 500 fold excess of p40 relative to IL-12 by normal monocytes.30 In addition, according to Heinzel et al., following LPS challenge, the p402 level increases significantly in the serum of mice.31 Moreover, presence of 20% to 40% of p40 in the form of homodimer in the serum of normal and endotoxin-treated mice suggests a possible biological role of p402.31 Secondly, in contrast to IL-12p35 knockout, the IL-12p40 knockout mice are resistant to EAE.1 Thirdly, Th1 differentiation was augmented in allografted IL-12p35 deficient mice but not in allografted IL-12p40 deficient mice.4 Lastly, it has been found that IL-12p35 deficient mice produce normal level of p40.4 These evidences clearly suggest that IL-12p40 alone has important biological activity in the form of both monomer and homodimer which may be critical for physiological function as well as disease pathogenesis.
The IL-12p40 in EAE and MS
Clinical and Pathophysiological Evidences
Several lines of evidences demonstrate severely augmented expression of IL-12p40 in the CNS of many demyelinating diseases such as MS, animal model of experimental autoimmune encephalomyelitis (EAE), Guillain-Barre syndrome and neuritis.32-34 According to several reports mRNA level of p40 in the CNS of MS patients is significantly higher than the CNS of control patients whereas the p35 level was almost comparable or sometimes even lower in the CNS of MS patients that of control subjects.32, 35, 36 Similarly, in EAE, an animal model of MS, the p40 mRNA level but not p35, significantly increases in the CNS and spinal cord. These evidences strongly suggest that IL-12p40 either in the form of monomer or homodimer or both is potentially involved in the pathogenesis of EAE or MS. However, the underlying molecular mechanism behind the role of IL-12p40 in pathogenesis of EAE or MS is not understood. In the following sections we will highlight some recent studies which were attempted to explore possible molecular mechanisms that may underlie behind the role of IL-12p40 in the disease pathogenesis.
IL-12p40 Homodimer Induces iNOS and Production of NO via Activation of NF-κB
Because induction of iNOS and subsequent production of NO are potentially harmful in EAE and MS, in our earlier studies we attempted to explore the role of p402 in induction of iNOS (Figure 2). Previous studies clearly showed that p402 dose-dependently induced expression of iNOS and subsequently the nitrite production in BV-2 glial cells, mouse primary microglia and peritoneal macrophages.3 Further, it was demonstrated that p402 induced activation of NF-κB as evidenced by its increased DNA-binding and transcriptional activity and this activation of NF-κB was essential for induction of iNOS.3 Inhibition of p402-mediated activation of NF-κB as well as the induction of iNOS and nitrite production by SN50, a synthetic peptide that specifically blocks nuclear translocation of NF-κB suggests that activation of NF-κB is necessary for p402-mediated induction of iNOS and nitrite production.3 In addition, our unpublished observation suggests that IL-12p40 monomer and homodimer markedly upregulates expression of iNOS in human primary astrocytes thus indicating a strong role of IL-12p40 in CNS inflammation of MS.
However, it is not clear yet, how p402 activates NF-κB. It is likely that p402-mediated activation of NF-κB involves only IL-12Rβ1 but not IL-12Rβ2 and that activation of IL-12Rβ1 by p402 transmits signals to activate NF-κB (Jana and Pahan, unpublished observation).
