

Abstract
A structure-activity relationship study of dorsomorphin, a previously identified inhibitor of SMAD 1/5/8 phosphorylation by bone morphogenetic protein (BMP) type 1 receptors ALK2, 3, and 6, revealed that increased inhibitory activity could be accomplished by replacing the pendent 4-pyridine ring with 4-quinoline. The activity contributions of various nitrogen atoms in the core pyrazolo[1,5-a]pyrimidine ring were also examined by preparing and evaluating pyrrolo[1,2-a]pyrimidine and pyrazolo[1,5-a]pyridine derivatives. In addition, increased mouse liver microsome stability was achieved by replacing the ether substituent on the pendent phenyl ring with piperazine. Finally, an optimized compound 13 (LDN-193189 or DM-3189) demonstrated moderate pharmacokinetic characteristics (e.g. plasma t1/2 = 1.6 h) following intraperitoneal administration in mice. These studies provide useful molecular probes for examining the in vivo pharmacology of BMP signaling inhibition.
Bone morphogenetic proteins (BMPs) are a group of > 25 protein ligands that comprise a subset of the transforming growth factor β (TGF-β) family. BMPs modulate a multitude of biological processes, including bone and cartilage formation during embryogenesis.1a However, they are also intimately involved with numerous nonosteogenic developmental and physiological processes throughout adulthood as well as several pathological conditions.
BMPs bind to two classes of cell surface bone morphogenetic protein receptors (BMPR-I and BMPRII).1a The BMPR-I receptor class consists of three receptor types, activin receptor-like kinase-2 (ALK-2 or ActR-IA), ALK-3 (BMPR-IA) and ALK-6 (BMPR-IB). The BMPR-II receptor class is comprised of three receptor types, BMPR-II, ActR-IIA and ActR-IIB. Binding of BMPs results in the formation of heterotetrameric complexes containing two type I and two type II receptors. In addition to an extracellular binding domain, each BMP receptor contains an intracellular serine/threonine kinase domain. Following binding of BMPs, constitutively active type II receptor kinases phosphorylate type I receptor kinase domains that in turn phosphorylate BMP-responsive SMADs 1, 5, and 8, which can enter the cell nucleus and function as transcription factors.1b Phosphorylation of these specific SMADs results in various cellular effects, including growth regulation and differentiation. Signaling via BMP receptors may also activate other pathways, including mitogen activated protein kinase (MAPK).1c
Several diseases are known to arise from inborn defects in the BMP signaling pathway, including idiopathic pulmonary arterial hypertension,2 hereditary hemorrhagic telangiectasia syndrome and juvenile familial polyposis syndrome,3 all of which involve loss-of-function mutations in BMP receptors or co-receptors. Acquired defects in the BMP signaling pathway are thought to contribute to metastasis of prostate carcinoma4 and renal cell carcinoma.5 Other disorders, such as fibrodysplasia ossificans progressiva (FOP)6 and anemia of chronic disease7 may result from increased BMP signaling. For conditions where increased BMP signaling contributes to disease pathogenesis, inhibitors may offer therapeutic benefit.
Inhibition of BMP signal transduction could be envisioned to occur through various mechanisms, including antagonizing BMP binding to BMPRs or inhibition of the intracellular BMP receptor kinase activity.8 Numerous endogenous protein antagonists that sequester BMP ligands preventing engagement with BMP receptors are known, including noggin, follistatin, chordin and gremlin. Small molecule antagonists of the BMP ligand-receptor interaction have not been identified, possibly due to difficulties antagonizing this protein-protein interaction.9 In addition, the structural diversity of BMP receptors and ligands, and functional redundancy of both systems might pose a challenge for effective blockade of extracellular domains. However, inhibition of SMAD phosphorylation by BMPR-I intracellular kinase domains with small molecules may provide more efficient signal transduction pathway inhibition. This latter approach has been used to identify inhibitors (i.e. SB-431542) of the TGF-β1 receptor kinase ALK5.10
Recently, dorsomorphin, 1,7a, 11, 12 was discovered as an inhibitor of SMAD 1/5/8 phosphorylation by BMP type 1 receptors (ALK2, 3, and 6) utilizing a phenotypic screen to identify compounds that perturb embryonic dorsoventral axis formation. Furthermore, this inhibition was shown to decrease BMP-regulated hepatic hepcidin gene transcription, leading to increased iron levels in vivo.7a However, 1 only demonstrated moderate inhibition of SMAD 1/5/8 phosphorylation by BMPR-I with an IC50 ~ 0.5 µM and lacks metabolic stability. Herein, we describe the results of a structure-activity relationship (SAR) study to optimize BMP signaling inhibition of SMAD 1/5/8 phosphorylation. In addition, we addressed the metabolic stability of this compound series and report a pharmacokinetic study for an optimized derivative.