IL-12p40 Homodimer and Monomer and IL-12p70 Induce Tumor Necrosis Factor-α in Microglia and Macrophages
Activation of microglia, the CNS resident professional macrophages is implicated in pathogenesis of several neurodegenerative diseases including MS.37 The microglia upon activation secretes several proinflammatory neurotoxic molecules including tumor necrosis factor-α (TNF-α), a major player in inflammation and demyeliantion of MS.35 Although according to recent clinical trial reports, antagonists of TNF-α worsens clinical symptoms of MS, analysis of CSF of MS patients shows increased level of TNF-α compared to normal subjects and this increased level of TNF-α also correlates with the disease severity of MS.38, 39 The most predominant role of TNF-α in MS is that it triggers oligodendrocyte death. Active MS lesion shows selective expression of both p55TNFR and p75TNFR on oligodendrocytes and several studies have shown that TNF-α alone can kill oligodendrocytes in cultures.40-42
Considering the importance of TNF-α in pathogenesis of MS, it is necessary to understand the possible molecular mechanisms underlying the induction of TNF-α. IFNγ can induce TNF-α in microglia, but microglia isolated from adult human has been poor source of TNF-α in response to IFNγ. 43 Because both IL-12p40 mRNA and TNF-α were significantly upregulated in the CNS of MS patients, studies were undertaken to explore whether IL-12p40 plays any role in induction of TNF-α. Recently, we demonstrated that IL-12p40 monomer and homodimer and IL-12p70 markedly upregulated the expression of TNF-α in BV-2 glial cells, mouse primary microglia and mouse peritoneal macrophages (Figure 2).44 Previous studies further suggest induction of TNF-γ by IL-12p40 monomer requires activation of both NF-κB and C/EBPβ as demonstrated by increased DNA-binding and transcriptional activity of NF-κB and C/EBPβ induced by IL-12p40 monomer.44 When explored further upstream signaling it was revealed that induction of TNF-α by IL-12p40 monomer requires activation of both p38MAPK and MEK/ERK pathway, however MEK/ERK pathway leads to activation of only C/EBPβ but p38MAPK activates both NF-κB and C/EBPβ.44
It is still unknown what receptor activation stimulates activation of TNF-〈 in response to IL-12p40 and IL-12p70. It may appear that IL-12p40 monomer and dimer transmits signal via IL-12Rβ1 whereas IL-12p70 activates IL-12R complex of IL-12Rβ1 and IL-12Rβ2 in order to transduce signal for induction of TNF-α. Taken together we may conclude that IL-12p40 and IL-12p70 contributes significantly to the induction of TNF-α and therefore may play a key role in CNS inflammation and demyelination in EAE and MS.
Down-Regulation of Tregs by IL-12p40 Homodimer via NO
Tregs play a key role in the maintenance of homeostasis between immune response and immune tolerance. Tregs are specific subset of T cells designated by CD4+CD25+Foxp3+ T cells. In autoimmune and several other chronic inflammatory diseases including MS, expression of Foxp3 is severely down-regulated.45 Because both increased level of IL-12p40 and decreased Foxp3 correlates with disease severity of MS, it seems likely that IL-12p40 might play a critical role in down-regulation of Tregs. Our recent unpublished observation strongly suggests that IL-12p40 homodimer down-regulates Tregs solely via IL-12Rβ1-mediated induction of iNOS as it does not require IL-12Rβ2 (Figure 3). These findings indicate that IL-12p40 apart from its pathogenic role in CNS may also act in the periphery and destabilize immune homeostasis.
Figure 3.
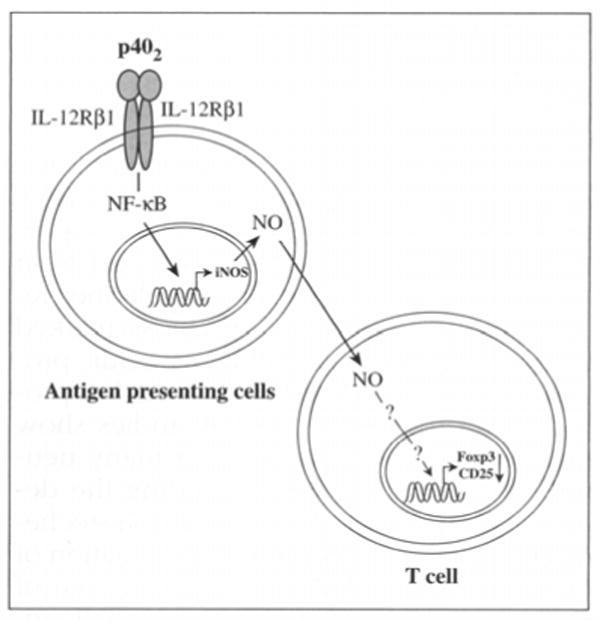
A hypothesis for down-regulation of tregs by IL-12 P402.
Involvement of NO in gliosis
Gliosis is one of the major hallmarks of MS and other neurodegenerative disorders. As it was observed that p402 and IL-12p70 have significant contribution in induction of glial iNOS and production of NO and that NO is a key mediator of glial activation or gliosis, these cytokines are expected to play an important role in gliosis. Here the role of NO in microgliosis and astrogliosis will be briefly delineated.
NO in Microgliosis
Activation of microglia has been a key pathophysiological step for almost all neurodegenerative diseases including EAE and MS. Although activation of microglia is necessary for phagocytic removal of dead cells and secretion of neurotrophic factors key to neuronal survival, overactivation of microglia produce several proinflammatory neurotoxic molecules including NO. Microgliosis is the consequence of overactivation of microglia associated with excessive production of NO that ultimately suppresses the beneficial effects of microglial activation. The upregulation of the surface marker CD11b is considered to be a hallmark for microgliosis and increased expression of CD11b corresponds to microglial overactivation and severity of many neurodegenerative diseases including EAE and MS.37, 46 Immunohistochemical studies showing upregulation of CD11b in the spinal cord of EAE indicate that microgliosis may be actively involved in the pathogenesis of MS.
Recent studies demonstrate that NO plays a key role in the upregulation of CD11b in mouse microglia (Figure 2).47 NO stimulates soluble guanylyl cyclase (sGC) as its immediate target which catalyzes the formation of cGMP that in turn activates PKG and subsequently stimulates upregulation of CD11b. Further downstream of CD11b activation requires phosphorylation of the transcription factor CREB followed by its recruitment to the CD11b promoter element.47 The PKG has been reported to phosphorylate CREB.47 Therefore understanding the NO-PKG-CREB-CD11b may be immensely helpful in designing therapeutic compound that can suppress this signaling and thereby the severity of the disease process of EAE.
NO in Astrogliosis
Astrocytes, the major glial cells of CNS, outnumber neurons by ten to one. Although astrocytes are important for maintenance of CNS homeostasis, in response to neurodegenerative insults they react vigorously leading to astrogliosis marked by upregulation of glial fibrillary acidic protein (GFAP) considered to be a marker protein for astrogliosis.48 Several studies show that astrogliosis is involved in many neurodegenerative diseases including the demyelinating disease EAE. Immunohistochemical studies demonstrating upregulation of both iNOS and GFAP in the spinal cord of EAE mice suggest that NO is potentially involved in astrogliosis and thus the pathophysiology of EAE.49
Similar to microgliosis, NO also has been shown to be essential for upregulation of GFAP expression in mouse primary astroglia and NO-mediated upregulation of GFAP follows sGC-cGMP-PKG pathway (Figure 2).50
Taken together, it is likely that IL-12p40 and p70 may function as potential candidates that ultimately trigger gliosis via NO-sGC-cGMP-PKG pathway. Understanding this mechanism will be beneficial for dissecting further the role p40 family of cytokines in pathogenesis of EAE and MS.
Role of NO in Demyelination
EAE and MS are CNS demyelinating diseases in which myelin and oligodendrocytes, the myelin producing cells are severely damaged. In MS plaques, oligodendrocytes exist with a dying and necrotic morphology with swollen nuclei and disrupted plasma and mitochondrial membranes.51 Involvement of reactive nitrogenous species has been potentially implicated in the pathophysiology of oligodendrocyte damage. Here we will briefly mention about possible biochemical mechanisms by which NO contributes to CNS demyelination.
Augmented production of NO may lead to conformational change in the structure of proteolipid protein (PLP), a critical component of myelin, directly via S-nitrosylation of cystein residues of PLP, resulting in myelin decompaction and axonal damage.52 However, most of the NO-induced myelin damage is caused by peroxynitrites (ONOO-). Peroxynitrites can cause nitration of tyrosine residues, known as protein tyrosination which imparts conformational change and subsequent loss of function of major myelin proteins ultimately resulting in oligodendrocyte death. Increased nitrotyrosine formation has been detected in spinal cord of EAE mice and considered as a footprint of pathologic activity of NO during CNS inflammation.53 Peroxynitrites can also promote oligodendrocyte death via glutamate-mediated excitotoxicity, DNA-strand break and activation of p38 MAPK leading to potassium efflux and cell shrinkage.54-56
At the end we can conclude that IL-12p40 and p70, the potentially strong inducers of glial NO, may also have significant contribution in CNS demyelination via production of glial NO.