The synthesis of substituted pyrazolo[1,5-a]pyrimidine derivatives was initially accomplished according to Scheme 1 (Method A). Arylacetonitriles, 2, were allowed to react with dimethyformamide dimethylacetal (DMFDMA) to give 3. In the case of pyridine or quinoline acetonitrile salts, an equivalent of triethylamine was also added. Cyclization of 3 in the presence of hydrazine hydrobromide gave 2-amino-1H-pyrazoles 4a. Condensation of 4a – c with various malondialdehydes in acetic acid and ethanol either under conventional or microwave (MW)13 heating yielded pyrazolo[1,5-a]pyrimidine derivatives 5a – c. In the case of 5c, palladium-mediated cross-coupling with arylboronic acids also gave 5a. This reaction was useful for preparing derivatives where the corresponding arylacetonitriles were not readily available. Dealkylation of the 3- or 4-methoxy ethers on the pendent phenyl rings was accomplished with hydrobromic acid in acetic acid with microwave heating to give 6. Finally, alkylation in one step with R2N(CH2)nCl or in two steps with Cl(CH2)nCl followed by amine addition gave 7.
Scheme 1.

(Method A). Reagents and conditions: (a) (MeO)2CHNMe2, Et3N (for pyridine and quinoline salts), DMF, 110 °C, 4 – 6 h, 100%; (b) NH2NH2·HBr, EtOH/H2O, 110 °C, 6 h, 45 – 80%; (c) ArCH(CHO)2, AcOH, EtOH, 110 °C, 6 h (or MW, 170 °C, 5 min); (d) ArB(OH)2, Pd2(dba)3, 2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl, K3PO4, n-BuOH, MW, 150 °C, 8 min, 84 – 90%; (e) HBr/HOAc, MW, 130 °C, 8 min, 65 – 86%; (f) R2N(CH2)nCl·HCl, Cs2CO3, NaI (cat), DMF, 60 °C, 3 h, (or MW, 140 °C, 6 min), 30 – 75% or Cl(CH2)nCl, K2CO3, DMF, MW, 140 °C, 6 min, then R2NH, NaI (cat), DMF, MW, 150 °C, 10 min, 30 – 60%.
Two other routes were subsequently developed for the synthesis of pyrazolo[1,5-a]pyrimidine derivative 1314 and other analogs that contained an amine on the 3- or 4- position of the pendent phenyl ring. The first alternate route, depicted in Scheme 2 (Method B), began in a similar manner as previously described starting with 815, except that 2-(4-bromophenyl)malondialdehyde was used to generate 11. Next, a palladium-mediated cross coupling with N-Cbz-piperazine yielded 12. Removal of the benzyl carbamate by hydrogenation (1 atm) in the presence of 5% Pd/C gave 13.
Scheme 2.

(Method B). (a) (MeO)2CHNMe2, 110 °C, 16 h, 100%; (b) NH2NH2·HBr, EtOH/H2O, 110 °C, 4 h, 80%; (c) 4-BrPhCH(CHO)2, AcOH, EtOH, MW, 170 °C, 5 min, 54%; (d) N-Cbz-piperazine, Pd2(dba)3, (2-biphenylyl)di-tert-butylphosphine, KOBu-t, DME, 100 °C, 20 h, 20 – 30%; (e) H2 (1 atm), 5% Pd/C (57% H2O), MeOH/CH2Cl2, rt, 4h, 86%.
The second alternate route to 13, depicted in Scheme 3 (Method C), began with 2-amino-1H-pyrazole, 4b, which was allowed to react with 2-bromomalondialdehyde to give 6-bromopyrazolo[1,5-a]pyrimidine, 15a. A palladium-mediated cross-coupling with 4-4-(tert-butoxycarbonyl)-piperazin-1-ylphenylboronic acid pinacol ester yielded 16. Next, a regioselective bromination of the C-3 carbon with N-bromosuccinimide (NBS) in dichloromethane at room temperature gave 17a in 79% yield. Palladium-mediated cross-coupling of this aryl bromide with quinoline-4-boronic acid produced 18a in a moderate 46% yield. Finally, deprotection of the tert-butyl carbamate with 4N HCl in a mixture of 1,4-dioxane and methanol gave 13 as the hydrochloride salt. This method was also used to prepare several other derivatives, including 18c that contains a C-2 substituent.
Scheme 3.

(Method C). (a) BrCH(CHO)2 (or 4-OMePhCH(CHO)2 for 15b) AcOH, EtOH, 80 °C, 7 h, 49%; (b) B(O[C(CH3)2]2O)-4-Ph-N-Boc-piperazine, Pd(PPh3)4, K2CO3, dioxane/H2O, MW, 150 °C, 8 min, 90% (or 110 °C, 3 h, 86%); (c) NBS, CH2Cl2, rt, 5 h, 79%; (d) quinoline-4-boronic acid, Pd2(dba)3, 2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl, K3PO4, n-BuOH, MW, 150 °C, 15 min, 46%; (e) 4 N HCl in 1,4-dioxane, MeOH, rt, 24 h, 95%; (f) HBr/HOAc, MW, 130 °C, 8 min, 81%; (g) Cl(CH2)2Cl, K2CO3, DMF, MW, 140 °C, 6 min, then N-Me-piperizine, NaI (cat), DMF, MW 150 °C, 10 min, 57%.
The synthesis of pyrrolo[1,2-a]pyrimidine derivatives is illustrated in Scheme 4 (Method D). 2-Trichloromethylketopyrrole, 19, was regioselectively brominated affording 20.16 Next, regioselective nitration with concentrated nitric acid gave 21.17 This compound was allowed to react with sodium methoxide in methanol producing the methyl ester 22. Palladium-mediated cross-coupling of this pyrrole bromide with quinoline-4-boronic acid generated 23 in 60% yield.12a Reduction of the nitro group with hydrogen (1 atm) in the presence of 10% Pd/C gave 24, which was used immediately without purification. Condensation with 2-(4-methoxyphenyl)malondialdehyde in acetic acid and ethanol yielded pyrrolo[1,2-a]pyrimidine derivative 25. Heating this material at 110 °C in aqueous sulfuric acid for 2 h gave 26 via ester hydrolysis and subsequent decarboxylation.18 Prolonged heating of 25 for 2 days resulted in ether hydrolysis producing 27. Finally, alkylation of the phenol afforded 28.
Scheme 4.

(Method D). (a) Br2, CHCl3, 0 °C, 57%; (b) HNO3 (70%), Ac2O, −40 °C to rt, 40 %; (c) NaOMe, MeOH, rt, 99%; (d) quinoline-4-boronic acid, Pd(PPh3)4, Na2CO3, 1,4-dioxane, reflux, 16 h, 60 %; (e) H2 (1 atm), 10% Pd/C, MeOH, rt, 0.5 h; (f) 4-MeOPhCH(CHO)2, AcOH, EtOH, reflux, 16 h, 73 %; (g) 40% aqueous H2SO4, 110 °C, 2 h, 91 %; (h) 40% aqueous H2SO4, 110 °C, 2 days, 71 %; (i) piperidyl-NCH2CH2Cl·HCl, 60 % NaH, DMF, rt, 24 h, 80 %.
The synthesis of pyrazolo[1,5-a]pyridine derivatives is outlined in Scheme 5 (Method E). A palladium-mediated cross-coupling of 3-bromopyridine, 29, with 4-methoxyphenylboronic acid produced 30 in 58% yield.19 This pyridine derivative was converted to the 1-aminopyridinium salt 31a utilizing O-(2,4-dinitrophenyl)hydroxylamine.20 Cyclization of 31a upon treatment with methyl propiolate gave regioisomers 32a and 32b in a 1:2 ratio and a combined yield of 33% over two steps.21 In a similar manner, 29 was converted to 33a and 33b (1:2 ratio) in 37% yield, via intermediate 31b. Compound 33a was further converted to 34 via a palladium-mediated coupling. Then, 32a and 34 were hydrolyzed with aqueous sodium hydroxide and the resulting carboxylic acids were subjected to metal-mediated decarboxylative coupling22 with 4-bromoquinoline in the presence of Pd(acac)2 and CuI producing 35 and 36, respectively, albeit in low yields (10 – 22%). Finally, exposure of 36 to 4N HCl in 1,4-dioxane resulted in removal of the tert-butyl carbamate yielding 37 as the hydrochloride salt.