The interleukin-23 (IL-23) and its role in EAE and MS
The IL-12p40 while forming heterodimer, not only associates with p35 but also with p19 to form a novel covalently-linked cytokine, the IL-23. The p19 is secreted only when it is associated with IL-12p40 and like p35 it is structurally homologous to IL-6 and G-CSF.57 The IL-23 receptor complex is made up of IL-12Rβ1 and IL-23R, a novel gp130-like chain.58 The p19 subunit appears to bind with IL-23R associated with Jak2 whereas the p40 subunit binds with IL-12Rβ1 associated with Tyk2. Like IL-12, IL-23 are produced by similar types of cells and their receptors are expressed on T cells and NK cells as well as phagocytic and dendritic hematopoietic cells.59
Initially IL-23 was only thought to play major role in maintenance and proliferation of Th1-committed memory cells.60 However later it has been proved that IL-23 is key player in the pathogenesis of autoimmune diseases like EAE and MS and even more devastating than IL-12 in the disease pathogenesis. As mentioned earlier, IL-12p40 and IL-12Rβ1 knockout mice where both IL-12 and IL-23 functions were abrogated, were resistant to EAE in contrast to IL-12p35 and IL-12Rβ2 knockouts where only IL-12 functions were abrogated, EAE was in fact aggravated. The striking difference between these two groups to susceptibility to EAE suggest that IL-23 could be critical for pathogenesis of EAE and this was finally confirmed in IL-23p19 deficient mice which were completely resistant to EAE.61 The IL-23 is known to be a proinflammatory cytokine and apart from its direct proinflammatory role, it also triggers differentiation of a novel subset of T cells, the T-helper-17 (Th17) cells characterized by production of IL-17 cytokine, a cytokine strongly implicated in the pathophysiology of various autoimmune diseases.60 In addition, IL-23 also stimulates T cells to produce IL-6 and TNF-α which play key roles in CNS inflammation and demyelination. However, interestingly, IL-23 is unable to drive differentiation of naïve T cells to Th17 cells as naïve T cells do not express IL-23R.58 Some recent studies suggest that IL-23 induces differentiation and proliferation of Th17 cells from a pool of activated memory T cells.62
IL-23 signaling has also similarity with IL-12 as binding of IL-23 to its receptor complex triggers autophosphorylation and transphosphorylation of receptor-associated Jaks and the Jaks in turn phosphorylate tyrosine residues located in the intracellular domain of the receptor subunits.14, 59 Human IL-23R contains three putative tyrosine phosphorylation sites located within SH2 binding sites. These phosphorylated tyrosine residues provide the docking sites for STAT family of transcription factors. However, unlike IL-12, in response to IL-23, STAT3/STAT4 heterodimer or STAT3 homodimer is probably predominantly activated instead of STAT4 homodimer.14, 59 The STAT3/STAT4 heterodimer or STAT3 homodimer might play important role in transcriptional activation of IL-17 but according to recent studies STAT family of transcription factors may not be critical for Th17 commitment.14, 59
Recently, a novel transcription factor RORγt has been identified and shown to be involved in generation of Th17 cells.63 Interestingly, RORγt deficient animals do not contain Th17 population of cells and transduction of naïve T cells with RORγt-encoding retrovirus induces IL-17 production. These observations suggest that RORγt may be a critical transcription factor for commitment to Th17 cells. However it requires further investigation to determine whether IL-23 activates RORγt or RORγt regulates transcriptional activation of IL-17.
Interestingly, recent studies suggest a possible role of TGFβ and IL-6 in differentiation of Th17 cells.64 At steady state, TGFβ, produced locally by dendritic cells, naturally occurring Tregs and many other cell types, induce differentiation of adaptive Tregs and thereby promote immunosuppression and immune regulation. However, in proinflammatory milieu, TGFβ in combination with IL-6 transmits signal that suppress Foxp3, the Treg marker, but activates RORγt that in turn induces IL-17 production.64 Therefore, it appears that IL-23 even indirectly, via production of IL-6 may contribute to Th17 differentiation and subsequently IL-17 production. The IL-17 may act in a positive feed-back loop that further induces IL-6 and thereby ultimately suppresses Tregs while enriches Th17 population. As a result, the lack of Tregs results in vigorous activation of myelin-specific T cells and subsequent CNS inflammation and demyelination.
Conclusions
In this comprehensive review, we have precisely discussed how p40 family of cytokines modulates immune signaling particularly with relevance to the pathophysiology of multiple sclerosis. Despite having a shared subunit (p40) and a common receptor subunit (IL-12Rβ1), these cytokines induce distinct signaling cascades critically important for immune regulation and glial function. Although optimal level IL-12 and IL-12-derived IFNγ have many protective functions, overproduction of these cytokines are severely deleterious as they directly or indirectly lead to immune-dysregulation (Th1 hyperpolarization), CNS inflammation, glial activation and ultimately demyelination (key features of MS pathophysiology). According to recent studies, implication of IL-12p40 homodimer and IL-23 in the pathophysiology of MS is even more significant than IL-12. IL-12p40 homodimer is considered as a potentially strong candidate for triggering CNS inflammation and glial activation via induction of iNOS. In addition, the IL-12p40 homodimer may activate immune response of myelin-specific T cells via downregulation of Tregs. The IL-12p40 monomer also has been reported to contribute CNS inflammation via upregulation of TNF-α in microglial cells. However, further studies are required to explore the precise role of IL-12p40 monomer in the pathophysiology of MS. The evolution of IL-23 has led to develop Th17 hypothesis that may eventually supersede Th1/Th2 paradigm for understanding the pathophysiology of MS. IL-23 downregulates Tregs and promotes differentiation of Th17 cells credited for sustained CNS inflammation and tissue damage.