Scheme 5.

(Method E). (a) 4-MeOPhB(OH)2, Pd(PPh3)4, K3PO4, 1,4-dioxane, 100 °C, 18 h, 58%; (b) 2,4-di-NO2PhONH2, CH3CN, 40 °C, 20 h; (c) HC≡CCO2Me, K2CO3, DMF, rt, 33 – 37% over two steps (32a:32b and 33a:33b ~ 1:2); (d) B(O[C(CH3)2]2O)-4-Ph-N-Boc-piperazine, Pd(PPh3)4, K2CO3, 1,4-dioxane/H2O, 110 °C, 5 h, 73%; (e) NaOH, EtOH/H2O (6:1), Δ, 3 h; (f) 4-bromoquinoline, Pd(acac)2, CuI, K2CO3, 1,10-phenanthroline, 4Å MS, NMP, 165 °C, 24 h, 10 – 22% (over two steps); (g) 4N HCl in 1,4-dioxane, MeOH, rt, 24 h.
Evaluation of BMP4-induced phosphorylation of SMAD1/5/8 was performed using a sensitive cytoblot cellular ELISA assay in the presence of varying concentrations of test compounds.23a,b Functional IC50 values were calculated for the inhibitory effects of test compounds on phosphorylation of SMAD1/5/8 and are shown in Table 1 - Table 3.23c
Table 1.
IC50 determinations for inhibition BMP4-induced phosphorylation of SMAD1/5/8.
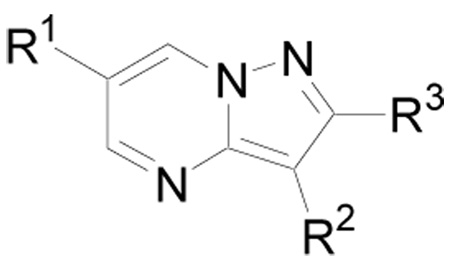 | |||||
|---|---|---|---|---|---|
| Compd | R1 | R2 | R3 | Method | IC50 (µM) |
| 1 | Pip-CH2CH2O-4-Ph | 4-Py | H | --- | 0.43 |
| 38 | 4-MeO-Ph | 4-Py | H | A | 6.5 |
| 39 | Morph- CH2CH2O-4-Ph | 4-Py | H | A | 2.0 |
| 40 | Et2N-CH2CH2O-4-Ph | 4-Py | H | A | 0.50 |
| 41 | NMP-CH2CH2O-4-Ph | 4-Py | H | A | 0.45 |
| 42 | NMP-CH2CH2O-3-Ph | 4-Py | H | A | 4.5 |
| 43 | NMP-CH2CH2O-4-Ph | 4-Py | Me | C | > 20 |
| 44 | Pip-CH2CH2O-4-Ph | H | H | A | > 20 |
| 45 | Pip-CH2CH2O-4-Ph | 3-Py | H | A | > 20 |
| 46 | Pip-CH2CH2O-4-Ph | Ph | H | A | > 20 |
| 47 | NMP-CH2CH2O-4-Ph | 3-F-4-Py | H | C | 3.3 |
Pip = piperidinyl; Morph = morpholinyl; NMP = N-methylpiperazinyl; Py = pyridyl.
Table 3.
IC50 determinations for inhibition BMP4-induced phosphorylation of SMAD1/5/8.
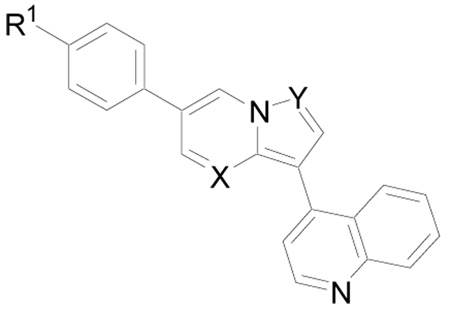 | |||||
|---|---|---|---|---|---|
| Compd | R1 | X | Y | Method | IC50 (µM) |
| 25 | MeO | N | CCO2Me | D | > 20 |
| 26 | MeO | N | CH | D | > 20 |
| 28 | Pip-CH2CH2O | N | CH | D | 2.6 |
| 35 | MeO | CH | N | E | 0.15 |
| 37 | Piperazinyl | CH | N | E | 0.005 |
Pip = piperidinyl
Introduction of an aminoether at the 4-position of the pendent phenyl ring on the pyridine derivatives (e.g. 1 and 39 – 41 vs. 38) increased activity three to fifteen fold in addition to improving aqueous solubility (Table 1). Introduction of a substituent on the 2-position of the pyrazolo[1,5-a]pyrimidine ring (43) abolished activity. In addition, activity was dramatically affected by the nature of the substituent on the 3-position of the pyrazolo[1,5-a]pyrimidine ring. Removal (44) or replacement of the 4-pyridyl in 1 with 3-pyridyl (45) or phenyl (46) resulted in complete loss of activity. Replacement of the pyridine ring with 3-fluoro-4-pyridyl (47) likewise resulted in reduced activity.
Due to these significant substituent effects on the 3-position of the pyrazolo[1,5-a]pyrimidine ring and the demonstrated influence of heterocyclic substituents on other TGF-β receptor type inhibitors,24 quinolines attached through various positions were examined (Table 2). In compound 52, where the pyrazolo[1,5-a]pyrimidine ring is connected to the 4-position of the quinoline, a significant increase in activity was observed. Introduction of an aminoether to the 4-position of the pendent phenyl ring (53) increased potency as was previously observed for the pyridine derivatives (vida supra). Replacing the aminoether with piperazine (13) was also well tolerated. However, transposing this substituent to the 3-position of the pendent phenyl (58) resulted in significant loss of activity. Likewise, introduction of a chloride to the 7-position of the quinoline ring (59 vs. 52 and 60 vs. 13) resulted in decreased activity.
Table 2.
IC50 determinations for inhibition BMP4-induced phosphorylation of SMAD1/5/8.
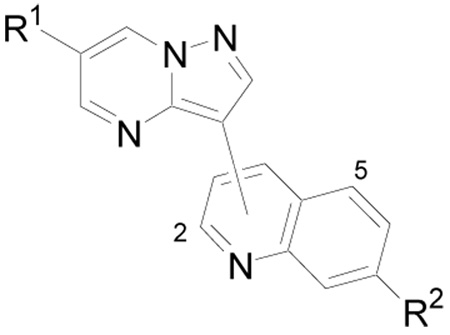 | |||||
|---|---|---|---|---|---|
| Compd | R1 | Position | R2 | Method | IC50 (µM) |
| 48 | 4-MeO-Ph | 6 | H | C | > 20 |
| 49 | 4-MeO-Ph | 8 | H | C | > 20 |
| 50 | 4-MeO-Ph | 5 | H | C | 3.0 |
| 51 | 4-MeO-Ph | 3 | H | C | > 20 |
| 52 | 4-MeO-Ph | 4 | H | C | 0.055 |
| 53 | NMP-CH2CH2O-4-Ph | 4 | H | A | 0.010 |
| 54 | Pip-CH2CH2O-4-Ph | 4 | H | A | 0.090 |
| 55 | H | 4 | H | A | 5.0 |
| 56 | Ph | 4 | H | A | 0.75 |
| 57 | HO-4-Ph | 4 | H | A | 0.25 |
| 13 | Piperazinyl-4-Ph | 4 | H | A,B,C | 0.0049 |
| 58 | Piperazinyl-3-Ph | 4 | H | C | 20 |
| 59 | 4-MeO-Ph | 4 | Cl | C | 0.50 |
| 60 | Piperazinyl-4-Ph | 4 | Cl | C | 0.35 |
Pip = piperidinyl; NMP = N-methylpiperazinyl
Next, the contributions of the nitrogen atoms in the 1- and 4-positions of the pyrazolo[1,5-a]pyrimidine ring were examined (Table 3). The importance of the N-1, but not the N-4, nitrogen atoms was previously demonstrated for KDR kinase inhibition by pyrazolo[1,5-a]pyrimidine derivatives.25 Similarly, in the present series of compounds the N-1 (28 vs. 54) appears necessary for potent inhibition of BMP4-induced phosphorylation of SMAD1/5/8, whereas the N-4 was not essential (37 vs. 13).