As MS is an autoimmune demyelinating disease characterized by CNS inflammation and gliosis, and the p40 family of cytokines significantly modulates immune regulation, CNS inflammation and glial activation, understanding the distinct molecular cascades triggered by these cytokines would be immensely helpful in designing therapeutic strategy to prevent multiple sclerosis.
Acknowledgments
This work was supported by grants from National Institutes of Health (NS39940 and NS48923).
References
- 1.Becher B, Durell BG, Noelle RJ. Experimental autoimmune encephalitis and inflammation in the absence of interleukin-12. J Clin Invest. 2002;110:493–7. doi: 10.1172/JCI15751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–40. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pahan K, Sheikh FG, Liu X, Hilger S, McKinney M, Petro TM. Induction of nitric-oxide synthase and activation of NF-kappaB by interleukin-12 p40 in microglial cells. J Biol Chem. 2001;276:7899–905. doi: 10.1074/jbc.M008262200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gately MK, Renzetti LM, Magram J, Stern AS, Adorini L, Gubler U, et al. The interleukin-12/interleukin-12-receptor system: role in normal and pathologic immune responses. Annu Rev Immunol. 1998;16:495–521. doi: 10.1146/annurev.immunol.16.1.495. [DOI] [PubMed] [Google Scholar]
- 5.Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. 2003;3:133–46. doi: 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- 6.Bacon CM, McVicar DW, Ortaldo JR, Rees RC, O'Shea JJ, Johnston JA. Interleukin 12 (IL-12) induces tyrosine phosphorylation of JAK2 and TYK2: differential use of Janus family tyrosine kinases by IL-2 and IL-12. J Exp Med. 1995;181:399–404. doi: 10.1084/jem.181.1.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zou J, Presky DH, Wu CY, Gubler U. Differential associations between the cytoplasmic regions of the interleukin-12 receptor subunits beta1 and beta2 and JAK kinases. J Biol Chem. 1997;272:6073–7. doi: 10.1074/jbc.272.9.6073. [DOI] [PubMed] [Google Scholar]
- 8.Constantinescu CS, Goodman DB, Hilliard B, Wysocka M, Cohen JA. Murine macrophages stimulated with central and peripheral nervous system myelin or purified myelin proteins release inflammatory products. Neurosci Lett. 2000;287:171–4. doi: 10.1016/s0304-3940(00)01184-8. [DOI] [PubMed] [Google Scholar]
- 9.Zipris D, Greiner DL, Malkani S, Whalen B, Mordes JP, Rossini AA. Cytokine gene expression in islets and thyroids of BB rats. IFN-gamma and IL-12p40 mRNA increase with age in both diabetic and insulin-treated nondiabetic BB rats. J Immunol. 1996;156:1315–21. [PubMed] [Google Scholar]
- 10.Ogawa M, Tsutsui T, Zou JP, Mu J, Wijesuriya R, Yu WG, et al. Enhanced induction of very late antigen 4/lymphocyte function-associated antigen 1-dependent T-cell migration to tumor sites following administration of interleukin 12. Cancer Res. 1997;57:2216–22. [PubMed] [Google Scholar]
- 11.Balabanov R, Strand K, Goswami R, McMahon E, Begolka W, Miller SD, et al. Interferon-gamma-oligodendrocyte interactions in the regulation of experimental autoimmune encephalomyelitis. J Neurosci. 2007;27:2013–24. doi: 10.1523/JNEUROSCI.4689-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin W, Bailey SL, Ho H, Harding HP, Ron D, Miller SD, et al. The integrated stress response prevents demyelination by protecting oligodendrocytes against immune-mediated damage. J Clin Invest. 2007;117:448–56. doi: 10.1172/JCI29571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu C, Wang X, Gadina M, O'Shea JJ, Presky DH, Magram J. IL-12 receptor beta 2 (IL-12R beta 2)-deficient mice are defective in IL-12-mediated signaling despite the presence of high affinity IL-12 binding sites. J Immunol. 2000;165:6221–8. doi: 10.4049/jimmunol.165.11.6221. [DOI] [PubMed] [Google Scholar]
- 14.Watford WT, Hissong BD, Bream JH, Kanno Y, Muul L, O'Shea JJ. Signaling by IL-12 and IL-23 and the immunoregulatory roles of STAT4. Immunol Rev. 2004;202:139–56. doi: 10.1111/j.0105-2896.2004.00211.x. [DOI] [PubMed] [Google Scholar]
- 15.Morinobu A, Gadina M, Strober W, Visconti R, Fornace A, Montagna C, et al. STAT4 serine phosphorylation is critical for IL-12-induced IFN-gamma production but not for cell proliferation. Proc Natl Acad Sci U S A. 2002;99:12281–6. doi: 10.1073/pnas.182618999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lu HT, Yang DD, Wysk M, Gatti E, Mellman I, Davis RJ, et al. Defective IL-12 production in mitogen-activated protein (MAP) kinase kinase 3 (Mkk3)-deficient mice. EMBO J. 1999;18:1845–57. doi: 10.1093/emboj/18.7.1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bö L, Dawson TM, Wesselingh S, Mörk S, Choi S, Kong PA, et al. Induction of nitric oxide synthase in demyelinating regions of multiple sclerosis brains. Ann Neurol. 1994;36:778–86. doi: 10.1002/ana.410360515. [DOI] [PubMed] [Google Scholar]
- 18.Liu JS, Zhao ML, Brosnan CE, Lee SC. Expression of inducible nitric oxide synthase and nitrotyrosine in multiple sclerosis lesions. Am J Pathol. 2001;158:2057–66. doi: 10.1016/S0002-9440(10)64677-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Taylor BS, de Vera ME, Ganster RW, Wang Q, Shapiro RA, Morris SM, Jr, et al. Multiple NF-kappaB enhancer elements regulate cytokine induction of the human inducible nitric oxide synthase gene. J Biol Chem. 1998;273:15148–56. doi: 10.1074/jbc.273.24.15148. [DOI] [PubMed] [Google Scholar]
- 20.Saha RN, Pahan K. Regulation of inducible nitric oxide synthase gene in glial cells. Antioxid Redox Signal. 2006;8:929–47. doi: 10.1089/ars.2006.8.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saha RN, Pahan K. Signals for the induction of nitric oxide synthase in astrocytes. Neurochem Int. 2006;49:154–63. doi: 10.1016/j.neuint.2006.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xie QW, Whisnant R, Nathan C. Promoter of the mouse gene encoding calcium-independent nitric oxide synthase confers inducibility by interferon gamma and bacterial lipopolysaccharide. J Exp Med. 1993;177:1779–84. doi: 10.1084/jem.177.6.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martin E, Nathan C, Xie QW. Role of interferon regulatory factor 1 in induction of nitric oxide synthase. J Exp Med. 1994;180:977–84. doi: 10.1084/jem.180.3.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fujimura M, Tominaga T, Kato I, Takasawa S, Kawase M, Taniguchi T, Okamoto H, Yoshimoto T. Attenuation of nitric oxide synthase induction in IRE-1-deficient glial cells. Brain Res. 1997;759:247–50. doi: 10.1016/s0006-8993(97)00264-3. [DOI] [PubMed] [Google Scholar]
- 25.Ferber IA, Brocke S, Taylor-Edwards C, Ridgway W, Dinisco C, Steinman L, et al. Mice with a disrupted IFN-gamma gene, are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE) J Immunol. 1996;156:5–7. [PubMed] [Google Scholar]
- 26.Willenborg DO, Fordham S, Bernard CC, Cowden WB, Ramshaw IA. IFN-gamma plays a critical down-regulatory role in the induction and effector phase of myelin oligodendrocyte glycoprotein-induced autoimmune encephalomyelitis. J Immunol. 1996;157:3223–7. [PubMed] [Google Scholar]
- 27.Zhang GX, Gran B, Yu S, Li J, Siglienti I, Chen X, et al. Induction of experimental autoimmune encephalomyelitis in IL-12 receptor-beta 2-deficient mice: IL-12 responsiveness is not required in the pathogenesis of inflammatory demyelination in the central nervous system. J Immunol. 2003;170:2153–60. doi: 10.4049/jimmunol.170.4.2153. [DOI] [PubMed] [Google Scholar]
- 28.Tarrant TK, Silver PB, Wahlsten JL, Rizzo LV, Chan CC, Wiggert B, et al. Interleukin 12 protects from a T helper type 1-mediated autoimmune disease, experimental autoimmune uveitis, through a mechanism involving interferon gamma, nitric oxide, and apoptosis. J Exp Med. 1999;189:219–30. doi: 10.1084/jem.189.2.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.O'Sullivan A, Chang HC, Yu Q, Kaplan MH. STAT4 is required for interleukin-12-induced chromatin remodeling of the CD25 locus. J Biol Chem. 2004;279:7339–45. doi: 10.1074/jbc.M309979200. [DOI] [PubMed] [Google Scholar]