Both the original lead compound 1 and a more potent derivative 53 demonstrated poor metabolic stability in mouse liver microsomes (1: half-life (t1/2) of 10.4 min and intrinsic clearance (CLint) of 133 ± 6.6 µL/min/mg protein; 53: t1/2 of 13.3 min and CLint of 104 ± 3.4 µL/min/mg protein).23b, 26 However, replacement of the ether on the pendent phenyl ring with piperazine resulted in a significant increase in mouse liver microsome stability. For example, 13 demonstrated a t1/2 of 82 min and CLint of 16.9 ± 5.6 µL/min/mg protein. Based on the potency and metabolic stability of 13, it was selected for in vivo pharmacokinetic analysis following a single bolus intraperitoneal (ip) administration of 3 mg/kg in male and female C57B16 mice.23b The results of this study are shown in Table 4. The pharmacokinetics of 13 was similar in both male and female mice. The average maximal plasma concentrations were slightly higher in males (1.54 µM) than in females (1.29 µM) and were reached quickly (> 5 min) following administration. The plasma half-life (1.6 h) and the average AUC∞ values (994 and 1030 ng·h/mL) were similar in male and female mice.
Table 4.
Pharmacokinetic analysis of 13 in plasma following bolus intraperitoneal administration in mice (N = 3 / sex)
| Sex | Dose | Cmax | tmax | t1/2 | AUC∞ |
|---|---|---|---|---|---|
| mg/kg | µM | min | h | ng·h/mL | |
| male | 3.0 | 1.54 | < 5 | 1.6 | 994 |
| female | 3.0 | 1.29 | < 5 | 1.6 | 1030 |
In conclusion, a structure-activity relationship study of dorsomorphin, 1, a previously identified inhibitor of SMAD 1/5/8 phosphorylation by BMP type 1 receptors ALK2, 3, and 6, revealed that increased inhibitory activity could be accomplished by replacing the pendent 4-pyridine ring with 4-quinoline. The nitrogen atom in the 1-position of the pyrazolo[1,5-a]pyrimidine ring was determined to be necessary for inhibitory activity. However, the nitrogen atom in the 4-position was not vital. In addition, increased mouse liver microsome stability was achieved by replacing the ether substituent on the pendent phenyl ring with piperazine. Finally, an optimized compound 13 (LDN-193189 or DM-3189) demonstrated moderate pharmacokinetic characteristics (e.g. plasma t1/2 = 1.6 h) following intraperitoneal administration in mice. Evaluation of this inhibitor in various animal disease models in which BMP signaling has been hypothesized to play a role, such as FOP and anemia of chronic disease, are currently on-going.
Supplementary Material
Figure 1.

BMP signaling inhibitor of SMAD 1/5/8 phosphorylation.
Acknowledgments
The authors thank Partners Healthcare for financial support. This work was also supported in part by NIH grants HL079943 (PBY), HL074352 (KDB), and HL079267 (RTP), the Harvard NeuroDiscovery Center (GDC, XX and JKL), a Pulmonary Hypertension Association Mentored Clinical Scientist Award (PBY) and a grant from the GlaxoSmithKline Research & Education Foundation for Cardiovascular Disease (PBY). The pharmacokinetic study was performed by Absorption Systems (Exton, PA) and the microsome stability experiments were performed by Cyprotex (Macclesfield, UK).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary data
Supplementary data associated with this article can be found, in the online version, at ().