- 30.D'Andrea A, Rengaraju M, Valiante NM, Chehimi J, Kubin M, Aste M, et al. Production of natural killer cell stimulatory factor (interleukin 12) by peripheral blood mononuclear cells. J Exp Med. 1992;176:1387–98. doi: 10.1084/jem.176.5.1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heinzel FP, Hujer AM, Ahmed FN, Rerko RM. In vivo production and function of IL-12 p40 homodimers. J Immunol. 1997;158:4381–8. [PubMed] [Google Scholar]
- 32.van Boxel-Dezaire AH, Hoff SC, van Oosten BW, Verweij CL, Dräger AM, Adèr HJ, et al. Decreased interleukin-10 and increased interleukin-12p40 mRNA are associated with disease activity and characterize different disease stages in multiple sclerosis. Ann Neurol. 1999;45:695–703. doi: 10.1002/1531-8249(199906)45:6<695::aid-ana3>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 33.Podlaski FJ, Nanduri VB, Hulmes JD, Pan YC, Levin W, Danho W, et al. Molecular characterization of interleukin 12. Arch Biochem Biophys. 1992;294:230–7. doi: 10.1016/0003-9861(92)90162-p. [DOI] [PubMed] [Google Scholar]
- 34.Wang X, Wilkinson VL, Podlaski FJ, Wu C, Stern AS, Presky DH, et al. Characterization of mouse interleukin-12 p40 homodimer binding to the interleukin-12 receptor subunits. Eur J Immunol. 1999;29:2007–13. doi: 10.1002/(SICI)1521-4141(199906)29:06<2007::AID-IMMU2007>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 35.Constantinescu CS, Goodman DB, Hilliard B, Wysocka M, Cohen JA. Murine macrophages stimulated with central and peripheral nervous system myelin or purified myelin proteins release inflammatory products. Neurosci Lett. 2000;287:171–4. doi: 10.1016/s0304-3940(00)01184-8. [DOI] [PubMed] [Google Scholar]
- 36.Fassbender K, Ragoschke A, Rossol S, Schwartz A, Mielke O, Paulig A, et al. Increased release of interleukin-12p40 in MS: association with intracerebral inflammation. Neurology. 1998;51:753–8. doi: 10.1212/wnl.51.3.753. [DOI] [PubMed] [Google Scholar]
- 37.González-Scarano F, Baltuch G. Microglia as mediators of inflammatory and degenerative diseases. Annu Rev Neurosci. 1999;22:219–40. doi: 10.1146/annurev.neuro.22.1.219. [DOI] [PubMed] [Google Scholar]
- 38.Wiendl H, Hohlfeld R. Therapeutic approaches in multiple sclerosis: lessons from failed and interrupted treatment trials. Biodrugs. 2002;16:183–200. doi: 10.2165/00063030-200216030-00003. [DOI] [PubMed] [Google Scholar]
- 39.Sharief MK, Hentges R. Association between tumor necrosis factor-alpha and disease progression in patients with multiple sclerosis. N Engl J Med. 1991;325:467–72. doi: 10.1056/NEJM199108153250704. [DOI] [PubMed] [Google Scholar]
- 40.Bonetti B, Raine CS. Multiple sclerosis: oligodendrocytes display cell death-related molecules in situ but do not undergo apoptosis. Ann Neurol. 1997;42:74–84. doi: 10.1002/ana.410420113. [DOI] [PubMed] [Google Scholar]
- 41.D'Souza SD, Bonetti B, Balasingam V, Cashman NR, Barker PA, Troutt AB, et al. Multiple sclerosis: Fas signaling in oligodendrocyte cell death. J Exp Med. 1996;184:2361–70. doi: 10.1084/jem.184.6.2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Raine CS. Multiple sclerosis: TNF revisited, with promise. Nat Med. 1995;1:211–4. doi: 10.1038/nm0395-211. [DOI] [PubMed] [Google Scholar]
- 43.Becher B, Dodelet V, Fedorowicz V, Antel JP. Soluble tumor necrosis factor receptor inhibits interleukin 12 production by stimulated human adult microglial cells in vitro. J Clin Invest. 1996;98:1539–43. doi: 10.1172/JCI118946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jana M, Dasgupta S, Saha RN, Liu X, Pahan K. Induction of tumor necrosis factor-alpha (TNF-alpha) by interleukin-12 p40 monomer and homodimer in microglia and macrophages. J Neurochem. 2003;86:519–28. doi: 10.1046/j.1471-4159.2003.01864.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huan J, Culbertson N, Spencer L, Bartholomew R, Burrows GG, Chou YK, et al. Decreased FOXP3 levels in multiple sclerosis patients. J Neurosci Res. 2005;81:45–52. doi: 10.1002/jnr.20522. [DOI] [PubMed] [Google Scholar]