References and notes
- 1.(a) Chen D, Zhao M, Mundy GR. Growth Factors. 2004;22:233. doi: 10.1080/08977190412331279890. [DOI] [PubMed] [Google Scholar]; (b) Zwijsen A, Verschueren K, Huylebroeck D. FEBS Lett. 2003;546:133. doi: 10.1016/s0014-5793(03)00566-0. [DOI] [PubMed] [Google Scholar]; (c) Nohe A, Keating E, Knaus P, Petersen NO. Cell Signal. 2004;16:291. doi: 10.1016/j.cellsig.2003.08.011. [DOI] [PubMed] [Google Scholar]
- 2.(a) Eickelberg O, Morty RE. Trends Cardiovasc. Med. 2007;17:263. doi: 10.1016/j.tcm.2007.09.003. [DOI] [PubMed] [Google Scholar]; (b) Yu PB, Beppu H, Kawai N, Li E, Bloch KD. J Biol. Chem. 2005;280:24443. doi: 10.1074/jbc.M502825200. [DOI] [PubMed] [Google Scholar]
- 3.Liu D, Wang J, Kinzel B, Müeller M, Mao X, Valdez R, Liu Y, Li E. Blood. 2007;110:1502. doi: 10.1182/blood-2006-11-058594. [DOI] [PubMed] [Google Scholar]
- 4.Ye L, Lewis-Russell JM, Kyanaston HG, Jiang WG. Histol. Histopathol. 2007;22:1129. doi: 10.14670/HH-22.1129. [DOI] [PubMed] [Google Scholar]
- 5.Kim IY, Lee DH, Lee DK, Kim BC, Kim HT, Leach FS, Linehan WM, Morton RA, Kim SJ. Clin. Cancer Res. 2003;9:6046. [PubMed] [Google Scholar]
- 6.(a) Billings PC, Fiori JL, Bentwood JL, O’Connell MP, Jiao X, Nussbaum B, Caron RJ, Shore EM, Kaplan FS. J. Bone Miner Res. 2008;23:305. doi: 10.1359/JBMR.071030. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shore EM, Xu M, Feldman GJ, Fenstermacher DA, Cho TJ, Choi IH, Connor JM, Delai P, Glaser DL, LeMerrer M, Morhart R, Rogers JG, Smith R, Triffitt JT, Urtizberea JA, Zasloff M, Brown MA, Kaplan FS. Nat. Genet. 2006;38:225. [Google Scholar]
- 7.(a) Yu PB, Hong CC, Sachidanandan C, Babitt JL, Deng DY, Hoyng SA, Lin HY, Bloch KD, Peterson RT. Nat. Chem. Biol. 2008;4:33. doi: 10.1038/nchembio.2007.54. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Babitt JL, Huang FW, Xia Y, Sidis Y, Andrews NC, Lin HY. J. Clin. Invest. 2007;117:1755. doi: 10.1172/JCI31342. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Weiss G, Goodnough LT. N. Engl. J. Med. 2005;352:1011. doi: 10.1056/NEJMra041809. [DOI] [PubMed] [Google Scholar]
- 8.Gazzerro E, Minetti C. Curr. Opin. Pharmacol. 2007;7:325. doi: 10.1016/j.coph.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 9.Whitty A, Kumaravel G. Nat. Chem. Biol. 2006;2:112. doi: 10.1038/nchembio0306-112. [DOI] [PubMed] [Google Scholar]
- 10.Callahan JF, Burgess JL, Fornwald JA, Gaster LM, Harling JD, Harrington FP, Heer J, Kwon C, Lehr R, Mathur A, Olson BA, Weinstock J, Laping NJ. J. Med. Chem. 2002;45:999. doi: 10.1021/jm010493y. [DOI] [PubMed] [Google Scholar]
- 11.(a) Also know in the literature as “compound C”; This compound was initially reported as a AMP-activated protein kinase (AMPK) inhibitor, see: Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, Musi N, Hirshman MF, Goodyear LJ, Moller DE.J. Clin. Invest 20011081167.
- 12.For similar compounds reported as KDR kinase inhibitors see: Fraley ME, Hoffman WF, Rubino RS, Hungate RW, Tebben AJ, Rutledge RZ, McFall RC, Huckle WR, Kendall RL, Coll KE, Thomas KA. Bioorg. Med. Chem. Lett. 2002;12:2767. doi: 10.1016/s0960-894x(02)00525-5. Fraley ME, Rubino RS, Hoffman WF, Hambaugh SR, Arrington KL, Hungate RW, Bilodeau MT, Tebben AJ, Rutledge RZ, Kendall RL, McFall RC, Huckle WR, Coll KE, Thomas KA. Bioorg. Med. Chem. Lett. 2002;12:3537. doi: 10.1016/s0960-894x(02)00827-2.
- 13.Daniels RN, Kim K, Lebois EP, Muchalski H, Hughes M, Lindsley CW. Tetrahedron Lett. 2008;49:305. [Google Scholar]
- 14.13: 1H NMR (DMSO-d6) δ9.75 (d, J = 2.2 Hz, 1H), 9.40 (br s, 1H), 9.29 (d, J = 5.9 Hz, 1H), 9.28 (d, J = 2.2 Hz, 1H ), 9.07 (s, 1H), 8.70 (d, J = 8.4 Hz, 1H), 8.51 (d, J = 5.9 Hz, 1H), 8.47 (d, J = 8.4 Hz, 1H), 8.21 (t, J = 7.6 Hz, 1H), 7.99 (t, J = 7.6 Hz, 1H), 7.88 (d, J = 8.8 Hz, 2H), 7.19 (d, J = 8.8 Hz, 2H), 3.51 – 3.58 (m, 4H), 3.20 – 3.30 (m, 4H); HRMS m/z 407.1979 (calc for C25H23N6, MH+, 407.1979).
- 15.(a) Prepared from quinoline-4-carboxaldehyde in three steps: i) reduction (NaBH4, MeOH, 0 °C to rt); ii) conversion of the alcohol to a chloride (2 equiv. SOCl2, DCM, 0 °C to rt); iii) conversion of the chloride to a nitrile (NaCN, DMF, rt); For an alternate procedure see: Engler TA, Furness K, Malhotra S, Diefenbacher C, Clayton JR.Tetrahedron Lett 2003442903
- 16.Kitamura C, Yamashita Y. J. Chem. Soc., Perkin Trans. 1997;1:1443. [Google Scholar]
- 17.Marques MA, Doss RM, Urbach AR, Dervan PB. Helv. Chim. Acta. 2002;85:4485. [Google Scholar]
- 18.Loeber S, Hueber H, Utz W, Gmeiner P. J. Med. Chem. 2001;44:2691. doi: 10.1021/jm015522j. [DOI] [PubMed] [Google Scholar]
- 19.Kondolff I, Doucet H, Santelli M. Synlett. 2005:2057. [Google Scholar]
- 20.Legault C, Charette AB. J. Org. Chem. 2003;68:7119. doi: 10.1021/jo034456l. [DOI] [PubMed] [Google Scholar]
- 21.Tamaru Y, Sumida Y, Miki Y, Ikeda M. J. Chem. Soc., Perkin Trans. 1975;1:406. [Google Scholar]
- 22.(a) Baudoin O. Angew. Chem. Int. Ed. 2007;46:1373. doi: 10.1002/anie.200604494. [DOI] [PubMed] [Google Scholar]; (b) Forgione P, Brochu M-C, St-Onge M, Thesen KH, Bailey MD, Bilodeau F. J. Am. Chem. Soc. 2006;128:11350. doi: 10.1021/ja063511f. [DOI] [PubMed] [Google Scholar]; (c) Goo□en LJ, Deng G, Levy LM. Science. 2006;313:662. doi: 10.1126/science.1128684. [DOI] [PubMed] [Google Scholar]
- 23. Takata M, Filippov G, Liu H, Ichinose F, Janssens S, Bloch DB, Bloch KD. Am. J. Physiol. Lung Cell Mol. Physiol. 2001;280:L272. doi: 10.1152/ajplung.2001.280.2.L272. (b) See supplementary data for additional details; (c) Average values based on two to four independent experiments.
- 24.(a) Sawyer JS, Beight DW, Britt KS, Anderson BD, Campbell RM, Goodson T, Jr, Herron DK, Li H-Y, McMillen WT, Mort N, Parsons S, Smith ECR, Wagner JR, Yan L, Zhang F, Yingling JM. Bioorg. Med. Chem. Lett. 2004;14:3581. doi: 10.1016/j.bmcl.2004.04.007. [DOI] [PubMed] [Google Scholar]; (b) Gellibert F, Woolven J, Fouchet M-H, Mathews N, Goodland H, Lovegrove V, Laroze A, Nguyen V-L, Sautet S, Wang R, Janson C, Smith W, Krysa G, Boullay V, de Gouville A-C, Huet S, Hartley D. J. Med. Chem. 2004;47:4494. doi: 10.1021/jm0400247. [DOI] [PubMed] [Google Scholar]
- 25.Wu Z, Fraley ME, Bilodeau MT, Kaufman ML, Tasber ES, Balitza AE, Hartman GD, Coll KE, Rickert K, Shipman J, Shi B, Sepp-Lorenzino L, Thomas KA. Bioorg. Med. Chem. Lett. 2004;14:909. doi: 10.1016/j.bmcl.2003.12.007. [DOI] [PubMed] [Google Scholar]
- 26.Baranczewski P, Stańczak A, Sundberg K, Svensson R, Wallin A, Jannson J, Garberg P, Postlind H. Pharmacol. Rep. 2006;58:453. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.