- 46.Ling EA, Wong WC. The origin and nature of ramified and amoeboid microglia: a historical review and current concepts. Glia. 1993;7:9–18. doi: 10.1002/glia.440070105. [DOI] [PubMed] [Google Scholar]
- 47.Roy A, Fung YK, Liu X, Pahan K. Up-regulation of microglial CD11b expression by nitric oxide. J Biol Chem. 2006;281:14971–80. doi: 10.1074/jbc.M600236200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eng LF, Ghirnikar RS. GFAP and astrogliosis. Brain Pathol. 1994;4:229–37. doi: 10.1111/j.1750-3639.1994.tb00838.x. [DOI] [PubMed] [Google Scholar]
- 49.Dasgupta S, Zhou Y, Jana M, Banik NL, Pahan K. Sodium phenylacetate inhibits adoptive transfer of experimental allergic encephalomyelitis in SJL/J mice at multiple steps. J Immunol. 2003;170:3874–82. doi: 10.4049/jimmunol.170.7.3874. [DOI] [PubMed] [Google Scholar]
- 50.Brahmachari S, Fung YK, Pahan K. Induction of glial fibrillary acidic protein expression in astrocytes by nitric oxide. J Neurosci. 2006;26:4930–9. doi: 10.1523/JNEUROSCI.5480-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Parkinson JF, Mitrovic B, Merrill JE. The role of nitric oxide in multiple sclerosis. J Mol Med. 1997;75:174–86. doi: 10.1007/s001090050102. [DOI] [PubMed] [Google Scholar]
- 52.Bizzozero OA, DeJesus G, Howard TA. Exposure of rat optic nerves to nitric oxide causes protein S-nitrosation and myelin decompaction. Neurochem Res. 2004;29:1675–85. doi: 10.1023/b:nere.0000035802.27087.16. [DOI] [PubMed] [Google Scholar]
- 53.Koprowski H, Spitsin SV, Hooper DC. Prospects for the treatment of multiple sclerosis by raising serum levels of uric acid, a scavenger of peroxynitrite. Ann Neurol. 2001;49:139. doi: 10.1002/1531-8249(200101)49:1<139::aid-ana28>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 54.Bossy-Wetzel E, Talantova MV, Lee WD, Schölzke MN, Harrop A, Mathews E, et al. Crosstalk between nitric oxide and zinc pathways to neuronal cell death involving mitochondrial dysfunction and p38-activated K+ channels. Neuron. 2004;41:351–65. doi: 10.1016/s0896-6273(04)00015-7. [DOI] [PubMed] [Google Scholar]
- 55.McDonald JW, Althomsons SP, Hyrc KL, Choi DW, Goldberg MP. Oligodendrocytes from forebrain are highly vulnerable to AMPA/kainate receptor-mediated excitotoxicity. Nat Med. 1998;4:291–7. doi: 10.1038/nm0398-291. [DOI] [PubMed] [Google Scholar]
- 56.Pitt D, Werner P, Raine CS. Glutamate excitotoxicity in a model of multiple sclerosis. Nat Med. 2000;6:67–70. doi: 10.1038/71555. [DOI] [PubMed] [Google Scholar]
- 57.Oppmann B, Lesley R, Blom B, Timans JC, Xu Y, Hunte B, et al. Novel pl9 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 2000;13:715–25. doi: 10.1016/s1074-7613(00)00070-4. [DOI] [PubMed] [Google Scholar]
- 58.Parham C, Chirica M, Timans J, Vaisberg E, Travis M, Cheung J, et al. A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rbeta1 and a novel cytokine receptor subunit, IL-23R. J Immunol. 2002;168:5699–708. doi: 10.4049/jimmunol.168.11.5699. [DOI] [PubMed] [Google Scholar]
- 59.Trinchieri G, Pflanz S, Kastelein RA. The IL-12 family of heterodimeric cytokines: new players in the regulation of T cell responses. Immunity. 2003;19:641–4. doi: 10.1016/s1074-7613(03)00296-6. [DOI] [PubMed] [Google Scholar]
- 60.Bettelli E, Oukka M, Kuchroo VK. T(H)-17 cells in the circle of immunity and autoimmunity. Nat Immunol. 2007;8:345–50. doi: 10.1038/ni0407-345. [DOI] [PubMed] [Google Scholar]
- 61.Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–8. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- 62.Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem. 2003;278:1910–4. doi: 10.1074/jbc.M207577200. [DOI] [PubMed] [Google Scholar]
- 63.Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–33. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 64.Stockinger B, Veldhoen M, Martin B. Th17 T cells: Linking innate and adaptive immunity. Semin Immunol. doi: 10.1016/j.smim.2007.10.008. [DOI] [PubMed] [Google Scholar]